
Abstract
Introduction
O
Both epigenetic dysregulation and genetic defects in epigenetic regulators have been uncovered in a plethora of human diseases, including cancer, developmental syndromes, neurodegenerative diseases, and neural and metabolic disorders. Mutations in epigenetic regulators underlie monogenic diseases and have also been found to modify the severity or penetrance of more complex disorders (10, 53). In addition, some regions of the genome, termed metastable epialleles, show high variability of gene expression both cell-to-cell and between individuals under normal circumstances (124). The activity of these regions is highly dependent on the epigenetic state established during development (44, 124). Metastable epialleles in mice have been found to be particularly sensitive to environmental factors such as diet, which can strongly influence the ultimate epigenetic state (166). In humans, metastable epialleles have been found for more than 1000 genes, many of which are associated with human diseases and disorders, and have the potential to play an etiologic role in individual disease development and progression (64).
In this review, we focus on the epigenetic mechanisms involved in facioscapulohumeral muscular dystrophy (FSHD). FSHD is an autosomal dominant myopathy with a strong epigenetic etiology involving mutations in epigenetic regulators, roles for epigenetic modifiers, and a disease locus with characteristics of a metastable epiallele. FSHD provides an outstanding model to investigate broadly applicable epigenetic mechanisms of gene regulation and how these are dysregulated in disease.
FSHD Genetics and Clinical Presentation
FSHD is the third most prevalent of the nine myopathies classified as muscular dystrophies (115, 117). It is classically considered an autosomal dominant disease characterized by progressive weakness and atrophy of specific muscle groups. Muscles of the face and upper body are typically affected first, followed by muscles of the lower extremities; however, the range and severity of affected muscles is highly variable and often asymmetric (117, 156). FSHD is characterized by a wide variability, both between and within families, in disease onset, progression, and severity, which ranges from asymptomatic to clinically severe (117, 148, 150). Disease symptoms are generally late onset with patients usually developing noticeable weakness in their second or third decade, while some genetically characterized FSHD individuals may not develop clinical symptoms until much later in life, if at all. Overall, ∼20% of FSHD-affected individuals eventually become wheelchair bound (85). This high variability within the clinical spectrum suggests that multiple genetic, epigenetic, developmental, and environmental factors likely play integral roles in the development and progression of FSHD pathology.
The most common form of the disease, FSHD1 (OMIM 158900), is linked to contractions of the polymorphic D4Z4 macrosatellite repeat array in the subtelomere of chromosome 4 at 4q35 (159, 167, 168). In the general healthy population, this repeat array varies between 11 and 100 D4Z4 repeats on both 4q chromosomes, whereas in FSHD1 patients, the array is contracted to 1–10 repeats on one 4q chromosome with a requirement for at least one D4Z4 unit to develop disease (Fig. 1) (158, 168). Only contractions in cis with specific disease-permissive haplotypes of the 4qA distal subtelomere are associated with FSHD1, indicating that the deletion itself is merely permissive and not necessarily pathogenic (94, 96, 98, 99). Not surprisingly, these large deletions of subtelomeric macrosatellite DNA in FSHD1 correlate with epigenetic changes at 4q35, discussed next, which appear to be essential for developing disease (40, 160). It is interesting to note that chromosome 10q26 contains a subtelomeric D4Z4 macrosatellite that is highly homologous to the array at 4q35 (5, 41); however, FSHD1 is linked only to contractions on chromosome 4 and D4Z4 contractions at 10q26 are non-pathogenic (99, 100, 130, 181). Thus, in combination with the clinical diagnosis, the genetic diagnosis for FSHD1 is a contraction at 4q35 to 1–11 D4Z4 repeats, in cis with a permissive 4qA subtelomere.
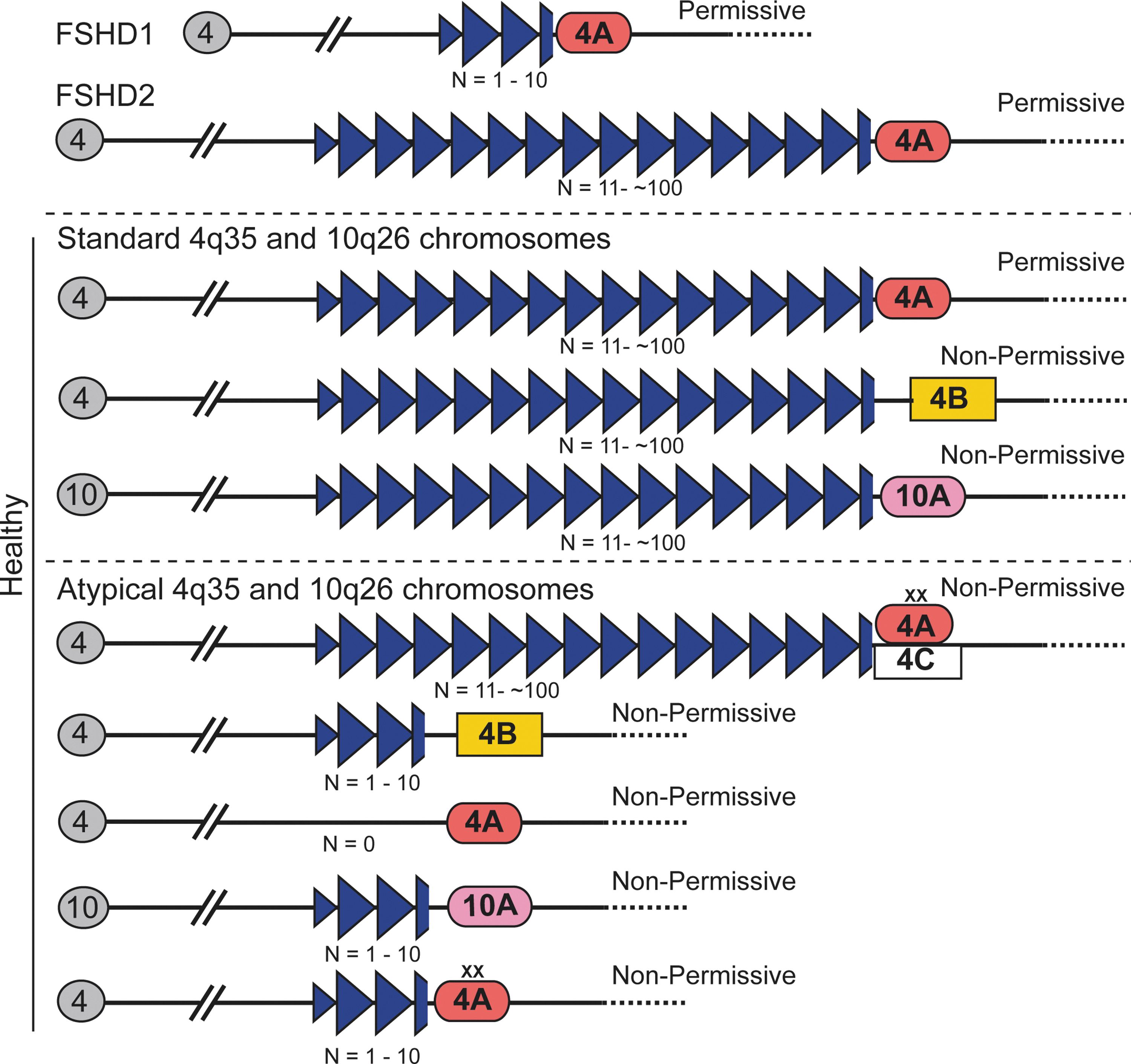
Early indications that FSHD may have an epigenetic component came from investigating correlations between repeat size and disease severity. While there is no linear relationship, there is an imperfect correlation among the extremes of pathogenic sized arrays, as FSHD1 subjects with 1–3 repeat units tend to be clinically severe cases while subjects with 8–10 repeats often present with milder symptoms or can be asymptomatic (104, 127, 148, 160, 170). In addition, the current accepted genetic requirements for FSHD are present in ∼1–3% of the general population, typical of a common genetic variant and two orders of magnitude higher than the reported incidence of FSHD, highlighting that these genetic conditions are merely disease permissive (128, 140). These seemingly healthy individuals who do not recognize any muscle weakness in themselves are considered FSHD1 asymptomatic and it is not clear whether they truly lack pathology, have pathology but no noticeable weakness due to compensatory muscles, or whether pathology is merely delayed and they will develop the disease later in life. Similarly, FSHD family studies have identified some striking examples of asymptomatic FSHD1 cases, even at advanced ages, with the added caveat that a first-degree relative with the same contraction is clinically affected (e.g., severely affected 66 year-old and asymptomatic 69 year-old brothers) (78, 170). Even multiple cases of monozygotic twins with discordant FSHD phenotypes have been reported (60, 149, 154). Thus, there is more to developing clinical FSHD1 than the known diagnostic genetic lesion and overall, the FSHD1 clinical data are suggestive of a strong epigenetic component to disease onset, progression, and severity (3, 58, 78, 127, 128, 136, 140, 153, 170, 178).
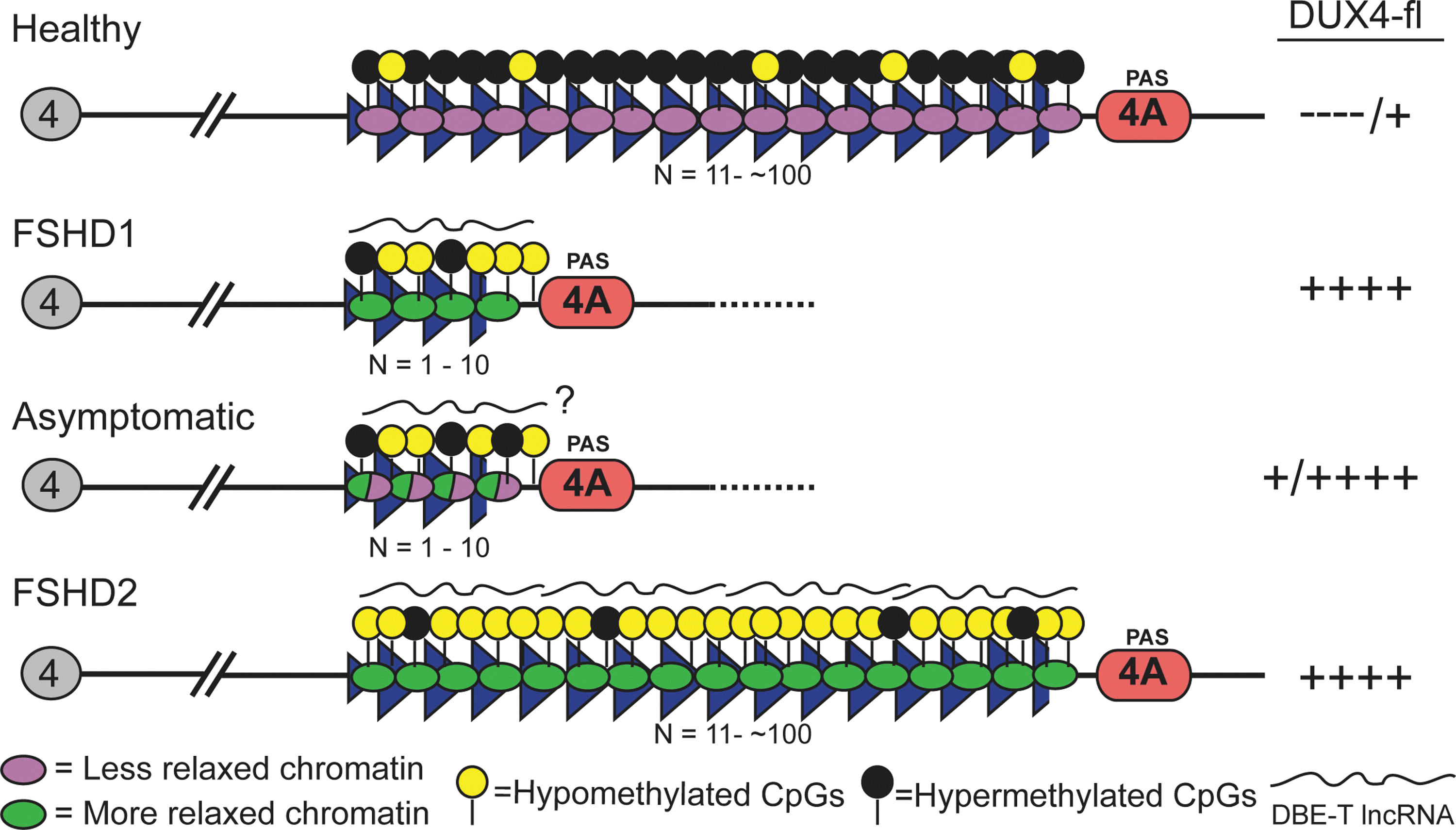
Representing the remaining ∼5% of cases is contraction-independent FSHD, or FSHD2 (OMIM 158901), which is clinically indistinguishable from FSHD1 (39). Although there is no contraction of either chromosome 4q35 D4Z4 array (Fig. 1), FSHD2 is still genetically linked to the 4q35 region in that all FSHD2 patients carry at least one permissive 4qA distal subtelomere (40, 56, 95, 96, 100). FSHD2 is also epigenetically linked to 4q35 via epigenetic dysregulation that is common to all forms of FSHD (Fig. 2).
A third recognized class of FSHD is the infantile form, IFSHD, which is clinically more severe and progresses more rapidly than adult-onset FSHD. IFSHD has the same genetic diagnosis as FSHD1, but is generally associated with very short (n=1–3) D4Z4 arrays (22, 24, 85). IFSHD is distinguished by an early age of clinical onset with facial weakness apparent before 5 years, and shoulder weakness before 10 years of age (22). Muscle weakness is often accompanied by extramuscular manifestations, including high-frequency hearing loss, retinal vasculopathy, and cognitive impairment as well as occasional cardiac and respiratory symptoms (24, 30, 85). This severe form of FSHD1 further supports the existence of genetic or epigenetic modifiers of disease severity, as discussed next. Thus, all forms of FSHD share the genetic and epigenetic linkage to 4q35 D4Z4 and a requirement for a permissive 4qA haplotype (96, 98).
The DUX4 model of FSHD pathogenesis
With no obvious mutations in any protein-coding gene, the pathogenic defect in FSHD1 is likely regulatory. DNA repeats can play regulatory roles for nearby genes in cis and in trans, express regulatory non-coding RNAs (ncRNAs), and, as is the case for D4Z4, encode protein-coding genes within the repeat (21, 28, 46, 52, 155). Numerous candidate genes have been proposed for mediating FSHD pathogenesis based on differential expression between FSHD-affected and healthy myogenic cells, with little consensus (2, 26, 27, 43, 50, 86, 125, 129) (71). Many genes are misexpressed in FSHD (55, 123), and models for pathogenesis should also take into account the linkage of both FSHD1 and FSHD2 to the disease permissive 4qA haplotype. To date, one of the only genes consistently found to be misexpressed in both FSHD1 and FSHD2 myogenic cells is DUX4, a retrogene located within each D4Z4 repeat unit (Fig. 3) (48, 52, 78, 96, 143). In the DUX4 model of FSHD, although each D4Z4 repeat encodes the entire DUX4 open reading frame, only DUX4 transcribed from the distal-most D4Z4 unit produces a mature mRNA that is stabilized by splicing to a downstream polyadenylation signal present only in 4qA disease-permissive subtelomeres (Fig. 3), thus explaining the linkage of both FSHD1 and FSHD2 to 4qA (96, 143, 151). This DUX4 model was essentially confirmed independently using a large collection of myogenic cells and biopsies from FSHD family cohorts of first-degree relatives; however, DUX4 expression was also found in some asymptomatic subjects, at similar levels to those in affected subjects, and in a few healthy subjects, at significantly lower levels than in affected subjects (78). This expression of DUX4 in the absence of clinical symptoms indicates that in addition to modifiers of DUX4 expression, modifiers of DUX4 function also likely exist.

DUX4 encodes two different protein isoforms generated through alternative mRNA splicing: a non-pathogenic short form of unknown function (DUX4-S) that is often expressed in healthy somatic cells and a full-length form (DUX4-FL) which is generally not expressed in healthy adult somatic cells (143). Although DUX4-S regulates expression of a much smaller set of genes (55), both isoforms contain the same double homeobox DNA binding domain and are thought to function as transcription factors (43, 55, 175). However, only expression of the DUX4-FL isoform is linked to FSHD (78, 96, 143) and DUX4-FL-specific target genes, which include genes expressed in the germline and in early development, immune mediators (e.g., β-defensin 3), and retroelements (e.g., MaLRs), are misregulated in FSHD (55, 175). Therefore, FSHD involves both an increase in DUX4 gene transcription and a switch in DUX4 alternative splicing. With increasing evidence that alternative splicing can be regulated by DNA methylation, histone post-translational modifications (PTMs), and small interfering (si)RNAs, all of which are differentially represented in FSHD at the 4q35 locus (1, 87, 102, 103, 135, 141), epigenetic mechanisms may be involved in regulating both the expression levels of DUX4 and the pathogenic switch from DUX4-s to DUX4-fl mRNA isoforms in FSHD, as discussed next.
DUX4 is thought to have originated after a gene conversion event in the DUXC macrosatellite array that occurred in the primate and Afrotheria lineages, and subsequent translocation to 4qter in primates (31, 92). Although generally silent in adult somatic tissues, DUX4-fl is expressed in testis and in pluripotent stem cells (143), and a recent report describes expression of DUX4-fl in both muscle and non-muscle somatic tissues of FSHD1 and healthy fetuses (23). Thus, the DUX4 retrogene has likely evolved to play a normal role strictly during primate development, but on loss of epigenetic silencing in FSHD, abnormal DUX4-fl expression in adult skeletal muscle has pathological consequences. Low levels of DUX4-FL are highly cytotoxic when expressed in somatic cells or during vertebrate development (20, 89, 111, 164, 171), and DUX4-FL expression in myogenic cells disrupts differentiation and causes the atrophic myotube phenotype found in FSHD myotubes (19, 162). Although the mechanisms are still unclear, it is thought that aberrant expression of DUX4 targets (immune mediators, germline genes, and the products of DUX4-activated retroelements) leads to muscle pathology (55, 175). Two myogenic enhancers proximal to D4Z4 were recently identified and shown to regulate DUX4 (70), providing a potential explanation for the relatively muscle-specific pathology seen in FSHD. Whether aberrant DUX4 expression occurs in FSHD muscle satellite (stem) cells has not yet been addressed. It has been suggested that DUX4 expression in satellite cells might lead to a progressive loss of muscle regenerative capacity over time (20), resulting in the late onset of clinical symptoms.
Thus, increased DUX4-fl expression in FSHD skeletal muscle is consistent with both FSHD1 and FSHD2, accounts for the permissive A-type subtelomere requirement, is detrimental to myocytes, and induces gene expression profiles found in FSHD muscle biopsies. Furthermore, the overall low frequency of DUX4-FL expression correlates with the sporadic muscle involvement seen in FSHD patients (78, 143). Together, these findings make increased DUX4-FL expression in skeletal muscle a prime mechanism for generating FSHD pathology.
FSHD Is an Epigenetic Disease
Epigenetic disruption of the 4q35 D4Z4 array is associated with all forms of FSHD
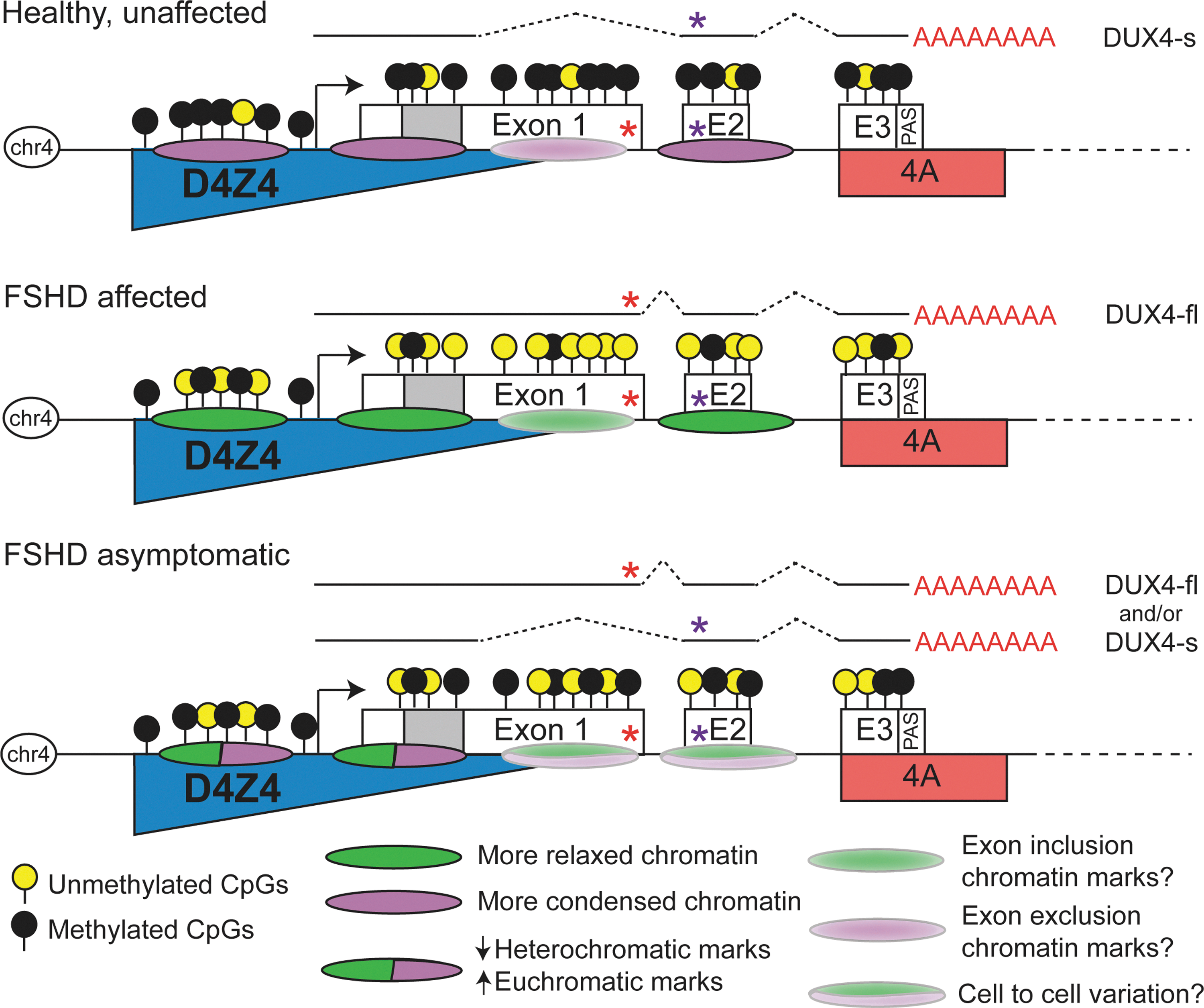
Chromatin is a highly complex and organized nucleoprotein structure that enables the ∼2 linear meters of the human genome to be packaged into a somatic nucleus which is ∼10 μm in diameter. Composed of DNA, histones, associated non-histone proteins, and RNAs, chromatin is a highly dynamic structure that not only serves to compact and package DNA into the nucleus, but is also involved in many nuclear processes, including gene and genome regulation. Through changes in its content, regulated and reversible modifications of its core components, and changes in nuclear location, chromatin provides an additional layer of regulation above the underlying DNA sequence that is capable of integrating with and responding to signals from the environment. Although dynamic, chromatin content and organization, once established for a locus, can also be highly stable and heritable, which has important consequences for maintaining gene expression patterns over the long term and across generations (67, 112). This has profound implications for FSHD, as many aspects of the 4q35 chromatin environment are different between FSHD-affected, asymptomatic, and healthy individuals (Figs. 2, 4, and 5).

Each D4Z4 repeat in the macrosatellite consists of ∼3300 bp of DNA (>15 nucleosomes); in the healthy population, the tandem arrayed copies number from 11 to more than 100 repeats, but on average 25–35 copies on both 4q arms (130, 138). Thus, FSHD1-sized D4Z4 contractions result in the absence of hundreds of nucleosomes containing GC-rich repetitive sequence, which significantly alters the chromatin content of 4q35 and likely affects establishment of the proper epigenetic state during development. This was demonstrated in a study using healthy and FSHD1-derived induced pluripotent stem cells (iPSCs) (143). Pluripotency led to a general relaxation of the D4Z4 chromatin and activation of DUX4 expression, but on myogenic differentiation only the FSHD1-derived iPSCs failed to establish heterochromatin at D4Z4 and repress DUX4-fl expression.
Highlighting the importance of epigenetic dysregulation at 4q35 in FSHD, contraction-independent FSHD2 shares a similar epigenetic profile with FSHD1, albeit on both 4q chromosomes, despite maintaining normal D4Z4 repeat lengths. In FSHD2, the epigenetic lesion is not caused by the physical deletion of chromatin, but by mutations in the proteins that are responsible for establishing and/or maintaining compaction of the arrays. The most commonly mutated gene in FSHD2 is SMCHD1 (structural maintenance of chromosomes hinge-domain protein 1; OMIM 614982), a GHKL family ATPase required for repressing repetitive elements and establishing DNA methylation at certain loci in plants and vertebrates (4, 95, 113). Interestingly, the murine Smchd1 is a modifier of metastable epialleles and is involved in X-inactivation, and the Arabidopsis orthologous SMCHD1 complex, composed of two proteins (the GHKL ATPase DMS11+the SMC hinge domain protein DMS3), is involved in RNA-directed DNA methylation (RdDM), highlighting the integral role of SMCHD1 orthologs in epigenetic regulation (4, 13, 101). Thus, the key shared genomic features of FSHD1 and FSHD2 are the changes in epigenetic status of the 4q35 D4Z4 array in a permissive 4qA haplotype that result in a more relaxed and less repressive chromatin state (40, 157).
This leads to the current favored model in which FSHD is caused by a disruption of epigenetic regulation at the usually silent D4Z4 macrosatellite, the consequences of which are changes in gene expression locally (e.g., increased DUX4-fl expression), regionally, and potentially globally (Fig. 5) (12, 38, 157). Importantly, the two key epigenetic regulatory systems found in vertebrates, DNA methylation and Polycomb Group (28, 77, 139), show disease-specific changes at D4Z4, providing strong support for the epigenetic model of FSHD. Additional FSHD-specific epigenetic alterations include changes in D4Z4 chromatin modifications, D4Z4 insulator activity, nuclear organization, and potential trans interactions between D4Z4 arrays (Fig. 5) (145). ncRNAs have recently been recognized as key epigenetic regulators of the genome; accordingly, the expression of ncRNAs, both proximal to and within the D4Z4 array, is altered in FSHD. Finally, the subtelomeric localization of the 4q35 array provides the potential for telomeric regulation that is altered in disease and during aging (7). In fact, telomeric effects have been reported to modulate nearby gene expression, and telomere length impacts gene expression differentially on the shortened FSHD1 arrays compared with healthy controls (144). Thus, many epigenetic mechanisms, discussed in greater detail next, are involved in regulating the healthy and disease states of the FSHD-associated 4q35 locus.
The similarities between FSHD1 and FSHD2 support a model in which epigenetic dysregulation is required for FSHD pathology. It is more difficult to explain the existence of asymptomatic individuals with a diagnostic FSHD1 genetic deletion and disease permissive haplotype, but no apparent muscle weakness. Presumably, the epigenetic status of these subjects would correlate with the lack of disease symptoms, despite the genetic diagnosis. Interestingly, an initial analysis of asymptomatic subjects shows that DNA methylation levels at the pathogenic distal D4Z4 repeat unit of short 4q35 alleles are higher than in FSHD-affected relatives possessing the same diagnostic deletion (Fig. 4) [Jones et al., Unpublished observation; (79)]. This is reminiscent of metastable epialleles, for which different epigenetic states are probabilistically established during embryogenesis and stably maintained, leading to different gene expression patterns (124). A characteristic of metastable epialleles is variable expression in the absence of genetic heterogeneity, including variable expression between cells of an individual, and variable expression and phenotypic mosaicism between individuals (44, 124). All these are characteristics of FSHD, suggesting that the 4q35 D4Z4 array functions as a metastable epiallele in this disease. Overall, with regard to clinical FSHD, the epigenetic components more closely correlate with disease presentation and noticeable muscle weakness than the known genetic component; thus, FSHD is clearly an epigenetic disease.
D4Z4 DNA methylation
Symmetrical methylation of DNA at cytosine in CpG dinucleotides is a key epigenetic mechanism involved in regulating and maintaining gene expression patterns in vertebrates (77). Approximately half the genes in the human genome contain regulatory regions that are enriched with CpG dinucleotides, termed CpG islands (CGIs). CGIs associated with active genes are usually unmethylated, while CGI methylation is associated with stable, long-term transcriptional silencing (72). Interestingly, each D4Z4 repeat is highly GC rich (73%) with characteristics of a CGI. However, each D4Z4 unit also contains GC-rich DNA repeat sequences (LSau and the hhspm3 repeats) that are usually associated with heterochromatin (68). It should be noted that due to the highly repetitive nature and high GC content of the FSHD region, and the existence of numerous other D4Z4 loci in the human genome, analyzing the 4q35 region, particularly with PCR-based assays, is fraught with difficulties which are not usually encountered when analyzing more typical regions of the genome (Box 1).

Initial analyses of D4Z4 arrays in healthy individuals showed that the arrays at 4q and 10q have characteristics of heterochromatin (9, 68, 105, 169). However, FSHD1 cells assessed for DNA methylation using methyl-sensitive restriction enzymes covering a few CpG dinucleotides in the proximal 4q D4Z4 repeat indicated hypomethylation compared with healthy controls (40, 160, 161). This hypomethylation was restricted to the contracted 4q D4Z4 array, suggesting that the physical loss of D4Z4 chromatin is the primary determinant of the hypomethylated state, likely due to a failure to establish heterochromatin at the short D4Z4 during development in FSHD1 patients (40, 143). Considering that FSHD1-sized D4Z4 contractions are polymorphic between patients and the disease severity and onset are quite variable, one might expect a close correlation between DNA methylation levels and clinical manifestation. There is, in fact, an imperfect correlation in which severely affected patients with very short alleles show pronounced hypomethylation, but methylation profiles at the other end of the FSHD1 contraction spectrum are less consistent and show great individual variation [Jones et al., Unpublished observations; (79, 160)]. Surprisingly, there does not appear to be a direct correlation between overall D4Z4 DNA methylation levels on the contracted 4qA allele and DUX4-fl expression levels in genetically defined FSHD1 myogenic cells [Jones et al., Unpublished observations; (78, 79)]. This likely indicates that specific sites of methylation as well as other chromatin modifications are important for regulating DUX4-fl expression and alternative splicing.
In contrast to FSHD1, FSHD2 patients exhibit pronounced hypomethylation of both 4q arrays as well as both D4Z4 arrays on 10q, none of which are contracted, indicating a disruption of the mechanism(s) establishing or maintaining DNA methylation at all D4Z4s (40). Bisulfite sequencing to assess DNA methylation at three regions across the D4Z4 repeat in FSHD2 cells confirmed a general hypomethylation on both 4q and 10q D4Z4s compared with controls; however, there was a dramatic focal demethylation at the most proximal region, which likely corresponds to an uncharacterized regulatory element (65). Thus, although the FSHD-associated D4Z4 array is hypomethylated, this demethylation is not uniform across the D4Z4 repeat. Local changes could have a large impact by affecting the binding of regulatory factors that are sensitive to DNA methylation status, such as methyl-CpG binding proteins, CTCF, or Kaiso (25). In fact, there are multiple consensus DNA recognition sites for many of these factors within each D4Z4 repeat. This is particularly relevant, as the most important D4Z4 repeat for both forms of FSHD, the distal-most unit, which contains the specific DUX4 gene that is transcribed, alternatively spliced, polyadenylated, and translated, has not been specifically analyzed. This underscores the need for a comprehensive methylation analysis of the locus, distinguishing the distal 4qA repeat from all others and analyzing the gene body in addition to the promoter and other regulatory regions.
Hypomethylation of D4Z4 repeats is not restricted to FSHD. Many repetitive regions, including D4Z4, are generally hypomethylated in cancer (47), and hypomethylation of the D4Z4 repeat has been found in leukemia cells (49); however, there is no reported link between cancer and FSHD. Similarly, homozygous loss-of-function mutations in DNMT3B (DNA methyltransferase 3B), which encodes the de novo DNA methyltransferase for D4Z4 repeats, lead to immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome (62, 173). ICF patients have extreme hypomethylation of all D4Z4 repeats, including the 4q35 macrosatellite, but show no indications of muscle weakness; however, the majority of ICF patients die before the typical age of onset in FSHD. Interestingly, as with Smchd1, mutations in Dnmt3b were identified in the same screen for modifiers of metastable epialleles (36). In future studies, we may find that mutations in DNMT3B, similar to SMCHD1, result in FSHD2 or modify the severity of FSHD1.
Overall, the DNA methylation data support the model that FSHD is caused by a decrease in local epigenetic repression, mediated in part by DNA hypomethylation, and the aberrant relaxation of the D4Z4 region, including increased sporadic expression of DUX4-fl. The dramatic changes in the D4Z4 DNA methylation profiles in FSHD are accompanied by numerous other changes in chromatin content, discussed next, and an overall disruption of the epigenetic state, essentially creating a metastable epiallele at 4q35.
D4Z4 chromatin
In concert with DNA methylation, PTMs of histones, histone variants, and chromatin-associated proteins represent another, more dynamic mechanism for regulating gene expression (11, 45, 83, 88, 132). Together, these modifications present unique interaction surfaces, in addition to DNA sequence, which are differentially recognized by the nuclear machinery to translate the information into a wide variety of dynamic and heritable states (132). Genome-wide mapping as well as single gene studies have shown that certain combinations of histone PTMs and chromatin-associated proteins tend to be associated with particular expression states and regions of the genome (32). Combinations of chromatin marks can be used to predict transcriptionally active, poised, or repressed promoters/enhancers, transcribed or unexpressed gene bodies, alternatively spliced exons, and transcriptionally silent regions. Similar to the changes in DNA methylation observed between FSHD and healthy genomes, there are also marked changes in histone PTMs and associated proteins (6, 26, 95, 180); however, due to challenges in analyzing the FSHD-associated array (Box 1), the story is still incomplete.
Initial chromatin immunoprecipitation (ChIP) experiments investigating histone H4 acetylation levels and immuno-fluorescent in situ hybridization analysis of the 4q35 D4Z4 array failed to find clear differences between FSHD and healthy controls; however, the region displayed characteristics of unexpressed euchromatin rather than constitutive heterochromatin (75, 174). A more thorough ChIP analysis of the 4q35 and 10q26 D4Z4 arrays investigating the repressive histone mark H3K9me3 (lysine 9 tri-methylation) and its associated histone reader, heterochromatin protein 1 (HP1) showed that H3K9me3 was decreased at the D4Z4 array in both FSHD1 and FSHD2 patients and this correlated with a loss of HP1γ and cohesin recruitment to the array (180). Importantly, H3K9me3, HP1γ, cohesin, and DNA methylation levels are not diminished at other D4Z4 homologs in the genome in FSHD cells, indicating that epigenetic alterations at D4Z4 in FSHD are specific to 4q and 10q (179). This underscores the importance of analyzing only the relevant regions in sequence-based assays (Box 1). Similarly, a recent analysis of chromatin compaction (ratio of the repressive H3K9me3 to the active H3K4me2 mark) at the DUX4 promoter in each 4q/10q D4Z4 repeat confirmed that this region has a relaxed chromatin environment in both FSHD1 and FSHD2 cells compared with healthy controls (6). The DUX4 promoter exhibited even less compaction and much less variability in FSHD2 versus FSHD1 cells, perhaps reflecting an overall chromatin relaxation at all four arrays, consistent with the results of DNA methylation studies (40). Interestingly, despite the correlation with the presence or absence of disease, there was no correlation between the amount of chromatin compaction and clinical severity. This mimics the DNA methylation results and suggests that additional determinants of disease progression lie outside the D4Z4 locus (6).
Analysis of another well-studied repressive histone mark, H3K37me3, which is mediated by the Polycomb repressive complex 2 (PRC2) histone methyltransferase (HMT) EZH2 and associated with long-term repression (28), showed no FSHD-specific difference in levels within the D4Z4 array (179, 180), although H3K27me3 levels decrease during differentiation of both FSHD and control myoblasts, consistent with the increased expression of genes in the region (DUX4, FRG1, and FRG2) during muscle differentiation (17, 51, 70, 78, 129). By contrast, there is an FSHD-specific reduction in H3K27me3 levels and PRC binding, and a corresponding enrichment of the transcriptional activating HMT Trithorax group protein ASH1L, in the region immediately proximal to the array. This region is in close proximity to the FSHD-specific DBE-T long non-coding RNA (lncRNA) promoter, discussed next (17, 26).
In addition to mediating activity of promoters and enhancers, localized chromatin marks and transcription factors can affect gene expression via regulation of alternative splicing (102, 103, 135). For example, CTCF binding promotes inclusion of weak upstream alternatively spliced exons, and a number of histone marks have been shown to correlate with exon inclusion splicing decisions (135, 141). In FSHD, not only do levels of DUX4 expression increase, but there is also a change in the mRNA isoform produced due to alternative splicing, specifically a change in 5′ donor splice site usage (143). DNA methylation, H3K9me3, CTCF, and HP1 play roles in the regulation of alternative splicing (135, 141) and there is evidence that each of these is differentially represented in FSHD-affected versus healthy arrays. It is conceivable, for example, that DNA hypomethylation in the DUX4 gene body could allow for CTCF binding to its consensus motif in exon 1, thus mediating a switch from the innocuous DUX4-s to the pathogenic DUX4-fl mRNA. While this type of analysis has yet to be reported, likely due to technical challenges as discussed (Box 1), targeting alternative splicing with small molecules is a viable therapeutic approach, thus underscoring the need to understand mechanisms of alternative splicing in FSHD (8, 146).
ChIP-seq data released from the ENCODE consortium (32) (
The 4q35 D4Z4 chromatin architecture
D4Z4 arrays on chromosomes 4q35 and 10q26 each consist of >100 kb of GC-rich repeat subtelomeric DNA in healthy individuals. Perturbations in these regions have the potential to affect intra-chromosomal interactions, nuclear localization of the respective chromosomes, and global nuclear architecture. FSHD-specific changes in gene expression and chromatin architecture, and whether these translate to pathogenesis, are still being investigated and debated. Numerous gene expression studies on adult myocytes and muscle biopsies have generally failed to come to a consensus on which gene(s) outside of DUX4, if any, are subject to misexpression in FSHD (55, 71, 86, 123). However, expression analysis of 4q35-localized genes and genes that are important for myogenesis in human fetal muscle supports an FSHD-specific disruption in gene regulation both in the region and globally during development, at a time when the epigenetic state is still being established (23).
At the other end of the age spectrum, telomere length has been found to play an FSHD-specific regulatory role in 4q35 gene expression, which could be mediated through changes in intrachromosomal interactions, epigenetic state during aging, or nuclear positioning (144). FSHD myoblasts with short telomeres exhibit a distance-dependent increase in expression of 4qter genes, with a large effect on DUX4, a moderate effect on FRG2 (∼40 kb proximal to D4Z4), and no effect on FRG1 (∼130 kb proximal to D4Z4) (144). This study suggested that age-dependent shortening of telomeres may cause increased expression of DUX4 in FSHD muscle cells, due to a loss of D4Z4 insulator function against telomeric heterochromatin (116) in a contracted allele that may be mediated, in part, by changes in nuclear localization. In this model, myoblast proliferation during one's lifetime results in telomere shortening, leading to increased toxic DUX4-fl expression and the progressive cycles of degeneration and regeneration observed in FSHD. This provides one potential explanation for the delayed onset of FSHD pathogenesis. In addition, intrinsic variability in telomere length may also play a role in the variable manifestation of the disease, although this remains to be demonstrated.
Unlike most telomeres, 4qter is localized to the nuclear envelope and the nuclear lamina component lamin A/C is required for this localization (108). Interestingly, several other neuromuscular disorders (Emery–Dreifuss muscular dystrophy, limb-girdle muscular dystrophy 1B, and dilated cardiomyopathy) are caused by defects in the nuclear envelope, either through a disruption of nuclear structural integrity or through an alteration of signaling pathways and gene expression (107). In one model, contractions in D4Z4 and the presence of the β-satellite in 4qA might lead to altered recruitment of transcription factors or chromatin modifiers at the nuclear envelope, resulting in a dystrophic phenotype (108). In support of this model, each D4Z4 repeat in the 4q35 array contains several potential binding sites for CTCF, a multifunctional protein that can mediate long-range chromatin interactions, associate with the nuclear matrix, act as an enhancer-blocking insulator, or function in gene activation and repression (63, 110, 116, 177). In transfection assays, a single D4Z4 unit behaves as a CTCF and lamin A-dependent transcriptional insulator that is capable of enhancer blocking and barrier activity, and these activities are lost on multimerization (116). In this model, FSHD deletions, DNA hypomethylation, and subsequent CTCF binding lead to formation of a gain-of-function insulator at contracted D4Z4 repeats, which, in turn, blocks the normal repression of the proximal genes mediated by the 4q distal subtelomere and telomere (116).
The region proximal to the array contains multiple potential CTCF-binding sites as well as a nuclear scaffold/matrix attachment region (S/MAR) that can function as an enhancer blocking boundary element (119). The association of this S/MAR with the nuclear matrix is regulated by its epigenetic status and diminished in FSHD myogenic cells compared with controls (84, 120). The D4Z4 region and neighboring genes usually lie in two chromatin loops, whereas loss of nuclear matrix attachment in FSHD resulted in formation of a single loop due to loss of methylation of a single CpG and the presence of H3K9 acetylation (84, 120). In this study, loss of this functional boundary element in FSHD cells allowed a transcriptional enhancer located within D4Z4 to effect de-repression of the proximal genes (119).
CTCF binding may also mediate changes in long-range chromosome interactions in FSHD. The 4q35 region interacts in cis with other regions of chromosome 4 and likely in trans with other loci. Chromosome conformation capture demonstrated a number of interactions in the 4qter region in both normal and FSHD myoblasts (121). In particular, an FSHD-specific association between the region distal to D4Z4 (4qA/B) and proximal genes ANT1, FRG1, and DUX4c was observed (121). A similar study showed that the D4Z4 repeat interacts with two proximal genes, TUBB4q and FRG1, in both FSHD and control myoblasts (17). When the cells were induced to differentiate, the association with FRG1 was greatly reduced, whereas the association with TUBB4q was maintained (17). Although there is no consistent evidence that any of these genes are misregulated in FSHD adult myocytes, these studies demonstrate that the 4qter region makes long-distance contacts which are altered in FSHD cells or during muscle differentiation.
The 4q35 ncRNAs
Bidirectional transcription of tandem repeats leading to formation of dsRNAs is a regulatory mechanism for mediating heterochromatin assembly in cis that is conserved from fission yeast and plants to flies and vertebrates (93, 118, 122, 133, 163). These dsRNAs can function in either transcriptional gene silencing (TGS) or post-transcriptional gene silencing (PTGS) (133). Characteristic of macrosatellite repeats, the 4q35 D4Z4 array is actively transcribed in both directions, generating ncRNAs with transcripts originating both from within and outside the array (16, 26, 142). Along with the two protein-coding DUX4 isoforms, the D4Z4 array at 4qA generates a plethora of sense and antisense transcripts, including siRNA and microRNA (miRNA)-sized fragments, in both FSHD and normal muscle cells (142, 143). Although the functional significance of these ncRNAs has not been addressed, it is tempting to speculate that they may act to modulate transcription of DUX4 or to modify chromatin structure either in cis, as in the case of D4X4 repeats (29), or in trans. Keeping in mind that D4Z4 is an invasive retrotransposon element, si-RNAs derived from within the array could be functioning in RdDM to mediate silencing of the array (82).
New data from Arabidopsis are particularly intriguing in this regard. Heterochromatic (het)siRNAs direct DNA methylation and TGS of transposons (91). However, transposons are also targeted by a PTGS mechanism that is responsive to changes in DNA methylation levels. Reduced DNA methylation on transposons during reprogramming in the germline or due to mutations in the DNA methylation machinery leads to their transcriptional reactivation, but the RNAs generated are subsequently targeted by miRNAs to generate epigenetically activated siRNAs (easiRNAs) for effective PTGS of the transposon (33). Although neither of these mechanisms has been validated in vertebrates, TGS via RdDM occurs in humans, and similar types of regulation and genome protection may be functioning in the D4Z4 region (82).
The 4q35 region and D4Z4 array can also be regulated by at least one lncRNA (26). Each D4Z4 repeat contains a region, termed the D4Z4-binding element (DBE), that is capable of recruiting the PRC2 components YY1 and EZH2, and the Polycomb recruiter protein HMGB2, to mediate transcriptional silencing (17, 42, 51). In myogenic cells from FSHD1 patients, the DBE is transcribed using a D4Z4-proximal promoter to produce the DBE-T lncRNA (26). DBE-T binds to the D4Z4-proximal 4q35 region in cis, thus only on the contracted allele, and can mediate de-repression of genes in the 4q35 region, including DUX4 (26). One function of lncRNAs is the localization of effector complexes to specific sites in chromatin (81) and DBE-T is proposed to function in a similar capacity (26). De-repression of the 4q35 region takes place through recruitment of the TrxG protein ASH1L, which places the activating histone marks H3K4me3 and H3K36me2 (59, 147, 176). Conversely, in cells from healthy subjects, the non-contracted D4Z4 arrays show increased PcG binding and no transcription of DBE-T. Collectively, these results support a model in which the presence of long D4Z4 arrays in healthy subjects enables extensive binding by PcG proteins, resulting in DNA methylation, histone deacetylation, and a repressive chromatin environment.
Epigenetic modifiers
Metastable epialleles are inherently variable in their expressivity, and the associated diseases display great individual variability and phenotypic mosaicism. Importantly, environmental and genetic factors can influence and skew the probabalistic epigenetic state established during development (4, 14, 44, 76). Indeed, environmental factors have been shown to drive major epigenetic changes, as in the case of fetal alcohol syndrome, in which alcohol alters developmental patterns of DNA methylation (126), and maternal diet, which can affect the methylation and expression status of a metastable epiallele in mice (166). As discussed, the FSHD-associated 4q35 D4Z4 array has the epigenetic and gene expression characteristics of a metastable epiallele in both forms of FSHD. The clinical phenotypes, ranging from asymptomatic to severe, even within families and between siblings, as well as the common asymmetry and apparently random muscle weakness, support a vital role for epigenetic modifiers.
In FSHD, the high variability in clinical phenotype, even between monozygotic twins (60, 149, 154), suggests that epigenetic mechanisms—perhaps determined in part by genetic and/or environmental factors—are likely to be a major determinant of penetrance and severity. In a study of seven FSHD1 cohorts, some gene expression patterns were found to differ significantly between families (71), underscoring the importance of analyzing paired cohorts in studies of disease gene expression. DUX4-fl expression levels are also variable among families (78), and different families display differential methylation of the DUX4 promoter and gene body [Jones et al., Unpublished observation; (79)]. A general hypomethylation of these regions correlates with the DUX4 expression observed in FSHD1 and FSHD2 myocytes, with the gene body showing significantly less methylation than the promoter; conversely, hypermethylation of these regions is consistent with the extremely rare DUX4 expression in unaffected myocytes and non-muscle cells [Jones et al., Unpublished observation; (78, 79, 143)]. Although the mechanisms are still unclear, methylation of promoters results in altered binding of some transcriptional regulators, whereas methylation within the gene body can modify patterns of alternative splicing (77, 109, 141). Thus, epigenetic mechanisms might lead to differences in DUX4-fl expression or stability, resulting in differential activation of downstream targets and, consequently, variable pathology.
A recent breakthrough in the field was the identification of SMCHD1, the gene most commonly mutated in FSHD2, as a modifier of disease severity in several severe cases of FSHD1 (95, 134). In this study, three out of six unrelated patients with borderline FSHD1 alleles (8–10 D4Z4 units) and marked D4Z4 hypomethylation (indicative of FSHD2) also had mutations in SMCHD1 (134). In general, D4Z4 repeat length tends to correlate inversely with clinical severity (57, 104, 127, 148); however, the three patients with mutations in SMCHD1 exhibited very severe clinical phenotypes, despite having borderline alleles (nine D4Z4 units each) (134). Knocking down SMCHD1 protein in FSHD1 myotubes led to increases in both DUX4-fl mRNA and DUX4 target gene expression, suggesting that the modifying role for SMCHD1 in determining FSHD1 severity was at the 4q35 locus (134). However, SMCHD1 regulates more than D4Z4 arrays; it is required for maintenance of DNA methylation on the inactive X chromosome, regulates autosomal genes with monoallelic expression, and has enhanced binding to long telomeres, where it likely plays a role in establishing silent chromatin (13, 54, 61, 113). It is possible that a reduction in SMCHD1, in addition to mediating increased DUX4-fl expression, contributes to the extreme disease phenotype by affecting expression of many other genes not typically misexpressed in FSHD (e.g., imprinted genes). Thus, severe forms of FSHD may actually represent a complex disorder with multiple etiologies. Considering the number of asymptomatic but genetically FSHD1 individuals in the general population, in FSHD1-affected individuals, the presence of modifying mutations that affect other genetic loci may be somewhat common, at least for severe cases.
Additional FSHD2 genes, and thus candidate epigenetic modifiers of FSHD1, should exist. Whole-exome sequencing of FSHD2 families revealed that while 15 out of 19 families had mutations in SMCHD1, four families had no changes in SMCHD1 (95). In addition, 5 out of 26 FSHD2 patients with a mutation in SMCHD1, D4Z4 hypomethylation, and a permissive 4qA allele were, nonetheless, asymptomatic (95). These data strongly implicate the existence of additional FSHD modifiers. Since the only known modifier of FSHD functions as an epigenetic regulator establishing repressive chromatin, it is reasonable to suspect that other factors with similar roles are good candidates for modifiers of FSHD.
Smchd1 was originally identified in an elegant forward genetic screen for dominant modifiers of murine metastable epialleles (MommeD), performed in the Emma Whitelaw lab (14). The identities of many of the underlying mutations have been recently reported and, not surprisingly, many occur in genes with known functions in DNA methylation and chromatin modification (4, 13, 14, 36, 37). The screen revealed both enhancers and suppressors of epigenetic variegation, including DNMTs (Dnmt1 and Dnmt3b), HMTs (Setdb1, Suv39h1), a histone deacetylase (Hdac1), components of chromatin remodeling machines (Smarca4/BRG1, Smarca5, Smarcc1/BAF155, Pbrm1/BAF180, and Hdac1), epigenetic regulators (Smchd1, Uhrf1, Trim28/KAP1/TIF1β, and WIZ), telomeric proteins (Rif1, Smchd1), chromatin-dependent transcriptional regulators (Brd1, Rlf, and Baz1b), and the translation initiation factor eIF3h.
Interestingly, many of the identified proteins are either components of the same complex or work together in linked pathways to establish repressive chromatin. For example, Trim28/KAP1 is an E3 SUMO protein ligase (73). Several heterochromatin proteins are known targets of SUMOylation, including SMCHD1 and HP1α, which localize to the expanded D4Z4 arrays (106, 152). SUMOylation promotes targeting of HP1 and recruitment of the NuRD chromatin-remodeling complex, which recruits the SETDB1 HMT (73). This, in turn, enhances the repressive H3K9me3 mark, leading to increased HP1 deposition and enhanced heterochromatinization. UHRF1 is a multifunctional E3 ubiquitin ligase that also acts as a negative regulator of transcription, recruiting chromatin proteins to regions enriched for H3K4me0/K9me3 (80, 114). Of particular interest, UHRF1 interacts with DNMT1, acting as a link between H3K9me3 and maintenance of DNA methylation patterns (131).
It is evident from the Whitelaw modifier screen that disruption of these processes tips the balance in determining epigenetic states at metastable epialleles, toward being either more euchromatic (mutations in suppressors) or more heterochromatic (mutations in enhancers) (14). These epigenetic modifiers provide insights into the types of proteins involved in establishing and maintaining repression at vertebrate metastable epialleles such as the FSHD locus, and it is reasonable to suspect that inactivating, hypomorphic, or dominant negative mutations in such factors could modify FSHD1 or be causal for FSHD2.
Several repeat expansion disorders, including Fragile X Syndrome, myotonic dystrophy type I (DM1), Friedrich's Ataxia, and amyotropic lateral sclerosis, are characterized by the expanded repeat triggering DNA methylation and repressive histone modifications, which lead to decreased gene expression (66, 172). Although it remains to be shown, it is certainly possible that the range in phenotypic severity for each disease, which is particularly striking in DM1, is not just a function of repeat length, but dependent on epigenetic modifiers. Defects in epigenetic regulators are responsible for a growing number of genetic disorders (10), and such regulators are prime candidates for modifiers of FSHD and other complex diseases.
Therapeutic approaches to FSHD
While therapeutic approaches for many myopathies and other diseases have focused on the replacement, correction, or reactivation of the mutated disease gene, FSHD presents a different problem: a disease locus and gene product that need to be silenced. There are numerous potential therapeutic targets for FSHD (Fig. 6), and many of these avenues are actively being investigated. For example, small-molecule activation of the Wnt/β-catenin signaling pathway suppresses DUX4 expression in FSHD myotubes (15) and small-molecule pharmacological inhibitors of DUX4-FL myoblast toxicity have been reported (18). FSHD would seem to be an ideal candidate disease for dsRNA and antisense oligonucleotide therapies, and successful proof-of-principle experiments have already been performed (162, 165). However, although DUX4-fl appears to be an excellent therapeutic target, there is a lack of clear evidence that DUX4-fl expression in adult skeletal muscle is sufficient to cause disease or that simply blocking its expression would be therapeutically beneficial. Any mouse model of FSHD suffers from the caveat that DUX4 and a number of DUX4-FL gene targets are primate specific (55, 175), and the first published DUX4-fl transgenic mouse appears quite healthy despite expressing DUX4-FL and a number of its downstream targets in skeletal muscle (90). It is not clear whether this is due to the lack of true pathogenic targets in the mouse, a failure to recapitulate the spatiotemporal pattern of DUX4-fl expression that takes place during human development, or whether the low levels of expression in adult muscle are simply not enough to generate pathology in mice. It is encouraging that expression of DUX4-fl in another mouse model causes pathology, albeit in multiple tissues (35); presumably, a useful FSHD-like mouse model can be generated with proper regulatory mechanisms in place to ensure correct levels and timing of DUX4-fl expression. However, the fact that some asymptomatic subjects express levels of DUX4-fl similar to those of affected patients strongly suggests that FSHD requires more than a simple elevation of DUX4-fl expression in adult skeletal muscle (78).

A viable alternative therapeutic approach for FSHD involves targeting the aberrant epigenetics and epigenetic modifiers in the region to restore a healthy, nonpathogenic gene expression profile (Fig. 6). This type of approach would not necessarily be DUX4 dependent and could simultaneously address additional factors involved in pathology. Fortunately, the D4Z4-2.5 and D4Z4-12.5 mouse models have recapitulated much of the epigenetic status of the human locus and will be useful for testing epigenetic-based therapies (90).
There are several reasons to specifically target epigenetic dysregulation for therapy of FSHD. Affected and asymptomatic individuals in FSHD1 families exhibit highly variable levels of DUX4-fl expression in cultured myogenic cells that do not correlate with disease manifestation (78). However, FSHD1-affected subjects differ epigenetically from asymptomatic and healthy individuals using DNA methylation or epigenetic stability of D4Z4 as a readout [Jones et al., Unpublished observation; (79)]. Thus, as opposed to expression levels of DUX4-fl or of DUX4-FL target genes, there is an epigenetic difference that correlates well with disease manifestation. In addition, when considering the role of SMCHD1 in FSHD1 and FSHD2, it should be noted that SMCHD1 is also required for the monoallelic expression of some imprinted genes and the clustered protocadherins (113), suggesting that alterations in SMCHD1 could also modify genes that cause diseases of imprinting and neural circuit assembly. While the FSHD focus is on the D4Z4 region and DUX4 expression, other regions of the genome are similarly affected by mutations in epigenetic modifiers. Targeting epigenetic dysregulation could potentially correct this more complex issue.
Conclusion
Although genetic conditions are required for pathology and numerous unresolved questions remain (Box 2), it is clear that FSHD is primarily an epigenetic disease. Many epigenetic mechanisms function at the disease locus, either to maintain repression in healthy and asymptomatic individuals or to create a permissive environment for gene expression in FSHD patients. The 4q35 D4Z4 macrosatellite has multiple epigenetic states both among the healthy and affected populations as well as within families, displaying cell-to-cell variegation and individual variability. Thus, FSHD provides an opportunity to investigate a human disease locus with characteristics of a metastable epiallele. In addition, the FSHD locus is subject to the action of multiple epigenetic modifiers, mutations that may be causal for disease, protect from disease, or affect disease severity. It is clear that FSHD serves as an excellent model to investigate epigenetic gene regulation and epigenetic modifiers in the context of human disease.

Footnotes
Acknowledgments
This work was financially supported by the National Institute of Arthritis, Musculoskeletal, and Skin Diseases grant number 1R01AR062587. The authors thank Jennifer Burgess, Chris Carrino, and the Chris Carrino Foundation for FSHD, and Daniel P. Perez and the FSH Society for their support of the authors' FSHD projects.