
Abstract
Introduction
G
Glucose 6-phosphate dehydrogenase (G6PD) deficiency is associated with drug-induced hematological disorders. We used diamide to mimic drug-induced oxidative stress, and applied a metabolomics approach to study its effect on metabolism of G6PD-deficient red blood cells (RBCs). The impact of oxidant on metabolism of normal RBCs is much less than that of G6PD-deficient cells. Several metabolic pathways, such as GSH metabolism and methionine cycle, are differentially affected in G6PD-deficient cells. Adenosine triphosphate (ATP) depletion and adenosine monophosphate (AMP) accumulation activate AMP protein kinase (AMPK), leading to enhanced glycolysis. However, inhibition of pyruvate kinase thwarts RBCs from replenishing their energy reserve. Our findings suggest that G6PD-deficient cells mount futile metabolic signaling response to maintain redox balance on oxidative stress.
G6PD deficiency is associated with hematological disorders. The clinical manifestations range from neonatal jaundice, drug- or infection-mediated hemolytic crisis, favism, and, less commonly, chronic nonspherocytic hemolytic anemia (9), and profound hemolysis may lead to hemoglobinuria and acute kidney injury. The inability of G6PD to produce a reduced form of nicotinamide dinucleotide hydrogen phosphate (NADPH) and to maintain redox homeostasis may underlie the pathogenesis of symptoms associated with G6PD deficiency.
Mature red blood cells (RBCs) lack cellular organelles, and are deficient in cellular functions such as de novo synthesis of proteins and lipids. They are highly adapted to transport of oxygen, carbon dioxide, and metabolites (19). RBCs are particularly susceptible to oxidative damage, as they are rich in heme iron and oxygen (48), and oxidative damage of cytosolic and membrane components occurs despite a highly efficient defense system to maintain a proper redox status. Oxidative damage of erythrocytic membranes and other macromolecules leads to RBC removal by reticuloendothelial system, or hemolysis in case of acute stress (54). Particularly, oxidation of membrane lipids and proteins affects membrane structural integrity and deformability, and renders the red cells less flexible to pass capillaries and their removal from circulation (55).
Glutathione (GSH) and enzymes, such as glutathione peroxidases, are essential components in all cells, for detoxification of reactive oxygen species (ROS), as well as for reduction and repair of oxidatively damaged cellular components. GSH buffer is maintained through activity of glutathione reductase (GR), which catalyzes the conversion of oxidized glutathione (GSSG) to GSH in an NADPH-dependent manner. This depends on proper functioning of pentose phosphate pathway. G6PD deficiency severely compromises this GSH-centered antioxidant system (63). Biological mechanisms underlying the pathophysiology of G6PD deficiency are incompletely understood. There are variations in clinical features of individuals with the same G6PD mutation. G6PD enzymatic activity does not correlate very well with clinical severity (8), and is not a predicator for the severity of hemolysis. Moreover, it has been shown that disruption of glutathione peroxidase (GPx-1) gene does not significantly affect hemoglobin oxidation in murine RBCs on exposure to oxidant. The interplay between G6PD, GSH, ROS, and hemolysis is more complicated than previously thought. These findings highlight the necessity to re-examine the metabolism of G6PD-deficient RBCs and its roles in diseases. Metabolite profile is an appropriate indicator of pathophysiology of cells and organisms, as metabolites are downstream products of transcriptional and translational processes (21). In light of the lack of active transcription and translation in mature RBCs, metabolite profiling helps track the metabolic responses of cells to stress. Complete profiling of metabolites in red cells contributes to an understanding of oxidant- and drug-induced red cell damage and hemolytic crisis.
Some antimalarial or antibacterial drugs, such as primaquine and dapsone, are metabolized into species that induce oxidative stress and GSH depletion (11). Quinones can be generated from menadione, primaquine, and doxorubicin, and undergo redox cycling to induce oxidative stress. Being a Michael acceptor, quinones react readily with sulfur-containing nucleophiles, leading to GSH depletion (10). The study of the metabolite profile of the drug-treated red cells is complicated by biotransformation of drugs into pharmacologically active species. To simplify the analysis, we treated red cells with diamide (DIA), which mimics oxidative stress associated with and GSH-depleting effect of these drugs. DIA is an oxidant that reacts specifically with intracellular thiols, including both low-molecular-mass thiols and protein sulfhydryls, and has been shown to result in increased toxicity in G6PD-deficient cells (25).
In this study, we studied the changes in global metabolism and compensatory responses of RBCs in response to DIA. There are a few differences in metabolite profile between untreated normal and G6PD-deficient cells. DIA-treatment of normal RBCs causes modest reversible changes in metabolism. In contrast, DIA treatment of G6PD-deficient RBCs induces significant changes in GSH-related pathways, resulting in appearance of unusual metabolites such as ophthalmate. Apparently, the flux through methionine cycle increases to provide GSH precursors. Such metabolic shift is accompanied by significant changes in purine metabolism, including the depletion of adenosine triphosphate (ATP) and accumulation of adenosine monophosphate (AMP). AMP accumulation activates AMP protein kinase (AMPK), leading to accelerated glycolysis. Changes in RBC functionality, which are apparently reversible in normal cells, become irreversible in G6PD-deficient cells. This inability to repair oxidative damage, together with energy depletion, may underlie the aberrant glycolysis in G6PD-deficient cells, which is, in part, attributed to oxidative inactivation of pyruvate kinase (PK). These findings suggest that metabolic dysfunction exacerbates oxidant-induced anomalies of G6PD-deficient RBCs.
Results
Subject characteristics
G6PD activities in whole blood specimens of G6PD-deficient subjects (6.6±4.8 U/1012 RBCs) were significantly reduced (p<0.001) as compared with those of a control group (137.9±7.7 U/1012 RBCs) (Fig. 1A). All G6PD deficient subjects enrolled in this study fall in the class 2 G6PD deficiency according to the criteria of the World Health Organization (24), and carry the 1376 G→T mutation (Fig. 1B), the most common allele that causes G6PD deficiency in Taiwan and mainland China (60).

Enhanced susceptibility of G6PD-deficient RBCs to oxidant-induced damage
Since the susceptibility of RBCs to DIA is indicative of their redox status (35), it is expected that G6PD-deficient cells are more sensitive to DIA and oxidative crosslinking of membrane-associated cytoskeletal proteins. Treatment with DIA of G6PD-deficient RBCs induced the appearance of high-molecular-weight protein aggregates in membranes (Fig. 2A) and RBC protein glutathionylation (Fig. 2B). The appearance of oxidatively modified proteins was accompanied by depletion of low-molecular-weight proteins. The high-molecular-weight protein aggregates disappeared when the samples were treated with dithiothreitol (Fig. 2A), suggesting their origin as sulfhydryl-crosslinked proteins. Control RBCs did not show appreciable protein aggregation during DIA treatment. These findings suggest that DIA preferentially causes crosslinking and glutathionylation of RBC proteins in G6PD-deficient RBCs.
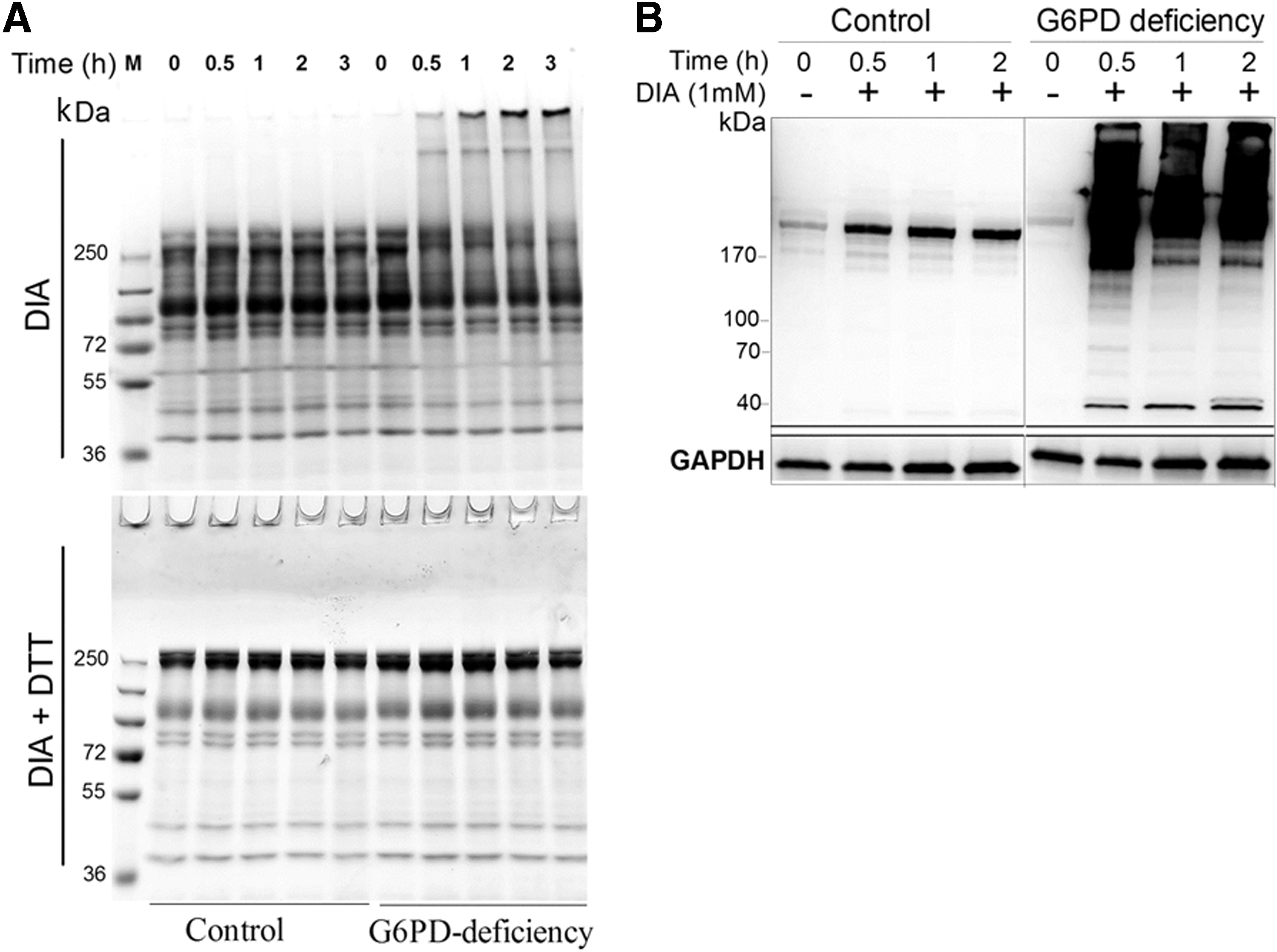
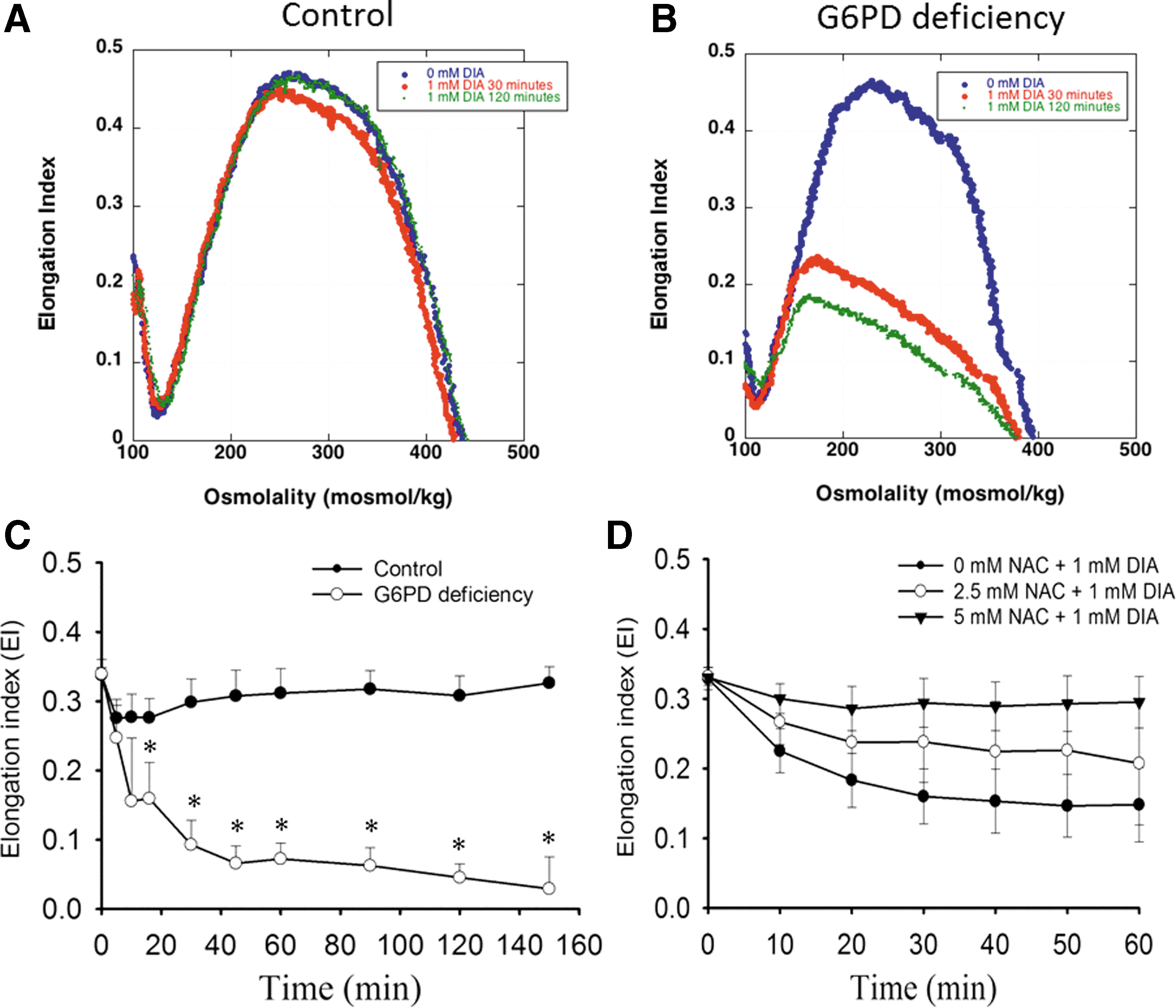
DIA-induced crosslinking of proteins is associated with changes in function of RBCs. Deformability, an index of RBC function, was measured by ektacytometry. In the absence of DIA treatment, the ektacytometric osmotic deformability profile of RBCs from G6PD-deficient individuals was not different from that of control. DIA treatment induced a significant decrease in RBC deformability. At 1 mM DIA, the deformability of normal RBCs but not that of G6PD-deficient cells was restored after an initial decrease (Fig. 3A, B). It is likely that DIA-induced damage can be repaired in normal RBCs, but not in G6PD-deficient RBCs. These findings were consistent with the data on RBC deformability measured with microfluidic ektacytometer Rheoscan. Elongation indices (EIs) of RBCs from untreated G6PD-deficient and normal individuals were nearly the same. On DIA treatment, EI of normal RBCs showed a reversible change, whereas that of G6PD-deficient cells did not (Fig. 3C). Pretreatment with various doses (2.5 and 5 mM) of N-acetylcysteine protected G6PD-deficient RBCs from DIA-induced reduction in erythrocytic deformation (Fig. 3D).
Distinct metabolic profile of G6PD-deficient RBCs on oxidative stress
To study how cellular metabolism changes in response to oxidative stress, we analyzed the changes in metabolomes of G6PD-deficient and control RBCs in response to DIA. The cells were treated with 1 mM DIA for a period ranging from 30 min to 3 h, and the hemolysates were analyzed by RRLC-ESI-TOF-MS operating in positive-ion mode. Principal component analysis of the metabolomic dataset showed only minor differences between untreated control and G6PD-deficient RBCs (Fig. 4A). DIA treatment caused little change in global metabolism of normal RBCs. In contrast, DIA treatment resulted in distinct differences between G6PD-deficient and normal RBCs (Fig. 4A and Supplementary Fig. S1; Supplementary Data are available online at
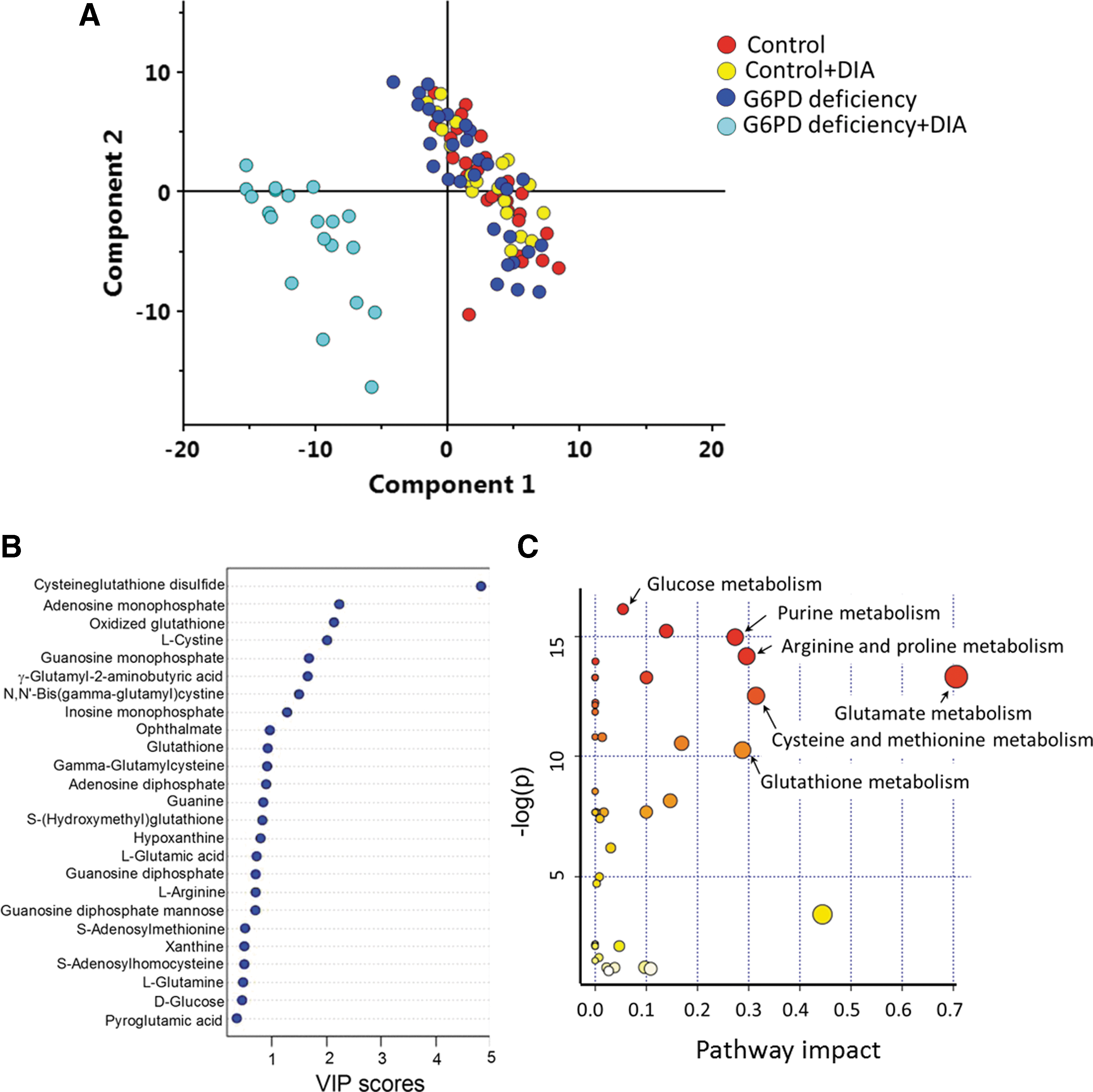
A total of 254 distinct metabolites showed significant changes in their abundance as indicated by analysis of variance (ANOVA) analysis (Supplementary Table S1). These metabolites were searched against HMDB's databases, and the identities of 52 metabolites were validated by LC-MS/MS (Supplementary Table S2). These 52 metabolites were further subject to analysis of their variable importance in the projection (VIP) scores (Fig. 4B) and pathway impact analysis (Fig. 4C). Notably, cysteineglutathione disulfide and GSSG had a very high VIP score. AMP having the second highest contribution to the difference in metabolic profiles between DIA-treated G6PD-deficient and normal RBCs was unexpected. Ophthalmate, which has not been previously reported in human RBCs, made a major contribution to this difference (Fig. 4B). Pathway impact analysis showed that glutathione metabolism, cysteine and methionine metabolism, glucose metabolism, and purine metabolism were differentially affected in control and G6PD-deficient RBCs after DIA treatment (Fig. 4C).
Altered GSH and methionine metabolism in G6PD-deficient RBCs on DIA challenge
As previously reported (25), DIA causes oxidation of GSH to GSSG. Level of GSH was maintained in normal cells treated with DIA. On the contrary, GSH was depleted in G6PD-deficient cells after 0.5 h of treatment, and it was accompanied by the accumulation of GSSG (Fig. 5). Level of γ-glutamylcysteine, the precursor to GSH, was greatly reduced in G6PD-deficient cells on DIA treatment, reflecting rapid DIA-mediated GSH exhaustion. Interestingly, ophthalmate, an analog of GSH with its thiol group replaced with a methyl group, is found in human RBCs. Accumulation of ophthalmate under oxidative stress has been reported in hepatic cells (58) but never in human RBCs under any conditions. Ophthalmate and an intermediate in its synthesis γ-glutamyl-2-aminobutyric acid accumulated dramatically in DIA-treated G6PD-deficient RBCs, whereas levels of these metabolites rose mildly in treated normal cells (Fig. 5). Level of GSH precursor γ-glutamylcysteine was greatly reduced in G6PD-deficient cells on DIA treatment, suggesting a shift of GSH synthesis toward ophthalmate synthesis in RBCs under stress.

GSH oxidation is accompanied by altered methionine metabolism. The levels of S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH), intermediates in methionine catabolism, significantly increased in these RBCs. This may represent an increase in the flux of the first step in methionine cycle catalyzed by s-adenosyl-methionine-synthetase. GSH-related metabolites showed a strong correlation with the RBC deformability (Table 1). The ratio of glutamine to glutamate level decreased in G6PD-deficient cells exposed to DIA, but remained relatively constant in normal cells (Supplementary Fig. S2A). It appears that glutamine/glutamate ratio correlates positively with EI of RBCs (Supplementary Fig. S2B). These results suggest a functional correlation between GSH-related metabolites and RBCs deformability.
FDR, false discovery rate.
Concomitant increases in ATP consumption and adenosine nucleotide metabolism in G6PD-deficient RBCs on DIA challenge
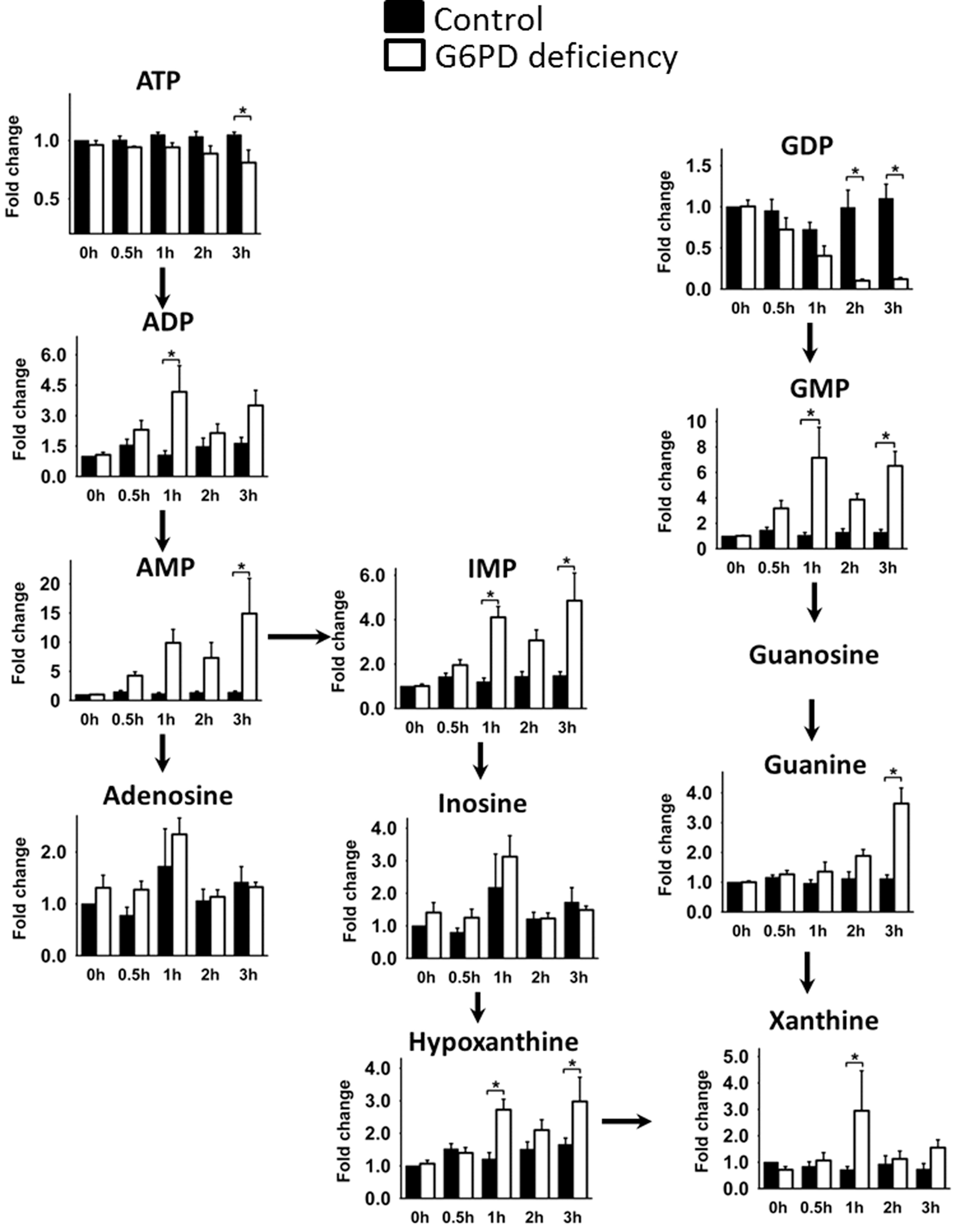
Since both γ-glutamylcysteine synthetase and glutathione synthetase catalyze ATP-requiring biochemical reactions, oxidative stress is likely to deplete the energy reserve. As expected, the amount of ATP significantly decreased in G6PD-deficient but not in normal RBCs on DIA treatment. This drop in ATP was accompanied by increases in the levels of adenosine diphosphate (ADP), AMP, inosine 5′-monophosphate (IMP), hypoxanthine, and xanthine (Fig. 6). Guanosine diphosphate (GDP) was gradually depleted during treatment, and guanosine monophosphate (GMP) accumulated during the same period.
Dysregulated glycolysis and impaired pentose phosphate pathway in G6PD-deficient RBCs on DIA challenge
The fall in ATP level and the rise in AMP level in DIA-treated G6PD-deficient cells is suggestive of energy deficit. To address how DIA affects energy metabolism in RBCs, we quantified the level of glycolytic and pentose phosphate pathway metabolites using LC-MS/MS (Fig. 7). Energy depletion was accompanied by an up-regulation of glycolysis, as evidenced by a steady increase in glucose levels and a decline in glucose 6-phosphate (G6P) and fructose 6-phosphate (F6P) levels (Fig. 7C). These findings indicate an accelerated turnover of glycolytic intermediates in G6PD-deficient RBCs during DIA treatment. In both normal and G6PD-deficient cells, the levels of 3-phosphoglycerate/2-phosphoglycerate (3PG/2PG) and phosphoenolpyruvate (PEP) increased initially after treatment. However, the kinetics of the changes in 3PG/2PG and PEP was different for these cells. The levels of 3PG/2PG and PEP in normal cells reached a maximum at 0.5 h after DIA treatment, and returned to their original values afterward. In contrast, 3PG/2PG increased to a much higher level in G6PD-deficient cells at 2 h after DIA treatment. The PEP level in these cells increased within the first 0.5 h and remained elevated for approximately 3 h. However, the increase in these intermediates was not accompanied by changes in pyruvate. The level of pyruvate was lower in G6PD-deficient RBCs than in normal cells (Fig. 7C). These findings imply that the entry of glucose into glycolysis is enhanced to a greater extent in G6PD-deficient RBCs than in normal cells in response to DIA. The conversion of PEP to pyruvate is inhibited in G6PD-deficient cells, suggesting dysregulated glycolysis in these cells under these conditions.

Pentose phosphate pathway is specifically affected by DIA. Levels of ribose 5-phosphate (R5P) and sedoheptulose 7-phosphate (S7P) significantly decreased in DIA-treated G6PD-deficient cells (Fig. 7B). However, R5P and S7P levels increased transiently in normal cells before they returned to their original values. Such results suggest that pentose phosphate pathway flux increases in normal RBCs under oxidative stress.
Functional implications of ATP depletion
It is known that ATP depletion by starvation leads to a loss in RBC deformability (17). It is possible that DIA-induced changes in adenine nucleotide content are causally related to the change in RBC deformability. Consistent with such an idea, normal RBCs incubated in ATP-preserving buffer for 21 h retained their ability to deform, while those depleted of ATP for 21 h showed a drop in deformability (Supplementary Fig. S3). The deformability partially recovered in ATP depleted cells under conditions that restored ATP. These findings suggest a functional correlation between adenine nucleotides and RBC deformability (Table 1).
Altered activities of AMPK and PK in G6PD-deficient RBCs on DIA challenge
Increased AMP levels in DIA-treated G6PD-deficient RBCs implies a change in AMP-activated signaling. Of such signaling pathways, AMPK can be activated by high AMP/ATP ratio and oxidative stress, and induces activation of glucose uptake (65) and glycolytic flux through translocation of GLUT1 to plasma membrane and regulation of key enzymes (66). To test such a hypothesis, we examined the activation status of AMPK as indicated by phosphorylation of Thr172. DIA treatment increased Thr172 phosphorylation of AMPKα in G6PD-deficient RBCs, but not in control RBCs (Fig. 8). It is possible that AMPK may be involved in a compensatory protective response of RBCs.
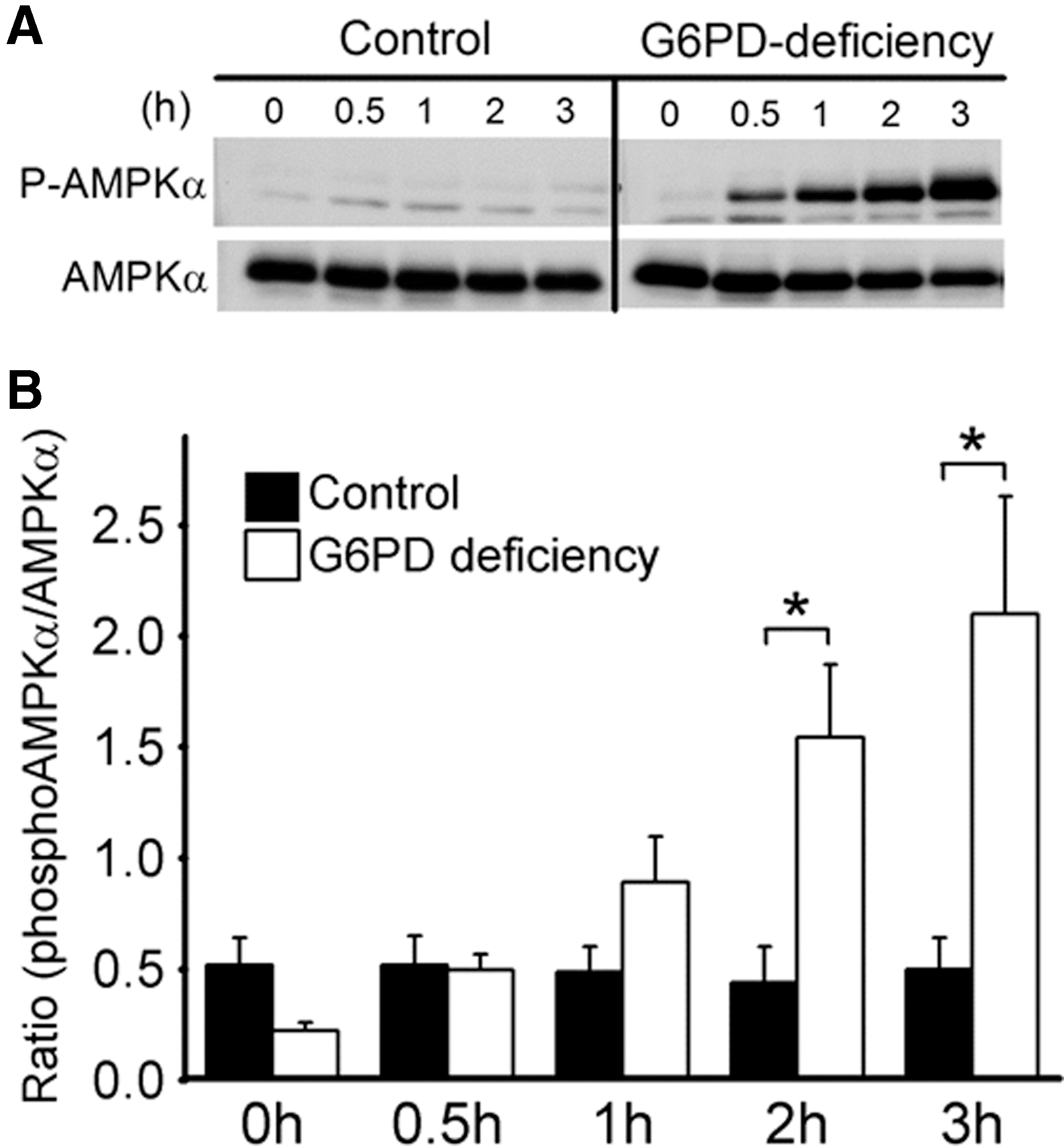
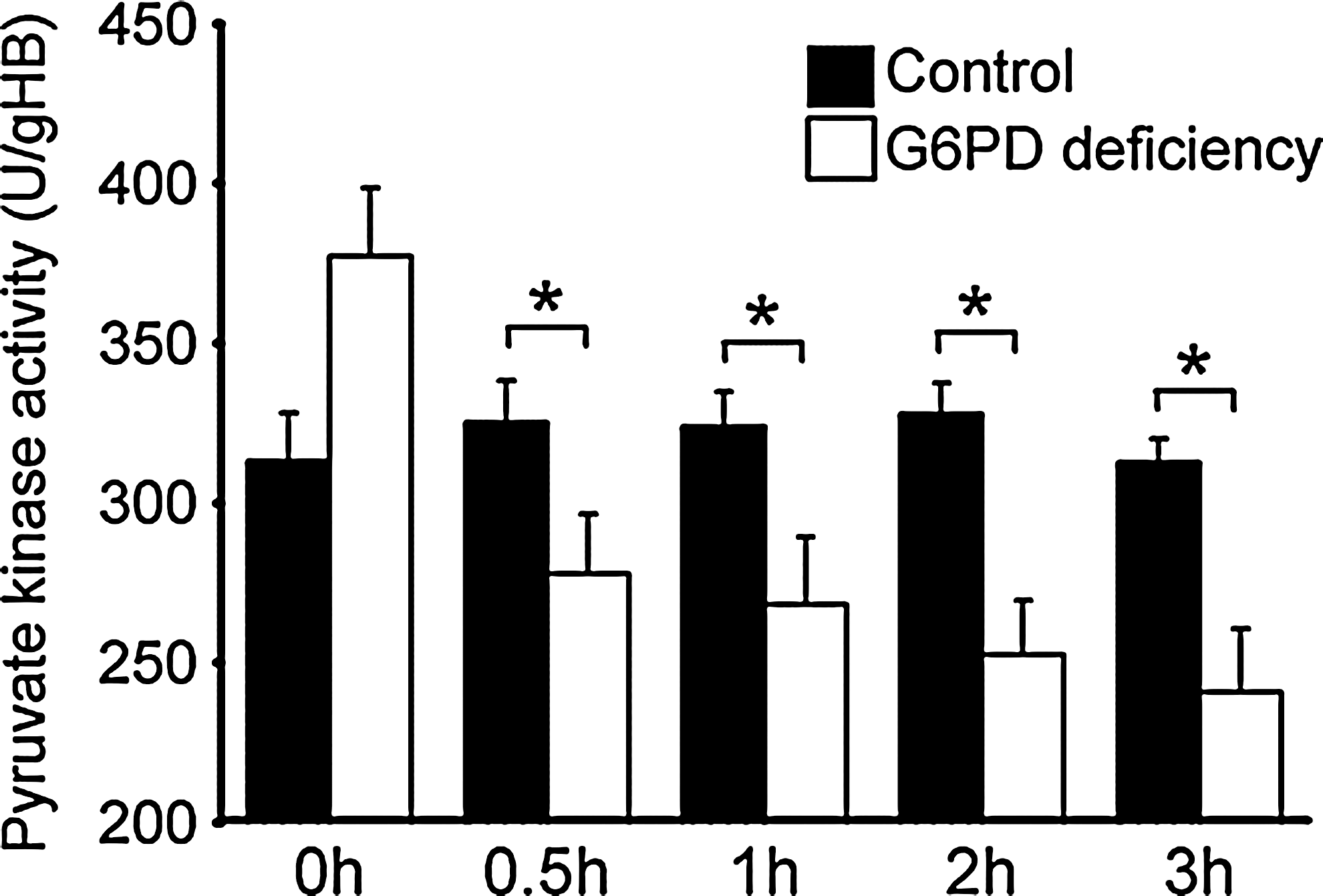
Enhanced glycolytic activity in DIA-treated G6PD-deficient cells was associated with PEP accumulation without an increase in pyruvate production (Fig. 7C). It is plausible that conversion from PEP to pyruvate is redox sensitive, as it is the case for redox-relatable PK M isoform (2). Our data show that PK activity was substantially reduced in DIA-treated G6PD-deficient RBCs as compared with normal RBCs (Fig. 9). This new finding may explain defective ATP production despite AMPK activation and enhanced glycolysis in DIA-treated G6PD-deficient RBCs.
Discussion
DIA reacts with thiols, and imposes oxidative stress on RBCs. There are significant changes in erythrocytic metabolism in G6PD-deficient but not in normal RBCs. GSH oxidation in the former is accompanied by increases in SAM synthesis and ATP utilization. Excessive ATP usage leads to buildup of ADP and AMP, and enhances glucose utilization by glycolysis. AMP accumulation is associated with AMPK activation. However, ATP utilization is unmet by ATP synthesis by glycolysis as a consequence of decrease in PK activity. Decreased ATP production, at least partially, contributes to a decrease in deformability of G6PD-deficient RBCs. These findings suggest that metabolic anomaly is an important causative factor of DIA-induced changes in RBC physiology.
RBC serves as a good model for studying the effect of oxidative stress on metabolism, as they are devoid of organelles such as nucleus and mitochondria. There is no complication in interpreting metabolic data due to changes in transcription and translation. During their physiological lifespan, RBCs act to transport oxygen and carbon dioxide; to protect their lipid membrane, cytoskeleton, hemoglobins from oxidative damage, which, if uncontrolled, would hamper their passage through blood vessels and oxygen-carrying capacity; to provide energy for maintenance of ion transport across cell membrane and other energy-dependent processes denaturation (62). To investigate the mechanism of drug-induced hemolysis of G6PD-deficient RBCs, we used an in vitro experiment with DIA treatment to mimic the oxidative stress generated by drugs under in vivo condition. DIA changes the oxidation state of thiols, in particular GSH. GSH is rapidly oxidized to its disulfide form, which can be reversibly reduced back to GSH by GR at the expense of NADPH. Besides, a number of low-molecular-weight disulfides, such as cystine, and mixed disulfides between protein sulfhydryl and low-molecular-weight thiols are formed (38). DIA-treated RBCs can be used as a model for studying how metabolism changes on oxidant treatment.
As mentioned earlier, the physiological states of RBCs correlate with their metabolic activities. Several metabolic pathways are differentially affected by DIA in normal and G6PD-deficent cells. These include GSH synthesis, adenine nucleotide metabolism, and glycolysis. DIA induces significant changes in GSH metabolism, especially in G6PD-deficient cells. We have demonstrated that G6PD deficiency renders hepatoma cells less efficient to generate GSH and more sensitive to DIA (25, 34). This study shows that DIA depletes cellular GSH pool and accrues GSSG in G6PD-deficient RBCs. Moreover, the disulfide form of cysteine (cystine) is rapidly formed in the presence of DIA. However, the thiol-depleting effect is minimal for RBCs from normal individuals. In G6PD-deficient RBCs, deficiency in NADPH supply may coerce cells to resort to the biosynthetic pathway. Despite the desperate effort of RBCs to increase GSH biosynthesis, the GSH level in DIA-treated G6PD-deficient cells diminishes due to the strong GSH-depleting ability of DIA. To meet the need for cysteine, G6PD-deficient RBCs ratchet up the fluxes of reactions in methionine cycle, as indicated by increases in SAM and SAH. The first step in methionine cycle is catalyzed by s-adenosyl-methionine-synthetase, leading to formation of SAM. As an intermediate in transsulfuration pathway, SAM acts as a precursor of cysteine and glutathione (42, 64). Increase in the flux through GSH biosynthetic pathway is paralleled by appearance of certain side reaction products. γ-glutamyl-2-aminobutyric acid and ophthalmate are synthesized via consecutive reactions catalyzed by γ-glutamylcysteine synthetase and glutathione synthase (50). A previous report showed that ophthalmate is synthesized under the condition of excessive GSH consumption, and this compound can serve as an oxidative stress biomarker (20). It is believed that excessive GSH consumption relieves the feedback inhibitory effect of GSH on γ-glutamylcysteine synthetase. Once activated, γ-glutamylcysteine synthetase synthesizes opthalmate from glutamate and 2-aminobutyrate.
The biochemical role of ophthalmate is currently unknown. It is perceived as a biomarker of oxidative stress and GSH depletion (20), and considered a better biomarker at low methionine concentrations (26). It has been shown that ophthalmate modulates the high-affinity GSH transport system of liver cells (4). Being a GSH analogue, ophthalmate may act as a competitive inhibitor of enzymes that depend on GSH as a substrate or cofactor. Accumulation of ophthalmate under oxidative stress may act to reduce the consumption of GSH by other GSH-dependent enzymes, and spare GSH for its antioxidative function.
Dysregulated GSH metabolism in G6PD-deficient RBCs is associated with a decline in energy production. Fall in ATP level and concomitant rises in AMP and ADP levels change cellular ATP/ADP ratio and fluxes of numerous energy-requiring and energy-producing reactions. Glycolysis, the major power-generating reaction in RBCs, is differentially affected in normal and G6PD-deficient cells. DIA-induced reduction in G6P level is suggestive of an increase in conversion of G6P to downstream products. The alternative interpretation that hexokinase-catalyzed production of G6P would have decreased is unlikely in light of the increases in levels of downstream products such as 3PG/2PG. Moreover, it has been previously reported that hexokinase is able to metabolize more glucose in the presence of oxidant than in its absence (44). These findings suggest that the rate of conversion of G6P to its downstream products exceeds its rate of production. Apparently, G6P is utilized at a higher rate in G6PD-deficient cells than in normal cells. The fate of glucose differs in these cells at this point. In normal red cells treated with DIA, the level of fructose 6-phosphate remained relatively unchanged, but the levels of R5P and S7P increased significantly and transiently. There was also a transient modest increase in 3PG/2PG level after treatment. These findings suggest that glucose utilization increases in response to DIA treatment, and more glucose is diverted to pentose phosphate pathway than to glycolysis. In G6PD-deficient red cells, the flux through pentose phosphate pathway is greatly reduced, and G6P is metabolized by glycolytic pathway. The excessive utilization of ATP for GSH and ophthalmate syntheses and the resultant increases in AMP and ADP can activate AMPK and up-regulate glycolysis. However, the robust increases in 3PG/2PG and PEP levels without the corresponding changes in pyruvate level are attributed to a reduction in PK activity. This actually adversely affects the rate of ATP production via glycolysis. It is in agreement with the recent finding that the PK isoform M is inhibited through oxidation of a critical cysteine residue (2). It is currently unclear whether PK isoform L/R in RBCs is inhibited in a similar manner. In addition, a number of metabolites involved in purine nucleotide metabolism change after DIA treatment. Levels of IMP, adenosine, inosine, hypoxanthine, and xanthine, catabolic products of AMP, increase. It is probable that AMP accumulation favors its degradation by adenylate deaminase (28). Furthermore, levels of GMP, guanine, and xanthine, catabolic products of GMP, are elevated. Intriguingly, cellular content of GDP and GMP change in a reciprocal manner after DIA treatment. GDP levels decrease with time of treatment. It is apparent that GDP reacts with ADP to form GMP and ATP. It has been previously shown that a form of guanylate kinase exists in erythrocytes (1). This may represent a mechanism to maintain a cellular ATP pool at depletion of GDP. These findings suggest that energy production and metabolism of purine nucleotide are dysfunctional in DIA-treated G6PD-deficient RBCs.
DIA-induced changes in energy production are associated with AMPK activation. AMPK is a heterotrimeric serine threonine kinase composed of catalytic α subunits, and regulatory β- and γ-subunits. It serves as a major energy sensor molecule that regulates various cellular activities (68). AMPK can be activated by an increase in cellular AMP/ATP ratio under metabolic stresses that interfere with ATP production or accelerate ATP consumption (29). Moreover, it can be allosterically activated by ADP and NAD (30, 53, 67). It has been recently found that AMPK can be regulated indirectly via reactive nitrogen species-mediated activation of CaMKKb and LKB1, or possibly via S-nitrosylation or S-glutathionylation of cysteine residues of AMPK α subunit (13, 57). DIA-induced increases in AMP and ADP, possibly together with increased oxidative stress, activate AMPK in G6PD-deficient RBCs. The intracellular glucose level increases in them, probably as a result of AMPK activation. It is known that AMPK acts to enhance glucose intake via Glut1 (5), which is the predominant form of glucose transporter in erythrocytes (49). In addition, AMPK phosphorylates and stimulates phosphofructokinase 2 (46), which generates fructose 2,6-bisphosphate, a potent activator of glycolysis. These findings suggest that AMPK is involved in regulation of metabolic responses to DIA.
DIA-induced stress leads to protein oxidative damage, crosslinking, and aggregation. It has been long recognized that protein oxidative damage increases the rigidity of membrane and reduces deformability (27, 51). Moreover, DIA may oxidatively modify erythrocytic proteins and alter their activities. DIA has been recently reported to cause reversible S-glutathionylation of α- and β-spectrin, protein 4.2, and other cytosolic proteins (55). S-glutathionylation of α-spectrin has been found in poorly deformed sickle cells (61). Accumulation of S-glutathionylated erythrocytic proteins in DIA-treated G6PD-deficient RBCs implies that such modification affects their membrane stability and deformability. An alternative and nonexclusive view is that DIA-induced metabolic changes contribute to loss of deformability. RBC membrane is considered a metabolically regulated dynamic structure. Active metabolism is crucial to regulation of its static and dynamic characteristics (7). ATP is essential to maintenance of biconcave shape of RBCs (3), and to dynamic membrane fluctuations, which reflect membrane deformability (47). Diminution of ATP increases mechanical rigidity of red cell membrane, as is the case in DIA-treated RBCs or in PK-deficient cells (40). It has been hypothesized that the ATP dependence of membrane dynamics is related to protein phosphorylation and dynamic assembly of cytoskeleton (41, 52). Dynamic membrane fluctuation depends on the integrity of spectrin network and its connection to membrane via actin, glycophorin C, and protein 4.1R. Continual attachment and release of spectrin in normal cells generates a certain tension in membrane. Depletion of ATP pool may keep 4.1R protein in unphosphorylated state, and favors interactions between spectrin, actin, and 4.1R, leading to an increase in tension. This may increase the effective viscosity of the red cell membrane skeleton and reduce the cellular deformability (6, 7). Other proteins are involved as well. Phosphorylation of spectrin modulates the mechanical properties of RBC membrane (45). Adducin, a protein involved in spectrin-actin interaction, is phosphorylated in poorly deformable RBCs. It has been recently found that its phosphorylation increases in AMPKγ1-deficient RBCs with increased membrane rigidity; AMPKα1-deficient RBCs show lowered deformability (22, 23). This also highlights the importance of AMPK, and possibly AMP and/or other activators, in control of structural flexibility, elasticity, and integrity of RBCs. Furthermore, it has been recently found that Rac GTPases act as a regulator of membrane deformability (36). The change in guanine nucleotide metabolism may affect deformability in this way. These findings suggest that DIA-induced metabolic changes and oxidative protein modification contribute to loss of deformability. Oxidation and S-glutathionylation of proteins as well as metabolic perturbation occurred to a much higher extent in G6PD-deficient RBCs than in normal cells; this correlates with irreversible loss of deformability of G6PD-deficient RBCs.
This study may have interesting implications about long-term storage of donated blood, especially that from G6PD-deficient donor. There are significant changes in RBC during storage. Such changes, referred to as storage lesion, include depletion of energy source, membrane loss, rheologic properties, and increase in the propensity for microparticle formation (37). Oxidative stress/damage markers accrue during the long-term storage (39, 59), and probably contribute to storage lesion. Enhanced anomalies of RBCs from G6PD-deficient individuals under stress imply that these red cells may be prone to storage lesion. Further studies are needed to clarify this.
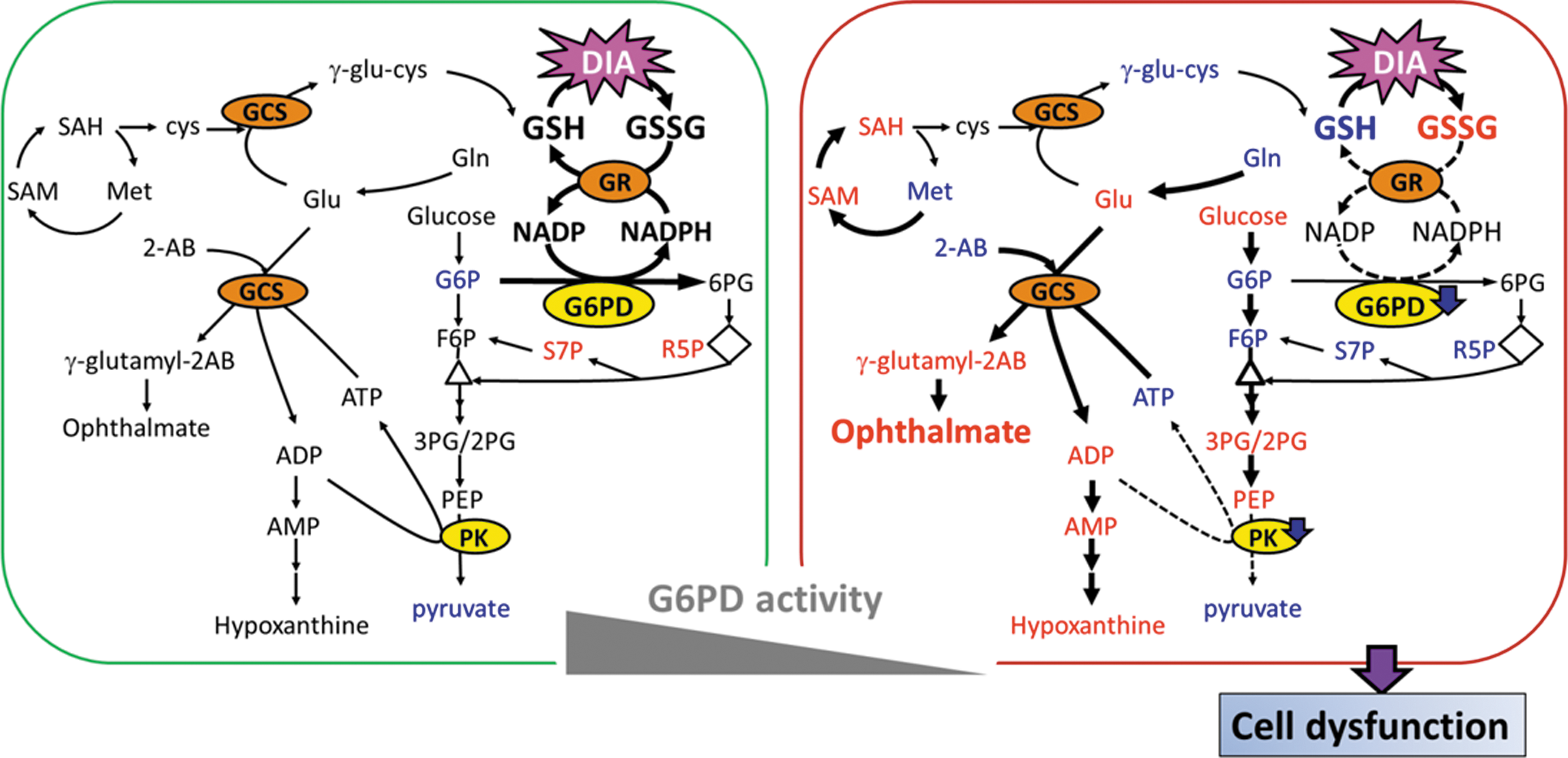
Based on our findings, we propose a model to explain the differential effects of DIA on metabolism of RBCs from normal and G6PD-deficient individuals (Fig. 10). In normal RBCs, pentose phosphate pathway and glycolysis are enhanced. G6PD is sufficient to produce NADPH efficiently for GSSG reduction and maintenance of GSH pool. The metabolism is perturbed to a lesser extent. The situation is different for G6PD-deficient cells. These cells can rely on the remaining G6PD activity for maintenance of GSH pool under normal circumstance. However, they are unable to generate enough NADPH under the condition of severe thiol depletion. GSH biosynthesis and methionine cycle are up-regulated at the expense of ATP, but fail to compensate for DIA-induced GSH depletion. A deficit of cysteine, caused by its oxidation to cystine, leads to accumulation of the side product ophthalmate. As a result of excessive ATP utilization, AMPK pathway is activated, and glycolysis is enhanced. However, certain glycolytic enzymes, such as PK, are inhibited, possibly due to oxidative modification. This leads to decline in cellular ATP content. Overall, such metabolic disturbance, together with protein oxidation, causes changes in RBC physiology, such as membrane deformability.
Materials and Methods
Sample collection and genetic characterization
Blood samples were collected from G6PD-deficient patients and age-matched healthy volunteers in EDTA-containing tubes and according to an IRB-approved protocol. The G6PD variant of the G6PD-deficient individuals was identified by restriction fragment length polymorphism as previously described (32). Whole blood was centrifuged at 1000 g for 10 min at 4°C. The supernatant and buffy coat were discarded, and the RBC pellet was washed thrice with phosphate-buffered saline (PBS) containing 5.5 mM glucose. After washing, the RBC count was adjusted using this buffer to 5×106 cells/μl, and 2×109 cells were incubated with 1 mM DIA for the indicated period.
Assay of G6PD activity
G6PD activity in whole blood was measured using the glucose-6-phosphate dehydrogenase activity assay kit (Sigma Aldrich) according to the manufacturer's instructions. The rate of NADPH formation is proportional to the G6PD activity and was measured spectrophotometrically at 340 nm (31).
Cytosol and membrane protein analysis
RBCs incubated with or without DIA for indicated times were treated with N-ethylmaleimide (NEM) to terminate the DIA reaction with thiol groups. In brief, the treated RBCs were hemolysed in a hypotonic phosphate buffer (2.5 mM NaH2PO4, 2.5 mM Na2HPO4, 1 mM EDTA, pH 8) containing 25 mM NEM, and centrifuged at 16,000 g to precipitate the ghosts. The membrane-bound cytoskeletal proteins in the ghost pellet were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), as described by Rossi et al. with modifications (55), and stained with coomassie brilliant blue.
The packed RBCs were lysed and analyzed by Western blotting with anti-phosphoAMPKα (Thr172) (1:1000 dilution, No. 2535; Cell Signaling), anti-AMPKα (1:1000 dilution, No. 2603; Cell Signaling), and anti-glutathione (1:1000 dilution, No. ab19534; Abcam).
Erythrocyte deformability
Control and G6PD-deficient RBCs, which had been treated with DIA for indicated times, were resuspended in a 10 mM PBS (pH 7.4) of 31 g/L polyvinylpyrrolidone (PVP) with NaCl for adjustment to the required osmolality. The osmotic deformability gradient of RBCs was determined with an ektacytometer by a method described by Clark et al. (18). In addition, the deformability was determined using a slit-flow ektacytometer (RheoScan-D system) (56) at a shear stress of 3 Pa in 31 g/L PVP at 290 mOsm/kg. In both methods, the laser diffraction pattern was used to calculate the EI and plotted against osmolality or shear stress. Deformability was assessed using EI measurement.
Global metabolite analyses
RBCs were incubated with or without DIA for indicated times. 4×108 RBCs were lysed in 200 μl of H2O containing 100 ppb of debrisoquine sulfate (Sigma Aldrich) as an internal control. The RBC lysate was extracted in 800 μl of methanol. The sample was incubated on ice for 15 min to precipitate proteins, and centrifuged at 16,000 g for 10 min at 4°C. The supernatant was dried under nitrogen gas, and dissolved in 400 μl of H2O containing 0.1% formic acid for LC-TOF-MS analysis. The procedure was carried out by the method of Cheng et al. (15). The details are provided in the Supplementary Data.
Targeted metabolite analysis of glycolytic intermediates
Hydrophilic metabolites were extracted and analyzed using Waters ultra-high-performance liquid chromatography coupled with Waters Xevo TQ MS. MS was operated in the negative ion and multiple reaction monitoring mode (43). Single analyte standard dissolved in a mixture of water/methanol 50:50 (v/v) were infused at a flow rate of 10 μl/min for tuning purposes. Major MS/MS fragment patterns of each analyte were determined. The optimized parameters were as follows: voltage at 25 V; capillary voltage at 1.3 kV; desolvation temperature at 500°C; source temperature at 150°C; and gas flow at 1000 L/h. The chromatographic separation was achieved on a BEH C18 (100×2.1 mm, particle size of 1.7 μm; Waters Corp.) at 25°C with eluent A (10 mM tributylamine with 15 mM acetic acid) and eluent B (50% acetonitrile containing 10 mM tributylamine and 15 mM acetic acid), and the flow rate was set at 0.3 ml/min. The gradient profile was as follows: linear gradient 99%–98% solvent B, 8 min; 12% solvent B, 2 min; 55% solvent B, 2 min; and 99% solvent, 2 min. The column was then re-equilibrated for 4 min. QC samples were analyzed during the analytical runs after every 15th samples.
Statistical analysis
The MS data were analyzed as described in the Supplementary Experimental Procedures section. For statistical analysis, multiple groups were compared using ANOVA with Tukey's post hoc test. Student's t-test was used for pairwise comparison. A p-value of less than 0.05 was considered significant. Correlation analysis between two ranked variables was then performed using the SPSS 20 to calculate Spearman's rank correlation coefficient, and false discovery rate analysis was applied for multiple-testing correction.
Footnotes
Acknowledgments
The authors are grateful to Prof. Arnold Stern of New York University, for his critical review of this article. This work was supported by grants from Chang Gung University (CMRPD190443, CMRPD1A0562, CMRPD1C0751, CMRPD1A0563, CMRPD190422, CMRPD190423, CMRPD1A0522, CMRPD391683, CMRPD1C0441, and CMRPD1C0761), the National Science Council of Taiwan (NSC99-2320-B-182-021-MY3, NSC101-2320-B-182-024-MY3, and NSC100-2320-B-182-010-MY3), and the Ministry of Education of Taiwan (EMRPD1C0271, EMRPD1D0871, and EMRPD1D0241).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.