
Abstract
Introduction
P
These results identify isolevuglandin-modified phosphatidylethanolamine (IsoLG-PE) as a potent macrophage activator that acts as a novel ligand for receptor for advanced glycation endproducts and that is significantly elevated during conditions associated with inflammation. Thus, these studies suggest that interventions that lower IsoLG-PE levels or its activity may have a therapeutic benefit in atherosclerosis and other inflammatory diseases associated with lipid peroxidation.
Initial studies focused on the potential effects of protein modification by IsoLGs, as the rapid reaction of synthetic IsoLGs (most typically 15-E2-IsoLG, as this represents one of the most abundantly formed regioisomers of IsoLGs) with purified proteins resulted in substantial protein adducts and loss of protein function (7, 8, 13, 21, 48, 58). Elevated levels of IsoLG protein adducts are found in a number of disease states, including cardiovascular disease (57), chronic kidney disease (57), Alzheimer's disease (14), hyperoxia (16), and allergic inflammation (65). In general, specific proteins that are modified during these conditions have not been identified, so the specific contributions of protein modification by IsoLGs to the pathogenesis of these diseases are largely unknown. Conceptually, modifications of proteins by IsoLGs that result in loss of function seem less likely to play a key role in disease than modifications of proteins or PE by IsoLGs that result in potent gain of function, since only a small percentage of the total number of copies of each protein or PE are modified.
Of particular relevance in this regard is our recent finding that PE modification by 15-E2-IsoLG converts PE to a biologically active molecule that induces cytokine expression and adhesion molecule surface expression in endothelial cells (28). In this case, relatively low levels (1–3 μM) of PE modified by 15-E2-IsoLG (IsoLG-PE) are needed for maximal response (28). Similarly, modification of PE by peroxidation of docosahexanoic acid (DHA) to form carboxyethyl pyrrole converts PE to a potent (low nM) activator of angiogenesis (66). Whether aldehyde-modified PEs exert direct effects on other cells that are important to inflammation, such as macrophages, is currently unknown.
Another reason that PE modification may be particularly relevant to the biological effects of IsoLGs is our finding that when exogenous 15-E2-IsoLG is added to cultured cells, more PE is modified than protein lysyl residues (64). Similarly, more PE than protein is modified when IsoLGs are generated within lipoproteins ex vivo by their exposure to myeloperoxidase (27). This suggests that sufficient amounts of IsoLG-PE could be formed in vivo during inflammatory conditions associated with lipid peroxidation to be biologically relevant. However, to date, there is only one published study that has examined the levels of IsoLG-PE formed in vivo (42). This study showed that IsoLG-PE were elevated in the liver of mice during chronic ethanol consumption (a model of alcoholic liver disease) and in the plasma of human patients with macular degeneration. Thus additional studies are clearly needed to examine the levels of IsoLG-PE generated in other highly relevant inflammatory conditions associated with lipid peroxidation.
One such condition is familial hypercholesterolemia (FH), an autosomal dominant disorder characterized by severely elevated levels of low-density lipoprotein cholesterol (LDL-C) (55). FH patients have markedly increased inflammatory macrophage activation in the sub-intimal space of large arteries, resulting in increased risk of premature atherosclerotic cardiovascular disease (55). Because patients with FH have significantly higher levels of circulating myeloperoxidase (54) and oxidized lipids (15), it seems likely that they also have elevated levels of IsoLG-PE that might contribute to their inflammation.
Similarly, obesity induces significant systemic elevation in lipid peroxidation and is associated with a host of inflammatory conditions, including atherosclerosis, nonalcoholic steatohepatosis, and arthritis (6, 30, 38). Lipid accumulation in the liver and resulting inflammation appears to be a major driver of both hepatic and systemic insulin resistance (45). We and others have shown that high-fat diet feeding increases macrophage infiltration and expression of inflammatory genes in the liver (12). It therefore seems likely that high-fat feeding might elevate liver IsoLG-PE levels, which could then induce inflammatory responses in the liver and contribute to the development of steatohepatosis.
In addition to understanding the conditions that lead to IsoLG-PE formation in vivo, it would be highly valuable to understand the molecular mechanisms whereby IsoLG-PE and other aldehyde-modified PE exert their effects. For instance, does IsoLG-PE act simply by perturbing membrane structure or does it act on specific receptors? Identification of specific receptor(s) that mediated cellular responses to IsoLG-PE could greatly facilitate identification of relevant biological activities and signaling pathways.
To address these critical questions, we examined whether levels of IsoLG-PE increased under conditions associated with inflammatory disease, whether IsoLG-PE could activate the inflammatory response of macrophages, and whether specific receptors mediate this activity.
Results
IsoLG-PE levels are elevated in conditions associated with inflammation
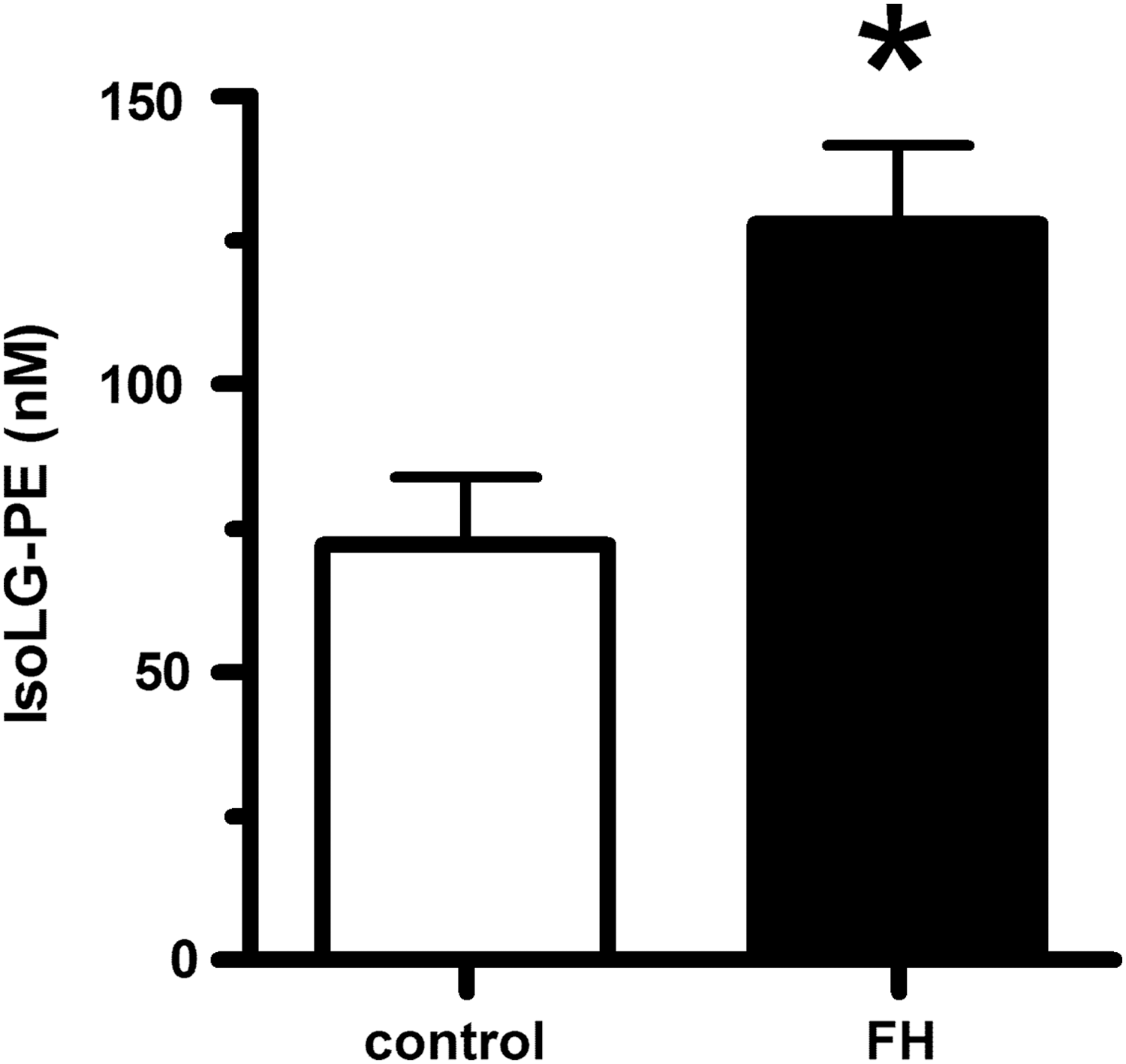
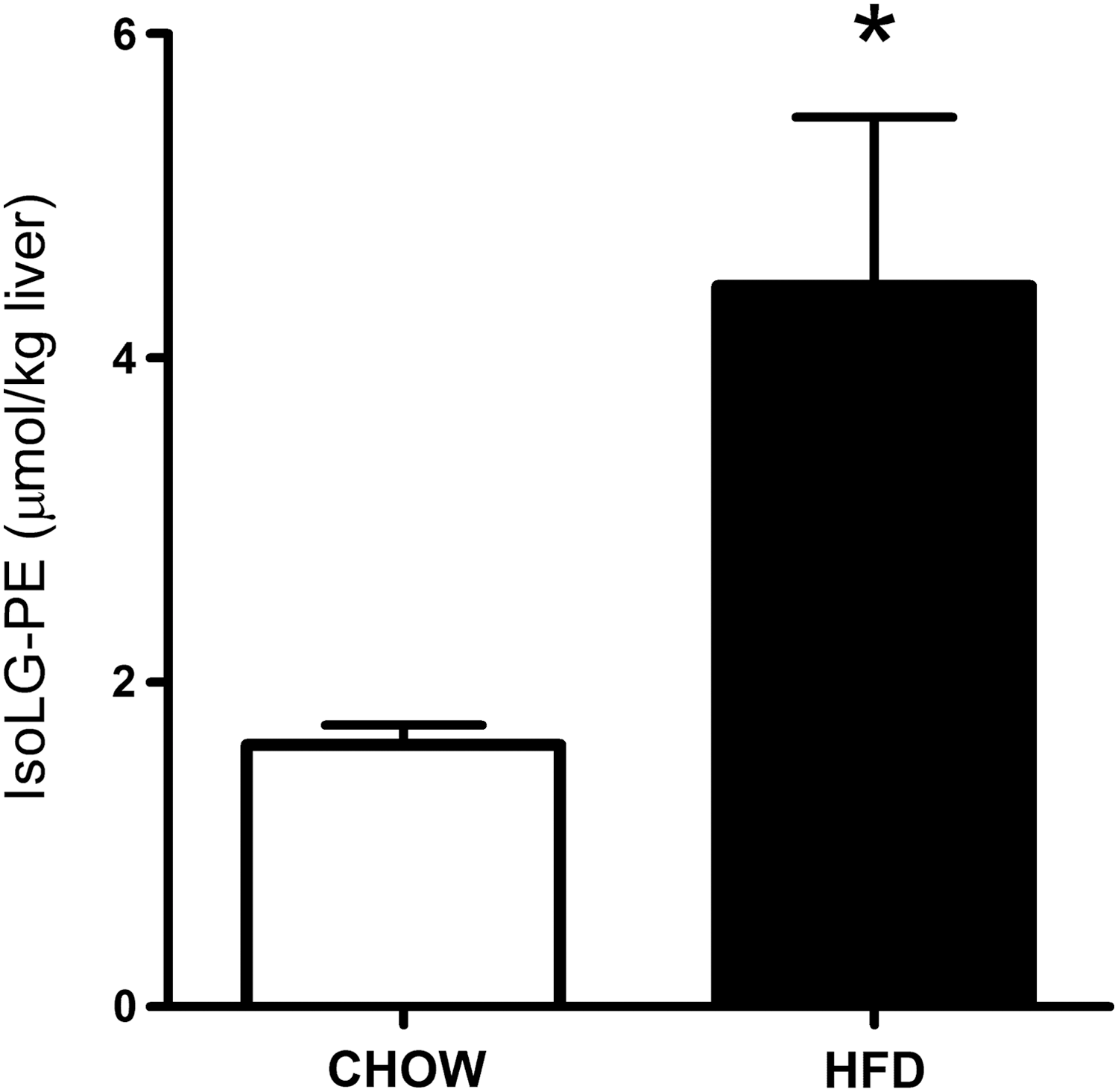
For PE modification by IsoLGs to play an important role in the inflammation and macrophage activation associated with lipid peroxidation, the levels of IsoLG-PE would have to be elevated during these conditions. We therefore measured IsoLG-PE levels in two such conditions: hypercholesterolemia and hepatosteatosis associated with obesity. To examine whether IsoLG-PE levels were elevated by high cholesterol levels, we isolated plasma from FH patients before their undergoing LDL apheresis and in healthy volunteers. We found that IsoLG-PE levels were increased 76% in FH patients compared with control subjects (Fig. 1). To examine whether IsoLG-PE were elevated during the hepatosteatosis associated with obesity, we measured IsoLG-PE in the liver of mice fed a high-fat diet (60% of total calories) for 9 weeks (12). The mice fed the high-fat diet had significantly higher liver weight (1.65±0.30 g) than mice fed the low-fat chow diet (1.22±0.03 g; p<0.05, two-tailed Student's t-test). Levels of IsoLG-PE per gram liver weight were 2.75-fold higher in mice fed the high-fat diet compared with those receiving the low-fat chow diet (Fig. 2). These results demonstrate that IsoLG-PE levels are increased during conditions associated with inflammation and macrophage activation.
IsoLG-PE induces NFκB activation and inflammatory cytokine expression in macrophages
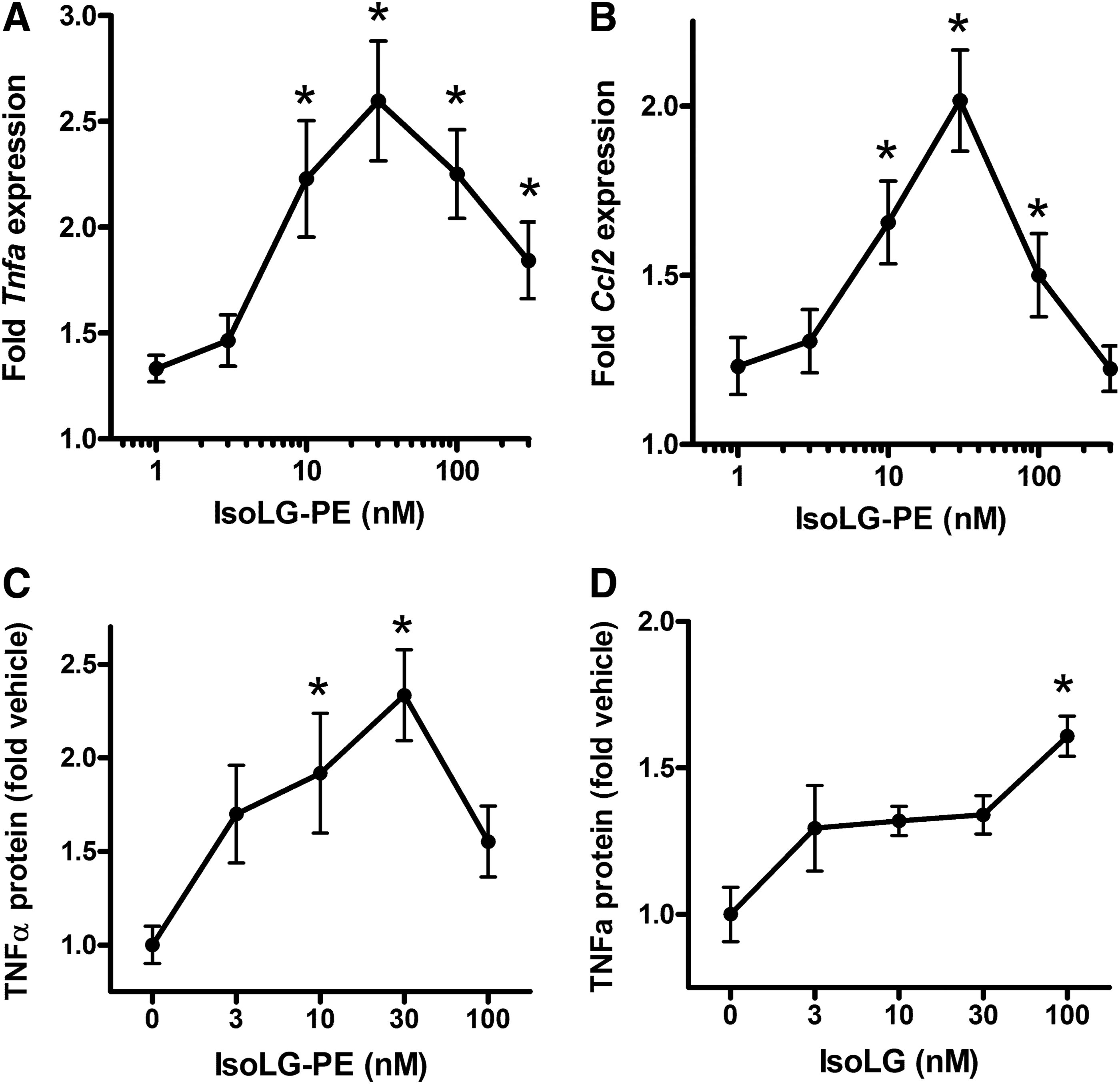
We have previously demonstrated that low micromolar concentrations of IsoLG-PE activated pro-inflammatory responses of endothelial cells, including increased expression of inflammatory cytokines and adhesion molecules (28). To examine whether IsoLG-PE also induced inflammatory gene expression in macrophages, we synthesized IsoLG-PE by reacting dipalmitoyl PE with 15-E2-IsoLG and treated mouse macrophage RAW264.7 cells with this IsoLG-PE. When we measured mRNA expression of the inflammatory cytokines Tnfa and Ccl2 (also known as MCP-1) by quantitative real-time PCR (qPCR), we found that 30 nM IsoLG-PE was sufficient to elicit maximal cytokine mRNA expression (Fig. 3A, B). To determine whether increased mRNA expression also increased protein expression, we then measured tumor necrosis factor-alpha (TNFα) protein levels in supernatant from RAW264.7 cells stimulated with IsoLG-PE, and again found that 30 nM IsoLG-PE was sufficient to elicit maximal TNFα protein expression (Fig. 3C). Addition of 15-E2-IsoLG itself was not as potent at eliciting TNFα protein expression as adding IsoLG-PE (Fig. 3D), consistent with the notion that modification of PE rather than modification of other targets by IsoLG drives this inflammatory response.
Expression of Tnfa and Ccl2 is typically downstream of nuclear factor kappa B (NFκB) activation, so we examined whether IsoLG-PE could elicit NFκB activation in RAW264.7 cells transfected with an NFκB luciferase reporter. We found that IsoLG-PE increased NFκB activity in a concentration-dependent manner with 30 nM IsoLG-PE being sufficient to elicit near maximal activation (Fig. 4A). A key step in the activation of NFκB is the phosphorylation of IκA and IκB by Iκ kinase β (IκKβ). Inhibition of IκKβ with 30 μM PS-1145 inhibited TNFα expression induced by IsoLG-PE (Fig. 4B). These results are consistent with IsoLG-PE inducing inflammatory cytokine expression via NFκB signaling.

IsoLG-PE does not require hydrolysis to IsoLG-ethanolamine for activity
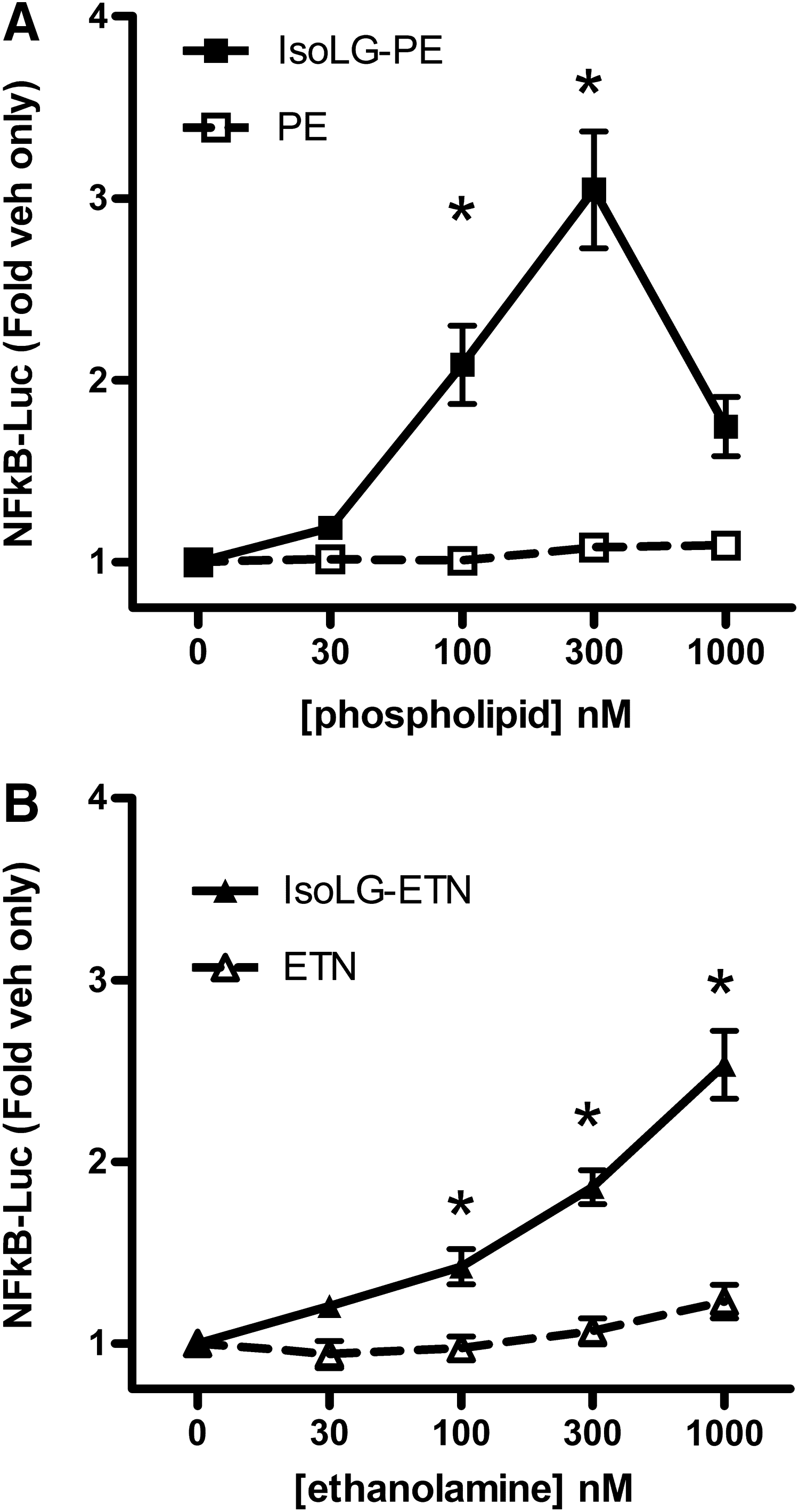
The ability of nanomolar concentrations of IsoLG-PE to activate macrophages suggested the possibility that IsoLG-PE acts as a ligand for receptor(s) on macrophages rather than by physical perturbation of membranes. Because we had previously found that IsoLG-PE is hydrolyzed to its ethanolamine analog (IsoLG-ETN) by the action of NAPE-PLD (29) and because NAPE-PLD is expressed in macrophages (75), we considered the possibility that hydrolysis of IsoLG-PE to IsoLG-ETN might be required to activate macrophages. If so, then IsoLG-ETN, rather than IsoLG-PE, would be the appropriate ligand to be used for receptor screening. We therefore compared the ability of IsoLG-PE and IsoLG-ETN to activate NFκB reporter expression. For these and subsequent experiments, we used passaged macrophages derived from bone marrow (BMDM) of mice where a luciferase reporter gene had been placed under the control of the NFκB-dependent promoter (nfkb-BMDM) (4). While both IsoLG-PE (Fig. 5A) and IsoLG-ETN (Fig. 5B) induced NFκB activation in a concentration-dependent manner, IsoLG-PE was more potent than IsoLG-ETN. We therefore used IsoLG-PE as a ligand for screening of the target receptor. Of note, IsoLG-PE stimulated NFκB reporter about 10-fold less potently in nfkb-BMDM than in the RAW264.7 cell line. The reasons for this difference are unknown but may be the result of decreased expression of the cognate receptor or increased expression of NAPE-PLD or other lipases.
Identification of receptor for advanced glycation endproducts as an IsoLG-PE receptor
Proinflammatory lipids induce NFκB signaling in macrophages both by G-protein coupled receptors (GPCRs) such as the prostaglandin receptors and via pattern recognition receptors such as receptor for advanced glycation endproducts (RAGE), CD36, and Toll-like receptors (TLR). To determine whether IsoLG-PE was a ligand for GPCRs, we used 1 μM IsoLG-PE to screen a commercial panel (DiscoveRx, Fremont, CA) of cells expressing 154 individual GPCR with known ligands. One GPCR, the thromboxane A2 receptor (TxA2R), came up as a weak hit (25% activity of known ligand) in this screen (Supplementary Table S1; Supplementary Data are available online at
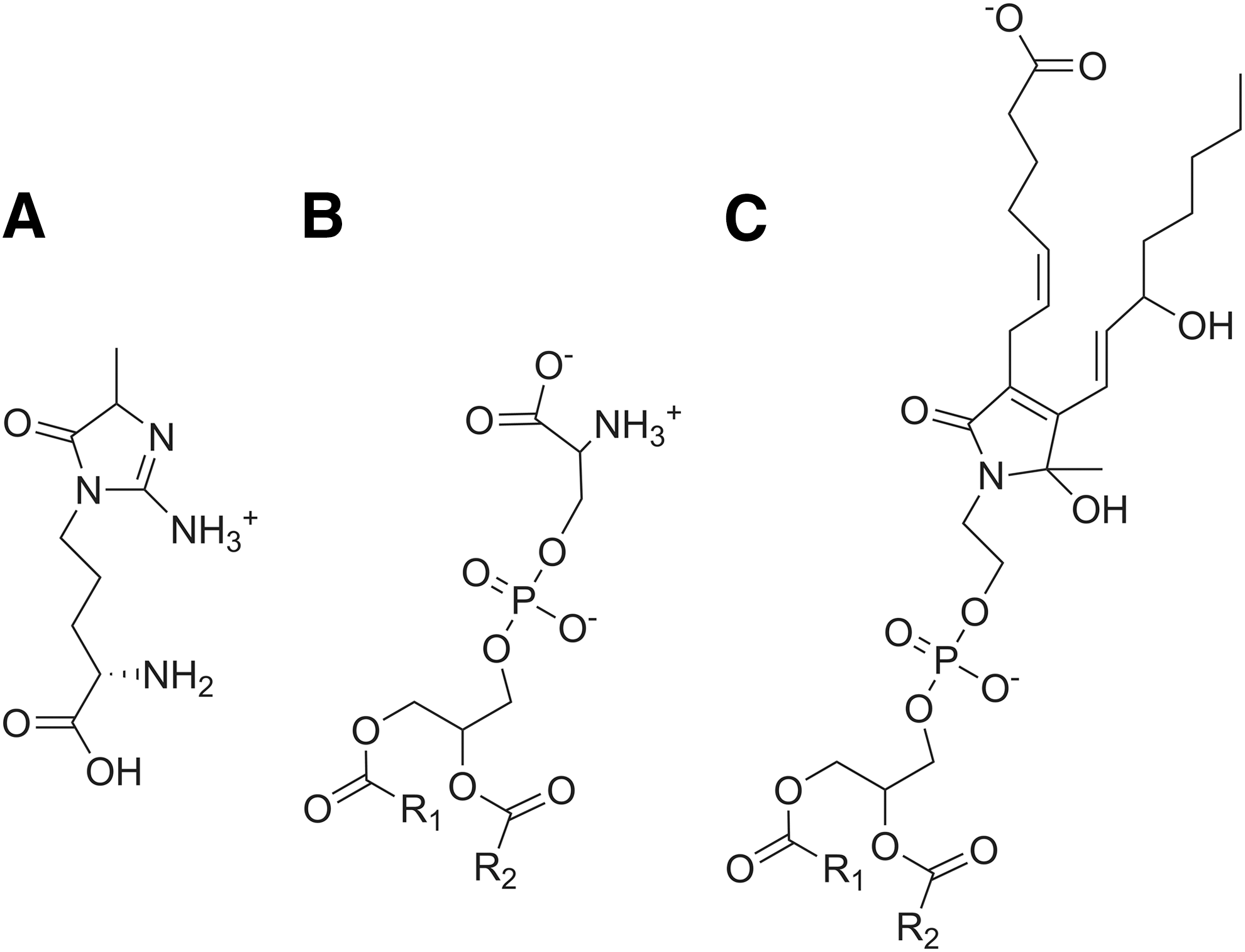
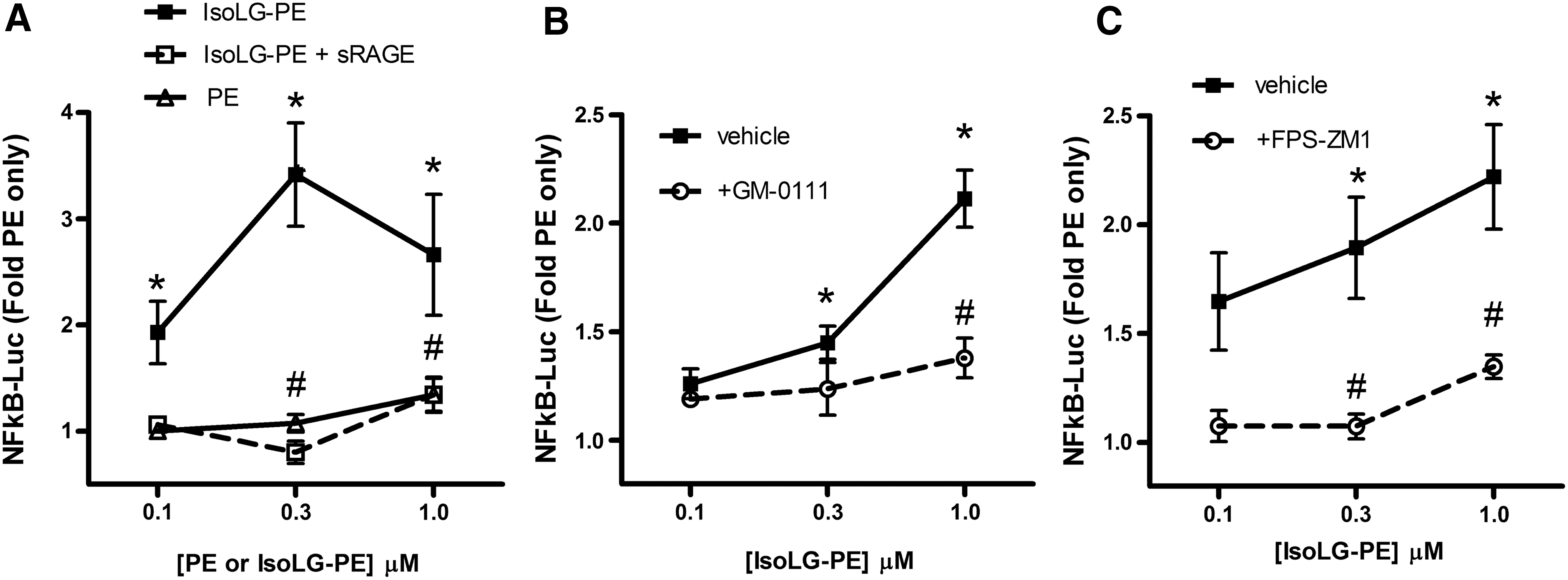
Our failure to identify a GPCR for which IsoLG-PE served as a robust ligand led us to consider whether IsoLG-PE might primarily act instead via pattern recognition receptors. In particular, we chose to focus on the RAGE, as this pattern recognition receptor initiates NFκB signaling in response to ligands such as aldehyde-modified peptides and anionic phospholipids that share some structural features with IsoLG-PE (Fig. 6). The soluble form of RAGE (sRAGE) acts as a decoy peptide to inhibit RAGE signaling, because sRAGE binds the same ligands as full-length RAGE but lacks its intracellular signaling domains (19, 23). We found that preincubation of IsoLG-PE with 2 mg/L of sRAGE markedly inhibited the ability of IsoLG-PE to induce NFκB activation (Fig. 7A). To test whether RAGE was required for IsoLG-PE-induced signaling, we examined the effect of two RAGE antagonists with differing modes of action on IsoLG-PE activation of macrophages. We found that GM-0111, a synthetic glycosaminoglycan previously shown to act as an RAGE antagonist (73), significantly inhibited IsoLG-PE-induced NFκB reporter activity (Fig. 7B). We also found that FPS-ZM1, a high-affinity antagonist that specifically binds the V domain of RAGE (18), markedly inhibited IsoLG-PE-induced NFκB reporter activity (Fig. 7C).
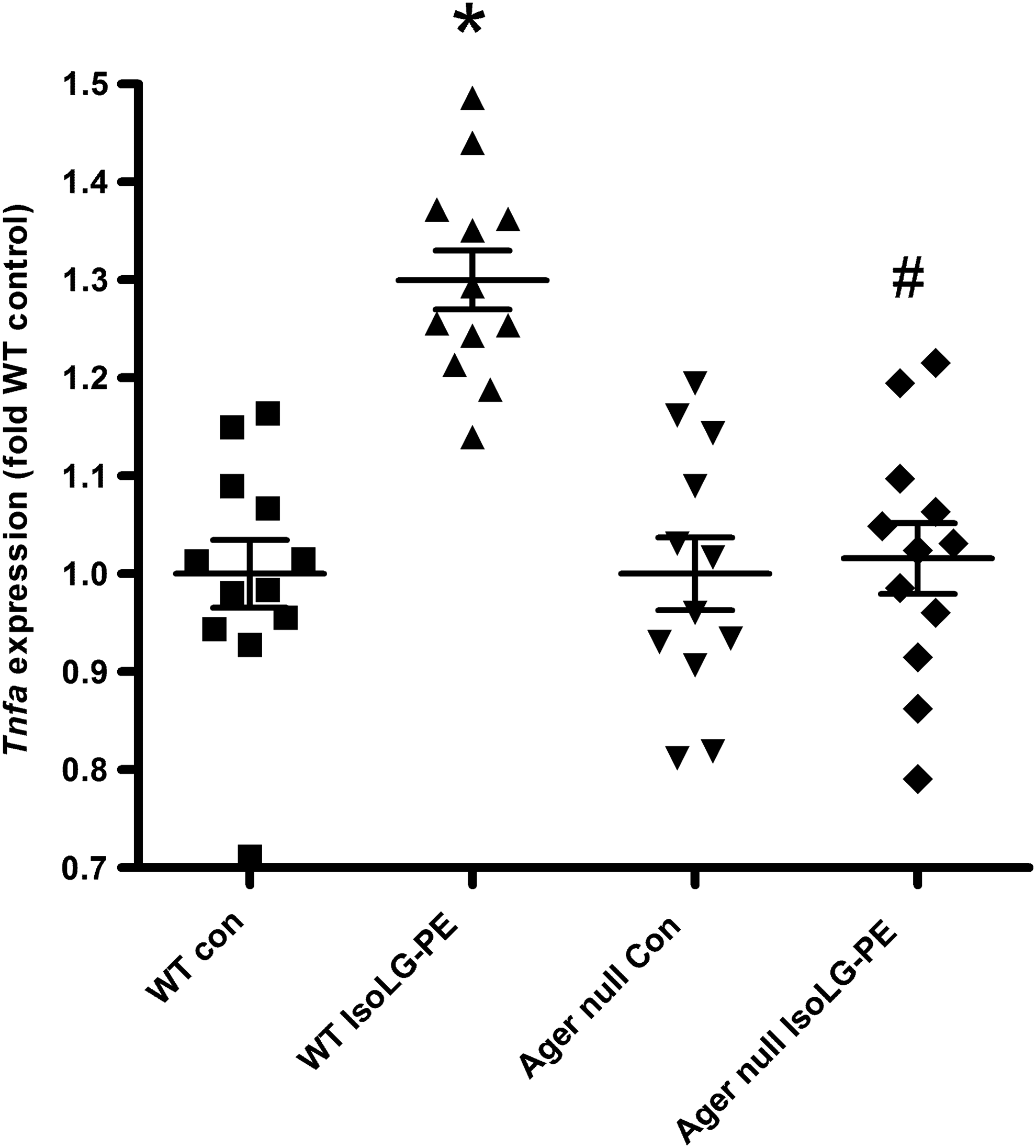
These experiments suggested that IsoLG-PE activated NFκB and inflammatory cytokine secretion via RAGE. To further examine the requirement for RAGE in IsoLG-PE activation of macrophages, we isolated macrophages from the bone marrow of wild-type (C57BL6J) mice and Ager null mice that lack RAGE and examined whether IsoLG-PE could activate cytokine expression in these macrophages. BMDM were chosen for these experiments, as they could be readily obtained from both wild-type and Ager null mice and BMDM have been widely used for studying the effects of pattern recognition receptors both in vitro and in vivo. In contrast to BMDM from wild-type mice, BMDM from Ager null mice failed to induce inflammatory cytokines in response to IsoLG-PE (Fig. 8).
Discussion
Our findings that IsoLG-PE levels are elevated in conditions associated with lipid peroxidation and inflammation, that IsoLG-PE activates NFκB and inflammatory cytokine expression at nanomolar concentrations, and that IsoLG-PE requires RAGE to induce macrophage activation implicate IsoLG-PE and RAGE as mediators of inflammation induced by lipid peroxidation.
A significant body of literature has previously demonstrated that risk factors for inflammatory diseases such as hypercholesterolemia and obesity increase lipid peroxidation (5, 15, 63). However, the extent to which IsoLG-PE levels are elevated during inflammatory conditions has been largely unknown, as only one previous study had measured IsoLG-PE levels in vivo. Li et al. found that feeding mice ethanol chronically, a condition that elevates lipid aldehydes including malondialdehyde and 4-hydroxynonenal (22), markedly increases hepatic levels of IsoLG-PE (42). They also showed increased plasma levels with macular degeneration, a condition previously established to increase levels of lipid aldehydes, including malondialdehyde and 4-hydroxynonenal (40).
Our findings suggest that a wide variety of conditions associated with lipid peroxidation are likely to elevate IsoLG-PE levels. For instance, we found that IsoLG-PE was markedly increased in plasma of FH patients. While future studies will be needed to elucidate the exact mechanisms underlying this increase in IsoLG-PE levels, we note that this inherited disorder is associated with markedly increased myeloperoxidase activity (54). Increased myeloperoxidase activity may be relevant, because Poliakov et al. previously showed that exposure to myeloperoxidase generated significant amounts of IsoLG protein adduct both in vitro and in vivo (53), and we showed that exposure of lipoprotein to myeloperoxidase in vitro generated IsoLG-PE (27). FH is associated with markedly increased inflammation (50) and risk for atherosclerosis (10). Our findings suggest that generation of IsoLG-PE and subsequent activation of macrophages by IsoLG-PE may be one mechanism underlying this inflammation, but future studies will be needed to test whether reducing IsoLG-PE levels also reduces inflammation in FH patients.
Our finding that IsoLG-PE levels were also elevated in the liver of mice fed a high-fat diet supports the notion that generation of IsoLG-PE may be a consistent feature of inflammation associated with lipid peroxidation. Obesity is a well-established risk factor for inflammatory diseases associated with increased lipid peroxidation such as steatohepatitis, atherosclerosis, and arthritis (2, 46, 67). The infiltration and activation of macrophages into the liver during development of obesity is considered a critical feature of metabolic syndrome and may play an important role in the development of insulin resistance and vascular disease associated with metabolic syndrome (6). Additional studies are needed to determine whether reducing IsoLG-PE levels in the liver will mitigate the effects of high-fat feeding on liver macrophage activation, insulin resistance, and liver injury. In this regard, it is important to note that while increasing NAPE-PLD activity could reduce IsoLG-PE levels, IsoLG-ETN was about threefold less potent than IsoLG-PE. Thus, accumulation of IsoLG-ETN could also contribute to inflammation, and it would be important to confirm that any potential therapeutic intervention markedly reduced both IsoLG-PE and IsoLG-ETN levels.
The potential biological significance of elevated IsoLG-PE levels is shown by our finding that IsoLG-PE potently activates NFκB and expression of Tnfa and Ccl2 in macrophages. Activation of macrophages is a key step in many inflammatory diseases such as atherosclerosis and steatohepatitis and elevated Tnfa and Ccl2 (MCP-1) levels in these diseases is well established (25, 32, 43). TNFα is a master regulator of inflammation and induces expression of cytokines, prostaglandins, platelet-activating factor, adhesion molecules, inducible nitric oxide synthase, and matrix metalloproteinases (52, 56, 74). The importance of macrophage secretion of TNFα in atherogenesis was shown by studies where transplantation of Apoe null/Tnfa null bone marrow into Apoe null receipt mice gave rise to lesions that were 83% smaller than transplantation of Apoe null-only bone marrow into Apoe null receipt mice (9). CCL2 is similarly important in atherogenesis, because mice lacking either Ccl2 or its receptor, Ccr2, develop lesions that are significantly reduced in size (17, 26). Thus, the ability of IsoLG-PE to activate Tnfa and Ccl2 expression by cultured macrophages suggests that IsoLG-PE may be an important inducer of macrophage TNFα and CCL2 secretion in FH and fatty liver disease.
Another key question our studies sought to answer was whether the effects of IsoLG-PE are mediated by specific receptors rather than simply due to membrane perturbations. At micromolar concentrations, IsoLG-PE markedly lowers the liquid crystalline to hexagonal phase transition temperature of liposomes and also induces endoplasmic reticulum (ER) stress responses in cultured endothelial cells (28), consistent with membrane perturbation exerting biological effects. However, the endothelial response can only be partially reversed by inhibition of ER stress, suggesting a role for other mechanisms besides membrane perturbation. Our finding that concentrations of IsoLG-PE (30 nM), which are well below that required to markedly perturb overall membrane structure, were sufficient to induce maximal inflammatory responses in murine macrophages strongly implicates receptor-driven mechanisms.
Our finding that soluble RAGE, RAGE antagonists, and genetic deletion of RAGE block the effects of IsoLG-PE in macrophages strongly implicate RAGE as an IsoG-PE receptor. Multiple ligands are known to activate RAGE, including advanced glycation endproducts (AGEs), S100 family proteins, high mobility group protein box-1 (HMGB1), amyloid β, and phosphatidylserine (19, 34). Common features of previously characterized RAGE ligands are a negative charge at neutral pH and an ability to drive RAGE oligimerization by forming multimers (19). Most RAGE ligands bind at a patch of positively charged residues formed by the V and C1 domains of RAGE and this binding stabilizes preassembled complexes of multiple RAGE monomers (39), allowing diaphanous-1 to bind and activate downstream signals, including ERK, JNK, p38, and NFκB (37). IsoLG-PE is also negatively charged and may form oligomers in the sense that closely adjacent PE are likely to undergo IsoLG modification. IsoLG-PE shares partial structural features with methylglyoxal-hydroimidazolones (MG-Hs) (Fig. 6), which bind RAGE with high-affinity (Kd 40 nM) (70) and are the most potent of the AGE class of RAGE ligands. IsoLG-PE also shares the same phosphatidyl moiety as the recently described RAGE ligand phosphatidylserine (34). Additional studies will be needed to identify the specific binding sites of IsoLG-PE on RAGE and the mechanisms underlying its activation of RAGE signaling complexes.
Our finding that IsoLG-PE activates RAGE on macrophages suggests that activation of RAGE may be responsible for the effects of IsoLG-PE or IsoLG seen in other cell types. For instance, IsoLG-PE activates the surface expression of adhesion molecules and expression of inflammatory cytokines in endothelial cells (28) and IsoLG added to platelets causes their activation and aggregation (3). RAGE is found on both of these cell types (20, 60), so that these activities might plausibly be mediated by RAGE. In particular, activation of RAGE on endothelial cells could explain our previous finding that IsoLG-PE induces the ER stress response of endothelial cells (28), similar to RAGE agonists such as HMGB1 that induce ER stress (44).
One feature of our findings that needs further investigation is why there are significant differences in the potency of IsoLG-PE between various cell types. For instance, IsoLG-PE induced NFκB activation about 10 times more potently in RAW264.7 macrophages (30 nM) than in bone marrow-derived macrophages (300 nM). For endothelial cells, 3 μM IsoLG-PE was required for maximal induction of inflammatory effects. These differences in sensitivity to IsoLG-PE might reflect differences between cell types in NAPE-PLD expression, RAGE surface expression, or interactions of RAGE with other receptors. Several RAGE ligands interact with other pattern recognition receptors such as CD36 and TLR4 (35, 36, 51, 69), and activation of these other receptors can increase RAGE expression. Because we found that the thromboxane receptor antagonists SQ29548 modestly inhibited IsoLG-PE activation of NFκB, we also cannot rule out that RAGE and the thromboxane receptor act synergistically in some manner to invoke responses in the macrophages.
Our identification of RAGE as the first known receptor for IsoLG-PE provides insight into ways in which IsoLG-PE might act in vivo. RAGE is an important mediator of inflammatory responses and has been implicated in atherosclerosis and inflammation associated with hypercholesterolemia and a high-fat diet. For instance, genetic deletion of Ager decreases atherosclerosis in both nondiabetic and diabetic Apoe null mice (31, 62). Ager null mice also show decreased obesity, adipose tissue inflammation, and insulin resistance in wild-type C57BL6 mice fed a high-fat diet (61). RAGE expressed on bone marrow-derived cells (e.g., monocyte/macrophages) specifically contributes to atherosclerosis, as transplantation of Ager null/Apoe null bone marrow into Apoe null mice results in markedly reduced atherosclerosis compared with those receiving bone marrow from Ager-positive Apoe null mice (47). Administration of sRAGE markedly decreases atherosclerosis and oxidative injury in animal models (11, 41, 68, 71, 72). Thus, activation of RAGE by IsoLG-PE could plausibly contribute to the development of atherosclerosis and liver disease associated with hypercholesterolemia or obesity. Future studies examining the effect of therapeutic interventions that lower IsoLG-PE levels in these diseases will be needed to investigate this potential contribution.
Materials and Methods
Materials
The following reagents were purchased commercially: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE; Avanti Polar Lipids, Alabaster, AL), ethanolamine (ETN; Sigma-Aldrich, St. Louis, MO), phospholipase A2 (PLA2) isolated from honey bee venom (Sigma-Aldrich), antibiotic-antimycotic mix (Gibco by Life Technologies, Rockville, MD), IQ SYBR Green supermix and iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA), SQ29548 (Cayman Chemicals, Ann Arbor, MI), and organic solvents, including methanol, chloroform, dichloromethane, and acetonitrile, were high-performance liquid chromatography grade (Sigma-Aldrich).
IsoLG-PE and IsoLG-ETN were synthesized by a reaction with DPPE and ETN with 15-E2-IsoLG as previously reported (28, 64). It is important to note that 15-E2-IsoLG is only one out of the eight regioisomers (5-E2-IsoLG, 5-D2-IsoLG, 8-E2-IsoLG, 8-D2-IsoLG, 12-E2-IsoLG, 12-D2-IsoLG, 15-E2-IsoLG, and 15-D2-IsoLG) that are potentially generated by peroxidation of arachidonic acid. Based on theoretical considerations, the 15- and 5-series IsoLGs are expected to be formed in significantly greater abundance than the 8- or 12-series IsoLGs. Levuglandin E2 (LGE2) formed nonenzymatically from prostaglandin H2 is chemically identical to 15-E2-IsoLG. For these reasons, 15-E2-IsoLG has been the most widely used regioisomer of IsoLG synthesized for use in studies. The synthesis of C17:0NAPE was adapted from (24) with a modified purification method (28).
Plasma from FH patients
Ethylenediaminetetraacetic acid (EDTA) plasma was isolated from blood of FH patients (n=5) before undergoing LDL apheresis. Two patients were FH homozygotes and three patients had severe heterozygous FH with LDL-C >200 mg/dl before each LDL apheresis session. As a control, EDTA plasma was isolated from blood of healthy volunteers (n=5). The study protocol was approved by the Vanderbilt University Medical Center institutional review board (IRB 101615). All participants gave informed consent.
Animal studies
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University. In a previously published study (12), we showed that prolonged feeding of a high-fat diet induced major hallmarks of hepatosteatosis, including triglyceride accumulation, macrophage infiltration, and expression of inflammatory genes [Figs. 4 and 5 of Ref. (12)]. In that same study, we fed 6-week-old male C57BL/6J mice a standard low-fat chow diet (Lab Diet 5001 13.5% kcal from fat, 60% kcal from carbohydrate, 28.5% kcal from protein; TestDiet, St. Louis, MO) or a high-fat diet (TestDiet® 58Y1, containing 60% fat by kcal; TestDiet) for 9 weeks and reported that the high-fat fed mice had significantly higher fasting glucose and insulin levels and impaired glucose tolerance than mice fed a low-fat chow diet [Fig. 11 of Ref. (12)]. After the 9 weeks of feeding, the mice were euthanized, the livers were harvested, and aliquots were stored at −80°C until analysis for this study. The mice fed the high-fat diet had higher body weight (37.52±0.74 g) and total body fat (12.14±0.45 g) than mice fed the low-fat chow diet (25.80±0.27 g body weight and 2.61±0.10 g body fat, respectively; p<0.05 for each parameter, two-tailed Student's t-test). Body weight was measured using a portable electronic scale. For body composition, mice were scanned by magnetic resonance imaging using a Bruker Minispec MQ10 NMR Analyzer to determine fat mass, lean mass, and free fluid.
Femurs from Ager null mice (two each from three individual mice, 6 months old) were a kind gift from Dr. Tim Oury, Department of Pathology, University of Pittsburgh and femurs collected from C57BL/6J male mice of a similar age were used as a control. For isolation of bone marrow-derived macrophages, femurs were harvested from euthanized mice and shipped in RPMI 1640 with 2% fetal bovine serum (FBS), 10 units/ml heparin, pencillin, and streptomycin at 4°C. After cutting off muscles, the ends of the femur were cut off and the bone marrow cells were carefully flushed out. Cells were resuspended by gentle pipetting and passed through a 70 μm filter into a 50 ml conical tube. Cells were collected by centrifugation and resuspended in 8 ml Dulbecco's modified Eagle's medium (DMEM) (per femur) with 10% FBS, 1×antibiotic-antimycotic, and 20 ng/ml macrophage colony-stimulating factor. One milliliter of cells was transferred to 1 well of a 12-well plate and incubated at 37°C in 5% CO2 atmosphere. Medium was changed every 2 days after first culture, and cells were used for experiments on day 7 and 10 when cells were 85%–90% confluent.
Measurement of IsoLG-PE in tissues
IsoLG-PE levels were measured by LC-MS/MS with a modified PLA2 assay (42). Plasma samples were vortexed with chloroform/methanol (2/1, v/v) containing 0.1% BHT. Liver tissues were homogenized manually in a tube with chloroform/methanol (2/1, v/v) containing 0.01% BHT. C17NAPE (0.1 nmol) was added as an internal standard. After incubation at 4°C for 1 h, a 1/3 volume of phosphate-buffered saline (PBS) was added, the mixture was centrifuged at 3000 rpm for 15 min, and the lower organic layer containing the lipids was collected and evaporated under a stream of nitrogen. After being re-dissolved in 1 ml of chloroform, lipids were loaded onto a Sep-Pak silica cartridge (Waters Corp., Milford, MA) pre-equilibrated with chloroform, the cartridge was washed with 8 ml of chloroform, and IsoLG-PE was eluted by 8 ml of 50% methanol/chloroform. The lipids were dried under nitrogen and stored at −80°C before hydrolysis and analysis. The lipid extract was re-dissolved in methanol (50 μl) followed by the addition of a PBS solution (10 mM, pH 7.4, 450 μl) containing CaCl2 (5 mM). Phospholipase A2 from honey bee venom (PLA2, 10 units/sample) was added, incubated under argon at 37°C overnight, and extracted with 1.5 ml of chloroform/methanol (2/1, v/v). Solvents were evaporated and the lipid was re-dissolved in methanol (50 μl) and analyzed by LC/MS/MS using a ThermoFinniganQuantum electrospray ionization triple-quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA) operating in negative-ion multiple reaction monitoring mode.
Mobile phase A consisted of 1 mM ammonium acetate in water/acetonitrile/methanol (1/1/3), and mobile phase B consisted of 1 mM ammonium acetate in ethanol. The lipids were chromatographed on a Zorbax XDB-C8 column (50×2.1 mm; Agilent Tech., Santa Clara, CA) with a constant flow rate of 400 μl/min. After a 0.5 min hold at 1% mobile phase B, the solvent was gradient ramped to 99% B over 6 min, held at 99% B for 1 min, returned to 1% B over 1 min, and held for 1 min before the next injection. The electrospray needle was used at −3300 V. The ion-transfer tube was operated at −35 V and 270°C. The tube lens voltage was set to −180 V. Mass transitions of m/z 704.3 → 255.1 for C17:0NAPE, m/z 798.3→153.0 for IsoLG-(C16:1)lysoPE, m/z 800.3→153.0 for IsoLG-(C16:0)lysoPE, m/z 824.3→153.0 for IsoLG-(C18:2)lysoPE, m/z 826.3→153.0 for IsoLG-(C18:1)lysoPE, and m/z 828.3→153.0 for IsoLG-(C18:0)lysoPE were monitored, with a collision energy of 50 eV. The chromatographic results were processed in Xcaliber software (Thermo Fischer Scientific, Waltham, MA) using nine-point Gaussian smoothing, and the peak heights in comparison to the internal standard were used to calculate the amount of each species present in the sample. Linear regression and correlation analysis was performed using GraphPad Prism (version 4.00 for Windows; GraphPad Software, San Diego, CA). For plasma, significant signal for IsoLG-(C16:0)lysoPE only was detected, so only this species was used for quantitation. For liver, significant signals for IsoLG-(C16:1)lysoPE, IsoLG-(C16:0)lysoPE, IsoLG-(C18:2)lysoPE, and IsoLG-(C18:1)lysoPE were detected, so the sum of these species in each sample was used for quantitation. Representative chromatographs from liver of chow and high-fat fed mice are shown in Supplementary Figure S2.
Cell culture
RAW264.7 macrophage cells (American Type Cell Culture, Manassas, VA) were cultured in 100 mm dishes with DMEM supplemented with 10% FBS and 1×antibiotic-antimycotic mix until 85%–90% confluent. Cells from confluent plates were trypsinized, transferred to six 24-well plates (1 ml each well), and cultured for 48 h before use. RAW264.7 cells transfected with NFκB reporter were a kind gift from Dr. Claus Schneider, Vanderbilt University. These cells were generated by transfection with plasmid from Takara Clontech containing an NFκB enhancer element 5′ to a minimal promoter driving expression of secreted Metridia luciferase reporter, as well as kanamycin/neomycin resistance gene. Transfected cells were selected with 1 mg/ml geneticin in DMEM supplemented with 10% FBS and 1×antibiotic-antmycotic mix.
Established NFκB reporter macrophages originally derived from bone marrow of transgenic mice expressing a reporter plasmid containing a total of eight NFκB-binding sites upstream of the herpes simplex virus minimal thymidine kinase promoter driving expression of Photinus luciferase (18) were a kind gift from Dr. Timothy S. Blackwell, Vanderbilt University. These passaged nfkb-BMDM were used for the majority of NFκB reporter experiments instead of transfected RAW264.7 cell line, because nfkb-BMDM were readily available and relatively easy to use to produce the large numbers of reporter cells needed for our assays, and because these nfkb-BMDM have been widely used in studies examining NFκB signaling, including studies examining the effects of reactive lipids on NFκB signaling (49). All cells were grown to 85%–90% confluence in 100 mm dishes containing DMEM containing 10% fetal bovine serum, 1×antibiotic-antimycotic mix, trypsinized, and cells from each plate were aliquoted to six 12-well culture dishes, cultured for 48 h, and incubated in the same medium with serum overnight before experiments.
Cytokine mRNA expression
RAW264.7 macrophage cells were stimulated with either PE (100 nM) or IsoLG-PE (1–300 nM) in Hanks' buffered saline solution (HBSS) with 0.1% human serum albumin and 0.2% dimethyl sulfoxide (DMSO) at 37°C for 4 h. The media was then removed, and macrophages were washed with cold PBS. RLT buffer was added, and the plates were incubated on ice for 30 min. The lysate was collected and RNA was extracted with RNeasy Mini kit (Qiagen, Valencia, CA) for PCR analysis. Levels of Actb (internal standard), Tnfa, and Ccl2 mRNAs were measured after conversion to cDNA (with iScript cDNA Synthesis Kit) by qPCR using IQ SYBR Green supermix. The primer pairs were purchased from Sigma-Aldrich as follows: (a) Actb: sense GAG CGC AAG TAC TCT GTG TG, antisense CGG ACT CAT CGT ACT CCT G; (b) Tnfa (Sigma-Aldrich): sense CCA TTC CTG AGT TCT GCA AAG, antisense GCA AAT ATA AAT AGA GGG C; and (c) Ccl2: sense ACT GAA GCC AGC TCT CTC TTC CTC, antisense TTC CTT CTT GGG GTC AGC ACA GAC. Results are shown as the average of 3 separate experimental days, four replicate wells per day, normalized to PE-treated cells. Bone marrow-derived macrophages from wild-type and Ager null mice were stimulated with either vehicle or 300 nM IsoLG-PE for 4 h, and Tnfa expression was measured by qPCR as described earlier.
TNFα protein expression
Supernatant from RAW264.7 cells treated with IsoLG-PE, IsoLG, or vehicle (HBSS with 0.1% human serum albumin and 0.2% DMSO for IsoLG-PE and 0.02% DMSO for IsoLG) overnight were collected and measured using a commercial mouse TNFα DuoSet ELISA assay kit (R&D Systems, Minneapolis, MN).
NFκB reporter activity
RAW264.7 NFκB reporter or nfkb-BMDM cells were washed twice with HBSS to remove serum and then treated with IsoLG-PE, IsoLG-ETN, PE, or vehicle (HBSS with 0.1% human serum albumin and 0.2% DMSO) for 30 min at 37°C. Cell media was then removed, Luciferase Assay Reporter Lysis Buffer (Promega, Madison, WI) was added, and the resulting bioluminescence was quantified and normalized to control.
For studies examining the effects of sRAGE, IsoLG-PE was preincubated with vehicle (HBSS with 0.1% HSA) or sRAGE (2 mg/L) for 37°C for 1 h before treatment of cells. For these experiments, 0.1 μM PE in vehicle was used as a control, and bioluminescence was normalized to 0.1 μM PE. For studies examining the effects of RAGE antagonists, cells were treated with either vehicle (HBSS), 0.2 μM FPS-ZM1 (Calbiochem, San Diego, CA), or 100 μM GM-0111 (GlycoMira, Salt Lake City, UT) along with IsoLG-PE or PE in HBSS with 0.1% human serum albumin and 0.2% DMSO at 37°C for 30 min and resulting bioluminescence was normalized to cells treated with 0.1 μM PE.
Screen of GPCR library
IsoLG-PE was synthesized as described earlier and purified from unreacted PE by HPLC with the fractions where IsoLG-PE eluded identified by LC/MS/MS and combined. The solvent was evaporated under nitrogen, and the IsoLG-PE was redissolved in DMSO at 100 μM for shipment to DiscoveRx for screening at 1 μM final concentration of IsoLG-PE using their proprietary functional Cell-Based GPCR Assay Panel with β-arrestin recruitment as the readout. In agonist mode for GPCR receptors with known ligands, the extent of GPCR activation by test compound is calculated based on maximal activation with known ligand.
Footnotes
Acknowledgments
This work was supported in part by grants from National Institutes of Health HL111945, HL116263, HL30568, HL34343, AT0007830, ES000267, and RR024975 and by the Laubisch, Castera, and M.K. Grey Funds at the UCLA. The authors have no other financial interests to disclose. They thank Dr. Tim Oury for the kind gift of femurs harvested from Ager null mice and Dr. Timothy Blackwell for established bone marrow-derived macrophages from NFκB reporter mice. They also thank Dr. Olivier Boutaud for helpful discussions about thromboxane receptor antagonists and activation.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.