
Abstract
Introduction
S
sCJD and PD are distinct disorders where a specific protein undergoes aggregation and accumulates in distinct regions of the brain, resulting in clinical symptoms typical of each disorder. The pathogenic mechanisms leading to protein aggregation are also different, and so are the neurotoxic pathways and the set of neurons affected by the pathology. In sCJD, the principal pathogenic event involves the accumulation of prion protein (PrPC) in the brain parenchyma in a detergent-insoluble and partially protease-resistant form termed PrP-scrapie (PrPSc) (127). The pathognomonic feature of PD brains, on the other hand, is the accumulation of Lewy bodies composed principally of α-synuclein in the substantia nigra (SN) pars compacta (113). The aggregation of PrPC occurs as a sporadic event, is initiated by exogenous PrPSc, or results from mutations in the PrPC gene (4). Triggers that initiate the aggregation of α-synuclein include sporadic events, environmental toxins, and mutations in the α-synuclein gene. Other gene products are also involved, some of which exacerbate the aggregation of α-synuclein (113). Despite these dissimilarities, mismetabolism of iron in diseased brains is a prominent feature of both conditions.
The diverse etiology and clinical presentation of sCJD, PD, and other neurodegenerative disorders associated with brain iron imbalance have led to the belief that iron accumulation is a result of massive neuronal death or nonspecific coprecipitation with protein aggregates. However, recent reports suggest otherwise (16, 23, 37, 62, 142, 150). In sCJD, the imbalance of brain iron metabolism is believed to result from the sequestration of iron in a biologically unavailable form, creating a phenotype of iron deficiency (146, 149, 154). In PD, increased expression of iron uptake proteins and downregulation of an iron exporter lead to the accumulation of excess iron in the SN. This creates an environment that promotes iron-mediated free radical generation. Since free radicals denature and aggregate proteins, it is likely that once the process is initiated, further accumulation of protein aggregates is facilitated by the redox-active nature of associated iron, which also exaggerates the toxic potential of these aggregates. It is therefore timely to review the available information on the role of redox-iron in protein aggregation and the contribution of impaired cellular clearance mechanisms in accentuating protein aggregate-mediated neurotoxicity in sCJD and PD.
Iron and Protein Misfolding in Prion Disorders
Prion disorders are a heterogeneous group of neurodegenerative conditions of humans and animals that result from the accumulation of protein aggregates in the brain parenchyma. These disorders are unique among neurodegenerative diseases of protein aggregation, in that, in addition to their sporadic and familial etiology, some forms are naturally transmissible. sCJD is the most common human prion disorder and comprises ∼80% of all reported cases. Animal prion disorders include scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in the deer and elk population. Scrapie and CWD are transmissible horizontally to the same species, whereas BSE has been transmitted to humans, raising significant public health concerns. The underlying cause is a change in the conformation of PrPC, a ubiquitous and evolutionarily conserved glycoprotein abundantly expressed on neuronal cells, mainly from an α-helical-rich to a β-sheet-rich PrPSc form. This change renders PrPSc insoluble in nonionic detergents and confers limited resistance to proteases, thus increasing its half-life significantly (127). Aggregates of PrPSc in the brain parenchyma are the principal cause of disease-associated neurotoxicity and transmissibility. The change in the conformation of PrPC to PrPSc is believed to occur as a spontaneous event in sporadic disorders, is facilitated by mutations in the PrPC gene in familial disorders, and results from direct contact with an exogenous source of PrPSc in transmissible disorders. Once initiated, the conversion of additional PrPC to the PrPSc form proceeds exponentially by the nucleation-mediated coaggregation phenomenon (Fig. 1) (3, 4, 21, 127, 153).

Much effort has gone into the identification of triggers that initiate or potentiate the conversion of PrPC to PrPSc, and the mechanism by which PrPSc induces neurotoxicity. Several important facts have emerged over the past years. Here, we will focus on the role of iron in these processes for two main reasons: (i) PrPC is involved in cellular iron metabolism (42, 128, 145, 147, 148, 150, 154) and (ii) iron plays a significant role in prion disease pathogenesis (25, 26, 62, 84, 86, 146). Since iron is highly toxic if mismanaged due to its ability to generate reactive oxygen species via the Fenton and Haber–Weiss reactions, it is important to understand the physiological and pathological interaction of PrPC and PrPSc with iron and the contribution of these processes to prion disease pathogenesis.
Experimental models have provided important insight into the factors that promote the aggregation of PrPC to PrPSc. Principal among these are amplification of PrPSc by the protein misfolding cyclic amplification (PMCA) reaction (20, 30, 115), cell models replicating PrPSc in culture (159), and transgenic mouse models lacking PrPC expression (15). PMCA has been instrumental in demonstrating that PrPC is the principal substrate for PrPSc, and if supplemented with certain cofactors, can drive the amplification of PrPSc seed by nucleation-mediated coaggregation (20). Recent studies have demonstrated spontaneous conversion of PrPC from brain lysates or recombinant PrP from bacteria to PrPSc by this method (20, 34), supporting the hypothesis that the initial conversion of PrPC to PrPSc is a sporadic event, and once initiated, the process proceeds exponentially. Minimal factors are required for this conversion, and iron does not appear to play a significant role in this in vitro reaction (172). Limited studies that have explored the role of other transition metals in the conversion of PrPC to PrPSc in vitro have reported variable results (85). It is interesting to note that several proteins in addition to PrPC aggregate when cell or brain lysates are subjected to the PMCA reaction, some of which are likely to coaggregate with PrPSc because of its hydrophobic nature (149). Among these, ferritin deserves a special mention not only because it forms detergent-insoluble and protease-resistant aggregates by the PMCA reaction (149), but it has been identified as one of the major contaminants of PrPSc purified from diseased brains despite the use of harsh purification conditions (104, 108, 136). Since ferritin is very rich in iron, the complex of PrPSc and ferritin is likely to be redox-active and cytotoxic unless degraded promptly by the cellular degradation machinery (65, 129).
Cell and mouse models expressing PrPC and mutant PrP forms or following infection with PrPSc have provided important information on the biogenesis, turnover, and the role of iron in PrPSc-mediated cytotoxicity. Exposure of neuroblastoma cells overexpressing PrPC (PrPC-cells) to ferrous ammonium citrate induces a change in the conformation of cell surface PrPC to an aggregated form that resembles pathogenic PrPSc in several characteristics, including insolubility in nonionic detergents and resistance to limited digestion by proteases (8). These aggregates are internalized and accumulate in the lysosomes in association with ferritin. When added to the culture medium of fresh cells, these aggregates undergo endocytosis and serve as a nucleus for the aggregation of additional PrPC in naive cells. Hemin, an iron-rich compound, induces a similar change in PrPC that is internalized and ultimately degraded in the lysosomes (94). Since ferritin is a major iron storage protein necessary for maintaining cellular iron homeostasis, coaggregation with PrPSc results in the sequestration of associated iron in a biologically unavailable form, creating a phenotype of cellular iron deficiency as indicated by the upregulation of iron uptake proteins, transferrin (Tf ) and transferrin receptor (TfR), and downregulation of cellular ferritin to reduce iron storage (8, 43, 44). Interestingly, ferritin isolated from CJD brains and scrapie-infected mouse brains and cell lines is insoluble in detergents and retains associated iron even after exposure to strong denaturing conditions, indicating a change in its biochemical characteristics due to coaggregation with PrPSc (104, 108). Colocalization of PrPSc and ferritin in lysosomal structures has been reported in scrapie-infected neuroblastoma cells and a hypothalamic cell line, supporting these observations (8, 146).
Cell lines expressing disease-associated mutations of PrP have provided important information on the pathogenic mechanisms underlying familial prion disorders (54 –60, 151, 167). Although some mutations destabilize the structure of mutant PrP and promote its aggregation (155), diverse cellular pathways are believed to contribute to the cytotoxicity associated with familial prion disorders (79, 104, 105). Human PrPC is a 209 amino acid glycoprotein linked to the outer leaflet of the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor. It contains two N-linked glycans at amino acids 181 and 197, and the two cysteines at residues 179 and 214 are linked by a disulfide bond. Several mutations in the coding region of PrPC gene segregate with familial prion disorders. Of these, three are in the GPI anchor signal peptide that is replaced by a preassembled GPI anchor within 5 min of synthesis (165). Most are in the relatively structured C-terminal region, although an insertion in the N-terminal octapeptide repeat region is also associated with a familial prion disorder. Most mutations alter the trafficking and/or turnover of the mutant protein in a manner that is specific to each mutation, and the associated cytotoxicity is influenced by the post-translational modifications, aggregation state, and the subcellular localization where the mutant protein accumulates (11, 54 –60, 181). Some of the mutations alter cellular iron metabolism, and specific instances are discussed below.
Recent reports suggest that PrPC enhances the uptake of Tf and non-Tf-bound iron by functioning as a ferrireductase (FR) (145). The N-terminal copper-binding octapeptide repeat region and the linkage of PrPC to the plasma membrane are necessary for this activity. Thus, the uptake of radiolabeled iron is significantly higher in cells that overexpress PrPC relative to cells that express mutant PrP forms that lack the octapeptide repeat region, GPI anchor, or carry mutations that inhibit transport of PrP to the plasma membrane (145). Consistent with these observations, the calcein-chelatable labile iron pool (LIP) and incorporation of radiolabeled iron in cytosolic ferritin is significantly higher in PrPC-cells relative to mutant PrP cell lines (145, 148). A similar pattern is noted in cell lysates, although the difference between PrPC and mutant cell lines is less perhaps due to the activity of other cellular FRs (114, 145, 177). Surprisingly, point mutation at residue 102 (P to L) associated with a familial prion disorder enhances the FR activity of mutant PrP. Consequently, cells expressing PrP102L have higher LIP and incorporate significantly more radioactive iron from the medium in cytosolic ferritin relative to PrPC-cells (145).
The functional role of PrPC in iron uptake is also reflected in mouse models. Transgenic mice carrying a deletion in the PrP gene and therefore lacking PrP expression in the brain and systemic organs (PrP knockout [PrP−/−]) show a phenotype of iron deficiency relative to matched (wild type [PrP+/+]) controls (147). The amount of total iron in the plasma, liver, spleen, and brain of PrP−/− mice is significantly lower, and the levels of Tf and TfR are significantly higher than PrP+/+ controls. When exposed to radiolabeled iron by gastric gavage, PrP−/− mice incorporate relatively more iron in red blood cells and the liver, the main iron utilization and storage compartments, respectively (147). Bone marrow macrophages of PrP−/− mice show minimal stainable iron, and when cultured in vitro, incorporate significantly less radiolabeled iron relative to PrP+/+ controls. There is no difference in the rate or amount of iron released from cultured macrophages, indicating that PrPC mediates uptake, not release of iron by these and other cells (145).
When considered with possible coaggregation of PrPSc with ferritin during prion disease pathogenesis (146), it is likely that the iron imbalance observed in CJD and scrapie-infected animal brains is due to the combined effect of the loss of normal function of PrPC in iron uptake and the gain of toxic function by the PrPSc-protein complexes due to their redox-active nature. The accumulation of redox-active iron in association with PrPSc plaques has been reported in variant CJD brains (123), and an increase in total iron and a paradoxical phenotype of iron deficiency as indicated by the upregulation of major iron uptake proteins, Tf and TfR, has been reported in CJD and scrapie-infected mouse brains (146). Levels of Tf increase with disease progression in scrapie-infected mouse and hamster brains and correlate directly with PrPSc levels, suggesting sequestration of iron in a biologically unavailable form in PrPSc–ferritin complexes. Transcriptional alteration of ferritin and iron regulatory proteins 1 and 2 in the hippocampus and cerebral cortex of scrapie-infected mice further supports these observations (84). Furthermore, the autophagosomal marker LC3 II is upregulated in prion disease-affected human and animal brains, indicating an attempt by the autophagosomal pathway to degrade such protein complexes (32). Figure 2 summarizes the different pathways leading to iron imbalance in sCJD brains.
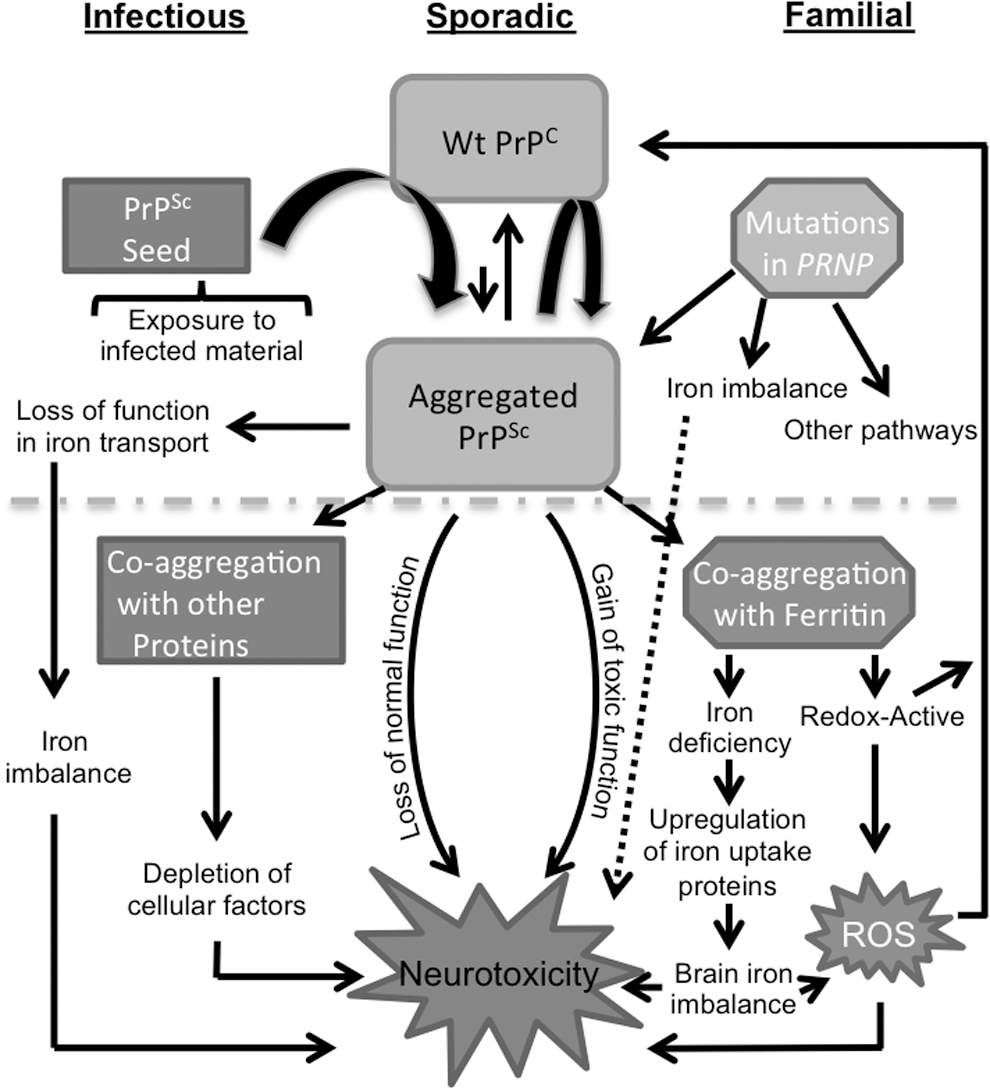
It is notable that the alteration of brain iron homeostasis in sCJD brains is reflected in the cerebrospinal fluid (CSF) much before end-stage disease. Levels of Tf are decreased, and the ferroxidase activity is increased in the CSF of CJD cases (62, 144). Surprisingly, the increase in CSF ferroxidase activity is restricted to a novel <3 kDa fraction, indicating that ceruloplasmin, the major brain and CSF ferroxidase, is not altered by CJD pathology (62, 76). Together, these biomarkers discriminate CJD from other neurodegenerative conditions with a sensitivity of 86.8%, specificity of 92.5%, and accuracy of 88.9%, indicating that the change in iron homeostasis triggered by prion disease pathology is disease-specific and can be used as a premortem diagnostic test for these disorders.
Although iron is not the principal factor that induces the aggregation of PrPC to the PrPSc form or disease-associated neurotoxicity, emerging evidence suggests a specific and significant role for this metal in sCJD and animal prion disorders. Future investigations on the mechanism(s) underlying iron-induced aggregation of PrPC and the accompanying change in brain iron homeostasis are necessary to develop therapeutic options based on restoring brain iron metabolism through chelation or other means.
Iron and Protein Misfolding in PD
PD is a progressive neurodegenerative disorder with selective loss of dopaminergic neurons in the SN pars compacta (113). Pathologically, surviving neurons accumulate intracytoplasmic inclusions called Lewy bodies composed principally of misfolded α-synuclein (161). Most cases of PD are sporadic in origin with no known underlying cause, although aging, genetic susceptibility, and environmental factors are believed to play a significant role in disease onset. A small percentage of PD cases are familial in nature and segregate with mutations in genes encoding α-synuclein, parkin, PTEN-induced putative kinase 1 (pink1), DJ-1, leucine-rich repeat kinase 2 (LRRK2), ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), and ATP13A2, a lysosomal type 5 ATPase (130). Mutations in α-synuclein and LRRK2 are associated with autosomal dominant PD, whereas those in DJ-1, parkin, pink1, and ATP13A2 segregate with autosomal recessive forms (13, 48, 51, 87, 126, 162).
Several observations suggest that the aggregation of α-synuclein is central to the pathogenesis of PD. First, overexpression of wild-type α-synuclein as a result of gene duplication or triplication is sufficient to cause PD in humans and animal models, suggesting a dose-dependent relationship between α-synuclein and PD (22, 47, 156). Second, point mutations in the α-synuclein gene are associated with autosomal dominant familial PD, in particular the three point mutations A53P, A30P, and E46K (90, 125, 182). Given the presence of aggregated α-synuclein in Lewy bodies and the propensity for mutant α-synuclein to misfold (38, 96), studies have examined specific factors that promote the aggregation of wild-type and mutant α-synuclein in cultured neuronal cells. When overexpressed, wild-type α-synuclein forms spherical punctate aggregates in the cytoplasm similar to those seen in the SN neurons of PD cases, suggesting self-aggregation in a concentration-dependent manner (47, 93, 156, 170). Gradually, these aggregates include other cellular proteins, especially ubiquitinated proteins targeted for degradation by the proteasomal machinery. In vitro studies with recombinant proteins indicate that both wild-type and mutant α-synuclein form amyloid-like fibrils similar to those isolated from Lewy bodies in a concentration-, temperature-, time-, and pH-dependent manner (29, 53, 66, 176). The rate of aggregation is higher in mutant α-synuclein forms, perhaps due to destabilization of the protein structure by the mutation. Intrastriatal inoculation of fibrillar α-synuclein into wild-type mice is sufficient to initiate Lewy body formation, dopaminergic cell death, and clinical symptoms of PD, reinforcing the central role of α-synuclein misfolding and aggregation in PD pathogenesis (75, 99).
This raises two questions. First, what triggers the aggregation of wild-type α-synuclein, and second, why, despite widespread expression in the central nervous system, are dopaminergic neurons of the SN susceptible to α-synuclein aggregation and toxicity. Although the answer to these questions is still unclear, other pathogenic processes associated with PD such as mitochondrial dysfunction, oxidative stress, impaired autophagy, and mismetabolism of iron in the SN have provided important insights (40, 63, 70, 174). It has now become increasingly clear that aggregation of α-synuclein and the metabolic pathways implicated in these pathogenic processes are closely related, and together, create an ongoing environment of oxidative stress that leads to further accumulation of misfolded α-synuclein and other proteins, creating a self-perpetuating feedback loop of redox-imbalance, protein aggregation, and neurotoxicity (Fig. 3) (113, 166).
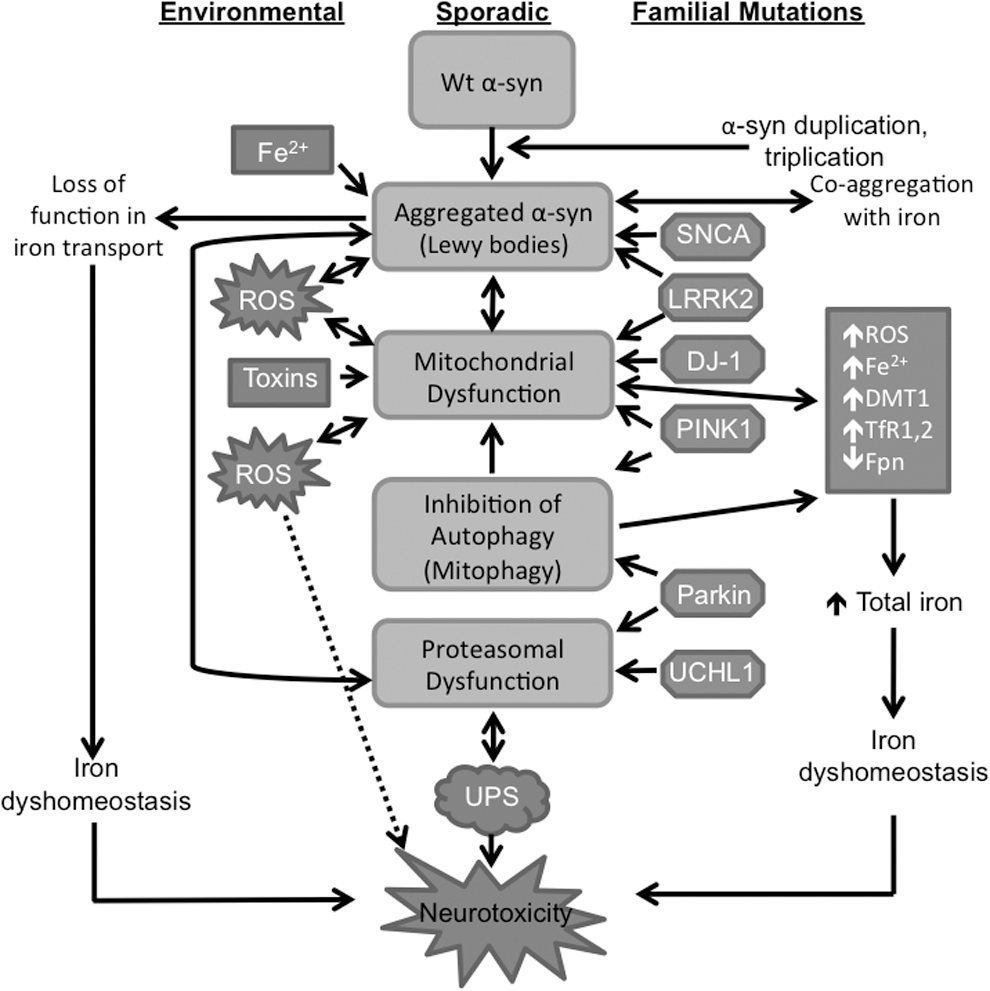
Several observations support the hypothesis that mismetabolism of iron in the SN promotes aggregation of α-synuclein. Levels of iron are significantly higher in the SN relative to other brain regions, increasing the susceptibility of this region to iron-mediated oxidative stress (36, 49, 101, 112, 179). Elevated levels of total iron and a shift in the equilibrium of iron to the oxidized state have been reported in the SN of sporadic and familial cases of PD (131, 158). Redox-active iron has been detected in association with α-synuclein aggregates in Lewy bodies (19), a phenotype that is likely to promote oxidization and further aggregation of α-synuclein and other proteins (52). Since iron is known to interact with the C-terminal region of α-synuclein, it is likely that under oxidizing conditions, it denatures α-synuclein and coprecipitates with the aggregates (10, 12, 89). Aggregation of α-synuclein is inhibited by the iron chelator desferrioxamine, supporting this hypothesis (64, 67).
Recent evidence suggests that iron regulates α-synuclein expression at the translational level through an iron-response element (IRE) in its 5′UTR (17, 41, 45). Conversely, α-synuclein modulates cellular iron homeostasis through its functional activity as a FR, converting Fe3+ iron to the biologically active ferrous iron (Fe2+) form (33). Accordingly, α-synuclein has greater affinity for Fe3+ relative to the Fe2+ form of iron (122). The FR activity of α-synuclein is particularly important in the SN where Fe2+ iron is required as a cofactor for tyrosine hydroxylase that catalyzes the rate-limiting step in dopamine synthesis. Surprisingly, α-synuclein colocalizes with heme synthesis enzymes in the mitochondrial membrane, suggesting that it may also be involved in the synthesis or metabolism of heme (138). Further investigations are necessary to understand the role of α-synuclein in cellular iron and heme metabolism.
Remarkably, the expression of divalent metal transporter 1 (DMT1), an iron import protein, is upregulated, and that of ferroportin, an iron export protein, is downregulated in the SN of PD cases, partly explaining the accumulation of iron in this region. Similar observations are noted in the SN and ventral mesencephalon of mouse models exposed to MPP+ (134). Exposure of the dopaminergic cell line MES23.5 to 6-hydroxydopamine (6-OHDA), another PD-inducing compound, increases intracellular levels of iron and, paradoxically, upregulates DMT1 containing an IRE sequence in its 5′UTR (DMT1+IRE) (78), suggesting misregulation of cellular iron metabolism. Paradoxically, the expression of ferroportin is downregulated, exacerbating cellular iron dyshomeostasis (160). Likewise, exposure of SH-SY5Y cells, a dopaminergic cell line, to MPP+ or the dopamine metabolite aminochrome increases the expression of iron uptake proteins DMT1 (+IRE) and TfRs 1 and 2, and downregulates ferroportin, thereby increasing the overall iron content of these cells (2, 18). The resultant phenotype of oxidative stress causes aggregation of α-synuclein, further compounding the phenotype of iron mismetabolism. It is interesting to note that polymorphisms and mutations in iron-modulating genes are associated with PD (14, 61), suggesting a critical role for iron in PD pathogenesis.
Although it is debatable whether excess iron is the cause or consequence of α-synucelin aggregation, once initiated, it creates a phenotype of continued oxidative stress that is fueled by several other pathological changes. Principal among these is mitochondrial damage, an invariable feature of PD pathology. Mitochondria of dopaminergic neurons of the SN are especially rich in α-synuclein, and its misfolding is believed to disrupt mitochondrial function by inhibiting Complex I of the electron transport chain, resulting in the loss of mitochondrial membrane potential and cell death (24, 35, 98). Similar observations are noted when α-synuclein is overexpressed in cell or mouse models, where it translocates to the mitochondria, impairs mitochondrial function, and increases oxidative stress (72, 117, 118, 139). Neuronal cell lines and mouse models expressing mutant forms of α-synuclein show a similar phenotype (9, 98, 163). When incubated in vitro with mitochondria, α-synuclein translocates to the mitochondria in a dose-dependent manner and inhibits the Complex I activity, supporting these observations (97). Immunocapture experiments and native gel electrophoresis indicate that α-synuclein interacts physically with Complex I, and the deletion of the proposed mitochondrial targeting signal from α-synuclein abolishes this effect. Toxins used for the induction of experimental PD such as MPP+ (active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), rotenone, 6-OHDA, and aminochrome (2, 91) also inhibit mitochondrial complex 1 activity, suggesting mitochondrial dysfunction as a prominent feature of PD pathogenesis (83, 120, 137).
Once damaged, mitochondria are removed from the cell by selective autophagy or mitophagy, a function necessary for cell survival (95). This function is mediated by a distinct set of proteins, among which parkin and pink1 play a major role. Not surprisingly, functional mutations in parkin and pink1 are associated with young onset, autosomal recessive form of PD partly because inhibition of mitophagy spares dysfunctional mitochondria that generate reactive oxygen species and induce further aggregation of α-synuclein and other vital proteins (110). Furthermore, increased production of reactive oxygen species exacerbates iron-mediated oxidative damage through reduced production of Fe-S clusters, a key component of the cellular iron regulatory mechanism (92, 102, 133). Downregulation of α-synuclein protects mitochondria from iron-induced toxicity, and conditional inhibition of autophagy and mitochondrial complex 1 activity causes age-dependent loss of dopaminergic neurons (6, 27), supporting the role of these processes in PD pathogenesis.
Together, the above observations suggest that iron chelation may provide therapeutic benefit to PD cases. Since iron levels increase in the SN before neurodegeneration, this approach may arrest further loss of dopaminergic neurons (112) and has shown promise in multiple PD model systems (64, 106, 135, 178). However, removing enough iron from susceptible tissues without negatively affecting systemic iron homeostasis remains a challenge for the use of this approach in PD cases (109).
Role of Autophagy in Protein Aggregation and Iron Imbalance
Neurons, like other cells, use two major pathways of protein turnover for maintaining a balance between biosynthetic and catabolic processes: the ubiquitin–proteasome system and the autophagy–lysosomal pathway. The latter degrades whole organelles in addition to dysfunctional proteins and is especially important for maintaining mitochondrial function.
The association of familial cases of PD with mutations in genes implicated in proteasomal and autophagosomal pathways underscores the role of cellular degradation machinery in PD pathogenesis (31). Thus, mutations in parkin, pink1, and UCH-L1 segregate with familial PD (113). Parkin is an E3 ubiquitin ligase that targets proteins for proteasomal degradation (107, 141, 183), pink 1 is a mitochondrial-targeted kinase (143, 168, 173), and UCH-L1 is an ubiquitin carboxy-terminal hydrolase. DJ-1 is a redox-dependent chaperone that inhibits the aggregation of α-synuclein (140). Overexpression of parkin and pink1 partially reverses the aggregation of α-synuclein (124), emphasizing the significance of these proteins in PD pathogenesis. Interestingly, aggregated α-synuclein inhibits proteasomal function, thereby sensitizing the cells to apoptosis (164) through decreased degradation of proapoptotic factors (77). Several PD-inducing toxins such as MPP+, rotenone, paraquat, NO, dopamine, and iron that cause aggregation of α-synuclein and mitochondrial dysfunction also alter the solubility of parkin, resulting in reduced proteasomal activity and accumulation of aggregated proteins (171). Since parkin modulates the degradation of DMT1 (73, 121), dysfunctional parkin spares DMT1 (50, 132) with resultant increase in iron uptake and worsening of iron-mediated oxidative stress. Functional mutations in DMT1 that impair iron import protect against toxin-induced PD in mouse models (134), supporting these observations and emphasizing the role of iron in PD-associated dopaminergic cell death.
Interestingly, dysfunctional pink1 induces a phenotype similar to parkin-associated PD (168). Thus, Drosophila with the deletion of either pink1 or parkin genes develop a muscle phenotype and mitochondrial impairment characteristic of PD. Although parkin overexpression rescues the phenotype of pink1 flies, the reverse is not true (28, 119). Pink1 also modulates mitophagy, an essential function necessary for cell viability. Normally, pink1 is translocated to the inner mitochondrial membrane where it is cleaved by proteases and eventually degraded by the proteasome system. Damaged mitochondria with decreased membrane potential are unable to translocate pink1, resulting in its accumulation on the outer mitochondrial membrane where it recruits parkin and targets mitochondria for autophagy. Disruption of this process due to functional mutations in pink1 causes accumulation of dysfunctional mitochondria, resulting in the generation of free radicals and aggregation of α-synuclein and other proteins (116).
The functional deficiency of DJ-1, a recessive familial PD gene, alters autophagy in murine and human cells (74). Wild-type LRRK2 and the G2019S mutant, a mutation associated with PD and known to increase its kinase activity (71), participate in the phosphorylation of α-synuclein and promote its aggregation and propagation to neighboring cells (88). Parkin also interacts with LRRK2 (157), and coexpression of parkin with mutant LRRK2 G2019S provides significant protection against neurodegeneration in Drosophila models (111), perhaps by degrading abnormally phosphorylated proteins through the E3 ubiquitin ligase activity of LRRK2. Dopaminergic cell loss can be attenuated by expressing pink1, DJ-1, or parkin in Drosophila models of LRRK2 mutations, supporting the above interactions (169). Moreover, mitochondrial membrane fragmentation induced by exogenous expression of α-synuclein can be rescued by coexpression of pink1, parkin, or DJ-1, not PD-associated mutations of these genes (81). Overexpression of pink1 enhances autophagy, which can be reduced by knocking down the gene encoding beclin1, a protein that interacts with pink1 and modulates its function. It is interesting to note that the W437X mutant of pink1 demonstrates impaired interaction with beclin1 and the ability to induce autophagy, whereas the kinase-deficient G309D mutant of pink1 does not interact with beclin1 and has minimal impact on autophagy (103). It is believed that the overexpression of α-synuclein induces mitochondrial dysfunction by inhibiting autophagy through disruption of Rab1a homeostasis (100, 175) and dysfunctional mitochondria interfere with microtubule-dependent transport of autophagosomes, further inhibiting autophagy (7).
In contrast to PD, the role of autophagy in prion disorders is less defined (69, 152). Limited studies suggest that inhibitors of autophagy increase intracellular accumulation of PrPSc, whereas stimulators of this function help in its clearance (1). Thus, lithium, trehalose, and rapamycin have been used to enhance the clearance of PrPSc from prion-infected cells through the induction of autophagy (1, 68). In prion-infected mice treatment with imatinib at an early phase of peripheral infection delays both the neuroinvasion of PrPSc and the onset of clinical disease (39, 180), suggesting a beneficial role of enhanced autophagy in prion disease pathogenesis.
Conclusions
Cumulative evidence from the literature leaves little doubt that mismetabolism of iron in sCJD and PD brains is intimately associated with the underlying pathogenic processes and therefore disease-specific (Fig. 4). Reflection of this phenotype in the CSF much before end-stage disease and the ability to discriminate sCJD from PD and other neurodegenerative conditions by specific changes in iron-modulating proteins with a high degree of accuracy leaves little doubt that brain iron imbalance is intimately associated with disease pathogenesis. In sCJD, sequestration of iron in PrPSc-protein complexes that include the iron-rich protein ferritin creates a phenotype of iron deficiency and is believed to be the principal cause of iron mismetabolism in diseased brains. The resultant alterations in the levels of total and redox-active Fe2+ iron, iron uptake proteins Tf and TfR, and the iron storage protein ferritin in the brain tissue and CSF are disease-specific and of diagnostic value. PD brains, on the other hand, accumulate iron in the SN due to diverse causes, and this phenotype is worsened by the upregulation of iron uptake proteins DMT1 and TfR and downregulation of the iron export protein ferroportin. Although iron chelation is an attractive therapeutic option, it is unlikely to succeed unless the pathogenic processes leading to altered iron metabolism are better understood. Future investigations are necessary to clarify these outstanding questions and develop effective therapies to restore iron homeostasis in diseased brains.
