
Abstract
Introduction
H
Excitation–Contraction Coupling
In diastole, cytosolic Ca2+ ([Ca2+]i) is low and there is little or no Ca2+ bound to the myofilaments. During systole, [Ca2+]i increases and binds to troponin C (TnC) to induce a strong interaction between TnC and troponin I (TnI). This will destabilize the interaction between TnI and actin causing a shift in the troponin–tropomyosin complex allowing myosin to bind actin resulting in myocyte force production (and/or shortening) (41). The rate and magnitude of myocyte force development are determined by systolic [Ca2+]. Thus, altering the force of cardiac contraction largely occurs by regulating systolic Ca2+ levels (contractility regulation) (summarized in Fig. 1).
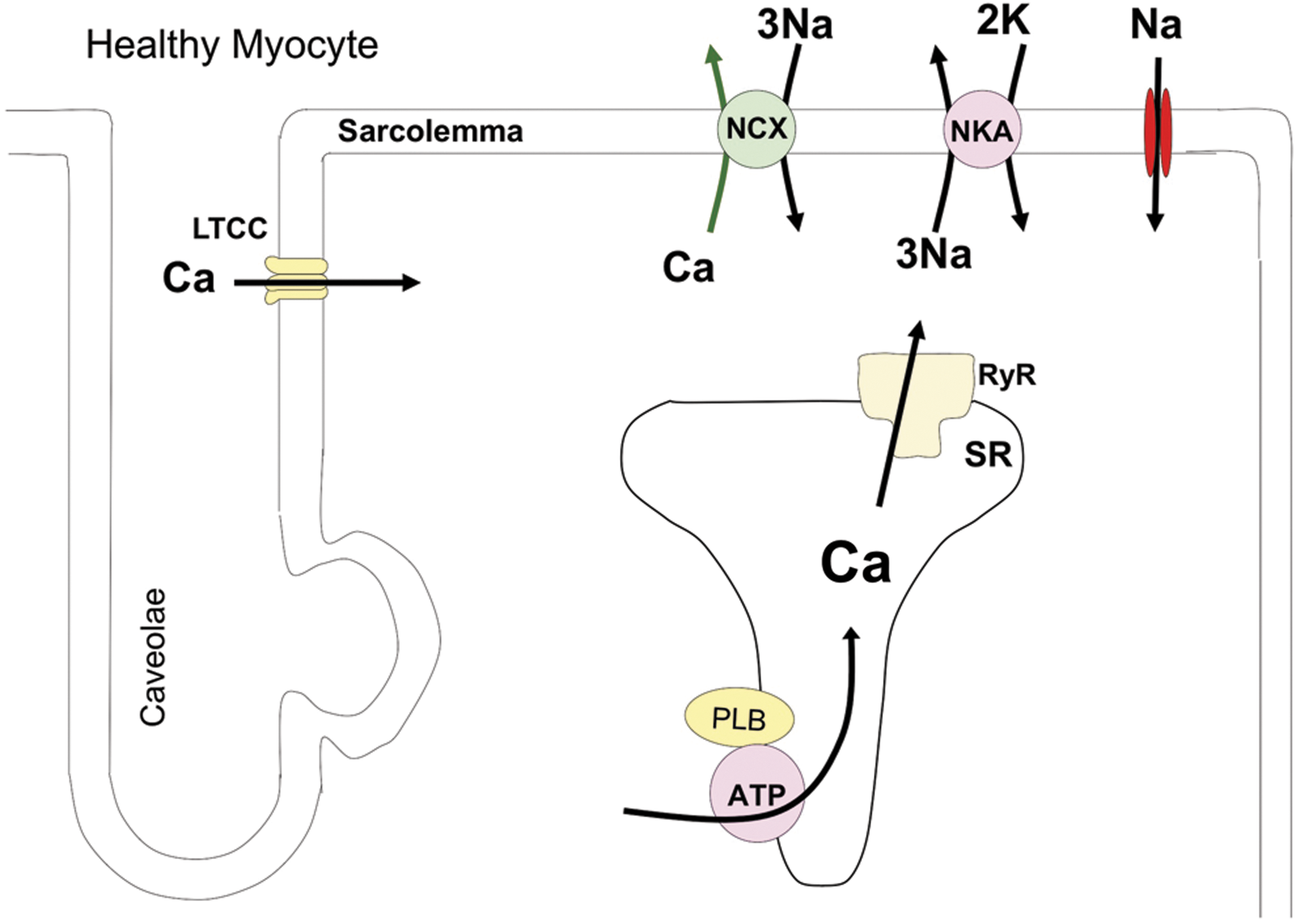
The increases in myocyte Ca2+ that initiate contraction occur via a process termed excitation–contraction coupling (ECC) (19). ECC is initiated with an action potential, in which the opening of Na+ channels causes membrane depolarization via the entry of positive charges (Na+ ions). This depolarization will activate voltage-gated Ca2+ channels causing the influx of Ca2+. In a process termed Ca2+-induced Ca2+ release, this Ca2+ influx will activate the Ca2+ release channel (ryanodine receptor [RyR]) in the junctional sarcoplasmic reticulum (SR). The resultant increase in [Ca2+]i is the sum of Ca2+ influx through L-type Ca2+ channels (LTCC) and SR Ca2+ release. The amount of Ca2+ influx and the amount of Ca2+ available for release from the SR (the SR Ca2+ load) are the major determinants of the amplitude of the Ca2+ transient amplitude (145) and thus myocyte contraction. Relaxation is initiated by a decrease in [Ca2+]i (i.e., the decline of the Ca2+ transient). This is primarily due to the SR Ca2+-ATPase (SERCA), which re-sequesters released Ca2+ back into the SR, and the sarcolemmal Na/Ca exchanger (NCX), which eliminates the Ca2+ that has entered with each heartbeat. In rodents, the SR Ca2+ uptake is responsible for >90% of the Ca2+ transient while Ca2+ efflux plays a significantly larger role in larger species, including humans.
The normal heart has the ability to increase its pump performance when needed, as during aerobic exercise (172). Increases in the amount of Ca2+ delivered to the contractile apparatus are essential for increases in cardiac contraction, termed contractility. The Ca2+ regulatory proteins responsible for physiological regulation of cardiac contractility are those that determine Ca2+ influx (LTCC), SR Ca2+ uptake (SERCA), and SR Ca2+ release (RyR). In the normal heart, changes in the activity of these proteins are largely caused by phosphorylation of these proteins via the activation of beta-adrenergic (β-AR) receptors (i.e., sympathetic regulation of cardiac function) (20).
A critical modulator of SERCA activity, and thus SR Ca2+ uptake, is the phosphoprotein phospholamban (PLB) (110). Under basal conditions, PLB phosphorylation is low and PLB in an unphosphorylated state slows SERCA-mediated SR Ca2+ uptake, and this results in a modest SR Ca2+ load and relatively slow SR Ca2+ uptake (i.e., prolonged Ca2+ transient decline). This is a low contractility state as might be seen in a person with low metabolic demands, such as occurring with sleep or during rest. With the activation of the sympathetic nervous system, there is activation of β-AR signaling pathway and a resultant activation of protein kinase A (PKA), with phosphorylation of PKA target proteins (20). PKA-mediated PLB phosphorylation at Serine16 disinhibits SERCA and results in an increase in the SERCA activity (131). Increased PKA activity also increases Ca2+ entry via LTCC. These phosphorylation changes cause greater Ca2+ influx, SR Ca2+ uptake, and SR Ca2+ load. The net result is an increase in the rate and amplitude of the systolic Ca2+ transient and a decrease in the duration of the transient. These PKA-mediated increases in systolic Ca2+ also lead to the activation of Ca2+-calmodulin-dependent protein kinase (CaMKII), which further enhances both Ca2+ entry and SERCA activity (PLB Threonine 17 phosphorylation) (111). PKA- and CaMKII-mediated phosphorylation of RyR can also have a minor effect on myocyte contractility by altering the Ca2+ dependence of RyR openings (58, 144, 173); however, this effect is not universally observed (185).
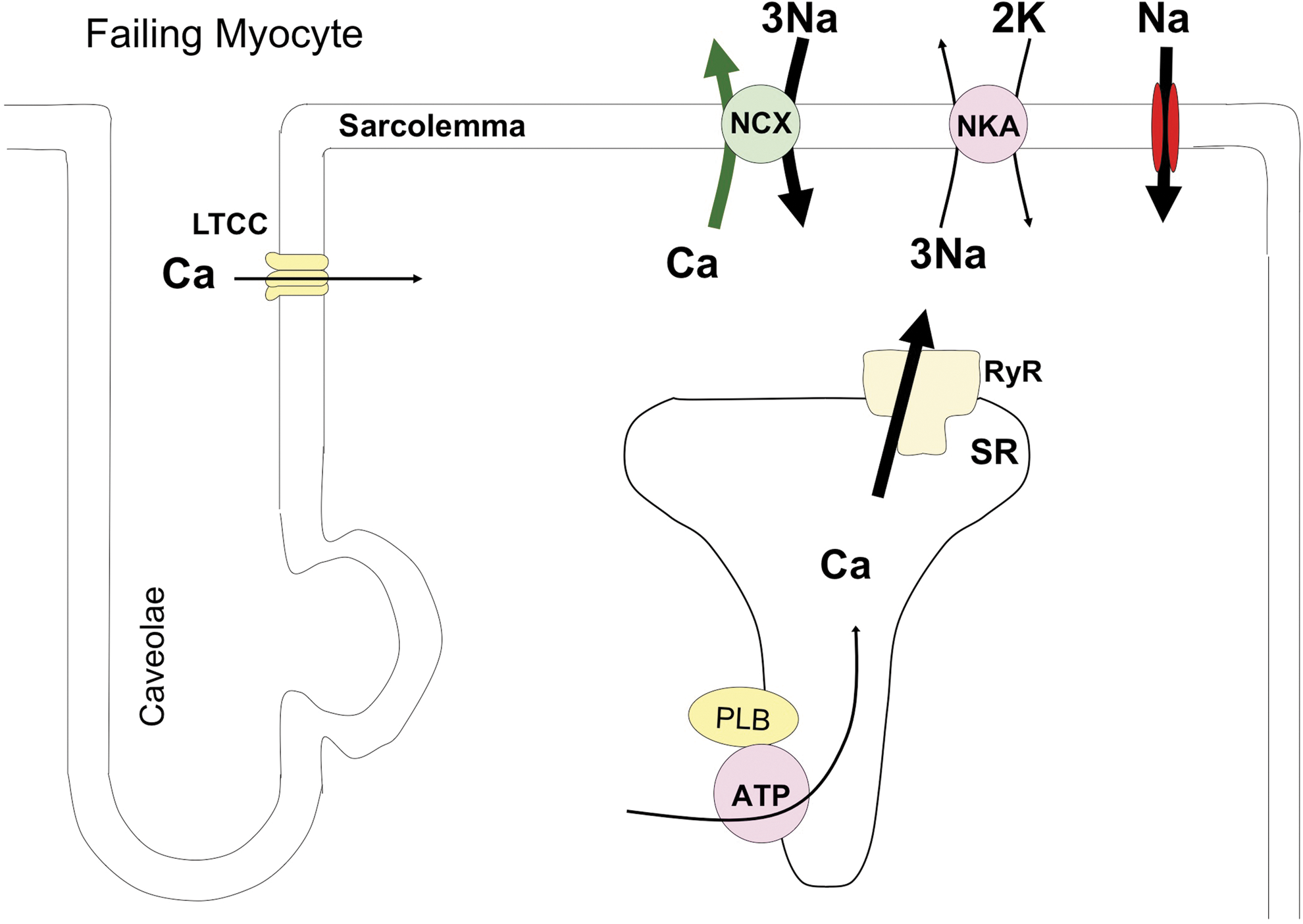
The in vivo contractile demands on myocytes within the failing heart are greater than in healthy hearts due to increased systolic wall stress, and these demands increase with HF progression. This persistent demand for enhanced myocyte function is associated with alterations in myocyte Ca2+ regulation (summarized in Fig. 2). LTCCs are hyperphosphorylated, but channel density appears to be reduced (32); SERCA abundance and PLB phosphorylation are reduced (70, 136, 141); NCX activity is increased (125); and RyRs are hyperphosphorylated (3, 112). These modifications limit the maximal Ca2+ entry, SR uptake and storage, and induce a more pronounced efflux of Ca2+ from the SR via RyR during diastole (i.e., diastolic Ca2+ leak). The sum of these changes limits the ability of failing myocytes to enhance systolic Ca2+ and thus contractility. Therefore, the failing heart must generate much higher than normal force (via activation of neurohormonal regulatory pathways) while the inherent ability of the resident myocytes to increase their contractility (contractility reserve) is reduced. The failing heart also loses its ability to increase force at higher heart rates (blunted force-frequency response [FFR]) (133) and has a depressed functional response to β-AR stimulation (193). Myocytes isolated from failing hearts and studied versus nonfailing myocytes under identical conditions have a decrease in SR Ca2+ load resulting in a decreased amplitude of the Ca2+ transient and a slowing of its decline (124). HF-associated changes in Ca2+ regulation also contribute to arrhythmias in HF. The increase in diastolic SR Ca2+ leak will activate electrogenic Ca2+ efflux via the NCX, and this causes afterdepolarizations that can trigger arrhythmias (36, 146). Restoring a normal Ca2+ regulatory system should improve cardiac function and is a therapeutic goal. However, those approaches that increase PKA signaling (which is already activated in HF) have been linked to arrhythmias and sudden death (119). Standard of care therapy for HF includes β-blockers, which are thought to prevent excessive PKA signaling and related arrhythmias (6). Therefore, strategies that can restore a more normal Ca2+ transient without activating PKA might increase contractility reserve without increasing arrhythmia risk. In the remainder of this review, we discuss the idea that abnormal post-translational modifications of Ca2+ regulatory proteins by redox reactions contribute to aberrant contractility in HF. If true, then restoring a normal redox state could improve myocyte contractility and cardiac pump function in HF.
Redox Signaling
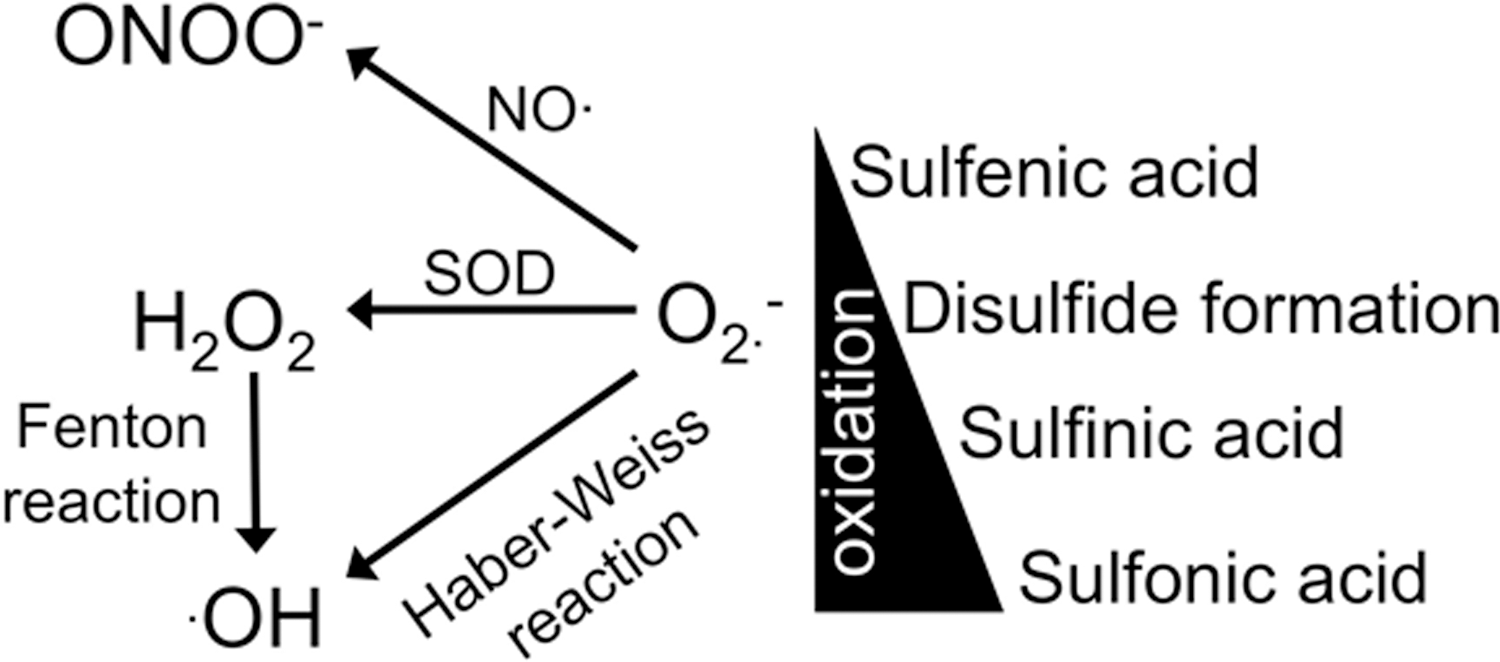
There is a precise regulation of reactive oxygen species (ROS) levels in which the production of ROS is counterbalanced by its degradation. Oxidative stress arises when there is an accumulation of ROS due to an increase in the production of ROS and/or a decrease in the degradation of ROS. The superoxide anion radical (O2 •−) is the main radical since it can be converted into other ROS (hydrogen peroxide [H2O2], •OH, peroxynitrite, etc.) via various enzymes and chemical reactions (Fig. 3). Within healthy cardiac myocytes, the major producers of O2 •− are the mitochondria (171), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (104), and xanthine oxidase (51) (Fig. 4). Production of O2 •− in the mitochondria occurs as a reactionary byproduct of respiration in the electron transport chain of complex I and III (77, 159). Unlike the mitochondria, the sole purpose of NADPH oxidases (NOX) is to produce O2 •− (163). Within ventricular myocytes, the NOX isoforms that are expressed are NOX2 and NOX4 (2, 15, 179). Xanthine oxidase is involved in purine degradation and produces O2 •− from the reduction of molecular O2 and is also expressed in ventricular myocytes (18, 90). The myocyte has an array of defenses to ascertain that ROS levels do not increase. These defenses include various enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase, and thioredoxin. The myocyte also has various compounds (e.g., vitamin E, ascorbic acid) and peptides (glutathione) that are also antioxidants. While there is a decline in the antioxidant defense with HF, it appears that oxidative stress is primarily due to an increase in ROS generation (Fig. 5). That is, mitochondrial O2 •− production is increased in HF patients due to state 3 respiration and dysregulation of cytosolic Ca2+ and Na+ (discussed further below in the [Na+]i regulation section) (83, 93). Furthermore, under these oxidative conditions, the mitochondria will also produce large amounts of H2O2 (9, 150). NOX activity is also increased with HF (75, 104, 107, 128). While there do not appear to be changes in NOX2 expression levels, post-translational modification and membrane translocation of subunits (e.g., p47phox, p67phox) are responsible for the increased activity. Unfortunately, it also appears that there is a connection between NOX activity and mitochondrial O2 •− production (47, 78). Thus, in the setting of HF, the increased mitochondrial O2 •− production may be further augmented via NOX. In addition, the dismutation of O2 •− to H2O2 can further augment production of O2 •− from neighboring mitochondria in cardiac myocytes termed ROS-induced-ROS release (8, 165, 194). Xanthine oxidase activity is also upregulated due to increased expression and altered modulation by NO (discussed further below in the Neuronal NOS section) (134). Unfortunately, the HF syndrome also results in additional ROS contributors within ventricular myocytes not observed under physiological states. Monoamine oxidases (MAO) are responsible for the oxidative deamination of neurotransmitters, such as epinephrine (epi) and norepinephrine (norepi). With the enhanced β-AR drive and increased epi/norepi in HF, MAO activity and expression (both MAO A and B) are increased resulting in enhanced H2O2 levels (86, 87). In addition, under certain conditions (as occurs in HF), NO production via NOS is interrupted resulting in the production of O2 •−, termed NOS uncoupling (154).


Oxidative stress occurs in HF, and many studies have focused on the effects of ROS on myocyte contraction. Acute exposure of myocytes to ROS (O2 •−, H2O2, •OH) results in decreased Ca2+ transient amplitude, slowed Ca2+ decline, and reduced SR Ca2+ load (60, 61, 64, 67, 100, 109, 121). These effects of ROS on myocyte Ca2+ handling are similar to what is observed in failing myocytes. These data suggest that oxidative stress contributes to the observed phenotype in failing myocytes (especially in which there are no changes in protein expression). Further studies have shown that scavenging of ROS, inhibiting the sources of ROS, or increasing antioxidants (e.g., glutathione peroxidase) in disease are able to reverse the contractile dysfunction (2, 54, 63, 65, 105, 128, 135, 148, 183). Hence, these studies unequivocally prove that oxidative stress does indeed contribute to the contractile dysfunction in failing hearts.
Studies have further investigated the molecular mechanisms for the altered Ca2+ handling induced by oxidative stress and found that all the major Ca2+ handling proteins (RyR, SERCA/PLB, and NCX) are modified. ROS is able to acutely modulate protein function via reversible post-translational modifications of the amino acids methionine and cysteine (i.e., redox signaling) (177). ROS are able to oxidize the sulfur atom in methionine to yield methionine sulfoxide, which is reversed via methionine sulfoxide reductase (24). Low levels of ROS will oxidize cysteine residues to yield sulfenic acid and then disulfide formation (which can occur between adjacent cyteines, between two proteins, or a protein thiol and glutathione, S-glutationylation). These post-translational modifications can be reversed back by thioredoxin and glutaredoxin (11, 17, 74, 155). With higher levels of ROS, there is further oxidation of cysteines to generate sulfinic acid and then sulfonic acid. These modifications are irreversible (although sulfiredoxin can reverse some of the sulfinic acid) (178). Thus, depending on the redox state of the cell, this range of cysteine modifications (Fig. 3) allows for the acute modulation of protein function during physiological stresses or can result in protein dysfunction during pathological stresses.
Ryanodine receptor
This large protein exists as a tetramer, with each monomer containing 89 cysteine residues. Thus, there can be an assortment of modifications (number and location of residues) resulting in varied effects. Indeed, this seems to be the case as ROS has a bidirectional effect of RyR activity (180, 184). Marginal levels of RyR oxidation seem to have little effect on activity, whereas low levels of oxidation result in an increase in activity (23, 89, 182, 189). Importantly, high levels of ROS have been reported to reduce or irreversibly increase RyR activity (50). Once again this may be due to distinctive cysteine modifications as well as time dependence of ROS exposure. However, with HF, it has been reported that RyR activity is increased due to ROS (113, 157). This ROS-mediated increase in activity has been shown to contribute to the generation of arrhythmogenic Ca2+ waves via increased diastolic Ca2+ leak from the SR. In addition to cause arrhythmias, this leak will empty the SR Ca2+ load (a hallmark of HF) and decrease contraction.
SR Ca2+-ATPase
Another cause of the decreased SR Ca2+ load in HF is decreased SR Ca2+ uptake via reduced SERCA activity. Many studies have shown that ROS inhibits SERCA function. This effect occurs via a direct attack on the ATP binding site, uncoupling Ca2+ uptake from ATP hydrolysis, and via sulfination at cysteine674 (101, 114, 139, 181).
Na/Ca exchanger
Since Ca2+ transport into the SR is reduced in HF, to maintain proper diastolic function, an increase in NCX activity could be beneficial by reducing [Ca2+]i (71). It has also been postulated that increased NCX activity (reverse mode) can increase SR Ca2+ load, which will occur when there is a concomitant increase in [Na+]i (see below in the [Na+]i regulation section) (124, 174). Studies have shown an increase in NCX activity (usually without changes in expression) in HF (125). Consistent with this increase in activity with HF, ROS has also been shown to increase NCX activity via oxidation (59, 100, 130). However, there is a negative consequence of this increased NCX activity, which is to increase the propensity to generate arrhythmias (7).
[Na+]i regulation
With NCX (and other pathways), there is a strong interdependence between [Ca2+]i and [Na+]i levels. Similar to Ca2+ handling, there are many proteins involved in the regulation of [Na+]i levels (Figs. 1 and 2). These proteins are also regulated by ROS. The voltage-gated Na+ channel (Nav1.5) inactivation is greatly impeded by oxidation of methionine residues (increasing Na+ entry) (56, 88). While NCX and Nav1.5 can increase the amount of Na+ influx into the myocyte, [Na+]i levels are largely determined by the Na+/K+-ATPase (NKA) (45). Studies have shown that NKA activity is reduced via ROS, in part via glutathionylation of Cysteine46 (55). Thus, with oxidative stress and direct protein modifications to NCX, Nav1.5, and NKA (along with the Ca2+ handling proteins), there will be a dysregulation of myocyte [Na+]i and [Ca2+]i homeostasis contributing to the contractile dysfunction and arrhythmias observed in HF. Furthermore, the dysregulation of [Na+]i and [Ca2+]i will alter mitochondrial dynamics resulting in the generation of more ROS setting up a vicious cycle (57, 93).
Kinase/phosphatase activity
While ROS can directly modulate ECC protein function via oxidation (i.e., redox signaling), it is also known that various kinases and phosphatases are redox sensitive. Thus, there may be an additional layer of complexity of ROS in which it may also regulate ECC protein function via changing phosphorylation status. As previously mentioned, PLB phosphorylation is decreased in HF. This decreased phosphorylation is partly due to the desensitization of the β-AR pathway via downregulation of β-AR receptors, increased Gαi, and increased GRK2 (53, 193). Another beneficial effect of β-blockers is to resensitize this pathway (27). However, protein phosphorylation levels will remain low due to an increase in phosphatase activity (22, 66, 118). Studies have shown that PP2a can be activated via various ROS to decrease PLB phosphorylation and blunt contraction (98, 99). It has also been shown that CaMKII can be activated via ROS (52). CaMKII can phosphorylate RyR at serine2814 to increase diastolic Ca2+ leak to empty the SR of Ca2+ and increase the propensity for arrhythmias (3, 36, 37, 162, 173). Furthermore, CaMKII phosphorylation of Nav1.5 will increase the late current resulting in [Na]i dysregulation and arrhythmias (166). Thus, oxidative stress manifests a “double whammy” on the ventricular myocyte by directly oxidizing and indirectly changing phosphorylation status of Na+ and Ca2+ handling proteins.
Antioxidant therapy
With the many studies considered above, one would expect that antioxidants could be used to treat HF. While antioxidant therapy (either scavenging ROS or inhibiting a source of ROS) has beneficial effects in small animal models of HF (4, 76, 117, 128, 147), clinical trials have been disappointing (21, 69, 108, 116). Thus, the field is reexamining redox signaling and why the failure in the treatment of HF. One likely reason is that, similar to other signaling pathways, redox signaling is compartmentalized. That is, redox signaling occurs with the production of specific species confined to spatially and temporally defined microdomains via specific antioxidant defenses (proteins, etc.). Thus, our current ways of antioxidant treatment may not be reaching or affecting these microdomains. Furthermore, by completely reducing ROS levels, our treatments may be inhibiting the beneficial effects of redox signaling. For example, studies have shown that angiotensin II and endothelin-1 stimulates ROS production via NOX to increase contraction (34, 43, 44). ROS production also plays an important role in the positive inotropic effects of β-AR stimulation (5). However, prolonged β-AR stimulation (>6 min) results in greater ROS production resulting in arrhythmogenic waves (25). It also appears that ROS (specifically H2O2 via NOX4) is important for vasodilation and lowering of blood pressure (129). This mechanism is also important during pressure overload to preserve capillary density to prevent cardiovascular dysfunction. Vasodilation also occurs via the cyclic GMP (cGMP)-dependent protein kinase (PKG), which can be activated via ROS independent of cGMP (29). In knock-in mice with a redox “dead” PKG, these mice were not able to vasodilate in response to acetylcholine and developed hypertension (127). We speculate that NOX4 signaling leads to vasodilation via activation of PKG; however, work needs to be done to confirm this. Another physiological function of NOX (specifically NOX2) is the stretch-dependent increase in contraction, termed X-ROS (126). Thus, with nonspecific antioxidants, we may be limiting the physiological effects of ROS, which can further deteriorate cardiac function during disease. In fact, this has been reported in some of the clinical trials (21, 108). Another important factor in redox signaling (specifically O2 •−) is its interaction with NO, known as the nitroso-redox balance (68).
Nitroso-Redox Balance
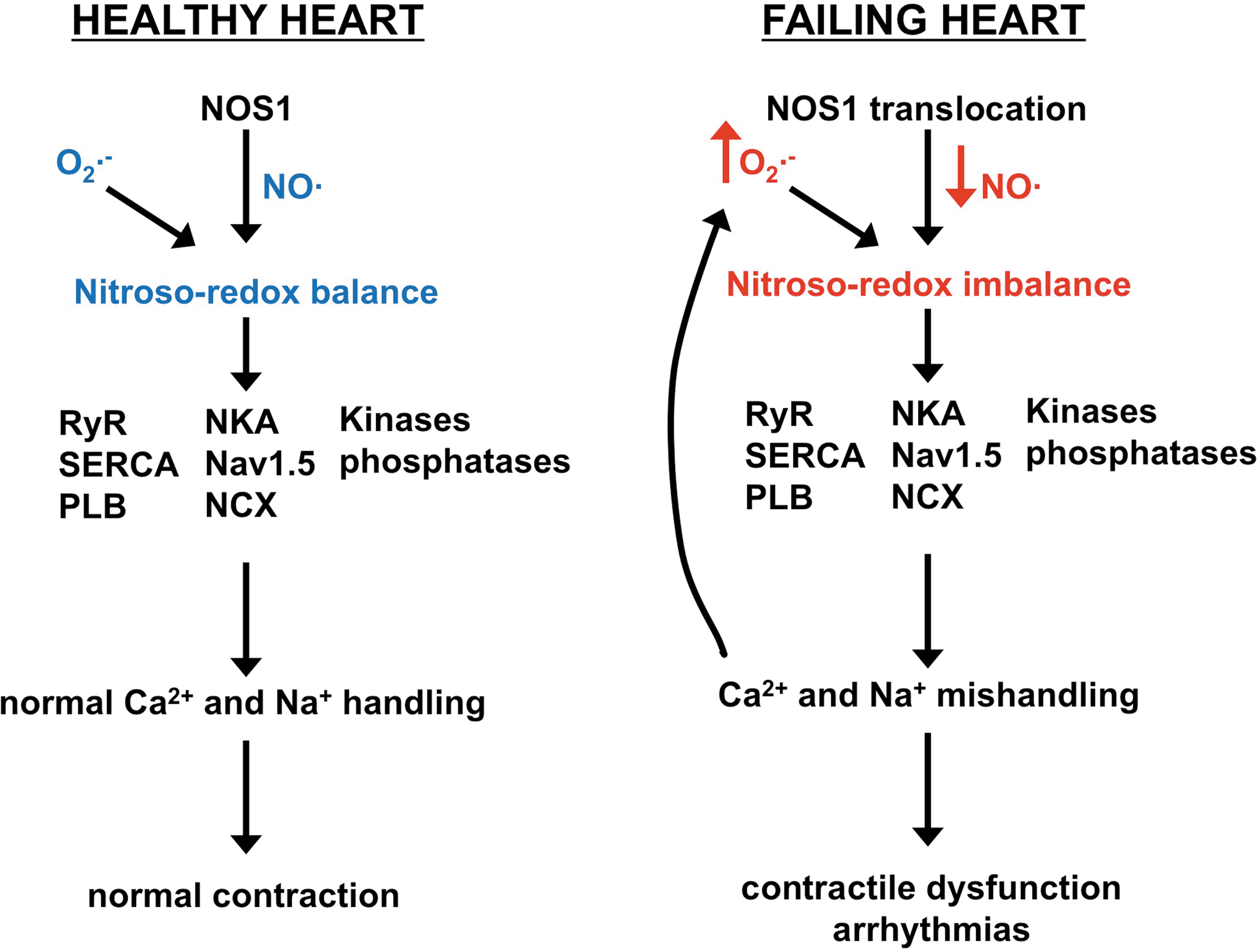
There is an interdependence between O2 •− and NO known as the nitroso-redox balance, in which the myocyte strives to keep NO levels greater than O2 •− (Fig. 4). The O2 •− and NO pathways communicate with each other via multiple means to make sure this nitroso-redox balance is maintained. One way is that NO acts as a buffer by reacting with O2 •− [one of the fastest reactions in nature (81)] to form peroxynitrite, which itself is a potent cardiac signaling molecule in health and disease (95). In addition, each signaling molecule can modulate the activity of enzymes on the other side of the balance. For example, NO is able to inhibit xanthine oxidase activity to limit O2 •− production (73, 82, 103). NO is produced via enzymes termed NOS (143). Within ventricular myocytes, the neuronal (NOS1) and endothelial (NOS3) NOS isoforms are constitutively expressed (191).
Endothelial NOS
While NOS3 activity can be modulated by ROS (e.g., O2 •− and H2O2) (31, 138), we do not believe that NOS3 contributes to the nitroso-redox balance that is involved in the regulation of myocyte Ca2+ handling. Our and others' previous data have shown that NOS3 signaling has no effect on basal myocyte Ca2+ handling and only modestly reduces β-AR-stimulated Ca2+ transient amplitude (10, 169). NO signals via the cGMP-independent (which is affiliated with the nitroso-redox balance) or the cGMP-dependent pathway (190). Our data demonstrated that NOS3 signals via the cGMP pathway in a spatially constricted microdomain to regulate caveolar LTCC (168). We believe that this is the basis for the minor role NOS3 plays in modulating contractile Ca2+. Furthermore, NOS3 colocalizes with SOD (26), an enzyme that reduces O2 •− levels, making NO more likely to signal via the cGMP pathway. This is further confirmed in studies that showed no change in myocyte ROS levels with NOS3 knockout (90). Thus, NOS3 signaling plays a minor role in the nitroso-redox balance that modulates myocyte Ca2+ handling. Our and others' data reveal that it is NOS1 that has this responsibility (90, 158).
Neuronal NOS
NOS1 is localized to the SR via binding to RyR (10) (Fig. 4). Unlike NOS3, NOS1 modulates ventricular myocyte contractile Ca2+ (192). That is, NOS1 signaling increases basal Ca2+ transient amplitude and accelerates Ca2+ decline (91, 167). In addition, physiological mediators of contraction such as the FFR and β-AR stimulation increase contraction and enhance lusitropy, in part, via altering Ca2+ handling (20, 85). Our and others' work has shown that NOS1 is a key protein involved in these pathways (10, 91, 167, 170). That is, in NOS1 knockout (NOS1−/−) mice, there is a blunted FFR and functional response to β-AR stimulation, which is similar to failing myocytes (i.e., decreased contractile reserve).
Similar to redox signaling, NO can also modify cysteine residues. This occurs by the addition of the NO moiety to a cysteine thiol to form an S-nitrosothiol (SNO) and is termed S-nitrosylation (74). There are also enzymes that reverse this reaction termed denitrosylation. One enzyme is the S-nitrosoglutathione reductase (106). However, this is not a direct denitrosylation as SNO proteins are not substrates. Instead, it reduces protein SNO levels by influencing the cellular redox environment. Another enzyme family, thioredoxin, has been found to be a direct denitrosylase (17). Thus, this cysteine modification (SNO) allows for the acute modulation of protein function, and we and others have found that NOS1 signals via the activation of this pathway (cGMP-independent) (167, 187). As with ROS, NO is also able to modulate various Ca2+ and Na+ handling proteins.
Ryanodine receptor
Since NOS1 localizes with RyR, studies have investigated the effects of NOS1 signaling on RyR function. We have found that NOS1 signaling increases RyR activity via S-nitrosylation (170). Specifically, by using various techniques to measure RyR activity that ranged from direct single RyR protein activity to indirect measurements in intact myocytes, we observed that NOS1−/− myocytes had reduced RyR activity associated with decreased S-nitrosylation. Additionally, when we added NO (SNAP), we could increase NOS1−/− RyR S-nitrosylation and activity. This is consistent with data obtained in mice with increased NOS1 activity (30). It should be noted that the Hare group has found the opposite results in which they report that NOS1−/− myocytes have increased RyR activity (62). We believe that our disparate results on RyR activity can be explained by nitroso-redox imbalance.
Nitroso-redox imbalance occurs in NOS1−/− mice via decreased NO bioavailability due to genetic deletion of NOS1 and increased O2 •− production via xanthine oxidase (90, 92, 158). Along with RyR, NOS1 colocalizes with xanthine oxidase. NO is able to inhibit xanthine oxidase and limit O2 •− production (73, 82, 103). Thus, with the loss of NOS1 and NO, xanthine oxidase is no longer inhibited and will produce an oversupply of O2 •−. It has also been shown that O2 •− production via NOX (most likely NOX2) is also increased in NOS1−/− myocytes (186). This loss of NO and S-nitrosylation will have two effects. First, the loss of SNO will have direct effects on protein function; second, S-nitrosylation of proteins protects them from oxidation (115, 152). Thus, RyR is now more susceptible to oxidation. Under low stress conditions, we hypothesize that loss of NO results in nitroso-redox imbalance leading to a decrease in RyR activity due to less RyR SNO. However, RyR in NOS1−/− myocytes is more susceptible to oxidative injury. We believe that under various stress conditions that will worsen the nitroso-redox imbalance by producing more O2 •− (e.g., FFR, β-AR stimulation, HF); there will be augmented oxidative injury to RyR. Data support this concept as restoring the nitroso-redox balance under low stress conditions increases RyR activity (158), whereas it decreases the activity under high stress conditions (48, 63). Thus, under low stress conditions, NOS1−/− myocytes have decreased Ca2+ transient amplitude due to decreased RyR activity, whereas high-stress (FFR, β-AR) NOS1−/− myocyte have decreased Ca2+ transient amplitude due to decreased SR Ca2+ load caused by excessive Ca2+ leak due to enhanced RyR activity. Furthermore, a similar situation was observed when investigating the effects of nitroso-redox imbalance, RyR, and arrhythmias (38). There were no observed arrhythmias with acute inhibition of NOS1. However, with the additional stress of elevated [Ca2+]i, the nitroso-redox imbalance now resulted in RyR-dependent arrhythmias.
SR Ca2+-ATPase
Unlike RyR, studies have not investigated if NOS1 is able to directly modulate SERCA activity. However, a study demonstrated that SERCA is S-nitrosylated (14), but the NO source responsible for this post-translational modification was not investigated. It has also been reported that nitroxyl (the 1 electron reduction product of NO) is able to modify SERCA via S-glutathiolation of cysteine674 (102). In addition to directly activating SERCA, we have shown that nitroxyl is able to target cysteines on PLB resulting in the formation of less monomeric PLB (the form that inhibits SERCA) (149). This may be important since it has been suggested that a product of NOS1 is nitroxyl (84, 140), and we have shown that the functional effects of nitroxyl are similar to NOS1 signaling (94). Thus, one would presume with NOS1 localized at the SR that it can directly activate SERCA activity via S-nitrosylation/glutathiolation, or indirectly by thiol modification of PLB, which would be consistent with the functional effects of NOS1 signaling. However, this remains to be established.
NCX and Na+ handling
Using an indirect method to measure NCX (decline of the caffeine-induced Ca2+ transient), we and others have found that there was no difference between WT and NOS1
Kinase/phosphatase activity
NOS1 can directly modulate ECC protein function via S-nitrosylation. But, similar to ROS, may also modulate the activity of various kinases and phosphatases to change the protein phosphorylation status. We have previously shown that a peroxynitrite donor (SIN-1) in a low dose was able to increase PLB Serine16 phosphorylation via activation of PKA (96, 97). Surprisingly, the peroxynitrite-mediated increase in PKA activity occurred in the absence of cAMP, and most likely occurs via S-nitrosylation (28). NOS1 signals, in part, via peroxynitrite and NOS1
Heart Failure
Oxidative stress contributes to the contractile dysfunction of HF, but antioxidant therapy has been ineffective. Since there is a strong connection between ROS and NO (nitroso-redox balance), one must also consider what occurs to NO with HF.
NOS in HF
Examining total NOS activity and NO bioavailability in HF, one finds that there is no difference or slightly decreased when compared with control. However, upon further evaluation, there is in fact a vast difference between HF and control when specifically examining NOS1 and NOS3. In animal models and humans with HF, it has been shown that NOS3 expression and activity are decreased (1, 39); however, NOS1 expression and activity are greatly increased (39, 40). Interestingly, NOS1 translocates from the SR to the plasma membrane and specifically to caveolin-3 after myocardial infarction (MI) and with HF. It has been hypothesized that this occurs to surmount the decrease in NOS3. NOS1 and NOS3 signaling are spatially compartmentalized due, in part, to their localization (191). Thus, with NOS3 localized to the caveolae, NO produced via NOS3 inhibits β-AR stimulated L-type Ca2+ current in a cGMP-dependent manner (as discussed above in the Endotheial NOS section). NOS1 localized to the SR augments SR Ca2+ handling to lead to positive inotropy, enhance the FFR, and enhance the functional response to β-AR stimulation via the nitroso-redox balance (as discussed above in the Neuronal NOS section). As expected, NOS1 translocation to the plasma membrane in HF now behaves like NOS3 to inhibit the β-AR stimulated Ca2+ current (16), which is protective. Nevertheless, there is a detrimental effect of this translocation and there is less NOS1 at the SR (Fig. 5).
Nitroso-redox imbalance in HF
We believe that this NOS1 translocation is akin to knockout NOS1. Ventricular myocytes from NOS1−/− mice exhibit slowed relaxation, a blunted FFR, and decreased response to β-AR stimulation. This is exactly what has been reported in failing myocytes. These data indicate that the translocation of NOS1 with HF results in nitroso-redox imbalance and depressed Ca2+ handling and thus contraction (Fig. 6). The significant role of the nitroso-redox imbalance in HF can be observed in NOS1−/− mice with MI. These mice had a larger nitroso-redox imbalance compared with WT mice with MI. Studies have shown that this massive nitroso-redox imbalance resulted in a faster and more severe ventricular remodeling, greater contractile dysfunction, and increased mortality (42, 137). Nitroso-redox imbalance was further investigated in an animal model of HF, the spontaneous hypertensive-heart failure (SHHF) rat (63). Similar to other HF models, this rat exhibited increased xanthine oxidase expression and activity with increased NOS1 expression and translocation. Similar to NOS1−/−, the SHHF rat displayed oxidative stress, which resulted in enhanced RyR oxidation and decreased S-nitrosylation. These RyR post-translational modifications increased RyR diastolic SR Ca2+ leak to reduce the SR Ca2+ load and blunt the FFR. Inhibition of xanthine oxidase with allopurinol decreased ventricular myocyte superoxide levels. The decrease in oxidative stress with allopurinol resulted in enhanced RyR S-nitrosylation. Since SNO and oxidative modifications can occur on the same cysteines, by decreasing ROS and thiol oxidation, these residues can now be nitrosylated given that NOS1 is still present (which is, although reduced at the SR, in the SHFF rat). Thus, the RyR SNO levels are highly determined by the level of ROS production by xanthine oxidase. With increased RyR S-nitrosylation and decreased oxidation, diastolic SR Ca2+ leak was reduced to increase SR Ca2+ load to improve the FFR. Interestingly, it has been shown in various skeletal muscle diseases (impaired exercise capacity, dystrophic muscle, environmental heat stroke, and sudden death) that the major contributor is hypernitrosylation of the skeletal muscle RyR isoform (RyR1) (12, 13, 49). Thus, maintaining proper RyR post-translational modifications appears to be vital to prevent pathological conditions. So, in HF, the decrease in RyR SNO due to increased ROS production via xanthine oxidase (nitroso-redox imbalance) will result in damaging oxidation to activate RyR and negatively impact contraction.
Correcting nitroso-redox imbalance: current status and perspectives
A previous study has shown that treatment with the xanthine oxidase inhibitor, allopurinol, was ineffective to increase contractility in a canine HF model in the presence of NOS inhibition (134). Thus, the effectiveness of antioxidants is highly dependent on NO bioavailability, suggesting that one needs to correct both NO (increase) and O2 •− (decrease) levels (i.e., restore nitroso-redox balance). Spin traps are used in the detection and identification of free radicals, including O2 •−, using electron paramagnetic resonance spectroscopy. Nitrones are a unique class of spin traps that produce NO as a byproduct after binding of O2 •− (164). We recently generated a novel spin trap that is able to permeate cell membranes (2-(2-ethoxy-2-oxoethyl)-2-(ethoxycarbonyl)-3,4-dihydro-2H-pyrrole 1-oxide [EMEPO]) (158). We hypothesized that EMEPO would be more beneficial for contraction and myocyte Ca2+ handling than antioxidants since it is both an O2 •− scavenger and NO donor. We tested the effects of EMEPO on NOS1−/− myocyte Ca2+ handling since these myocytes mimic the nitroso-redox imbalance observed in HF. As expected, EMEPO increased NO bioavailability and decreased O2 •− levels. Correcting the nitroso-redox balance resulted in an increased basal Ca2+ transient amplitude and enhanced the rate of relaxation and increased the FFR and functional response to β-AR stimulation. In fact, EMEPO significantly increased contraction greater than antioxidant treatment, which we believe is due to an additive effect of elevated NO and reduced O2 •− levels. We further showed that the improved Ca2+ handling was due to normalizing SR Ca2+ release and uptake. Specifically, EMEPO increased RyR activity and increased PLB Serine16 phosphorylation. The change in phosphorylation status suggests that we were able to reverse the harmful shift in the kinase/phosphatase balance in NOS1−/− myocytes. We also observed similar contractile effects of EMEPO on a disease model, in which acute treatment with EMEPO of ventricular myocytes from a canine MI model increased contraction by improving Ca2+ handling. Thus, designing a single therapeutic agent that can increase NO and simultaneously decrease O2 •− (i.e., correct the nitroso-redox imbalance) may be a promising approach for the treatment of the contractile dysfunction and arrhythmias associated with HF.
There have been clinical trials (HeFT and A-HeFT) that have examined the effects of isosorbide dinitrate (NO donor) and hydralazine (vasodilator) in HF patients (35, 156). These studies found that on top of standard care (β-blockers, digoxin, diuretics), the group treated with isosorbide dinitrate and hydralazine had decreased mortality, increased ejection fraction, and improvement in the quality of life compared with the placebo group. Although the molecular mechanisms of these beneficial effects of isosorbide dinitrate and hydralazine are unknown, it was suggested that this treatment increased NO bioavailability. However, we believe that a component of these beneficial effects of isosorbide dinitrate and hydralazine is by correcting the nitroso-redox imbalance and not just by increasing NO levels. Indeed, this was proven to be accurate (48). Once again, using the NOS1−/− mice as a model system of nitroso-redox imbalance, isosorbide dinitrate and hydralazine decreased O2 •− levels. This resulted in an increase in FFR due to improved Ca2+ handling and SR Ca2+ load via normalizing RyR activity and PLB phosphorylation. Interestingly, this treatment also increased myofilament Ca2+ sensitivity. It is known that sarcomeric proteins are also targets of redox signaling. However, this review is focused on Ca2+ handling, and the effects of ROS and reactive nitrogen species (RNS) on sarcomeric proteins has been reviewed elsewhere (151). Acute treatment with isosorbide dinitrate and hydralazine also increased the FFR in SHHF myocytes. Thus, it appears that the beneficial effects of isosorbide dinitrate and hydralazine are, in part, to restore the nitroso-redox balance by improving ventricular myocyte Ca2+ handling. While this combination of drugs works, in part, by correcting the nitroso-redox imbalance, cells may not equally uptake these drugs and the nitroso-redox imbalance will remain. We believe that a better approach will be the design of single compounds to correct the nitroso-redox imbalance.
Summary
A final common pathway of HF resulting in contractile dysfunction and arrhythmias is altered myocyte Ca2+ handling. It has been found that a major contributor to the altered Ca2+ handling in failing myocytes is oxidative stress targeting RyR, kinases, and phosphatases. Unfortunately, antioxidants have been clinically ineffective. We believe that the reason for the lack of effect of antioxidants is that they will only fix one side of the balance. That is, ROS (specifically O2 •−) is tightly connected with NO (nitroso-redox balance), which is reduced at the SR. NO also modulates RyR, kinases, and phosphatases. Recent data now demonstrate that it is important to simultaneously restore both NO and O2 •− levels to normalize Ca2+ handling in the setting of HF. Thus, reestablishing the nitroso-redox balance may be an important therapeutic target for the treatment of HF patients.
Footnotes
Acknowledgment
This research was supported by the National Institutes of Health (HL094692, M.T.Z.; HL33921, S.R.H.).