
Abstract
Introduction
S
The endocrine and exocrine tissues of the pancreas play central roles in systemic metabolism by secreting important hormones such as insulin, glucagon, somatostatin, and pancreatic polypeptide (PP), as well as several amylases and proteases. Both congenital and acquired pathologies lead to pancreatic dysfunction in adults, resulting in hormonal imbalances. Most striking is the systematic loss and/or impairment of pancreatic β-cell function that underlies chronic hyperglycemia and defines diabetes mellitus, affecting nearly 300 million people worldwide (121). While type 1 diabetes (T1D) is caused by autoimmune destruction of insulin-producing β-cells leading to absolute insulin deficiency, type 2 diabetes (T2D) is a condition characterized by insulin resistance and progressive decline in β-cell function. Despite the effectiveness of current strategies to manage blood glucose, both T1D and T2D patients can continue to develop chronic vascular complications (24, 74). Finding alternative strategies to regain metabolic homeostasis by endogenous insulin secretion is a major focus of current research.
Recent efforts to restore endocrine pancreas function by direct injection of pancreatic progenitors into diabetic mice have shown promising results for β-cell replenishment and glycemic control (54). Rapid developments in our understanding of transcriptional control in islet cells have led to novel strategies to generate functional β-like cells by direct delivery of endocrine factors. For example, the expression of endocrine transcription factors such as paired box 4 (Pax4) and pancreatic and duodenal homeobox 1 (Pdx1) can reprogram endogenous noninsulin-producing cells of pancreatic origin to insulin-producing “β-like” cells (2). This review addresses key concepts in pancreatic endocrine development and provides insights into the leveraging of developmentally critical transcription factors to efficiently reprogram α-cells to insulin-producing β-like cells. Several endocrine transcription factors are believed to play crucial roles in provoking α-to-β-like reprogramming, and the challenge now is to understand precise mechanisms of interplay between endocrine factors that regulate gene expression and epigenetic modifications in the production and secretion of insulin (2). To this end, we discuss potential roles for epigenetic modifiers such as histone acetyltransferase (HAT), histone deacetylase (HDAC), and histone methyltransferase (HMT) enzymes to enhance β-cell regeneration and insulin production in the diabetic pancreas.
Pathogenesis of Diabetes
The pancreas has complex anatomical specialization central to its endocrine and exocrine functions (Fig. 1). Almost 95% of the pancreas consists of pancreatic acini, the exocrine tissue that secretes a host of digestive enzymes such as amylases and proteases into the small and large intestines. The islet of Langerhans, the endocrine tissue, constitutes less than 5% of the pancreas. The β-cell population represents the major constituent (∼75%) of pancreatic islets. This tissue further comprises three additional cell types, α, δ, and PP, that secrete glucagon, somatostatin, and pancreatic-polypeptide hormones, respectively (Fig. 1).
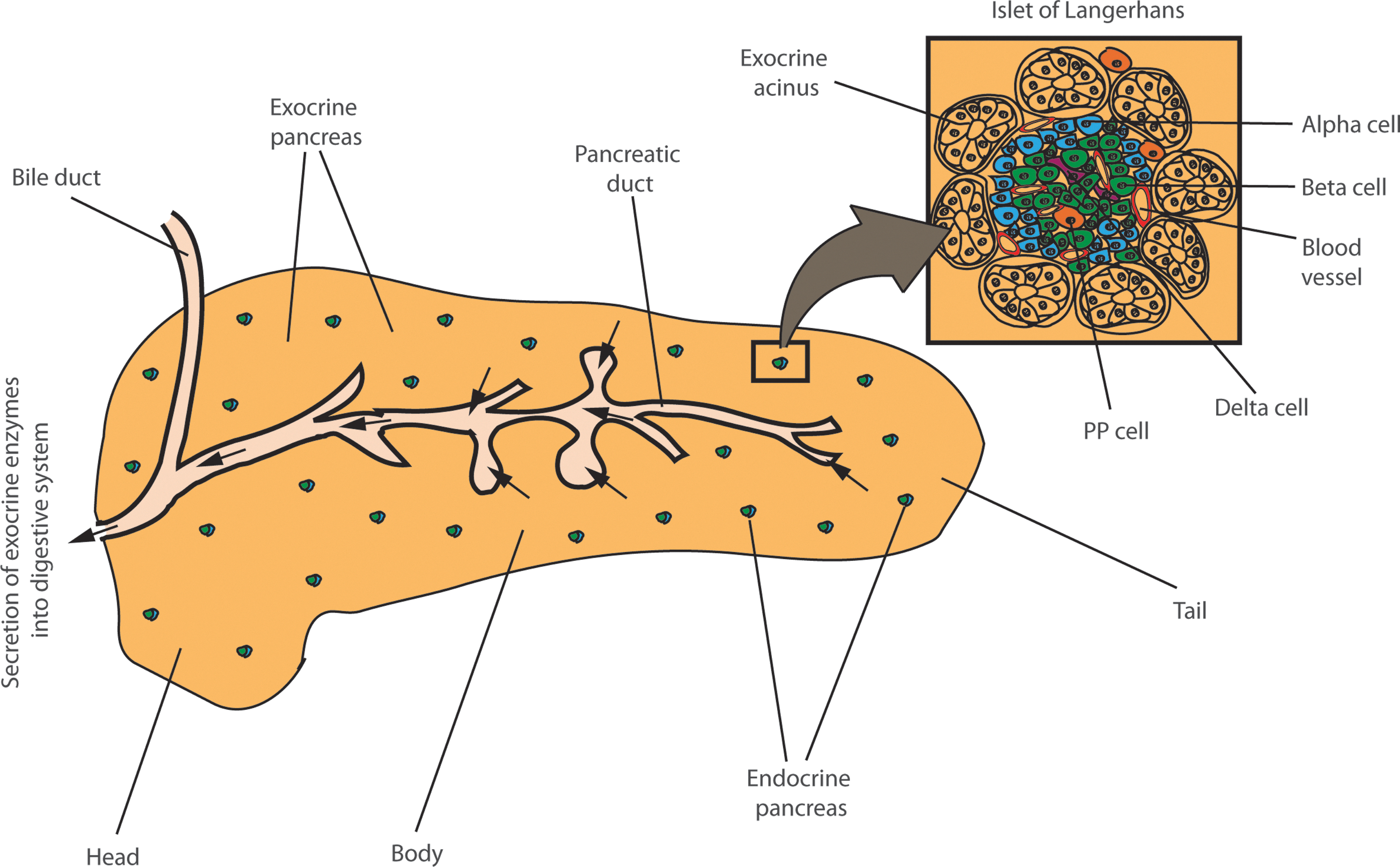
The β-cell is the primary cell type responsible for production and secretion of insulin into the blood stream (78). In T1D, several β-cell-specific auto-antigens such as GAD65 and islet antigen 2 (IA-2) are major targets of cell-mediated autoimmunity (10, 45). These autoantigens are currently believed to serve as epitopes for β-cell specific activation of CD4+ and CD8+ autoreactive T-cells, the primary initiators of β-cell destruction in the onset of T1D (34, 107). It is this autoreactive T-cell stimulation that leads to infiltration of pancreatic islets by mononuclear cells, resulting in inflammatory responses (5, 69). As a consequence, progressive decline in β-cell mass and insulin secretion accelerates over a period of years, causing chronic hyperglycemia. By comparing the long-term benefits of intensive and conventional therapy for hyperglycemia in T1D, the Epidemiology of Diabetes Intervention and Complications (EDIC) follow-up observational study to the Diabetes Control and Complications Trial (DCCT) identified that previous periods of poor glycemic regulation significantly increased the risk for developing chronic vascular complications (15, 38, 72). These data support the need to develop robust and novel strategies to restore β-cells and insulin production to more effectively treat hyperglycemia. Table 1 summarizes multiple approaches in cell reprogramming, including the recent emergence of innovative epigenetic strategies that have been successfully employed for reprogramming of distinct cell types.
Reprogramming the Diabetic Pancreas
A major restriction to repair damaged endocrine tissue in T1D is that the process of β-cell neogenesis from ductal-cell progenitors is limited after birth (99). Recent preclinical studies of diabetes show higher β-cell plasticity than previously appreciated (101). As opposed to neogenesis, recent findings implicate pre-existing β-cells as the predominant source of β-cell turnover in adult mice (22). In addition, hormones such as gastrin, glucagon-like peptide-1, and epidermal growth factor are believed to accelerate β-cell expansion (94). Recent clinical trials have shown that transplantation of human islets can alleviate insulin dependence in T1D (19, 85). However, the scarcity of organ donors combined with islet rejection have prompted research and development into alternative sources of functionally competent, insulin-secreting β-cells to meet the clinical need for transplantation therapy (13).
Both α and β-cells are marked by specific expression of endocrine transcription factors such as the Pancreas transcription factor 1 subunit alpha (Ptf1α), indicating a common developmental progenitor (30, 46). Remarkably, glucagon+ α-cell reprogramming to glucagon+/insulin+ β-like cells was recently found to drive increased insulin secretion and recovery from diabetes in animal models of toxin-induced acute selective near-total β-cell ablation (2, 101). These findings form the current rationale in diabetes research for transcriptional reprogramming of adult pancreatic exocrine and endocrine cells to produce insulin.
Gene Regulation in the Endocrine Pancreas
Recent advances in nucleic acid sequencing technologies enable accurate quantification of cell-specific and tissue-specific gene expression patterns with precise identification of novel splice variants to known and unknown genes. For example, gene expression profiling of pancreatic tissue highlight abundant RNA splicing (23). β-cell destruction in T1D is mediated by cytokine secretion, and recent data demonstrate that stimulation of β-cells by pro-inflammatory cytokines not only regulates gene expression but also results in mRNA splicing of several genes implicated in T1D (23). These new data suggest that regulation of genes implicated in diabetes occurs at multiple levels, such as post-transcriptional splicing in addition to transcriptional regulation.
Long intergenic noncoding RNAs (lincRNA) have recently emerged as key regulators of gene expression (66, 84). Hundreds of lincRNAs show β-cell specific expression (73, 84). Similarly, more than 5000 protein coding genes are differentially expressed in β-cells compared with the whole islet isolated from human pancreas (73). Tissue-specific gene expression is precisely controlled by coordinated actions of transcription factors and epigenetic enzymes that covalently modify histone proteins. Identification of precise interplay between endocrine factors, histone modifiers, and long noncoding RNAs (lncRNAs) that co-ordinate β-cell specific expression of coding and noncoding genes is likely to contribute to the development of novel therapies in diabetes research.
Direct Reprogramming by Transcription Factors
Identifying cell-specific transcription factors critical for lineage development is key to the ultimate success of regenerative medicine. Direct lineage conversion of endogenous cells by forced expression of defined transcription factors has proved successful in the regeneration of a variety of tissue types, including cardiac and renal, in preclinical disease-relevant models (42, 83). Recent reports have documented examples of several transcription factors that are responsible for pancreatic cell reprogramming (2, 101).
The Pax4 transcription factor is a critical regulator of pancreatic β/δ cell specification during early embryogenesis and throughout adult life (92). Ectopic expression of Pax4 can reprogram noninsulin producing cells such as endocrine precursors and mature α-cells into insulin-producing β-like cells (17). This was recently demonstrated in vivo, where regulated Pax4 misexpression-mediated reprogramming of mature α-cells to β-like cells significantly increased insulin production compared with control animals (2). Similarly, expression of Arx is essential for α-cell differentiation in the mammalian pancreas, and inactivation of this transcription factor efficiently triggered neogenesis and conversion of α-cells to functional β-like cells (16, 18, 111). In addition, the Pdx1 transcription factor has been shown to reprogram α-cells to bona fide β-like cells. Transgenic expression of Pdx1 sufficiently reprogrammed α-cells to β-like cells at early embryonic stages in mice (115). Furthermore, adenovector-mediated in vitro co-expression of pancreatic transcription factors Pdx1, Ngn3, and MafA efficiently reprogrammed exocrine pancreas and primary hepatocytes into insulin-producing β-like cells (1). In addition to α-cells, the adult duct-lining cells can be reprogrammed by forced expression of transcription factors in diabetic mice (2, 18).
Modulation of gene expression by ectopic expression of transcription factors is often supplemented with stimulatory factors to increase reprogramming efficiency (Table 1). A recent study exploited the role of ligand-bound thyroid hormone receptor α (TRα) in expansion of β-cell mass during postnatal development (28). Adenovirus-mediated expression of TRα in pancreatic acinar cells activated β-cell-specific transcription factors such as Hnf6, Foxa2, Nkx2.2, NeuroD1, Pdx1, Ngn3, and MafA when stimulated by thyroid hormone (28). These results suggest that identification of cell-specific markers within the endocrine system may be exploited to modulate gene expression by small-molecule compounds that drive direct reprogramming to insulin producing cells. Although the endocrine transcription factor Ngn3 is a marker of adult β-cell neogenesis, activation of Ngn3 is insufficient to reprogram pancreatic duct to β-like cells (95, 113). However, inhibition of delta-Notch signalling using gamma-secretase inhibitor L685,458 (L6) as well as coexpression of Myt1 along with Ngn3 showed moderate improvement in reprogramming efficiency of human duct cells into endocrine cells (95). Identifying key signaling pathways and master regulators mediating the complex interactions of networks that drive endocrine progenitors into α-cells that can then transdifferentiate into newly functional β-cells will greatly improve reprogramming efficiency and reduce off-target effects.
Epigenetics and Cellular Reprogramming
Epigenetic mechanisms involve chemical and structural modifications to the dynamic DNA-histone complex known as chromatin. Gene transcription is primarily controlled by the degree of chromatin accessibility, and epigenetic modulation is fundamental to nuclear processes and cellular homeostasis. Postreplicative addition or removal of methyl-groups at cytosine residues within the DNA sequence is classically associated with chromatin condensation and gene silencing. The methyl modification, as well as a plethora of other modifications such as acetylation also occurs at the N-terminal tails of core histone proteins to modulate chromatin architecture (11). Direct reprogramming by adenovirus-mediated ectopic expression of transcription factors influences gene expression through changes to chromatin structure and function (5, 117). More research is required to precisely understand the mechanism of exchange of transcription factors and co-regulatory complexes at promoter and enhancer regions of endocrine-specific genes. Epigenetic regulation of endocrine transcription factor genes presents a novel way of targeting endocrine-specific gene activation and repression. For example, the suppression of Pdx1, Pax4, MafA, and Nkx6.1 in cells of nonendocrine origin such as fibroblasts is driven by hypermethylation of promoter DNA and H3K9me3 modification, whereas expression of these transcription factors in the NIT1 pancreatic β-cell line is driven by DNA hypomethylation and H3K4me3 modifications (26). Insights into genome-wide binding of transcription factors come from interactions with co-activator and co-repressor complexes that alter chromatin structure and function. For example, tissue-specific transcription factors are associated in a context-dependent manner with epigenetic modifiers such as HMTs and HDACs (120). The molecular interplay between transcription factors and coactivators such as p300 and Set7 (also Set9 or Set7/9), as well as with co-repressors such as enhancer of zeste homolog 2 (Ezh2) are implicated in both gene activation and suppression (120).
Islet-specific expression of the Set7 lysine methyltransferase is required for the maintenance of gene expression implicated in islet physiology (20). Depletion of Set7 in insulinoma and primary mouse islet cells inhibited activation of genes involved in glucose-stimulated insulin secretion, including Ins1/2, Glut2 and MafA (20). Consistent with these findings, an islet-specific enhancer mapped to Set7 promoter exhibited Pdx1-dependent activation in β-cells (20). Previous experimental findings (27) demonstrate transcriptional repression of Insulin gene upon Pdx1 knockdown in βTC3 cells. In Pdx1-depleted βTC3 cells, a significant depletion of H3K4 methylation at the Insulin promoter indicates that Pdx1 and Set7 could synergistically promote insulin expression (Fig. 2) (27). Together, these data strongly implicate Set7 as a critical regulator of β-cell homeostasis and glycemia by modulating chromatin structure and function (20, 27). What remains unclear is the distinct role of Set7-mediated H3K4me2 in the β-cell, as opposed to its specific H3K4 monomethylase activity in other cell types. Furthermore, Set7 regulates gene expression by methylation of nonhistone proteins such as transcription factors in various tissues and conditions (48, 49). Defining Set7 function in the context of α- to β-cell transdifferentiation will further advance the prospects of cell-based therapies for diabetes.

Precisely controlled exchanges of co-regulatory determinants at gene promoters are often dependent on more than one covalent modification within the amino terminal of histone tails. For instance, H3K27me3 is believed to mark poised enhancers in undifferentiated human ESCs, whereas on differentiation, H3K27ac replaces the methyl-moiety to activate enhancers (81). Actively transcribed regions of chromatin are often enriched for H3K4me3 and H3K9/14ac (57). By contrast, nonpermissive chromatin regions are generally depleted of these modifications (57, 114). Combinatorial interactions between HMT, HAT, and HDAC enzymes are critical for robust gene regulation (120). For example, suppression of gene activity requires coordinated actions of histone deacetylation by HDACs and H3K27 methylation by Ezh2 (12). Interplay between different chromatin modifications and epigenetic modifiers is likely to perform a major role in regulating gene transcription in pancreas. Many genes in stem and progenitor cells are bivalently modified with two histone modifications—repressive H3K27me3 and the activating H3K4me3 mark. These genes are believed to be poised for activation or suppression (106). As cells differentiate and commit to a particular lineage, H3K27me3 modifications are dynamically replaced with H3K4me3 to drive gene activation. One extensive study compared gene expression and genome-wide H3K4me3 and H3K27me3 profiles in human pancreatic α- and β-cells, as well as exocrine cells (7). Remarkably, α-cells contained a greater proportion of bivalent genes (marked with both H3K4me3 and H3K27me3) compared with β-cells, which can be interpreted as a greater epigenetic plasticity for α-cells, allowing them to express properties associated with β-cells (Fig. 3). It is possible that this epigenetic landscape of α-cells is amenable to reprogramming, underscoring recent successes in direct conversion of α-cells to a β-like phenotype in preclinical models of diabetes. Remarkably, HMT inhibitors such as Adox can modulate the epigenetic landscape of α-cells to direct reprogramming to insulin-producing β-like cells (Fig. 4) (7). Evidence from studies of HDAC inhibition show cellular reprogramming is associated with de-repression of several genes (79). Targeting co-repressor complexes such as the polycomb repressive complex 2 (PRC2) and/or specifically Ezh2 could further enhance reprogramming efficiency. For example, inhibition of HDAC enzymes using small-molecule compounds depletes Ezh2 from chromatin, suggesting that HDACs and Ezh2 could operate together to suppress gene activity (14, 67, 105).
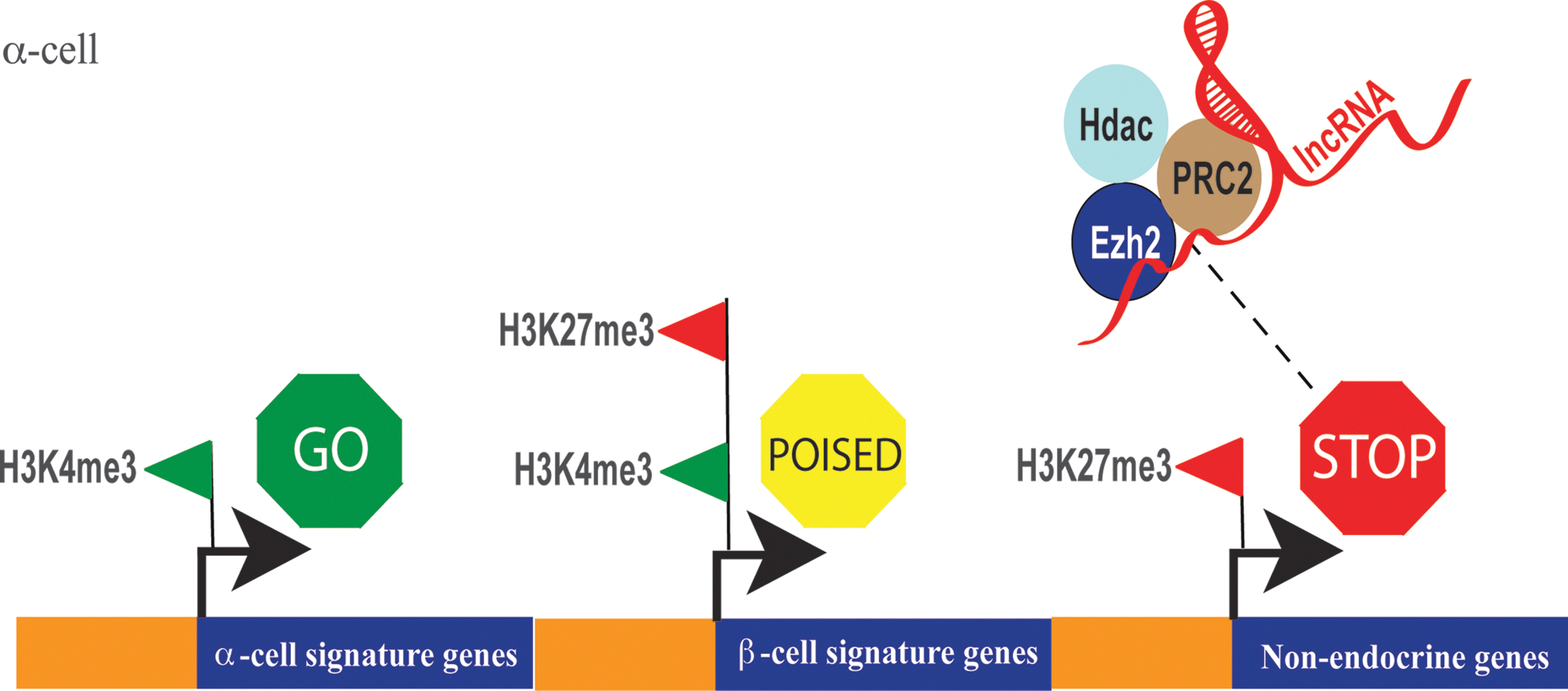
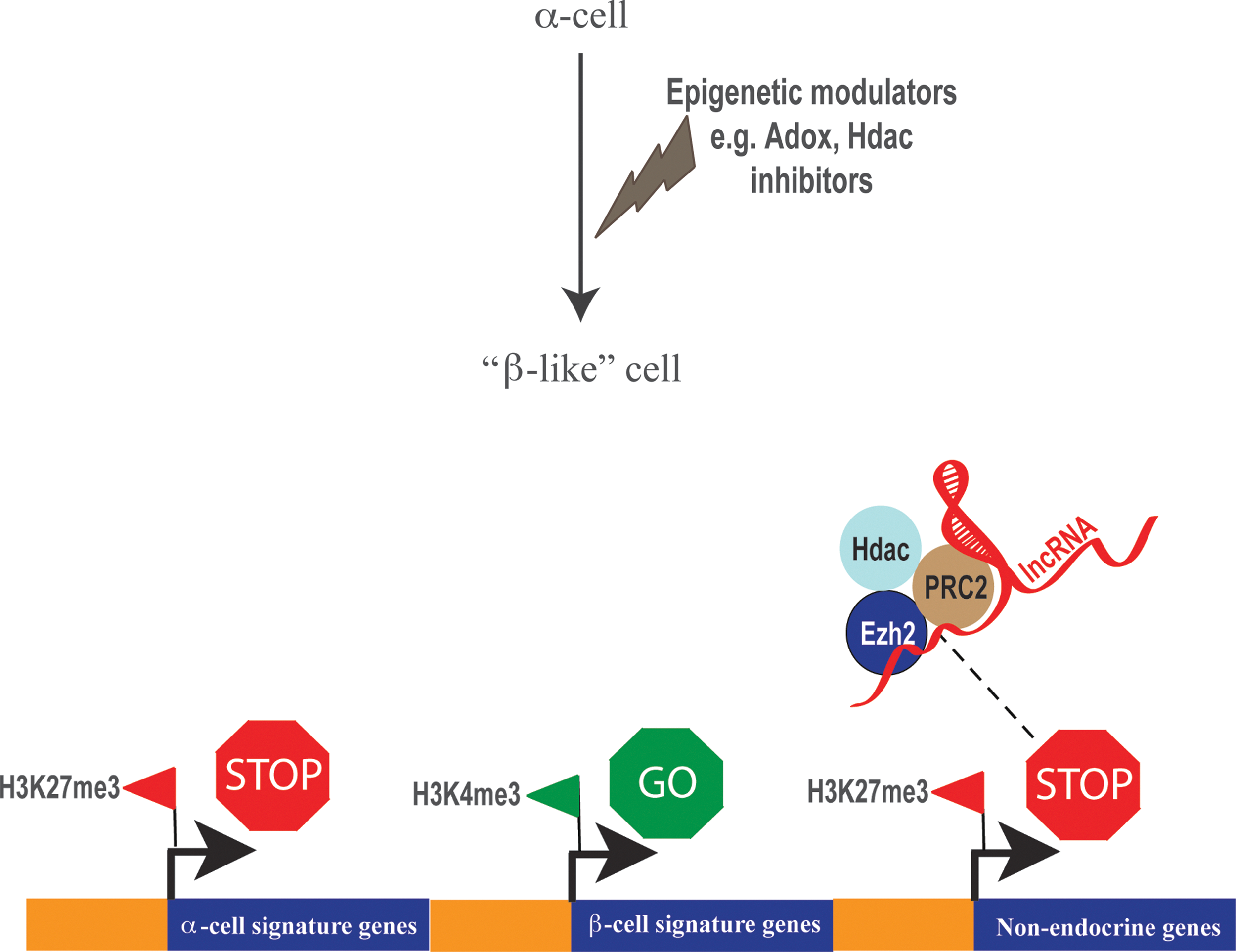
The Role of Ezh2 in Gene Suppression and Cellular Reprogramming
Integration of chromatin dynamics with transcription factor binding sites defines key signatures required for lineage commitment (37). More than 100 histone modifications have been identified, and more than 500 histone isoforms are believed to exist in human cells (43, 98, 103). PRC proteins deposit H3K27me3 marks on promoters to maintain tissue-specific gene expression patterns (65). PRC1 and PRC2 are the major repressor complexes that mediate silencing of hundreds of human genes by H3K27me3 (91). Throughout development, stage-specific H3K27me3 by Ezh2, a key component of PRC2, is critical for progenitor cell differentiation.
Long intergenic noncoding RNAs guide proteins to gene-specific binding, and their contribution to transcriptional reprogramming during cell differentiation is becoming increasingly appreciated (50, 109). For instance, the lincRNA-RoR (regulator of reprogramming) is specifically upregulated by pluripotent transcription factors that drive iPSC generation (59). PRC2 proteins, including Ezh2, harbor lncRNA-binding domains and are believed to regulate gene expression by specific interactions with lncRNAs expressed in a tissue-specific manner (65, 104). Although β-cell-specific lincRNAs are known, it has not yet been elucidated how lncRNAs and PRC2 integrate chromatin modifications to regulate gene expression that is critical for maintaining endocrine function and insulin production (73). Together, these recent data highlight the importance of epigenetic regulation and the potential to manipulate HMTs for efficient reprogramming of α- to β-like cells.
MicroRNAs in Islet Development and Reprogramming
Recent advances in deep sequencing of nucleic acids have uncovered numerous novel microRNA (miRNA/miR) genes in mammals (35). miRNA expression and regulation is central to embryonic development and cellular differentiation (68). Sequencing of the developing human pancreas identified several miRNAs specifically regulated during pancreatic lineage specification (87). The expression profile of regulated miRNAs is closely correlated with endocrine factor expression in the developing human pancreas (Fig. 5). For example, downregulation of Sox4 in pancreatic lineages was mediated by increased expression of miR-489. Specific downregulation of at least 19 miRNAs during pancreatic lineage specification permits the expression of genes such as NeuroD1, Foxa2, and Mafb, which are critical regulators of endocrine cell formation, as well as β-cell differentiation and maturation (87).

Disruption to pancreatic development by organ-specific deletion of the Dicer1 miRNA-processing enzyme was shown to significantly affect pancreatic β-cells (62). β-cell-specific deletion of Dicer1 leads to poor insulin secretion and impaired glucose metabolism followed by rapid progression to diabetes in mice (44). Furthermore, Dicer1-hypomorphic mice, whose Dicer1 expression was reduced 20% in all tissues, showed severe impairment of pancreatic function (69). By contrast, several other tissues were histologically normal after 4 weeks of age (69). These results suggest that miRNAs are vital for pancreatic development and endocrine signaling, and precise identification of miRNAs regulating developmental cascades has potential to more efficiently transdifferentiate α-cells to produce insulin (62).
Deep sequencing of mature α- and β-cells isolated from human pancreas revealed distinguishable gene expression patterns indicative of cell type-specific functions for several miRNAs (51). For example, miR-200c, miR-125b, and miR-182 were highly expressed in β-cells and believed to repress cMAF transcription factor required for glucagon expression in α-cells (51). Inhibition of miR-200c in β-cells as well as over-expression of miR-200c in α-cells resulted in upregulation and downregulation of cMAF, respectively, supporting the idea that miRNAs can modulate transcription factors to induce direct cell reprogramming.
Several miRNAs are required for normal glucose homeostasis and are often associated with the onset of diabetes. For example, miR-29 family (29a/b/c) is linked to β-cell death as well as with loss of insulin secretion (86). Similarly, miR-375 null mice developed hyperglycemia with severe loss of β-cell mass compared with control animals (80). By contrast, the population of α-cells in miR-375 null mice was increased, suggesting that miR-375 may be critical for maintaining a fine balance between α- and β-cell populations in pancreatic islets (80). Furthermore, over expression of miR-34a and miR-146 in diabetic mice is associated with reduced expression of vesicle-associated membrane protein 2 (Vamp2), a key component required for β-cell exocytosis (60). Exploring the functions of pancreatic miRNAs in diabetes could unravel novel miRNA targets that are linked to β-cell function and insulin production.
HDAC Inhibition in the Pancreas
In addition to methylation, acetylation of histone lysine residues is a key epigenetic regulator of gene expression (24, 25). High levels of histone acetylation deposited by HATs mark active promoters and open chromatin conformation (89). Conversely, HDACs remove this modification and, consequently, are associated with gene repression (89). There are four classes of HDACs: Class I (HDAC1, 2, 3, and 8; nuclear localization), class II, which is subdivided into class IIa (HDAC4, 5, 7, and 9; nuclear and cytoplasmic localization) and class IIb (HDAC6, 10; mainly cytoplasmic localization), class III (also known as sirtuins, Sirt1-7), and class IV (HDAC11, nuclear and cytoplasmic localization). Inhibition of HDACs has shown genome-wide acetylation and deacetylation changes to chromatin and is believed to efficiently induce genes implicated in cell reprogramming (47, 63, 82).
The current paradigm of cell reprogramming holds that chromatin condensation correlates with gene silencing and inhibitor-induced transcriptional reprogramming (40, 64). HDAC inhibitors such as valproic acid (VPA), trichostatin A (TSA), and sodium butyrate (NaB) are known to induce chromatin changes to an open conformation in which transcription is held in an active state that is associated with enhanced expression of genes involved in reprogramming of cell fate. For instance, preconditioning of ESCs using 5-azacytidine can effectively induce differentiation to cardiomyocytes in culture (100). In the context of cardiovascular disease, intramyocardial transplantation of epigenetically reprogrammed cardiomyocytes promoted cardiac repair by enhanced angiogenesis (100). Although the data are predominantly derived from in vitro studies, recent advances using a variety of tissue types, including the heart, lung, and brain, are showing promising therapeutic developments (8, 70, 119).
Preconditioning of explanted embryonic rat pancreas (E13.5) with inhibitors of Class I and Class II HDACs, TSA and NaB, resulted in an increase in the pool of β-cells, as well as elevated Pax4 expression (33). By contrast, Class I specific HDAC inhibitors, VPA and entinostat did not induce Pax4 expression, indicating a specific role for class II HDACs in the repression of Pax4 (52). Intriguingly, all four HDAC inhibitors induced Arx gene expression, suggesting class I HDAC-specific repression, which is essential for differentiation of glucagon-producing cells in the mammalian pancreas (16). Furthermore, knockdown of the class II HDACs, HDAC5, and HDAC9 in mice resulted in increased pools of β-cells (53). Moreover, the subsequent treatment of fetal pancreatic explants with a class IIa selective HDAC inhibitor (MC1568) increased Pax4 expression (53). While these results are limited to fetal pancreas, it is highly likely that a specific class IIa HDAC inhibitor such as MC1568 could be used to induce Pax4 in mature α-cells in the pancreas to initiate lineage reprogramming and β-cell regeneration (2).
Epigenetic Control of Pancreatic Programming
In Utero
The Developmental Origins of Health and Disease model proposes that predictive adaptive responses to the nutritional conditions in utero confer increased risk of chronic disease in adulthood (6). Epidemiological studies in human populations have associated intrauterine nutrition with increased susceptibility to adult onset of metabolic dysfunction and diabetes (36, 55, 102). For instance, studies reveal that the intrauterine nutritional environment can structurally remodel and functionally alter pancreatic islets, leading to impaired glucose tolerance and disease predisposition (88). Because chromatin marks established in utero can respond to local changes in nutrient availability, epigenetic variation introduced by gestational cues have strong capacity to define and direct persistent gene expression profiles of the developing pancreas (88).
The Pdx1 transcription factor is required for pancreatic development and β-cell maturation. Rodent models of intrauterine growth retardation (IUGR) exhibited strong reduction of fetal and neo-natal Pdx1 mRNA expression (93). This phenotype is exacerbated in adult IUGR rats, where Pdx1 expression is further decreased, concomitant with reduced β-cell mass and diabetes development (93). While no differences were observed at 2 weeks of age, methylation of a conserved CpG island at the proximal promoter and first exon of Pdx1, as well as Dnmt1 binding were significantly increased in IUGR rats compared with control animals (76). A cascade of histone modifications also paralleled the reduction of Pdx1 expression, characterized by age-dependent changes in H3K9 and H3K4 methylation. Furthermore, IUGR rats exhibited a progressive loss of histone acetylation, which were completely abolished by 6 months of age. These chromatin changes were associated with loss of upstream stimulatory factor-1 (USF-1) binding, as well as with recruitment of HDAC1 and the Sin3A co-repressor (76). Interestingly, HDAC inhibition, and not the DNA demethylating agent 5-azacytidine, was shown to normalize USF-1 binding and partially restore Pdx1 expression in IUGR islets at 2 weeks. These findings demonstrate the importance of histone modifications early in the progression of Pdx1 deregulation, offering a potential window for therapy before the establishment of stable DNA-methyl-dependent silencing. More recently, short-term administration of the glucagon-like peptide-1 (GLP-1) analogue exendin-4 to newborn IUGR rats was shown to permanently increase Pdx1 expression and prevent the development of diabetes (77). In addition to stimulating histone acetylation by increased recruitment of USF-1 and the PCAF acetylase, exendin-4 restored H3K4 methylation and prevented Dnmt1 binding and DNA methylation in IUGR islets (77).
Concluding Remarks and Future Directions
Even after near-total β-cell destruction in mice, α-cells can undergo spontaneous conversion to β-like cells, providing the basis for future treatment goals and strategies against T1D to effectively control endogenous insulin secretion (2, 56, 101). Often supplemented with drugs, viral-mediated forced expression of transcription factors and/or miRNAs remains the main strategy of cellular reprogramming. However, such virus-based gene transfer strategies influence global transcription factor binding mechanisms accompanied by potential dangers in tumor formation. For instance, recent evidence suggests that iPS cells are subject to genetic and epigenetic modifications during or after reprogramming (29, 58).
Success in achieving cell reprogramming using human models of development and disease compared with rodents are considerably limited. For example, reprogramming human fibroblasts is slower and less efficient compared with that observed in mouse fibroblasts partly due to additional stability and complexity of the epigenetic landscape in human cells. Our critical understanding of distinct lineage-specific chromatin landscapes in exocrine and endocrine differentiation is fundamental to drug-based cell reprogramming. While it has been known for years that epigenetic changes function as terminal steps in modulating gene expression, their role in early and late endocrine development, and promotion of direct reprogramming into functional insulin producing β-cells remains unclear.
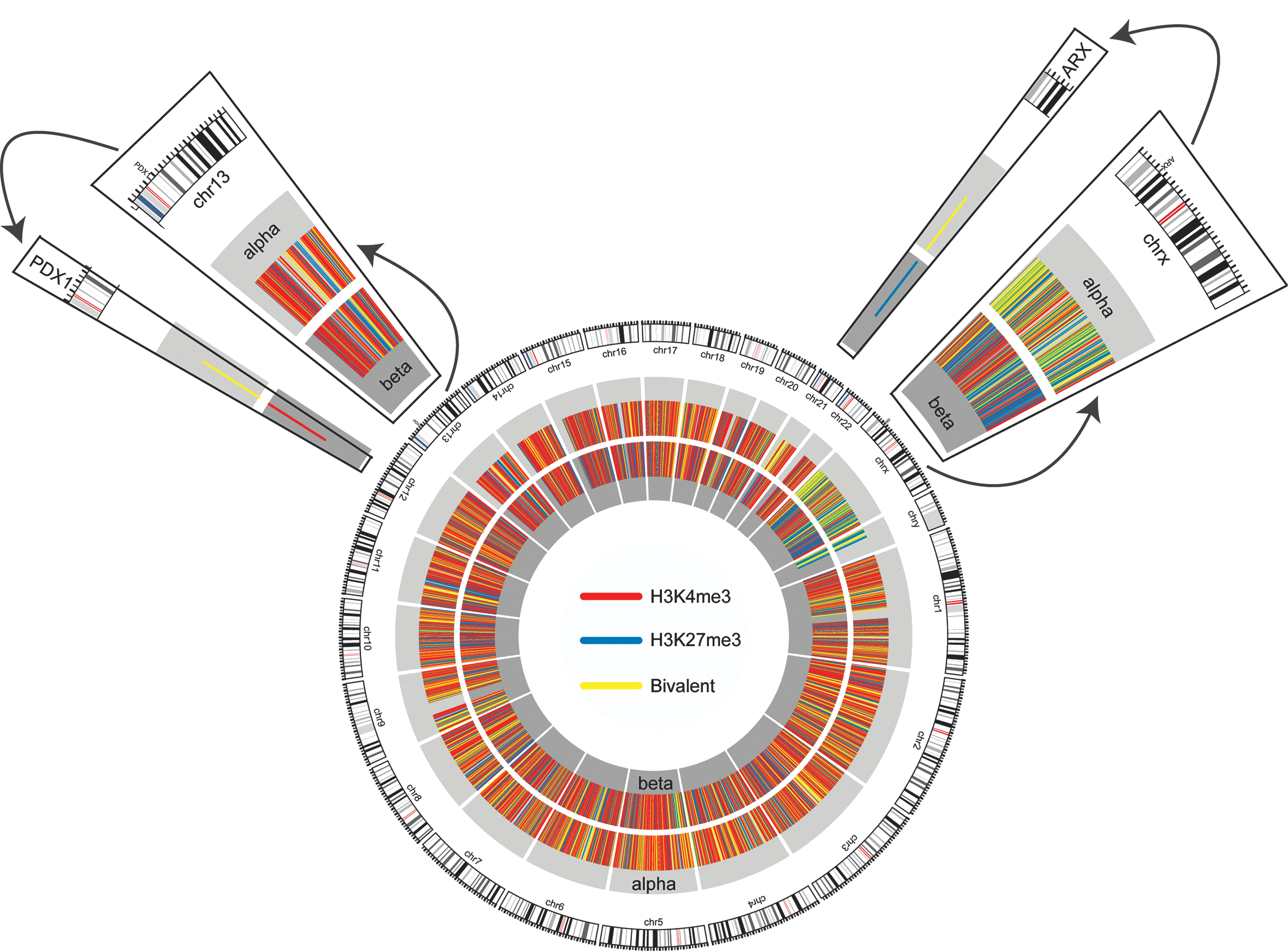
Epigenome and transcriptome profiling by massive parallel sequencing has greatly improved the knowledge of chromatin modifications that distinguish α-cells from β-cells in the human pancreas (Fig. 6) (7). A major limitation to successful reprogramming of one cell type to another is the lack of evidence in regulatory mechanisms underlying tissue-specific transcription factor genes. Comprehensive mapping of the epigenome by massive parallel sequencing for chromatin modifications and endocrine-specific gene expression patterns is beginning to reveal clues to the key regulatory events in the control of endocrine-specific pattern of gene expression (Fig. 6). Tissue-specific patterns of gene expression are precisely controlled by epigenetic mechanisms, and expression of two endocrine factor genes Arx and Pdx1 in α- and β-cells are medicated by specific histone modifications (81). The β-cell-specific expression of Pdx1 is mediated by gene-activating H3K4me3 modifications, whereas Pdx1 suppression in α-cells is mediated by bivalent chromatin represented by H3K4me3 and H3K27me3 modifications (Fig. 6). Clearly, deepening nucleic acid sequencing technologies as well as knowledge on α- and β-cell gene regulatory patterns hold the key to more targeted and efficient cell reprogramming strategies in the endocrine pancreas. Identification of chromatin regulators could lead to innovative therapeutic strategies, including the identification and validation of novel drug targets aimed at preventing, retarding, or reversing the long-term deleterious end-organ effects of chronic, ambient, and previous hyperglycemia.
It has yet to be determined whether small-molecule inhibitors of Ezh2 could enhance reprogramming of α-cells toward functional β-like cells by demethylation of the Pax4 promoter. Drugs such as TSA or class I specific HDAC inhibitors could be used to activate expression of genes repressed by the PRC2 alone, or in combination with Ezh2 inhibitor—the Ezh2 inhibitors act to remove an inhibitory mark, while the inhibition of HDAC1/2 leads to addition of an activating mark. Identification of histone modifications associated with endocrine gene transcription offers novel insights into gene-specific binding of HMTs such as Ezh2 and Set7, both of which could be strong candidates that enforce endocrine cellular reprogramming. In addition, combining lncRNA-based approaches to overcome barriers such as nonspecific genomic and epigenomic events can enhance reprogramming efficiency.
Indeed, potential pitfalls of pharmacological chromatin modulators should not be discounted. HDAC inhibitors are often toxic and poorly tolerated in preclinical and clinical studies (9). Promoter DNA hypermethylation is the hallmark of several cancers, and inhibitors of DNA methyltransferases have proved promising in several preclinical studies; however, they pose severe off-target concerns (61). Responses to pharmacological interventions vary between individuals due to epigenomic diversity within populations, and often individuals fail to respond to epigenetic drugs (61).
In addition to inhibitors of chromatin-modifying proteins are oligonucleotides designed to target endogenous miRNAs and fine tune target gene expression (75). However, such approaches fall short due to limited sequence specificity as well as due to off-target effects of antagomiRs or miRNA mimics and present challenges to successful clinical translation for the treatment of disease. Deep understanding of endocrine-specific miRNAs and their target regulation in α- and β-cells would greatly benefit reprogramming of these cells in the context of T1D.
Together, we envisage that successful reprogramming of noninsulin-producing cells of pancreatic origin to functionally competent, insulin-secreting β-cells will require different combinations of endocrine factors and epigenetic modulators to meet the clinical need for transplantation therapy.
Footnotes
Acknowledgments and Funding
The authors gratefully acknowledge the contribution of Haloom Rafehi (Human Epigenetic Laboratory, BakerIDI Heart and Diabetes Institute) for her assistance in generating ![]() .
.
The authors acknowledge grant and fellowship support from the Juvenile Diabetes Research Foundation International, the Diabetes Australia Research Trust, the National Health and Medical Research Council (NHMRC), and the National Heart Foundation of Australia. A.E.-O. is a senior research fellow supported by the NHMRC.