
Abstract
Introduction
N
Since the 1950s, oxidative damage not only in the brain but also throughout the body has been increasingly recognized as a major contributing factor to the process of normal aging, although the underlying causes (e.g., the decline of endogenous antioxidant defense mechanisms, or a general increase in ROS production) are not well understood and are likely to be complex in their interactions (54, 55, 92, 159). More recently, such oxidative damage is strongly implicated as an important feature of many major neurological (and other) diseases or conditions. Oxidative damage within the CNS is, for example, implicated in the inflammatory effects associated with infection, stroke, ischemia/reperfusion injury, and multiple sclerosis (53). Numerous neurodegenerative diseases also share commonalities such as metabolic and mitochondrial dysfunction, disruption of iron metabolism (inextricably linked with redox activity), and oxidative damage (42, 52, 68, 103, 182). In addition, while the major neurodegenerative diseases affect distinct neuronal types and regions of the brain [e.g., hippocampal neurons in Alzheimer's disease (91)], dopaminergic neurones in the substantia nigra for Parkinson's disease (PD) (9), striatum for Huntington's disease (HD) (32), and motor neurones primary motor cortex, corticospinal tract, and spinal cord for amyotrophic lateral sclerosis (ALS) (84); Figure 1, they share the additional commonality of featuring distinct aggregations of specific proteins (Fig. 1). Excessive levels of these proteins can promote increased ROS levels within cells, which can, in turn, have the positive feedback effect of increasing aggregation, as well as of contributing to neuronal loss (Fig. 1), primarily via apoptosis (21, 42). Although apoptosis is, doubtless, not the only pathway to neuronal death in these diseases, it is the primary focus in this article.

Interestingly, many of these neurodegenerative disorders are also associated with altered activity, function, or expression of K+ channels. Thus, numerous reports describe altered properties of diverse K+ channels in Alzheimer's disease (AD) (16, 123, 128), PD (93, 165) and HD (11, 167), ALS (72, 148), and ataxias (39, 131, 187). In this article, we discuss some key examples of oxidative modulation of K+ channels. Surprisingly, with the notable exceptions of Kv2.1 and BK channels (which are the main focus of our article), this field has not progressed dramatically ever since a previous, comprehensive review was published some 12 years ago (8), with regard to the pathological consequences of specific modification of this class of channel. However, a most recent review has provided a very valuable, state-of-the-art account of the molecular requirements of various K+ channels for redox sensitivity, focusing particularly on voltage-gated K+ channels in the CNS and elsewhere (136), and readers are directed to this article for such detailed information. In addition, redox modulation of KCNQ channels (which underlie M currents) is discussed elsewhere in detail in this Forum. Our aim in this article is to discuss redox modulation of specific K+ channels (Kv2.1 and BK channels) and how this relates to altered cellular function in the context of both normal cellular processes, including aging, and the development of neurodegenerative diseases. We also describe emerging evidence that K+ channels are major targets for modulation by the gasotransmitters carbon monoxide and hydrogen sulfide, and discuss whether their regulation by these gases may account for some of the protective effects of these agents against oxidative stress.
Oxidative Stress Within the CNS
Production of ROS or RNS is not restricted to any particular cell type within the CNS, and can arise from a number of different sources within a given cell, as schematically shown in Figure 2. Such sources include NADPH oxidases, xanthine oxidases, cytochrome P450-dependent oxygenases, and complexes I and III of the mitochondrial electron transport chain (ETC). The major ROS produced include free radicals; for example, superoxide (O2 •−), hydroxyl (•OH), and hydrogen peroxide (H2O2, which is not a free radical). In addition, there are RNS such as peroxynitrite (ONOO•−), which can contribute to cell death, although these are not considered further in depth. ROS can also be converted and/or degraded via a number of pathways (Fig. 2); for example, O2 •− is converted by superoxide dismutases (in mitochondria as well as in the cytosol) to H2O2 and reacts with nitric oxide (NO) to form ONOO•−. H2O2 itself can be degraded to water via catalase or glutathione peroxidase or can be partially reduced to form •OH in the presence of Fe2+ (Fenton reaction).

It should be noted that, although ROS are generally considered toxic and remain a major target for therapeutic and dietary interventions in the fight against neurodegenerative diseases (163), ROS are not always harmful: Much evidence has accumulated in recent years demonstrating that ROS act as important signaling molecules. For example, ROS are known to activate the extracellular receptor kinase (ERK) pathway and inhibit protein phosphatases and these actions, plus modulation of the activity of protein kinases A and C and calcium/calmodulin-dependent protein kinase II, are requirements for long-term potentiation (LTP), which is considered the major mechanism underlying learning and memory (63, 78, 97). Furthermore, there are examples at least in the periphery of ROS affording protection, for example, in the autoimmune Guillain Barre syndrome (110) and in rheumatoid arthritis (45). Thus, a one-sided perception of ROS as purely toxic is incorrect. However, imbalances arising from either excessive ROS production or compromised ROS buffering can switch the roles of ROS from physiological to pathological, and are implicated in neurodegenerative diseases. This raises the question of whether specific sources of ROS are of particular importance in the development of pathological conditions. The answer to such a question currently remains only partially answered, although some specific sources have been discounted. For example, cytochrome P450-dependent oxygenases in the endoplasmic reticulum (ER) are a source of ROS (108) but may not generate sufficient ROS to cause damage and so may be limited to physiological ROS signaling (90). By contrast, two major sources of ROS—NADPH oxidase (Nox) and mitochondria—have been implicated in oxidative stress and damage associated with neurodegenerative diseases.
The Nox family of enzymes consists of seven members: Nox1–5, which generate O2 •−, and dual oxidases 1 and 2, which generate H2O2. Each is a multiheteromeric protein complex, and subunits associate on activation to form the active complex at the plasma membrane. Nox1–4 are differentially expressed in the CNS and, of these, Nox2 and Nox4 appear to be of particular importance (56, 67). Thus, Nox2 and, under certain conditions, Nox4 are considered the major sources of ROS after traumatic brain injury and are proposed to play a major role in development of neurodegeneration in AD (150, 186). Indeed, there is evidence that the aggregating proteins associated with specific neurodegenerative diseases, including AD (Fig. 1), can lead to an increase in the expression of Nox isoforms (56), leading, in turn, to an increase in ROS. These mechanisms lead to a positive feedback loop with each of these aggregated proteins causing excessive ROS and neurodegeneration (61, 84, 98, 99).
The damage induced by activated astrocytes has received much attention over the last decade, but there is still controversy of the site of ROS production in these cells. It is clear that astrocytes express Nox isoforms and that these are upregulated when astrocytes become activated (2 –4, 67), and this has led to the suggestion that Nox enzyme inhibitors would provide a useful therapeutic strategy for the treatment of stroke and neurodegerative diseases (23, 152). Furthermore, many neuro-inflammatory conditions, for example, sepsis-associated encephalopathy, lead to long-term cognitive impairment due to glial activation, particularly in the hippocampus: It is likely that this is due to an increase in Nox2 expression and increased ROS production (57).
The most common sources of ROS within the CNS (and elsewhere) are NADH-ubiquinone oxidoreductase (Complex I) and ubiquinol-cytochrome c oxidoreductase (Complex III) of the ETC, located in the inner mitochondrial membrane (42). Electrons leaking from these complexes can react with oxygen to form O2 •−, and this can account for a small but significant (1%–2%) fraction of cellular oxygen consumption. Given this production of ROS, coupled to the fact that mitochondria provide energy in the form of ATP and are also pivotal in apoptosis, it is hardly surprising that they are the focus of much attention in the field of neurodegenerative diseases. Furthermore, key proteins associated with such diseases are known to interact with mitochondria; for example, β amyloid and presenilins of AD, α-synuclein, Parkin, DJ-1, and PINK1 of PD, huntingtin in HD, and SOD-1 in ALS (42). These proteins can directly or indirectly alter many aspects of mitochondrial function, and Figure 3 briefly summarizes the major mitochondrial defects common to these neurodegenerative diseases, in addition to increased ROS production. There is a vast literature describing specific details of mitochondrial defects associated with each of these major diseases, and it is apparent that most, if not all, aspects of mitochondrial function, movement, and turnover can be disrupted; several excellent review articles provide extensive detailed accounts of mitochondrial dysfunction in neurodegenerative diseases (25, 42, 51, 71, 80, 87, 112, 189). Here, the focus on mitochondria is purely as a source of ROS, which is clearly elevated in all of these diseases. Similarly, mitochondrial ROS are also regarded as an important factor in cellular aging, although this view is being re-evaluated in light of the growing awareness of physiological roles of ROS (89, 145).
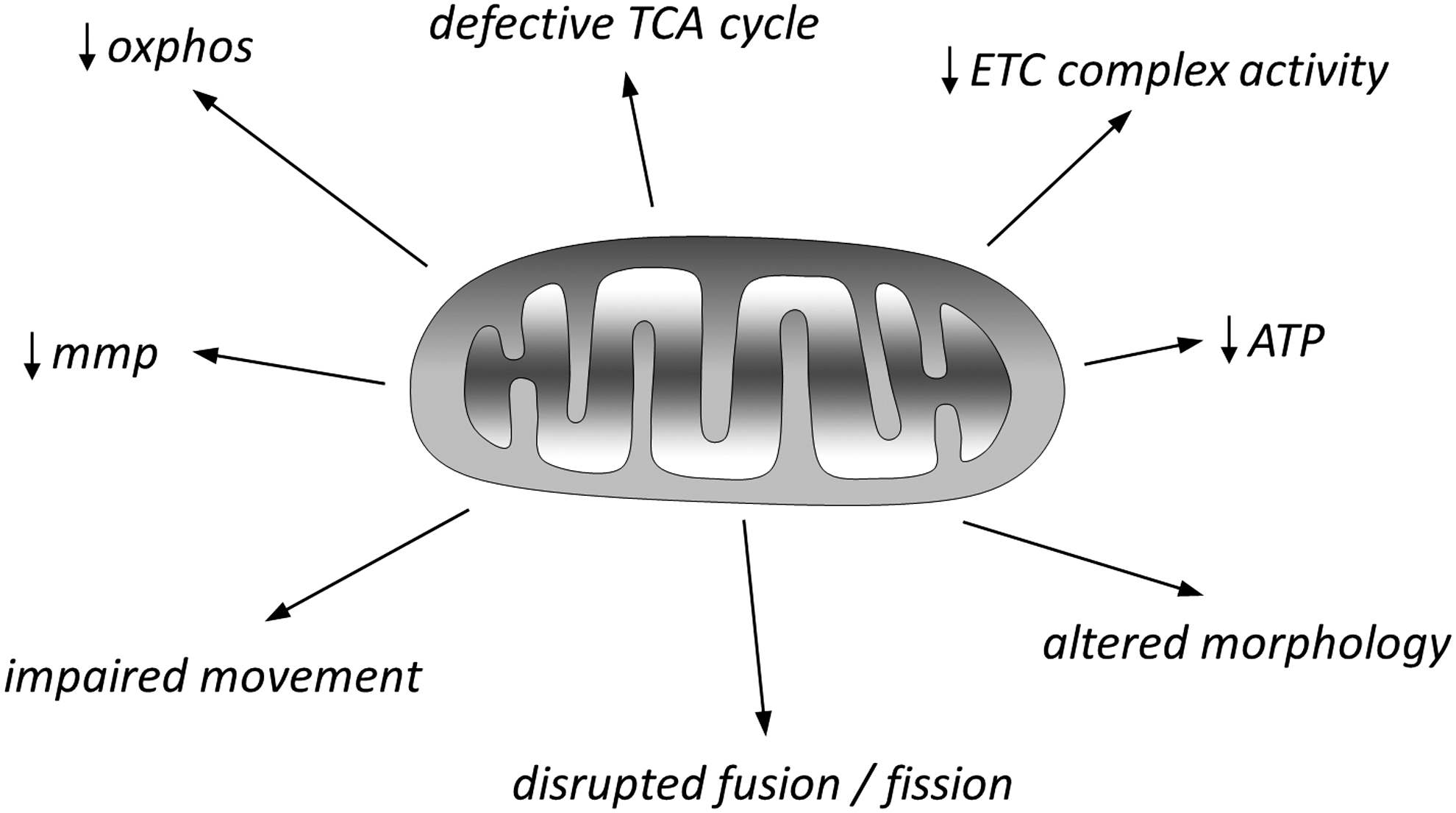
It is important to emphasize that, in considering processes of neurodegeneration, it should not be assumed that neuronal loss arises solely from autonomous ROS production or accumulation of misfolded proteins within neurons themselves, although this is doubtless important. It is clear that non-neuronal cells are important contributors to neurodegeneration observed in AD [e.g., astrocytes (see previous paragraph) and microglia (28)], PD [astrocytes, microglia, and oligodendrocytes (65, 175)], HD [astrocytes and microglia (138, 149)], and ALS [astrocytes, microglia, and cerebrovascular cells (65)].
Next, we consider evidence that specific K+ channels are targets for modulation by ROS, which may account for some of the deleterious effects of ROS in both neurodegenerative diseases and aging. In most cases, evidence that ROS modulation of K+ channels is causative in neurodegenerative disease progression is lacking. Although the wealth of reports of K+ channel modulation in such diseases points to a key role for this phenomenon, the significance and precise role for such effects awaits future investigation (see Summary section).
Redox Modulation of KVS-1 and A-Type Currents
One of the first and most compelling studies to indicate that oxidative modulation of a K+ channel, arising from physiological aging, leads to loss of sensory function and comes from studies of the K+ channel KVS-1 found in Caenorhabditis elegans worms. This channel is expressed in specific sensory neurons and influences chemotactic responses, which are vital for successful feeding behavior. KVS-1 gives rise to currents that are comparable to mammalian A-currents (i.e., currents displaying rapid activation and inactivation kinetics) when expressed in Chinese hamster ovary (CHO) cells, and oxidizing agents applied to CHO cells expressing KVS-1 caused dramatic slowing of current inactivation, which could only be reversed by reducing agents such as dithiothreitol (DTT) (22). Cai and Sesti demonstrated that chemotaxis, which usually declines with age, was much more preserved in aged worms that were engineered to express a C113S mutant form of KVS-1. This mutant channel was insensitive to modulation by oxidizing agents. Furthermore, reducing agents could restore chemotaxis in aged wild-type worms (22, 146). These findings, presented schematically in Figure 4, indicated that gradual oxidation of a specific K+ channel could lead to age-related modification of important organismal function.

As mentioned earlier, in mammalian tissues, A-type currents resemble the C. elegans KVS-1 channel kinetically, displaying rapid activation and inactivation. CNS A-type currents are composed of assorted combinations of channels from the Kv families Kv1–Kv4, and the diversity of this sub-family of K+ channels is increased further by association with a variety of auxiliary subunits (70, 146). Despite this structural variability, early recombinant expression studies revealed very similar modifications by oxidizing agents, that is, dramatic slowing or loss of inactivation, which could be reversed by DTT or reduced glutathione (GSH) (41, 135). Whether or not this modification in mammals is associated with normal aging, or indeed with oxidative modulation in neurodegenerative disease, remains to be determined.
Kv2.1 in Oxidative Stress
Kv2.1 (KCNB1) is a delayed-rectifier type of voltage-gated K+ channel that is expressed in numerous central neurones. It is the mammalian homologue of KVS-1 (31) and is particularly highly expressed in somatodendritic regions of hippocampal and cortical neurones where it strongly influences excitability during periods of high frequency firing (38, 95, 106, 113). Kv2.1 is unusual in that it is exquisitely regulated by phosphorylation/dephosphorylation at a large number of sites located primarily in the cytoplasmic C-terminal domain of the channel. Phosphorylation state is dynamically controlled by a number of factors, including neuronal activity, ischemia, and transmitters such as acetylcholine and glutamate. Regulation of phosphorylation state by these various factors often involves Ca2+/calcineurin-dependent dephosphorylation, and leads to important kinetic modifications of the channel, including dramatic shifts in the voltage dependence of activation (104, 105, 122). Thus, for example, glutamate-induced dephosphorylation led to a hyperpolarizing shift in the voltage dependence of activation, and a suppression of neuronal firing, functionally coupling neuronal activity to excitability (107).
Interestingly, associated with the phosphorylation state of Kv2.1 is its location: Misonou et al. reported that Kv2.1 channels were located in discrete clusters in the soma and somatodendritic regions of hippocampal neurones. However, glutamate exposure led to Ca2+-dependent dephosphorylation of the channels, which was associated with a loss of clustering and a more uniform distribution of the channels within the soma (105). However, the simple implication that increased phosphorylation is associated with clustering and a higher V0.5 for activation—and vice versa—is challenged by the studies of Tamkun and colleagues, who demonstrated that Kv2.1 channels located in clusters were largely nonconducting. Furthermore, experimental “declustering” of the channels did not lead to an anticipated increase in total Kv2.1 current (115). Instead, these authors provided evidence that Kv2.1 activity is directly determined by channel density in the plasma membrane regardless of clustering, so that at a high density (including, but not exclusively, in clusters) Kv2.1 channels become nonconducting (43). Furthermore, the clusters appear to act as microdomains at which new or recycled Kv2.1 channels (along with another channel, Kv1.4) arrive at the plasma membrane (36), indicating an important nonconducting role for this channel protein.
In addition to its roles in regulating activity-dependent excitability and trafficking, Kv2.1 also appears central to the process of oxidative neurotoxicity and to that of neuronal damage and death observed in AD. Thus, Sesti et al. have described in detail the effects of the oxidant H2O2 on Kv2.1 channels (146). They reported that oxidation led to a concentration-dependent increase in the formation of nonconducting Kv2.1 oligomers: Oligomerization arose from disulfide bridge formation between Kv2.1 subunits, involving cysteine residues in the N terminus (C73) and the C terminus (C710). The mutant channel Kv2.1C73A was incapable of forming oligomers. Notably, these authors found that H2O2 decreased whole-cell K+ currents in Kv2.1-expressing cells (consistent with the idea that oligomers are nonconducting), and this was associated with a modest (11 mV) hyperpolarizing shift of the V0.5 for activation. More importantly, channel oxidation correlated with oxidant-induced apoptosis, which was monitored by annexin V binding (which detects external exposure of phosphatidylserine as an early step in apoptosis). The authors found that exposure to another oxidant, dithiodipyridine (DTDP) induced apoptosis and this was exacerbated by overexpression of wild-type Kv2.1. Conversely, overexpression of the C73A (or nonconducting C73S) mutant decreased apoptosis. Thus, prevention of channel oligomerization—or aggregation—provided protection against apoptosis via a mechanism independent of its ability to conduct ions. The authors suggested that protein aggregation per se somehow increased cellular oxidative stress, and this was demonstrated using dichlorofluorescein (DCF) fluorescence.
Cotella et al. (31) also importantly demonstrated that Kv2.1 expression (determined by Western blot of brain samples) declined with age in mice. Using younger animals, they also demonstrated a striking increase in the proportion of Kv2.1 channels in the oxidized, oligomerized form in a mouse model of AD, the triple transgenic (3xTg AD) model, developed earlier by LaFerla and colleagues (116). In vitro exposure of neuroblastoma cells to exogenous amyloid β peptides (considered the primary toxic elements of AD) induced oxidative stress and Kv2.1 oligomerization. Subsequent studies revealed that oxidized, oligomerized Kv2.1 channels accumulated in lipid rafts in the plasma membrane, as they could not be efficiently internalized. Furthermore, this lipid raft accumulation was pro-apoptotic, because it activated c-Src tyrosine kinase, which, in the presence of additional oxidative stress, activated the c-Jun N-terminal kinase (JNK) (170). Thus, as described earlier, while oxidative stress is a feature of normal aging, it is also elevated from an early stage in the development of AD (40, 44, 87). Indeed, even patients with mild cognitive impairment (MCI), which often progresses to AD, show clear signs of oxidative stress [e.g., oxidized proteins and lipids, see Refs. (73, 129)], implicating such stress as an early feature of AD itself. Consistent with this, oxidative stress is a feature preceding amyloid plaque formation in murine models of AD (40, 130). The studies of Sesti and colleagues (shown schematically in Fig. 5) suggest that oxidative aggregation of Kv2.1 may be a key event in the progression of oxidative neuronal loss in both healthy aging and neurodegenerative diseases.

An Alternative Role for Kv2.1 in Oxidant-Induced Apoptosis?
A series of studies by Aizenman and co-workers have also provided a significant body of evidence to support a major role for Kv2.1 in oxidant-induced apoptosis, but there are significant discrepancies with the studies of Sesti and colleagues (31, 170). This alternative model, summarized schematically in Figure 6, incorporates Kv2.1 primarily as a K+ conducting channel rather than as a protein with multiple functions. Intracellular K+ has long been associated with cellular apoptosis (179), as it regulates mitochondrial and cellular osmolarity and volume as well as mitochondrial membrane potential and, crucially, caspase activation (179, 181). It is also well established that loss of intracellular K+ is a key early stage in apoptosis (19, 64). This is because a fall of intracellular K+ concentration triggers the apoptotic cascade, including cell shrinkage (due also to efflux of Cl−), mitochondrial cytochrome c release, and caspase activation (178, 181). Much evidence has indicated that K+ efflux occurs via K+ channels, and K+ channel inhibitors can protect against apoptosis triggered by a variety of insults, including oxidative stress (6, 20, 147).

The evidence that Kv2.1 is particularly important in regulating the K+ efflux that underlies neuronal apoptosis is substantial. Thus, for example, cortical neurons transfected with a dominant negative Kv2.1 constructs (and so lacking functional Kv2.1 channels) are protected against oxidant-induced apoptosis, and expression of Kv2.1 in CHO cells increases their susceptibility to apoptosis (118). Furthermore, pro-apoptotic agents cause a rapid increase in the surface expression of Kv2.1 channels (119). These key, early observations by Aizenman and colleagues have developed significantly over the past few years, and our current awareness of the mechanisms by which oxidative stress leads to increased Kv2.1 insertion into the plasma membrane is summarized in Figure 6. In brief, pro-apoptotic oxidant stresses stimulate separate rises of [Ca2+]i and [Zn2+]i; the former was due to release from the ER (101), whereas the latter arose from liberation from intracellular metal binding proteins (6). Zn2+ activates apoptosis signal-regulating kinase 1 (ASK-1), which, in turn, activates p38 MAPK to phosphorylate Kv2.1 at S800, an obligatory step in this process (10, 101, 133). Indeed, Kv2.1 also requires phosphorylation at the N-terminal Y124 in this pathway, and Zn2+ also acts to prevent dephosphorylation of this tyrosine residue (132). When phosphorylated at Y124 and S800, Kv2.1 can interact with syntaxin to enable insertion into the plasma membrane. This process is dependent on Ca2+/calmodulin-dependent kinase (CaMKII) activity, which is stimulated by the rise of cytoplasmic [Ca2+] after mobilization from the ER.
A central role of Kv2.1 in oxidant-induced apoptosis, thus, appears to have much experimental support. However, there are clear discrepancies in its potential role. Table 1 shows the key observations made under differential experimental conditions that have led to these apparent discrepancies. On the one hand, Sesti and colleagues indicate that oxidizing agents do not increase whole cell K+ currents (in fact, they lead to current inhibition), and instead trigger formation of nonconducting oligomers which themselves generate oxidative stress. Kv2.1 oligomer formation was induced by 12 h exposure to a high concentration (23 μM) of Aβ(1−42) and was observed in aged triple transgenic Alzheimer's mice, suggesting that this process was a part of the oxidative damage associated with neurodegeneration (31). By contrast, Aizenman and co-workers demonstrate that oxidizing agents, via mobilization of divalent metals, cause a profound increase in K+ currents carried by Kv2.1. Clearly, some reconciliation of these differences required before regulation of Kv2.1 activity can be considered a viable therapeutic means by which to decrease oxidative neuronal damage/loss arising as a result of aging or as a part of neurodegenerative disease progression. A major difference (Table 1) between these studies, as discussed most recently by Shah and Aizenman (147), is that oxidant-induced increases of Kv2.1 currents are recorded some 3–4 h after brief oxidant exposure (Table 1); whereas oxidant-induced current inhibition was recorded much more immediately. Thus, although oxidant-induced channel inhibition/oligomerization may be rapid, this does not necessarily exclude oxidant-induced increased membrane insertion, leading to a delayed increase in current amplitude. It is also noteworthy that many of these studies have employed neurones cultured from young animals and experimental conditions to induce apoptosis which are unlikely to reflect the slowly progressive nature of neurodegenerative processes. In addition, consideration should be taken regarding the major influence of K+ channel activity in general on membrane potential, and hence excitability and driving force for Ca2+ entry into neurones: Reduced K+ channel activity can lead to Ca2+ overload via voltage-gated Ca2+ entry, yet augmented K+ channel activity could also promote nonvoltage-gated Ca2+ entry into neurones, for example, via TRP channels (160). Thus, in addition to the proposed roles of Kv2.1 in apoptosis as reviewed here, effects on the driving force for Ca2+ entry and hence elevated cytosolic [Ca2+] should also be considered, as this is a major determinant of the progression of apoptosis (117, 188).
A comparison of key experimental details and observations made in studies of the effects of oxidation on Kv2.1, and its role in apoptosis. Studies reporting increased K+ current always also reported increased incidence of apoptosis in response to the same oxidant treatment.
CHO, Chinese hamster ovary; DTDP, dithiodipyridine.
Redox Modulation of BK Channels
BK channels (also often referred to as BKCa or maxi-K channels) are high conductance K+ channels regulated by both voltage and by [Ca2+]i. Splice variants of the pore-forming alpha subunits, along with association (or not) with a variety of auxiliary beta subunits ensures that the mammalian brain is endowed with BK channels showing a diverse range of activities (172, 183). Since BK channel activity is sensitive to [Ca2+]i, it is clear that these channels can be indirectly affected by oxidative stress, aging, and neurodegenerative diseases, as disruption of Ca2+ homeostasis is a central feature of each of these scenarios (14, 24, 47, 100). However, it is also apparent that they can undergo more direct redox modulation. Thus, generally speaking, oxidation of key methionine residues enhances BK activity (137); however, oxidation of cysteine residues causes channel inhibition by altering Ca2+ sensitivity (156). BK channels have approximately 30 of each residue type; however, oxidation of specific, identified residues can have profound effects on channel function (136). These studies were conducted using recombinant BK channels and specific, potent chemical oxidants, including chloramine-T, to target preferentially methionine residues. Native BK channels recorded in hippocampal neurones are preferentially activated by oxidizing agents (48), suggesting that cysteine modification may be functionally dominant in the CNS. Since BK channels control the fast after-hyperpolarization observed immediately after individual action potentials in, for example, CA1 hippocampal neurones, augmentation of their activity by oxidation suppresses electrical activity. This has been suggested to contribute to age-related loss of cognitive function (146); however, further studies are required to confirm this (e.g., assessment of oxidation on specific cell types within relevant neuronal circuits). However, sustained augmentation of BK channel activity in CA1 hippocampal neurones has been shown to occur after transient forebrain ischemia (49) and was attributable to direct oxidative modulation, so this mechanism of channel regulation is clearly of pathophysiological interest.
Redox modulation of BK channels is also likely to be important in AD: Ye et al. (176) reported depression of extracellularly recorded field excitatory postsynaptic potentials (fEPSPs) of the CA1 region of the hippocampus in a mouse model of AD, as compared with age-matched WT mice. Pharmacological interventions indicated that fEPSPs were mediated, at least in part, by presynaptic BK channel activity. The authors suggested a number of possible mechanisms to account for the disruption of this important physiological process, including redox modulation of BK channels, or disruption of Ca2+ homeostasis, both of which are associated with AD progression, as discussed earlier.
While our focus has been on the roles of Kv2.1 and BK channels in mediating apoptotic responses to oxidative stress and, where studied, in neurodegeneration, other K+ channels have also been proposed to be serving similar roles. Table 2 highlights some of these diverse K+ channel types that have been implicated in apoptosis induced by various experimental challenges, including ischemia/reperfusion injury where ROS are strongly implicated (27). In each of these studies, pharmacological or genetic inhibition of the specific K+ channel type reported to be augmented by the apoptotic challenge leads to inhibition of apoptosis. The implication from such studies is, as discussed earlier, that K+ channel modulation (which may in some circumstances arise from oxidative regulation) is a necessary step for apoptosis and, hence, neurodegeneration. However, direct causative evidence is lacking and the interpretation of many of these studies is complicated by experimental limitations; for example, excessively high concentrations of toxic agents, the use of strong oxidizing agents, and the maturity of the neurones.
Example studies investigating the role of neuronal K+ channels in apoptosis induced by diverse challenges. In each study, blockade of the identified K+ channel prevented or ameliorated the apoptotic response. Currents described as delayed rectifiers could include Kv2.1, but no molecular identification was made.
d.o., day-old.
Gasotransmitters: Cytoprotective Regulators of Ion Channels?
In recent years, the biological activity of endogenous “gasotransmitters,” particularly carbon monoxide (CO) and hydrogen sulfide (H2S), has emerged as a topic of great scientific interest with exciting translational possibilities for the treatment of a number of pathologies (75, 111, 154). No longer regarded as mere toxins, these agents are now known to be synthesized tonically in most, if not all, cell types via the specific activities of a limited number of enzymes: CO is generated primarily by the degradation of heme, catalyzed by heme oxygenases, while H2S is generated from a number of enzymatic sources associated with sulfur metabolism (85, 126). Both gasotransmitters have also, separately, been recognized as novel modulators of ion channels, acting via a number of different mechanisms to alter the activity of a range of different ion channel types, with important consequences for cell biology and pathology (124, 125, 168). In this section, we consider evidence that some of the cytoprotective effects of CO and H2S against oxidative stress in the CNS might arise due to their ability to modulate ion channels.
Carbon monoxide
A wealth of compelling evidence indicates that upregulation of heme oxygenase-1 (HO-1; the inducible enzyme capable of generating CO from heme degradation) in the CNS affords protection against oxidative and other stresses (5). HO-1 upregulation occurs in both neurones and glia after such insults (94). Its upregulation is also associated with progression of neurodegenerative diseases such as AD, but its role in this regard is debated: Despite the known neuroprotective properties of HO-1, degradation of heme liberates iron (Fig. 7) and this may compound the neurotoxic effects of iron accumulation (140). Furthermore, studies by Schipper et al. have indicated that astroglial HO-1 induction is detrimental, promoting oxidative stress and mitochondrial dysfunction (143). Contrasting with these findings are a number of studies suggesting that upregulation of HO-1 in astrocytes provides protection, via CO formation, for neighboring neurones (66) and can also protect astrocytes themselves (7). Furthermore, constitutive HO-2 activity is significant in many CNS regions (94), and appears to provide tonic protection against neurotoxicity; simple serum deprivation, or reduction in bathing K+ levels (maneuvers known to trigger death in cultured cerebellar granule neurons) led to far greater levels of apoptosis in cerebellar granule cultures taken from HO-2−/− mice, and in the penumbra surrounding infarcted brain tissue after transient focal ischemia in vivo (37). Glutamate toxicity can, in addition to upregulating HO-1, trigger a rapid, Ca2+-dependent increase in the activity of HO-2 (18).
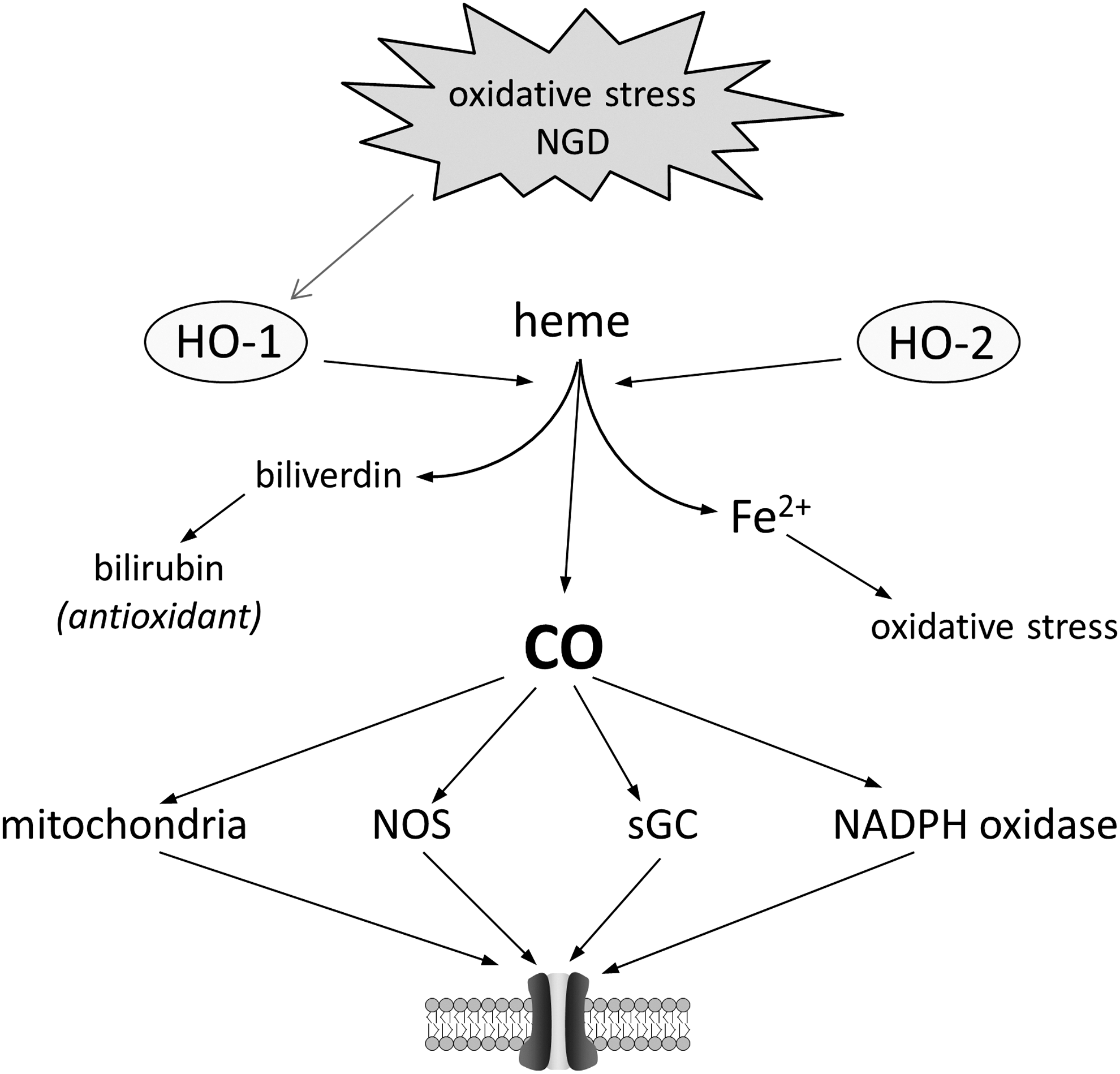
Much data indicate that CO may account for many of the beneficial effects of HO-1 and HO-2 (and may even account for beneficial vs. potential detrimental effects of HO-1 induction, as mentioned earlier). Thus, low (nontoxic) levels of CO have been shown to provide neuroprotection against oxidative stress both in vitro (161) and in vivo against the deleterious effects of focal ischemia (184). The possible mechanisms by which CO might afford this protection remain to be elucidated: CO has been proposed to react only with transitional metals (17), and cannot, therefore, be considered to have direct redox modulatory properties. Instead, via a transition metal interaction, it is capable of interacting with a number of signaling pathways: Those best established are shown in Figure 7, which illustrates that through direct modulation of redox intermediates (e.g., mitochondria, NADPH oxidase) ion channels may be end-point effectors mediating at least some of its protective, antioxidant effects. Interestingly, many of these pathways are known means by which ion channels can be regulated. It is clearly conceivable, therefore, that CO regulation of specific ion channels via these or alternative pathways may contribute to the protective effects of heme oxygenases against oxidative stress. In the case of the delayed rectifier channel, Kv2.1, the evidence is substantial.
Employing a recombinant expression system, Dallas et al. (34) demonstrated that overexpression of Kv2.1 increased susceptibility of HEK293 cells to oxidant-induced apoptosis. Oxidative stress was associated with an increase in the whole-cell K+ current amplitude (arising from increased insertion of recombinant Kv2.1 into the plasma membrane), in accordance with the studies of Aizenman and colleagues, as detailed earlier. In the presence of CO, however, K+ currents were suppressed and resistance to oxidative apoptosis was increased. Importantly, a similar response was observed in cultured hippocampal neurones, and the effects of CO were found to be selective for Kv2.1 among the numerous different K+ channel types expressed in these neurones (34). The mechanism by which CO inhibited Kv2.1 was not fully elucidated, but was partly mediated by ROS, and also required that the channel was tonically phosphorylated by protein kinase G. This nothwithstanding, the study provided direct evidence that some of the deleterious effects of oxidative stress which directly involved a specific K+ channel could be offset by the ability of CO to inhibit the channel and suppress apoptosis.
CO is also known to regulate BK channels in a number of different tissue types (60, 166, 169), although the consequences of such modulation have not been explored in depth in the CNS. It is interesting to note, however, that oxidation of native BK channels in hippocampal neurones leads to sustained channel activation (48). Since CO also increases BK channel activity (59), this would suggest additivity of CO with channel oxidation. However, there are also numerous reports of BK channels being inhibited via oxidation in response to a variety of oxidizing agents [indeed, there are a diverse range of effects reported in response to different oxidizing agents, as reviewed in Hou et al. (59)]. Thus, at present, it is unclear whether the ability of CO to activate BK channel activity would provide protection against oxidative stress in the CNS. Similarly, activity of the two pore domain K+ channel TREK-1 is known to be increased in the presence of low levels of CO (33). This channel is highly influential in the CNS, governing neuronal excitability and responding to a diverse array of regulatory factors, including pH, general anesthetics, and lipids (58). TREK-1 is believed to be activated in ischemia, providing neuroprotection via suppression of excitability (58), and CO may well also provide protection via activation of this channel, although no such studies have been provided to support such a suggestion to date.
Hydrogen sulfide
Cystathionine γ lyase (CSE) and cystathionine β synthetase (CBS) are the two major enzymes responsible for generating H2S from cysteine. More recently, mitochondrial 3-mercaptopyruvate sulfurtransferase (3MST) has also been shown to generate H2S in both the brain and the vasculature. 3MST generates H2S from 3-mercaptopyruvate, which is itself generated from cysteine aminotransferase (CAT) (74). H2S can also be mobilized from sulfur bound to cytosolic or mitochondrial proteins in a redox- or pH-sensitive manner (74). CBS is the dominant source of H2S in the CNS, although 3MST is also abundant (83); CSE is believed to generate the majority of H2S in the periphery, including the vasculature.
Numerous studies have provided evidence that H2S protects neurones against oxidative stress. Pioneered by Kimura and Kimura, such studies have indicated that H2S protects primarily via stimulating increased glutathione (GSH) levels (77). However, other mechanisms have been proposed, pertinent among which is the suggestion that protection of immortalized hippocampal HT22 cells by H2S involves activation of ATP-sensitive K+ channels (K-ATP channels) (76). K-ATP channels, which can be activated by oxidation in neurons (13), have long been known to be activated by H2S in the vasculature, an effect that accounts for the physiological effects of H2S on blood pressure (173) and arises as a direct modification of the channel protein by H2S known as sulfhydration [in which a thiol group, -SH is converted to –S-SH (114)]. Whether or not this specific mechanism of protein modification occurs in K-ATP channels of central neurones remains to be determined, but offers an exciting possibility by which this gasotransmitter may provide protection against oxidative stress (Fig. 8). In addition, vasodilation caused by activation of vascular K-ATP channels is likely to oppose progressive vascular damage that can contribute to some forms of neurodegenerative diseases.

As is the case for CO, H2S is also known to regulate BK channels. Telezhkin et al. (158) reported that H2S inhibits both recombinant BK channels and those expressed by type I cells of the carotid body, an effect which may account for the ability of this gas to excite this important chemoreceptor (86). Studies of the effects of H2S on BK channels in the CNS are lacking, and it is important to note that the same class of channel expressed in GH3 pituitary cells is reportedly activated by H2S via redox modulation of intracellular cysteine residues (151). It is, therefore, impossible at present to predict whether H2S can provide BK-mediated protection against oxidative stress within the CNS, but it is certainly a potential mechanism worthy of further exploration.
Two independent reports have shown that H2S can increase extracellular receptor kinase 1/2 (ERK1/2) activity in vascular smooth muscle cells (69, 174). It has yet to be demonstrated that H2S can act in a similar manner within the CNS, but it is noteworthy that the delayed rectifier, Kv2.1, can be tonically regulated by this kinase [which, interestingly, regulates its modification by CO (34)], thereby providing a mechanism by which H2S may regulate this channel in central neurones. However, as for the potential roles of H2S regulation of KATP and BK channels, any such effects on Kv2.1 remain to be determined.
Gasotransmitters in Neurodegenerative Diseases
Given (a) the potential for CO and H2S to provide protection against oxidative stress via ion channel modulation, and (b) the major role of oxidative stress in neurodegenerative diseases, as discussed earlier, it seems pertinent to consider the possible protective effects of these gases in neurodegenerative diseases. The ambiguous role of HO-1 in AD has been described earlier, but it is noteworthy that, experimentally, amyloid peptides (Aβ) of AD can induce HO-1 expression (121), consistent with the known HO-1 upregulation in AD patients (139, 141, 142). However, whether induction of HO-1 is a neuroprotective response, or contributes to iron toxicity, remains a subject to debate. Similarly, in other neurodegenerative diseases such as Parkinson's and multiple sclerosis, HO-1 induction is prominent (144). Interestingly, it has been shown that amyloid precursor protein (APP; from which amyloid peptides of AD are derived) binds to and inhibits HO-1, exacerbating amyloid-mediated oxidative damage in the brain. The inhibition of HO-1 was even greater for APP containing familial AD mutations (155). There is, therefore, much evidence in the literature that heme oxygenases and their products are intimately involved in the development and progression of AD.
Numerous reports implicate H2S as an important factor in AD. Thus, for example, levels of the CBS activator, S-adenosylmethionine are reduced in AD patients (109), and plasma H2S levels are reported to be reduced in AD patients (50), while homocysteine levels are elevated (30). H2S has been shown to decrease BACE-1 expression (one of the enzymes required for Aβ formation) and secretion of Aβ(1 –42) in vitro (185). Furthermore, H2S has anti-inflammatory and anti-apoptotic activity in the CNS [reviewed in Gong et al. (50)] that can improve cognitive function in MCI (102), promote induction of LTP (1), and protect neurones against oxidative stress (75, 77); thus, it has great potential to protect against AD via multiple means, and new evidence is emerging that this is the case, both biochemically and in behavioral murine tests (46, 171). Currently, the role for ion channel involvement in the protective effects of gasotransmitters against neurodegenerative diseases remains largely unexplored; however, it likely represents a field of great potential, given their central roles in responses to oxidative stress, and the known involvement of such stress in neurodegenerative disease. Future research will likely reveal channels as candidate targets for therapeutic development.
Summary
The evidence that ROS are of fundamental importance in the development and progression of neurodegenerative diseases is overwhelming. Focusing on two key K+ channels, Kv2.1 and BK channels, we have highlighted how their regulation can exert powerful influences on excitability and vulnerability to apoptosis. While some issues within these studies remain to be resolved, the underlying message is that such regulation of these channels is important in both aging and progression of neurodegenerative diseases. Clearly, developing a more comprehensive picture of the oxidative modulation of other K+ channels (and indeed other ion channels) within the CNS will allow us to better understand processes associated with healthy aging as well as distinct processes underlying progression of neurodegenerative diseases. Improved understanding is clearly required, as the role(s) of specific K channels in neurodegeneration appear to vary significantly between reports. Furthermore, the development of the emerging field of ion channel regulation by gasotransmitters, and how this interacts with redox modulation of K+ and other channels, will in the future enable us to determine the therapeutic value of these gases in future approaches for the treatment of such diseases. Presently, however, seemingly contradictory findings raise important questions. For example, how does neuroprotection arise from gasotransmitter activation of some K+ channels (e.g., BK, K-ATP channels) yet is a consequence of inhibition of other channels (Kv2.1)? Answers to such questions require further research, and may lie in the diversity of channel activity: The nonconducting properties of K+ (and other) channels (29) and the impact of such functions on cell activity and viability remains a largely unexplored field.
Future Directions
Research into the role of K+ channel modulation in neurodegeneration and aging is still at a relatively early stage, and a large number of important questions remain to be answered. The answers to these questions will depend on the development of new models and perhaps a re-assessment of the cellular targets of stress-inducing agents. The assumption that the K + channel target is on the plasma membrane is now clearly incorrect with recent work demonstrating an important role for mitochondrial K+ channels. Examples include modulation of mitoBKCa by hemin preventing the cytoprotective effects of K+ channel opening (12), BKCa channel openers reducing mitochondrial ROS production and increasing neuronal survival (79), and the KATP channel opener diazoxide protecting neurones from a variety of neurotoxic insults (162). Whether other subcellular organelles that have K+ channels in their membranes are similarly involved needs to be addressed. There is also a requirement to expand the models used beyond cell lines and primary cultures of neurones. This need is beginning to be met with more work being done on ex vivo preparations, such as organotypic brain slices and whole animal models of neurodegenerative diseases. However, there clearly needs to more uniformity in the experimental conditions used with concentrations of drugs, drug dosage, and animal models becoming more standardized in order to be able to compare results between laboratories. It is also clear that the isolation of neurones from astrocytic and microglial influence changes the response to a given set of stressors and so data from isolated neurones, should, where possible, always be compared with the response measured in more intact preparations, and ideally in animal models. One other important factor, that is sometimes not considered, is whether the modulation of K+ channel activity observed is the cause of neuronal damage or a downstream consequence of neuronal injury. Careful experimental design should allow this to be addressed. Addressing these questions and potential pitfalls should allow the field on K+ channel involvement in neurodegeneration and aging to progress more rapidly and hopefully lead to novel therapies for the treatment of neurodegenerative conditions.
Footnotes
Acknowledgments
The authors' own contributions to this field are supported by the Alzheimer's Society, Alzheimer's Research UK, The British Heart Foundation, and the Medical Research Council.
Author Disclosure Statement
No competing financial interests exist.