
Abstract
Introduction
L
Originally, a role for cytochrome P450 (CYP)-derived metabolites of arachidonic acid (AA) in EDHF-mediated responses was implied on the basis of the fact that CYP inhibitors, such as clotrimazole, miconazole, and 17-octadecynoic acid, markedly attenuated NO/PGI2-independent hyperpolarization and relaxation in various preparations. However, these conclusions were limited by the fact that the CYP inhibitors used could not discriminate between different CYP isoforms. Moreover, some directly interfered with the activation of the K+ channels thought to be the main cellular targets of EDHF (3). However, more selective epoxygenase inhibitors [e.g., 6-(2-proparglyloxyphenyl)hexanoic acid and N-methylsulphonyl-6-(2-propargyloxyphenyl)hexanamide] have since been developed (169), and these compounds have been reported to abolish NO/PGI2-independent vasodilatation of renal arterioles (68).
Data relying on techniques other than the pharmacological CYP inhibition have considerably strengthened the evidence that CYP activation is an integral component of the EDHF response. RT-PCR, Western blotting, and immunofluorescence techniques have been used to demonstrate that native coronary endothelial cells express CYP enzymes, including CYP2C8, CYP2C9, and CYP2J2 [for a review, see ref. (105)]. The functional relevance of CYP activity has been addressed by enhancing the expression of CYP enzymes, as well as by attenuating CYP expression. The AA metabolites thought to act as an EDHF are the epoxides or epoxyeicosatrienoic acids (EETs) generated by endothelial CYP enzymes (Fig. 1). These substances elicit the hyperpolarization of endothelial and vascular smooth muscle cells (VSMC) by activating calcium-dependent K+ (KCa) channels (23), as well as the Na-K-ATPase (134). Certainly, CYP inducers, such as β-naphthoflavone or nifedipine, were found to increase the synthesis of EETs by cultured and native endothelial cells, and to enhance the agonist-induced, EDHF-mediated hyperpolarization and relaxation of intact coronary artery segments (45, 47, 129). Perhaps the first convincing evidence obtained in support of the hypothesis that a CYP2C epoxygenase is intimately involved in the EDHF response in porcine coronary arteries was provided by the use of antisense oligonucleotides directed against the coding region of CYP2C8/9. Incubation of porcine coronary arteries with antisense, but not sense or scrambled, oligonucleotides markedly reduced CYP2C mRNA and protein. They also attenuated bradykinin-induced, EDHF-mediated hyperpolarization and relaxation without compromising responsiveness to endogenously produced NO or an NO donor (47). This inhibitory effect of antisense oligonucleotides provided the first non-pharmacological evidence that a CYP2C metabolite is an essential permissive factor for EDHF-mediated vascular responses. A similar approach was used to show that EDHF-mediated responses in isolated resistance arteries from hamster gracilis muscle can also be attributed to the activity of a CYP2C epoxygenases (16). In the meantime, a whole series of EET analogues have been developed which can act as agonists and antagonists of EET-mediated responses (39, 56, 184), indicating that one or more membrane EET receptor may exist. More recently, inhibitors of the soluble epoxide hydrolase (sEH), the enzyme that “inactivates” the EETs by metabolizing them to corresponding diols or dihydroxyeicosatrienoic acids (DHETs), have helped enormously elucidate the diverse effects of the epoxides and diols in biological systems as well as in cardiovascular disease (58).
CYP enzymes and CYP-generated metabolites of AA have also been implicated in the regulation of tone in VSMC—particularly in the myogenic response. Given that the focus of this review is on the endothelium, the reader is directed to excellent overviews on the CYP/sEH axis in VSMC (30, 67) for further information.
The CYP-Dependent EDHF in Humans
It is relatively easy to specifically target CYP enzymes in experimental animals or isolated arteries to demonstrate the importance of CYPs and the sEH for the regulation of vascular tone. However, the evidence for a vasodilator role of these epoxides in humans is less direct and relies on the use of CYP inhibitors that cannot be guaranteed to be completely selective. Compounds such as sulfaphenazole, which is one of the most selective inhibitors available for CYP2C9 (79), have revealed an important role for CYP metabolites in vascular homeostasis under certain conditions. Although several studies failed to demonstrate any effects of sulfaphenazole, on forearm vasodilatation in healthy subjects (105), components of the flow-induced vasodilatation of skeletal muscle arterioles (61) and the radial artery (12, 13, 43) were attenuated by the CYP inhibitor. Is there a way to explain this apparent discrepancy? Certainly, the CYP enzymes are not expressed in all endothelial cells and are the highest in coronary arteries and in small arterioles. Thus, the net effect of CYP inhibition depends on its expression profile and the contribution of CYP-expressing arteries/arterioles to the overall change in blood flow measured. In this regard, it is important to note that the studies reporting a significant effect of sulfaphenazole in healthy subjects looked at specific vessels and confirmed the expression of CYP2C protein in the tissue studied (43, 61). Clearly, however, disease can affect responses, as forearm vasodilatation to acetylcholine was blunted by CYP inhibitors in patients with essential hypertension (160), hypercholesterolemia (123), and primary hyperparathyroidism (166). Assessing the contribution of CYP enzymes to cardiovascular disease may, however, be hampered by the fact that several routinely prescribed cardiovascular pharmaceuticals, especially the Ca2+ antagonist, nifedipine, and the HMG-CoA reductase inhibitor, fluvastatin (45, 46), are metabolized by vascular CYP enzymes. Depending on whether or not the xenobiotic or AA is the preferred substrate for the enzymes, these substances may actually decrease epoxide production.
How do EETs exert their biological actions?
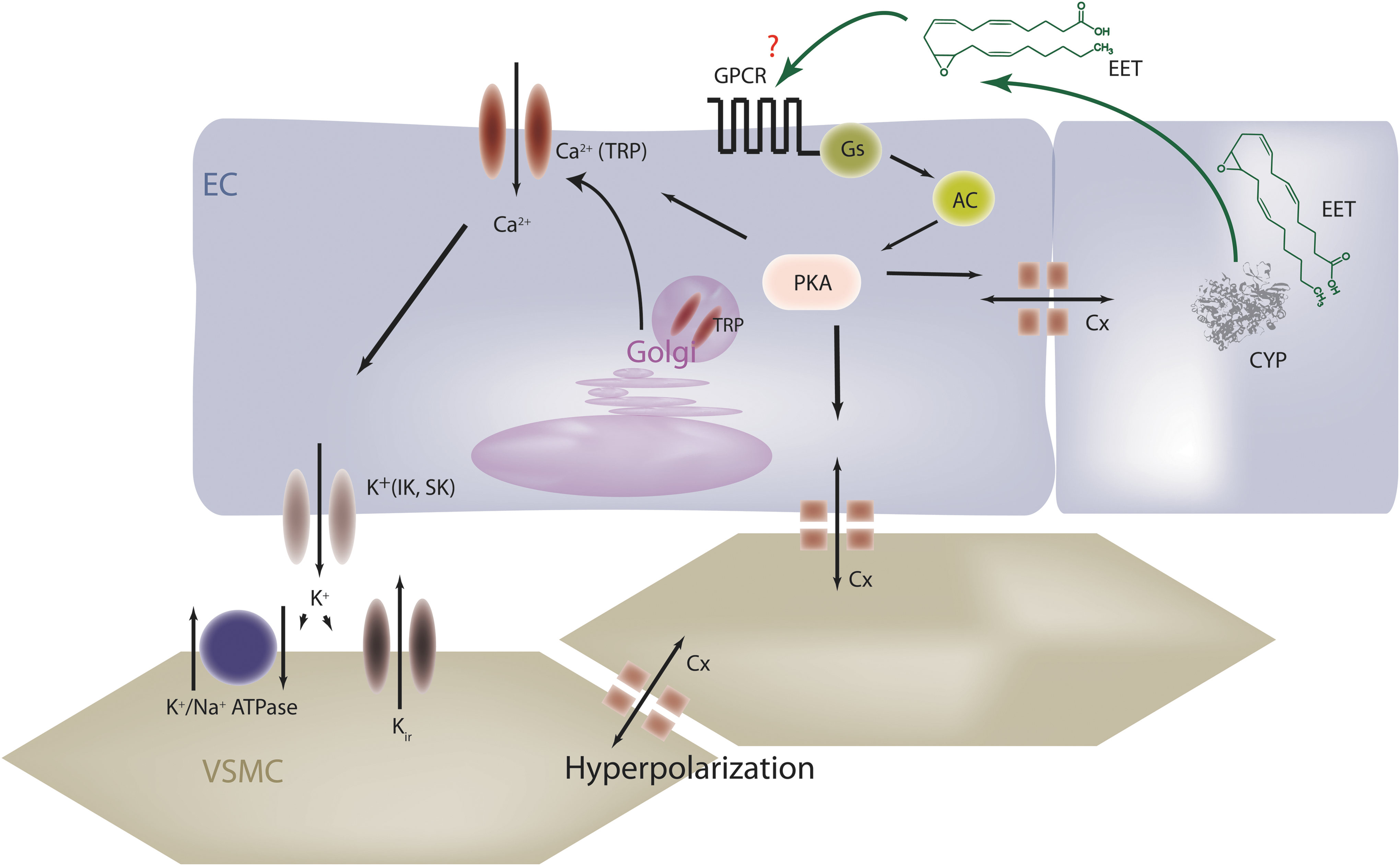
The realization that EETs, especially 11,12- and 14,15-EET, can activate large conductance KCa channels (BK) on VSMC to elicit hyperpolarization and relaxation led to their identification as a class of EDHF (23, 47). Back in the 1990s, the literature related to EDHF focused on an “it is” versus “it isn't debate” that centered on the sensitivity of responses to combinations of KCa channel toxins such as iberiotoxin, charybdotoxin, and apamin. Indeed, reports that EDHF-induced responses were mediated by small and intermediate conductance KCa channels (199) while EET-dependent responses were sensitive to iberiotoxin which targets BK channels (23), were taken to indicate that a factor other than an EET should regulate the EDHF response. However, the EDHF-mediated relaxation of porcine coronary arteries that was sensitive to the CYP2C9 inhibitor sulfaphenazole and the CYP2C antisense approach which was also insensitive to iberiotoxin and sensitive to charybdotoxin and apamin suggested that the role of EETs in the EDHF phenomenon may not simply be related to BK channel activation. Nowadays, the term EDHF has been recognized as an oversimplification, as three principal components seem to be linked to the EDHF phenomenon: (i) an increase in endothelial cell Ca2+ after cell stimulation that triggers KCa activation and the synthesis of a metabolite which is essential for the subsequent EDHF-mediated responses; (ii) K+, released from endothelial cells via KCa channels, induces smooth muscle hyperpolarization by activating inwardly rectifying K+ channels and/or the Na+/K+-ATPase on VSMC; and (iii) endothelial cell hyperpolarization is transmitted to the vascular smooth muscle via gap junctions. The strengths and weaknesses of the arguments for each of these specific types of EDHF have been discussed at length (20), but each of them appears to be valid in certain vascular beds. Interestingly, all of these mechanisms can be modulated by EETs.
EETs, Ca2+, and KCa channel activation
Despite early reports that EETs may act as Ca2+ influx factors in different cells (18, 57), it took a while to show that the EET-induced activation of KCa channels is preceded by an increase in intracellular Ca2+ levels that can be accounted for by an increased open probability of non-selective cation channels of the transient receptor potential (TRP) family. How this happens was initially attributed to the presence of an AA binding site in some of the TRP channels that can be activated by the parent lipid (174, 196) as well as by the EETs (167, 174). However, while relatively high concentrations of the EETs may affect TRP channels directly, more physiological concentrations activate TRP channels in a protein kinase A (PKA)-dependent manner that involves their translocation to caveolin-rich areas in the plasma membrane (35, 51, 95). There appear to be regioisomer-specific differences in EET-induced TRP channel translocation and activation as 5,6-EET but not 11,12-EET can activate TRPV4 in endothelial cells (167, 174)—a phenomenon that underlies the EDHF-dependent, flow-induced vasodilatation (95). On the other hand, 11,12-EET but not 14,15-EET or 5,6-EET enhances the bradykinin-induced capacitive Ca2+ influx in endothelial cells by stimulating the translocation of TRPC6 and TRPC3 (51). In fact, the EET-induced activation of TRP channels and the subsequent activation of KCa channels may well account for their role in the EDHF response.
EETs and gap junctional communication
The transfer of hyperpolarization via endothelial or myo-endothelial gap junctional communication appears essential for the maintenance of a functional syncytium in the smaller resistance-sized arteries where the EDHF phenomenon predominates (139, 140). This type of cell–cell communication requires different connexin proteins that can build hemichannels and intact gap junctional plaques which allow ions and small molecules to be transferred from cell to cell. EETs are able to affect the composition of the gap junctional plaque, that is, can recruit connexin proteins to the plaque. EETs can also affect the phosphorylation and thus conductance of individual connexin proteins (101, 130). Indeed, in murine resistance arteries, acetylcholine-induced, EDHF-like dilations were dependent on the expression of connexin 40 under isometric conditions (15). Similar to other actions of the EETs, the effects on connexin 43 were found to be dependent on PKA activation as well as on the ERK1/2 kinases (130).
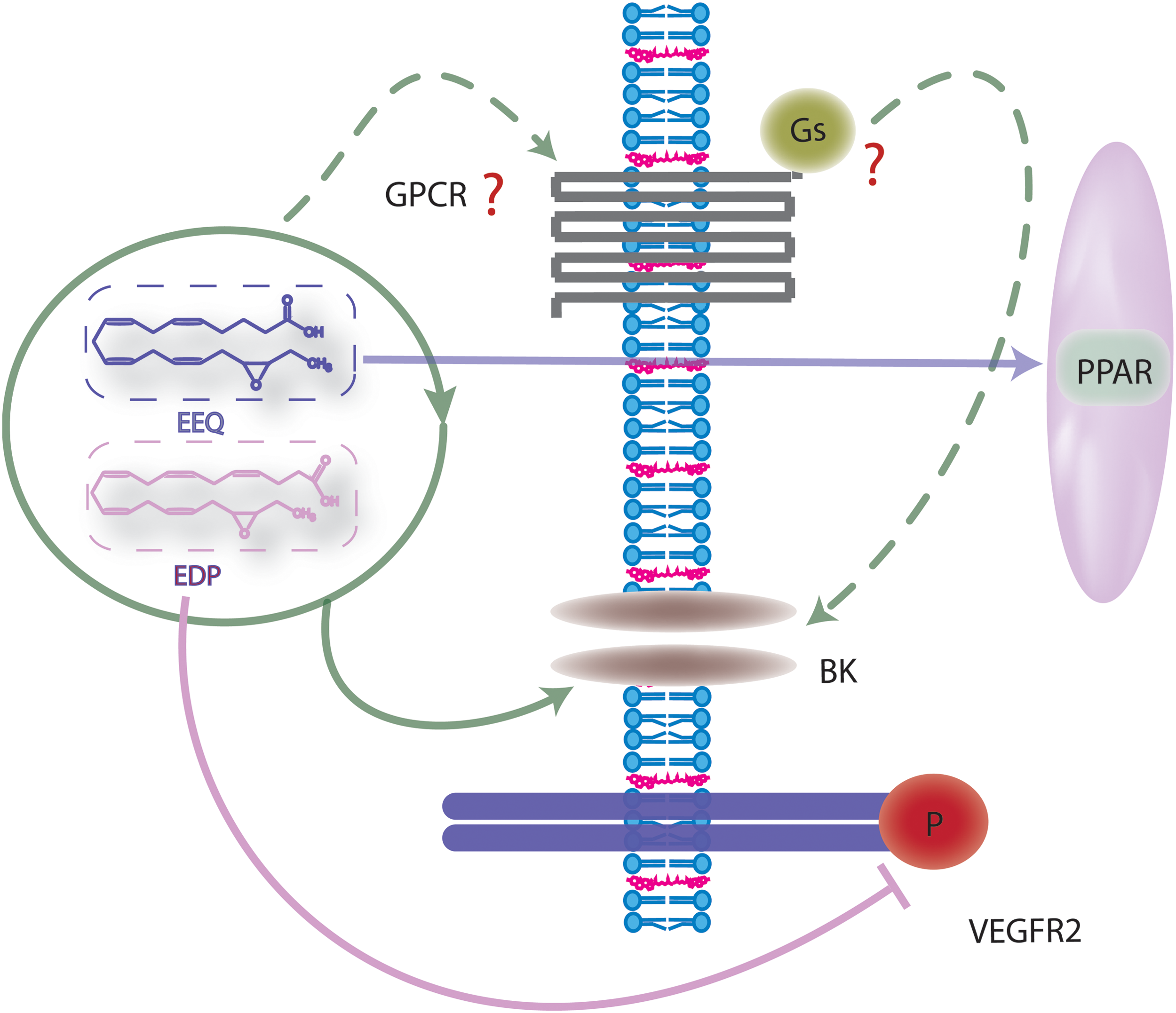
A G-protein-coupled EET receptor?
One characteristic of many EET-induced cellular responses, including cell proliferation, gap junctional communication, or TRP channel translocation, is the dependence on PKA activation. Given that high-affinity EET binding sites have been described on the surface of some cells, several groups have speculated that a specific EET membrane receptor may exist (128). This concept is supported by the fact that there are differences in responsiveness to different EET stereoisomers and not just the regioisomers. For example 11(R),12(S)-EET is a more potent activator of renal artery KCa channels (198) and rat airway electrical parameters (126) as well as endothelial cell TRP channel translocation and angiogenesis (35) than 11(S),12(R)-EET. This is, however, not a universal observation, as 11(S),12(R)-EET is reportedly more effective than 11(R),12(S)-EET in activating cardiac KATP channels (97).
Indirect evidence that strongly supports the concept of a membrane-bound EET receptor which recognizes defined structural components within the EETs is the fact that a series of stable and specific EET agonists and antagonists has been generated (39, 56, 184). The fact that the epoxides of AA increase intracellular Ca2+ may hint at an effect mediated by Gq/11 proteins, but the effects are indirect and involve TRP channels. Rather, placing the evidence for a protein receptor on cell membranes along with that indicating a reliance on cyclic AMP/PKA for EET-induced signaling—the existence of a Gs-coupled EET receptor has been postulated (70). Certainly, biochemical studies measuring GTP binding to G proteins in endothelial cells confirm the importance of a G protein and indicate that 11,12-EET increases GTPγ35S binding to Gs, but not Gi proteins (118). In addition, the 11,12-EET-induced activation of KATP channels is dependent on the Gs protein (187) and the activation of PKA (186), as are 11,12-EET-induced endothelial cell migration and angiogenesis (35). However, although many of the effects of the EETs rely on PKA activation, the EET-induced changes in cyclic AMP tend to be small and inconsistent—particularly in endothelial cells, indicating that the EET receptor may not turn out to be a classical Gs-coupled receptor.
Intracellular EET receptors?
Since AA epoxides are generated intracellularly, the highest concentrations of these mediators are expected to be reached in the cytosol—especially in cells that express CYP enzymes. Therefore, it is more than likely that these intracellular lipid mediators interact with intracellular receptor molecules such as the peroxisome proliferator-activated receptor (PPAR) family of nuclear receptors. There have been several reports of PPARα and PPARγ activation by the EETs and the consequences on vascular cell proliferation, migration, and inflammation (181). The importance of PPAR activation in mediating the effects of the EETs is, however, unclear—largely because some of the early sEH inhibitors that were initially found to result in PPARα activation did so via off-target effects (40).
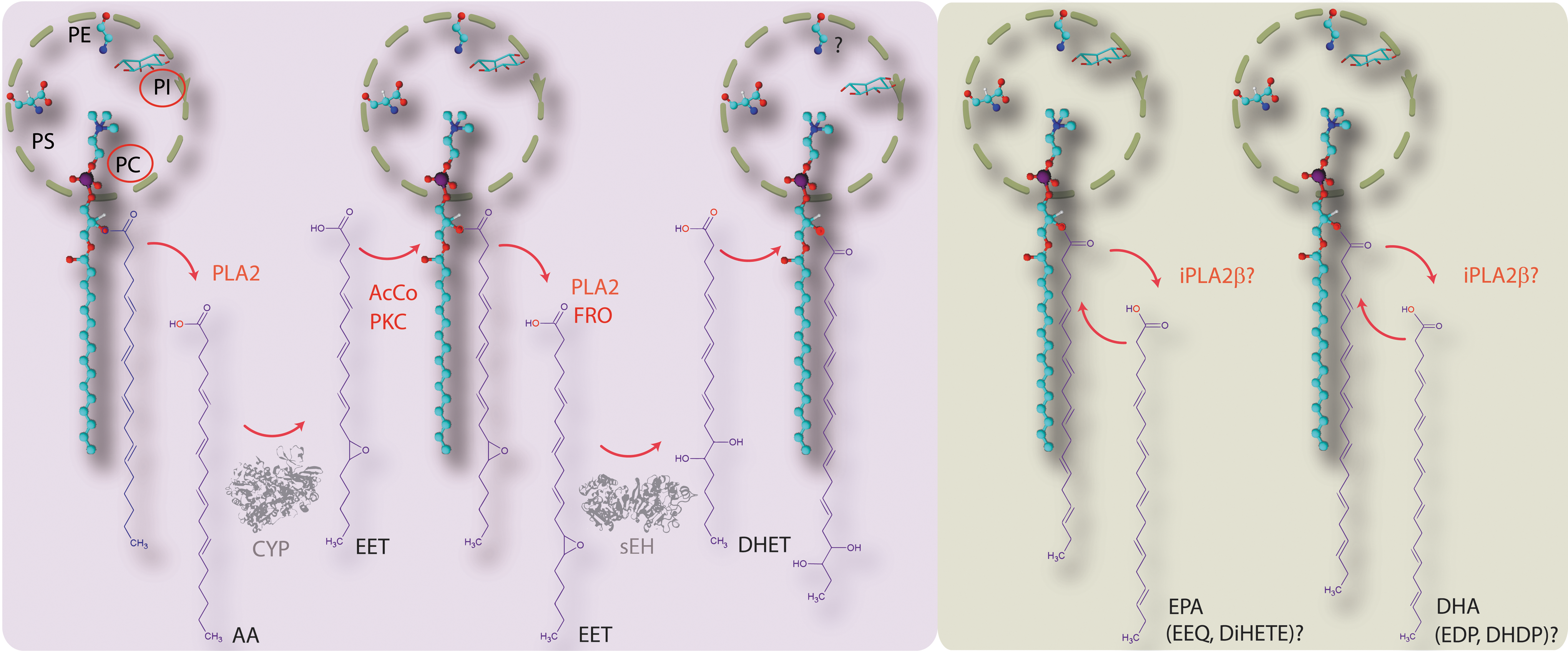
Phospholipid integration?
Lipids are not homogenously distributed within biological membranes but tend to be organized into domains of defined lipid composition that generate a suitable environment for protein function (93, 158). For some of the biological actions of the EETs, a cell surface receptor may not be essential as polyunsaturated fatty acid (PUFA)-derived epoxides and diols can incorporate into membrane phospholipids and potentially interfere with cell signaling by modulating the lipid microenvironment therein (84). There is evidence that EETs and DHETs can be incorporated into the sn-2 position of phospholipids, especially phosphatidylcholine and phosphatidylinositol phospholipids (24, 76, 113, 165) (Fig. 2). In endothelial cells, the incorporation of EETs into a phospholipid pool is reported to be catalyzed by acyl coenzyme synthase (176), and a similar protein kinase C-modulated phenomenon has been described in astroglial cells (150). This contrasts with the report that EETs can be generated nonenzymatically from EET-containing phospholipids by free radical oxidation (113) (Fig. 2). Although the physiological relevance of these processes remains to be determined, pre-loading isolated porcine coronary arteries with EET and DHET was found to enhance endothelium-dependent, but not endothelium-independent, relaxation. Such observations led to the suggestion that these esterified lipids are an intracellular storage form of EET, from which they can be recruited on stimulatory impulse, independently of CYP activity (176).
The sEH
Epoxide generation is thought to be determined by both the level of epoxygenase expression and the availability of the PUFA substrate. In the case of AA the latter is determined by the activity of phospholipases such as phospholipase A2. Intracellular levels of the epoxides are tightly regulated and metabolism occurs relatively rapidly by hydrolysis, β-oxidation, and chain elongation (66). The sEH is the most important epoxide-metabolizing enzyme that generates dihydroxy-fatty acids (or diols). The latter were considered for a long time to be less active than the parent epoxides—but recent evidence has challenged this assumption (see next). There are, of course, exceptions to every rule and some epoxides are poorer sEH substrates—the best-studied being 5,6-EET, which is more rapidly metabolized by cyclo-oxygenase (COX)2 (122). The development of sEH inhibitors and transgenic sEH mice—both of which increase tissue and circulating EET levels—has contributed immensely to the identification of the biological actions of the CYP epoxides and diols.
Surprisingly little is known about the mechanisms that regulate sEH activity. There have been a number of studies linking changes in sEH expression with inflammatory or hormonal stimuli (2, 115). Two tyrosine residues (Tyr383 and Tyr466) in the active site of the hydrolase are reportedly essential for enzyme activity (183), and these were recently shown to be nitrated by peroxynitrite both in vitro and in vivo in mouse models of type 1 and type 2 diabetes, leading to a decrease in sEH activity (10). It is currently only possible to speculate about the involvement of sEH tyrosine nitration in the amplification of inflammation associated with diabetes but at least one sEH polymorphism, which results in decreased enzymatic activity, has previously been associated with human insulin resistance (121). The sEH was also recently reported to be nitrosated in leptin-stimulated wild-type but not endothelial NO synthase knockout mice, suggesting that the effects of NO on PUFA metabolism may be partly related to the modulation of sEH activity (36).
Inhibition or deletion of the sEH increases tissue and circulating levels of the PUFA epoxides while simultaneously decreasing diol production and has pronounced effects on blood pressure (65, 74), inflammation (94), progenitor cell proliferation, angiogenesis, and vascular repair (54). Given that the sEH metabolizes PUFA epoxides to their corresponding diols, an increase in sEH expression or activity would be expected to decrease EET as well as eicosapentaenoic acid (EPA)- and docosahexaenoic acid (DHA)-derived epoxide bioavailability; to contribute to the blunting of endothelium-dependent vasodilatation; and to promote hypertension. In fact, there are several examples of hypertension being associated with elevated sEH expression and/or activity, including the spontaneously hypertensive rat (65, 188). Moreover, there have been numerous reports showing that specific sEH inhibitors are effective at preventing as well as reversing experimental hypertension [for a review, see refs. (58, 69)]. The particular effectiveness of sEH inhibitors against hypertension associated with activation of the renin–angiotensin system is most likely related to the fact that angiotensin II markedly increases sEH expression in vivo (2). Interestingly, hypoxia does the opposite and markedly downregulates sEH promoter activity and, thus, protein expression in the lung (77). There are other examples of hypertension being associated with elevated sEH expression and/or activity such as the spontaneously hypertensive rat. In these animals, elevated sEH expression is linked to an increase in the renal metabolism of EETs to DHETs and sEH inhibitors blunt the development of hypertension (65). Initial reports also documented that male sEH−/− mice have lower blood pressure and elevated EET levels than their wild-type littermates (151). However, the blood pressure phenotype now seems to be controversial as the loss of the hydrolase can be compensated by elevated concentrations of the pressor and vasoconstrictor eicosanoid, 20-hydroxyeicosatetraenoic acid, as well as increased lipoxygenase (LOX)-derived hydroxylation and prostanoid production (98). Several of the metabolites generated by the sEH, such as the EET-derived DHETs, are also biologically active, but generally less so than the parent epoxides. However, the DHETs are not as readily incorporated into membrane lipids as the EETs, and the latter are thought to be the form in which the majority of endothelium-derived EETs leave the cell (109).
EETs and Angiogenesis
Given the fact that the activation of KCa channels has been linked to endothelial cell proliferation (178) and EETs activate KCa channels, it would seem logical to assume that KCa activation would play a role in EET-induced proliferation. However, although the activation of KCa channels has been linked to endothelial cell proliferation induced by basic fibroblast growth factor (178), this mechanism appears not to be involved in the EET-induced proliferation of endothelial cells.
The first hint that EETs may affect cell signaling and proliferation was obtained in renal epithelial cells (27, 28) and soon afterward, “authentic EDHF” recovered from the luminal incubate of rhythmically stretched coronary arteries was found to activate a number of kinases, whose function was closely linked with endothelial cell proliferation (49). The activation of these MAP kinases could be inhibited by the treatment with CYP inhibitors as well as by antisense oligonucleotides directed against CYP2C and could be mimicked by the treatment of endothelial cells with 11,12-EET or by the overexpression of CYP2C8 (49). A more detailed analysis of the mechanisms involved revealed that CYP-derived metabolites of AA are able to transactivate the epidermal growth factor (EGF) receptor (29, 104). 14,15-EET was initially suggested to act as a second messenger after activation of the EGF receptor; however, it appears that 14,15-EET can also elicit the release of heparin-binding EGF-like growth factor from a renal epithelial cell line via a process involving the activation of matrix metalloproteases (29). Although the protease involved has not yet been identified, a very similar mechanism seems to be responsible for the transactivation of the EGF receptor in endothelial cells (104). The EET-mediated activation of the EGF receptor leads, in turn, to the activation of the kinase Akt and an enhanced expression of cyclin D1. All four EET regioisomers have been reported to elicit an increase in Akt phosphorylation and cell proliferation in murine endothelial cells but only the proliferative effects of 5,6- and 14,15-EET are reportedly sensitive to a phosphatidylinositol 3-kinase (PI 3-K) inhibitor. The 8,9- and 11,12-EET-induced increase in [3H]thymidine incorporation seems to be dependent on the activation of the p38 MAP kinase (133). In contrast, in bovine aortic endothelial cells 8,9-, 11,12- and 14,15-EET-induced cell proliferation can be attenuated by MEK, ERK, and PI 3-K inhibition (172). Other signaling pathways also contribute to the increase in cyclin D1 expression, including the MAP kinase phosphatase-1, which decreases JNK activity (132). Activation of Akt by EETs also induces the phosphorylation and therefore inhibition of the forkhead factors FOXO1 and FOXO 3a and, subsequently, a decrease in the expression of the cyclin-dependent kinase inhibitor p27kip1 (131). The involvement of this mechanism in the CYP2C9-induced endothelial cell proliferation could be demonstrated by the transfection of CYP2C9 overexpressing cells with either a dominant negative Akt or a constitutively active FOXO3a, both of which inhibit CYP2C9-induced endothelial cell proliferation (131). Although there is a precedent for the negative regulation of JNK after activation of Akt, inasmuch as Akt has been reported to phosphorylate and inactivate the kinase SEK1 and thus inactivate its substrate JNK (125), it remains unclear whether these pathways are linked to each other or are simply activated in parallel.
More convincing links between EETs and angiogenesis were made after studying co-cultures of astrocytes and endothelial cells. EETs released from astrocytes increased thymidine incorporation into endothelial cells and elicited the formation of capillary-like structures (112, 192). Moreover, the overexpression of CYP2C9 in and/or the application of 11,12- or 14,15-EET to monocultures of endothelial cells was associated with angiogenesis (102, 104). In vivo data rapidly followed to support these in vitro findings and EETs induced angiogenesis in the chick chorioallantoic membrane (104), as well as in EET-impregnated matrix plugs in adult rats (102) and in an ischemic rat hindlimb model in which the overexpression of different CYP isozymes, including CYP2C11 and 2J2, was found to increase muscle capillary density (172). In addition, tumor growth and metastasis can be increased by sEH inhibition in transgenic mice with high vascular EET levels, that is, animals which overexpress either the human CYP2C8 or human CYP2J2 specifically in Tie-2 expressing cells or that are treated with high concentrations of 14,15-EET (124). Thus, elevated EET production is linked with the promotion of angiogenesis. It is important to note that angiogenesis could be stimulated when EETs were generated by endothelial cells as well as when they were applied exogenously or generated from astrocytes. This means that the actions of the EETs cannot be restricted to an autocrine role but that a sufficient EET concentration should be able to leave the cell of origin to elicit paracrine actions on other cells.
More than just AA and EETs: other CYP substrates and their biological activity
The CYP-dependent metabolism of AA (Fig. 3) is often referred to as the third pathway for AA metabolism, that is, in addition to the COX and LOX pathways. However, this definition implies that the AA-derived epoxides and diols are the dominant CYP-derived products affecting physiology and pathophysiology (Fig. 4). Certainly, we know most about the actions of the AA products but this is simply because the appropriate tools are available for their study. However, CYP enzymes are able to metabolize a wide range of endogenous PUFAs and generate a wide variety of different signaling lipids, many of which are implicated in cardiovascular homeostasis.


At this stage, it is necessary to take a closer look at biological CYP enzymes and the substrates that can feed into the CYP/sEH axis. In general, the CYP enzymes are membrane-bound, heme-containing terminal oxidases that catalyze a broad spectrum of enzymatic reactions, generating epoxides, ω-terminal hydroxyls, or midchain dienols (190). Although the majority of CYP enzymes are highly expressed in the liver, several can be detected in the cardiovascular system (48, 190) as well as in inflammatory cells (55, 114, 149). The PUFAs that are substrates for the CYP enzymes can be provided directly via the diet or generated from another PUFA; for example, the ω-6 PUFA, AA, can be synthesized from the primary essential fatty acid linoleic acid (C18:2 ω-6) that is enriched in vegetable oils derived from sources such as corn and sunflower oil. Nowadays, linoleic acid is generally over-represented in most Western diets, which are, in turn, often deficient in ω-3 PUFAs such as the fish oils EPA (C20:5) and DHA (C22:6) (14). The latter PUFAs are required in brain tissue for normal cognitive function (80), as EPA and DHA are essential constituents of cell membranes. The shortest essential ω-3 PUFA, namely α-linoleic acid, is enriched in leafy green vegetables and can serve as a substrate for chain elongation and desaturation reactions to generate longer-chain ω-3 PUFAs such as EPA and DHA (92). However, ω-3 PUFAs cannot be interconverted by humans or rodents from the ω-6 fatty acids.
Just like AA, EPA as well as DHA can be metabolized by the LOX and COX pathways, even though both are regarded as rather poor substrates for the latter enzymes (73, 137). The products generated are a series of ω-3 prostaglandins (9, 168) and leukotrienes (143). CYP epoxygenases also generate a recently identified unique family of fatty acid mediators referred to as protectins and resolvins that can be summarized along with the maresins as pro-resolving mediators (145, 147). The latter have gained a lot of attention as potent mediators involved in the resolution of inflammation (32, 146).
It may well turn out that the metabolism of the fish oils by CYP enzymes and the sEH (Fig. 3) is biologically more important than their metabolism by COX and LOX, as numerous recent studies indicate that EPA and DHA are preferred substrates of the CYP enzymes (44). This is at the moment probably best demonstrated in a transgenic mouse that overexpresses a fatty acid desaturase from Caenorhabditis elegans that catalyzes the conversion of ω-6 (18- and 22-carbon chain length) substrates to ω-3 fatty acids (75). These animals demonstrate markedly enhanced ω-3 PUFA levels paralleled by reduced ω-6 PUFA levels and markedly increased production of the CYP-derived epoxides of EPA and DHA, particularly the EPA-derived epoxide 17,18-epoxyeicosatetraenoic acid (EEQ). Simultaneously, there were only minimal changes in levels of the LOX-derived EPA product, 5-hydroxyeicosapentaenoic acid and a decrease in the COX-derived AA metabolite bicyclo-PGE2 (6). Similar changes were reported in a recent study in which 20 human volunteers were given EPA/DHA supplements. This resulted in significant increases of plasma and urine levels of CYP-derived metabolites (44) and fits well with the fact that most CYP isoforms from different mammalian species accept the ω-3 PUFAs EPA and DHA as substrates for epoxidation as well as ω/(ω-1)-hydroxylation (86). Certainly, the human CYP2C8, CYP2C9, CYP2C18, CYP2C19, the rat CYP2C23, as well as the CYP2J family of enzymes accept EPA and DHA as substrates (86). Moreover, CYP enzymes that were previously defined, on the basis of their ability to metabolize AA, as classical hydroxylases have since been shown to possess epoxygenase activity as well. Two examples of this are the human CYP4A1 (89) and the murine Cyp4a12a, both of which metabolize EPA to its epoxide 17,18-EEQ (89, 111). The reason for this apparent difference may be the presence of additional double bonds in these PUFAs that are the preferential site of enzymatic action for most of the epoxygenases. The epoxides of EPA and DHA that are similar to the AA-derived EETs, in turn, also act as substrates for the sEH and, subsequently, are metabolized to the corresponding vicinal diols (i.e., dihydroxyeicosatetraenoic acids, dihydroxyeicosatetraenoic acid, and dihydroxydocosapentaenoic acids [DHDP]) (110) (Fig. 3). In fact, the ω-3 PUFA epoxides are even better sEH substrates than the EETs, with the exception of 19,20-EDP, which is converted at lower rates.
Biological Activity of CYP-Derived PUFA Epoxides and Diols
Although clinical studies indicated that dietary supplementation with fish oils was able to lower systemic blood pressure in patients suffering from hypertension several years ago (85), it is only relatively recently that interest in the signaling properties of the fish oil-derived epoxides and diols has emerged. In fact, the biological actions of the ω-3 PUFA epoxides seem to be similar to or even more potent than those of the EETs (60, 89, 185, 194). For example, epoxydocasapentaenoic acids (EDPs) seem to be more potent activators of BK channels than the EETs (185) and apart from the chemically unstable 5,6-EDP, all of the EDP regioisomers tested possess EC50 values in the pM range on the relaxation of porcine coronary arterioles after endothelin precontraction (185). Unlike the EETs, no regioselectivity has been reported for the DHA-derived CYP products to dilate vessels. To date, the CYP-derived ω-3 metabolites are similar to the EETs in that they display anti-inflammatory, vasodilator, or analgesic properties (110, 116, 185) but differ in that they appear to be mainly anti-angiogenic, anti-tumorigenic, and anti-metastatic (177, 193).
The mechanism(s) underlying the blood pressure-lowering effect of ω-3 PUFAs, similar to those of the EETs, may involve the activation of VSMC KCa channels, as both 19,20-EDP (88, 170) and 17,18-EEQ (89, 194) were found to activate BK channels. Indeed, the activation of BK channels by DHA was sensitive to the CYP inhibitor SKF525A (170) and could be directly mimicked by the EDP. There are fewer in vivo studies available, but it seems that blood pressure-lowering actions of ω-3 PUFAs are absent in mice lacking the gene which encodes the pore-forming β-subunit of the BK channel (63). While it is tempting to speculate that the effects described in wild-type mice in the latter paper could be attributed to the production of the epoxides from EPA and DHA, the effects were insensitive to SKF525A, which rather unselectively targets several different CYP enzymes, for example, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A (53), and can affect nicotinic receptor activation (157). An alternative means of increasing PUFA epoxide levels is to inhibit the sEH, and in a model of angiotensin II-induced hypertension where animals were supplemented with an EPA-/DHA-rich diet in combination with an sEH inhibitor—the antihypertensive effects of DHA/EPA were potentiated (163). Further studies have substantiated the antihypertensive effects of the ω-3 PUFA epoxides, in a murine model where Cyp1a1 was genetically deleted, the resulting hypertension went hand in hand with a reduced vasodilator response to ω-3 PUFA supplementation—potentially due to reduced production of 17,18-EEQ and 19,20-EDP (1).
Angiogenesis
As outlined earlier, the activation of the CYP/sEH axis that results in increased EET production has clearly been linked with the promotion of angiogenesis. However, the studies were somewhat artificial and focused on the products of AA metabolism, largely ignoring the biological actions of other lipids that feed into the same CYP/sEH axis.
It was partly to assess the role of the sEH in angiogenesis under more physiological conditions that the consequences of the global and induced deletion of the sEH as well as its pharmacological inhibition were studied in the vascular repair after ischemia and in the postnatal murine retina. The clear expectation from such studies was that the inhibition of the sEH should increase EET levels and increase angiogenesis—as earlier in the animal models where CYP enzymes were overexpressed. However, sEH deletion and inhibition resulted in markedly decreased angiogenesis (64) and vascular repair (54) and provided some of the first experimental data that linked the defect not to the accumulation of a PUFA epoxide but to the lack of diol production. To identify such lipids, liquid chromatography (LC)-mass spectrometry(MS)-based lipid profiling approaches were used to screen for the PUFA epoxides or diols most affected by the deletion of the sEH. Until now, biological activity has been attributed to the DHA-derived diol, 19,20-DHDP (64) and the linoleic acid-derived diol 12,13-dihydroxyoctadecenoic acid (12,13-DiHOME) (54). Interestingly, the signaling pathways targeted by the diols are distinct, as while the defective vascular repair in sEH−/− mice could be attributed to altered Wnt signaling followed by attenuated progenitor cell proliferation and mobilization (54) the defects in the retina could be linked to the translocation of presenilin-1 out of lipid rafts and the subsequent inhibition of the γ-secretase (64). This means that the ω-3/ω-6 profile of a particular tissue is likely to determine overall effects of CYP activation on angiogenesis.
Unlike some of the EET regioisomers, which have been attributed pro-proliferative and angiogenic properties (31, 104) and linked to an increased tumor angiogenesis and metastasis (124), 17,18-EEQ but not other EEQ regioisomers derived from EPA was found to inhibit cell proliferation in an immortalized endothelial cell line (34). In addition, the epoxides of DHA, mainly 19,20-EDP, display inhibitory effects on tumor growth and metastasis (193). The latter effect could be attributed to inhibition of the vascular endothelial cell growth factor (VEGF)-induced phosphorylation of the VEGFR2 (193), although the exact molecular mechanism (kinase inhibition, phosphatase inhibition, or interference with β1 integrin signaling) remains to be determined. Further analyses also suggested that EDPs were able to suppress VEGF-C- but not VEGF-A-induced responses. This is significant, as VEGF-C is an important mediator regulating the migration, proliferation, and outgrowth of lymphatic vessels (72), and hints at a possible role of EDPs in the inhibition of lymphangiogenesis, a key process in tumor metastasis (159).
CYP enzymes and the sEH have been linked with lymphatic development as in the zebrafish, where 19,20-EDP is the predominant PUFA epoxide, and sEH inhibition results in the prevention of parachordal vessel development or in paracordal vessel misguidance (54). In the zebrafish, the parachordal vessels usually give rise to lymphatic endothelial cells (82). Furthermore, in terms of angiogenesis, the sprouts from the axial vein that extend ventrally to form the caudal vein plexus were also affected and remodeled to a single luminal vessel (54). If 19,20-EDP contributes to the process, it is likely via a process other than VEGF signaling as although VEGF and its receptors drive the angiogenesis from the axial vessels (59, 179), interfering with the VEGF receptors results in much more severe defects. The latter include the incomplete segregation of the caudal vein and the dorsal aorta (59), and inhibition of the sprouting of the intersegmental arteries (90). However, sprouting from the axial vein that extends ventrally to from the ventral caudal plexus was not affected (179). In this context, it is important to note that lymphatic endothelial cell specification is attenuated by bone morphogenic protein 2, which is required for venous sprouting angiogenesis (179), via the negative regulation of Prox1 (37). Interestingly, for the topic of this review, other fatty acid epoxides such as 11,12-EET have already been linked with promoting venous differentiation in as much as (i) EphB4, a classical venous marker, is upregulated in native and cultured endothelial cells exposed to 11,12-EET and (ii) EET-induced angiogenesis in vivo can be attenuated by the siRNA-mediated downregulation of EphB4 (175). Such results emphasize the importance of higher resolution analytical methods to determine the ω-3/ω-6 PUFA profiles of cells or tissues and to unravel the different effects of the substrates and products of the CYP/sEH pathway.
Previous studies were able to assess the fatty acid composition of tissue and plasma using gas chromatographic techniques (7, 75), and while such studies provided estimates of the ω-3/ω-6 PUFA ratio they lacked detailed information about oxylipins, unesterified fatty acids, and more complex lipid species. To obtain the necessary detailed insights into the complex lipidome, a combination of different untargeted and targeted lipidomic approaches are necessary. With the help of targeted mass spectrometer-based methods available nowadays, a huge panel of oxylipins, especially those of COX, LOX, and CYP metabolites involved in inflammation and angiogenesis, can be reliably detected. A recent publication makes it clear that the integration of untargeted approaches can generate unbiased novel findings, for example, the replacement of the fatty acyl chains of cholesterol esters and phospholipids with ω-3 and ω-6 PUFAs (7). The latter study also highlighted that the most marked change associated with the higher ω-3 PUFA levels in the plasma of fat-1 mice compared with wild-type mice was an increase in EPA-derived epoxides and diols, that is, the metabolites of the CYP-sEH axis. The tracking of such fatty acid shuffling might provide further insights into the formation of more complex lipid structures, their impact on membrane composition, and their influence on membrane dynamics. While murine studies are not always applicable to the human situation, dietary supplementation with EPA and DHA directly modulates serum levels of EPA- and DHA-derived metabolites of the CYP/sEH pathway, making it tempting to suggest that potential beneficial effects of the fish oils on the cardiovascular system could be attributed to ω-3 epoxides (144). Certainly, in healthy human volunteers, EPA/DHA supplementation primarily resulted in a large increase of EPA-derived CYP-dependent epoxy-metabolites followed by increases of EPA- and DHA-derived LOX-dependent monohydroxy-metabolites, including the precursors of resolvin E and D families (44). A similar approach was also found to improve small peripheral artery function in a patient collective with intermediate to high cardiovascular risk (71).
Perhaps the best example of a tissue in which fatty acid profiling has helped understand biological responses is the retina, a tissue enriched in DHA (5, 52, 191). Long chain PUFAs were reported to be involved in pathological vascularization processes several years ago, and DHA/EPA levels were linked to the prognosis of age-related macular degeneration (8, 103, 154). Others demonstrated beneficial effects on vaso-oblitaration and retinal neovascularization in murine models either genetically modified to overexpress the C. elegans desaturase fat-1 (75) or exposed to hypoxia at the same time as dietary DHA/EPA supplementation (33). Studies in humans are less conclusive, but a DHA-rich diet was found to prevent the formation of acellular capillaries associated with diabetic retinopathy (162).
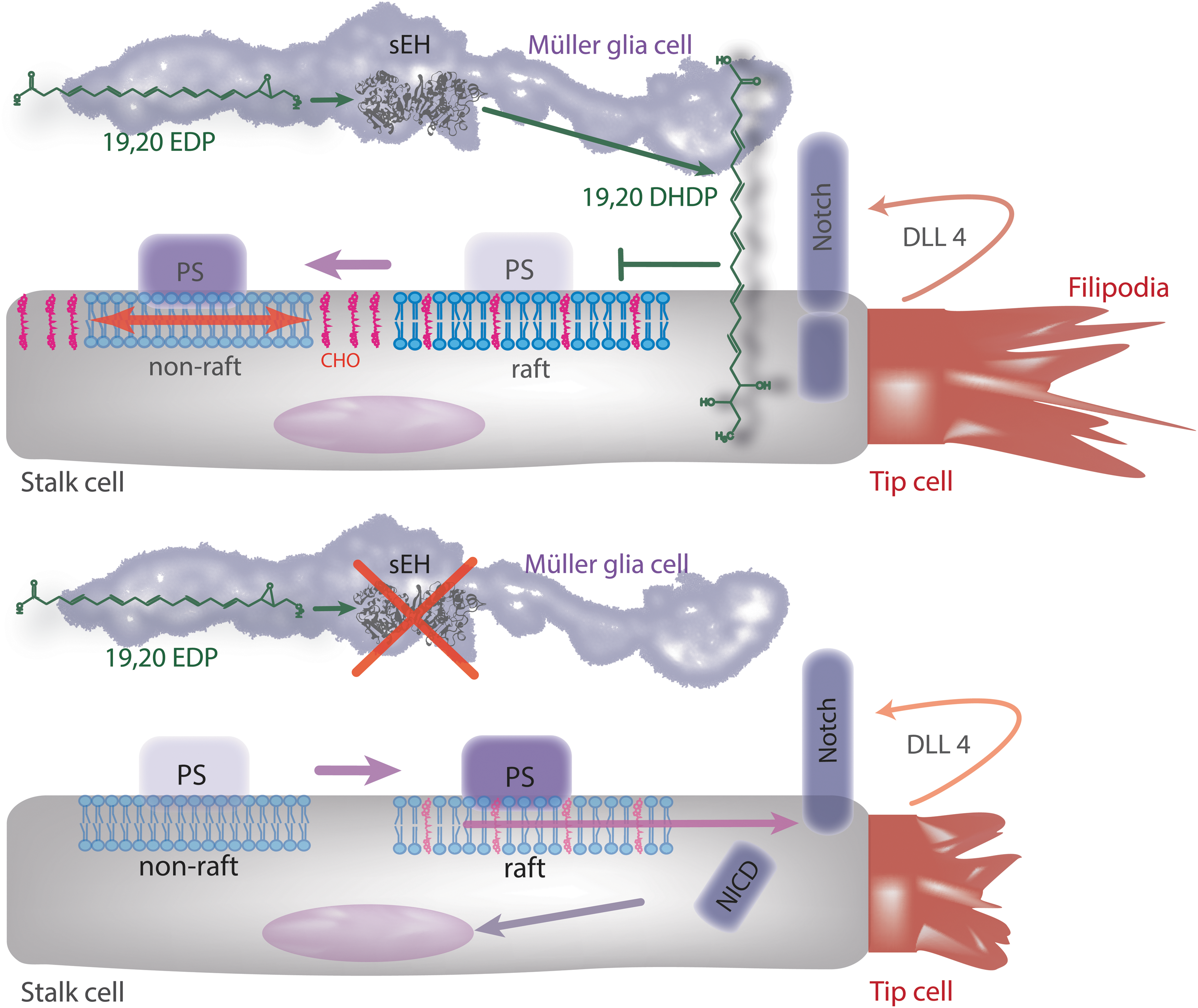
Given reports that epoxides of AA promote angiogenesis, the finding that the deletion of the sEH, which increases epoxide levels, inhibited retinal vascularization was unexpected. Surprisingly, in the postnatal murine retina, the cells expressing the highest amounts of endogenous sEH were not endothelial cells or even astrocytes but Müller glia cells (64). The latter cells span the retina and are in close contact with the vessels of the superficial layer as well as with the deeper capillaries. The deletion or inhibition of the sEH in mice resulted in the activation of retinal endothelial cell Notch signaling and thus attenuated sprouting angiogenesis (64). Mechanistically, these effects could be attributed to the lack of the sEH product and DHA diol, 19,20-DHDP (64). Furthermore, 19,20-DHDP but not the parent epoxide, 19,20-EDP, was able to rescue the attenuated angiogenesis in sEH−/− mice. This was also the case in retinas from animals lacking the Fbxw7 ubiquitin ligase—a downstream component in the regulation of the Notch signaling cascade that determines the half life of the activated form of the cleaved Notch receptor (153). Clearly, the targeted deletion of the sEH in Müller cells revealed a clear effect on endothelial cell proliferation as well as on Notch signaling and tip cell filopodia formation (64), suggesting that Müller cells are important contributors to retinal angiogenesis than generally expected and that their modulator action relies on the metabolism of DHA by CYP enzymes and the sEH.
A recent report also links the CYP/sEH pathway to pathologic neovascularization in mice (152) in a model of oxygen-induced retinopathy. This model reproduces the vaso-obliterative and neovascularization phases of retinopathy of prematurity that are a common complication of preterm birth that in the worst cases results in retinal detachment and subsequent blindness (127, 142). The transgenic overexpression of the human CYP2C8 in Tie-2-expressing cells in mice attenuated the pathologic neovascularization, an effect that was further enhanced by feeding an ω-3 PUFA-enriched diet (149). Although CYP2C8 is known to metabolize AA to generate 11,12-EET and 14,15-EET, the enzyme also accepts EPA and DHA as substrates to generate 17,18-EEQ and 19,20-EPD (4, 11, 42). The beneficial effects of CYP2C8 overexpression were attributed to its actions on Tie-2 expressing monocytes/macrophages (149). Also the fact that CYP2C expression in the retina is upregulated by hypoxia (106) while the expression of the sEH is decreased (78), means that an overall increase the retinal epoxide:diol ratio is expected. The upregulation at least at the mRNA level could also be demonstrated for Cyp2c55, the murine ortholog of the human CYP2C18. Almost all of the CYP enzymes, including CYPC18, that metabolize AA are also able to metabolize EPA and DHA (42), but the ability of Cyp2c55 to do so still remains to be determined. In contrast to the study of Zhang et al. (193), no difference in VEGF-C expression levels could be observed in Tie2-CYP2C8-Tg and Tie2-sEH-Tg retinas, but an increase in VEGF-A in the Tie2-CYP2C8-Tg and a decrease in Tie2-sEH-Tg retinas was reported (149).
How Do the ω-3 PUFA Epoxides and Diols Elicit Their Biological Actions?
Little is known about how the ω-3 products derived from the CYP/sEH axis initiate their biological effects (Fig. 4). The existence of specific membrane receptors cannot be ruled out, but so far, it is only known that some ω-3 PUFAs can act via GPR120 a receptor that recognizes several long chain ω-3 PUFAs (19, 120, 161) as well as the resolvins from the EPA-derived E and the DHA-derived D series (156). However, evidence for receptor-independent effects also exists, as the EDPs are able to activate BK channels in porcine arterioles and rat small arteries in an apparently GTP-independent manner (185) (Fig. 5). PPAR activation may also be possible, as 17,18-EEQ inhibits tumor necrosis factor α-induced inflammation in human bronchi, which is sensitive to PPARγ inhibition (108). In addition, similar to the epoxides and diols of AA, ω-3 PUFA epoxides and diols may initiate biological effects via incorporation into the gycerophospholipids of the cell membrane (155, 197). Certainly, the fatty acid microenvironment can directly influence the function of membrane proteins and EPA as well as DHA are readily incorporated into phospholipids of phosphatidylethanolamine, phosphatidylcholine, phosphatidylinositol, and phosphatidylserine species (81, 116) (Fig. 2). The resulting polyunsaturated phospholipids are able to infiltrate lipid rafts as well as form non raft domains (180). DHA, from which 19,20-DHDP is derived, is particularly interesting in this regard, as it is incorporated more efficiently into lipid rafts than EPA (180). In addition, because of its high level of unsaturation, it is extremely flexible and able to achieve different conformational states (173), potentially resulting in incompatibility with the steric ordered acyl chains and cholesterol (136, 148). However, the exact consequences of DHA incorporation and whether or not the ω-3 PUFAs disrupt raft-like structures or may have an organizing influence is unclear (83, 180, 189). Since lipid rafts seem to be important for the formation of an active γ-secretase complex (164), it is interesting to note that 19,20-DHDP was found to inhibit γ-secretase activity and subsequent Notch signaling by the exclusion of presenilin-1 and cholesterol from lipid rafts, an effect that could not be elicited by the parent epoxide 19,20-EDP (64). To date, it is not clear whether this effect on raft microdomains is the sole mechanism by which 19,20-DHDP interferes with γ-secretase activity, as high concentrations of such lipids may be necessary to alter the membrane dynamics and microdomain composition. Moreover, it is unclear why 19,20-EDP does not elicit a similar effect, as both compounds have comparable numbers of double bonds (Fig. 6).
Inflammation and Resolution of Inflammation
Interest in the role of the PUFA epoxides as anti-inflammatory mediators really took off after 11,12-EET or the overexpression of CYP2J2 was reported to attenuate the upregulation of vascular cell adhesion molecule-1 induced by a series of inflammatory mediators both in vitro and in vivo (117). At the molecular level, these effects were attributed to the inhibition of the IκB kinase and the subsequent degradation of IκBα, meaning that the NFκB subunit Rel A remained bound to IκBα and was, thus, unable to translocate to the nucleus. Interestingly, these effects were selectively induced by 11,12-EET while 14,15-EET was ineffective—potentially indicating that the effects may be EET-receptor mediated. Since the initial report, the overexpression of CYP2J2 has been linked with an impressive list of anti-inflammatory activities that includes; the activation of PPARα (182), inhibition of cardiac apoptosis and cardiac dysfunction (195), the inhibition of endoplasmic reticulum stress in heart failure (171), and the initiation of a protective autophagic response limiting mitochondrial dysfunction and reducing cellular death (138). However, not all CYPs were created equal, as while the overexpression of CYP2J2 (for which AA is a relatively poor substrate) attenuates adhesion molecule expression, the overexpression of CYP2C8 or CYP2C9 (which more readily metabolize AA) in endothelial cells results in exactly the opposite, that is, increased adhesion molecule expression (50). This may imply that PUFA mediators derived from lipids other than AA may account for some of the anti-inflammatory effects observed, especially in in vivo models, or that there are other major differences in enzyme activity which are of physiological consequence. There is evidence to support both of these possibilities. Certainly, the generation of reactive oxygen species by CYP2C8 and CYP2C9 is able to override any direct anti-inflammatory action of the EETs generated at the same time—at least as far as NFκB activation and adhesion molecule expression is concerned (50).
When talking about inflammation and its resolution, it is important to consider that monocyte/macrophages also express CYP enzymes and generate a range of CYP metabolites, including fatty acid epoxides. CYP2J2 and CYP2C8 were identified as EET-generating CYP enzymes in monocytes/macrophages (21, 114), but whether or not these are the most relevant enzymes remains unclear, as does the biological consequences of CYP activation. A recent study that used a proteomic approach to study CYP expression in monocyte-derived macrophages identified CYP2S1 in microsomes prepared from freshly isolated and cultured CD14+ human blood-derived monocytes (55). The major substrates for human CYP2S1 have not been fully established but preliminary reports suggested that the recombinant CYP2S1 can metabolize retinoic acids, AA, linoleic acid, and EPA (55). At the functional level, macrophage CYP2S1 expression was increased by lipopolysaccharide and interferon-γ (to generate classically activated or M1 polarized macrophages) and oxidized low-density lipoprotein (LDL) but not by interleukin-4 and interleukin-13 (to generate alternatively activated or M2 macrophages). In agreement with these findings, inflamed human tonsils that should contain predominantly M1 polarized macrophages showed a high level of CYP2S1 and CD68 colocalization while alternatively activated macrophages in breast cancer metastases in the human lung expressed no detectable CYP2S1. Oxidized LDL also significantly increased CYP2S1 levels in vitro, and the enzyme was present in macrophage foam cells recovered from ApoE−/− mice fed a high fat diet as well in aortae from similarly treated animals but not in wild-type animals. The latter findings fit well with the previous report that treating monocytes with minimally modified LDL elicited a significant increase in 11,12-EET formation (62).
How could the expression of CYP2S1 alter macrophage function? While the inhibition and downregulation of CYP2S1 led to an increased phagocytic uptake of bioparticles, inhibition of the sEH tended to decrease phagocytosis. The anti-inflammatory effects associated with CYP2S1 expression and activity could have been due to the ability of the enzyme to generate an anti-inflammatory factor (such as EET's) or the metabolism/downregulation of an immune modulator factor. One such factor is PGE2 and CYP2S1 was reported to metabolize the PGE2 precursors PGG2 and PGH2 to 12(S)-hydroxyheptadeca-5Z, 8E, and 10E-trienoic acid (17, 99). Thus, perhaps from the macrophage polarization point of view, the most relevant CYP2S1 substrates may well be PGG2 and PGH2, as the subsequent decrease in the immunomodulator PGE2 would certainly be expected to result in a macrophage subtype with attenuated angiogenic potential.
ω-3 versus ω-6 PUFAs
On the whole, shifting the dietary intake ratio of ω-3/ω-6 PUFAs in the direction of ω-6 has been linked with enhanced inflammation. The ω-6 PUFA, linoleic acid is the biological precursor of AA and can be metabolized by CYP enzymes to generate the linoleic epoxides, that is, 9,10- and 12,13- epoxyoctadecenoic acid. Both of these epoxides are, in turn, substrates for the sEH, which catalyzes the generation of 9,10-DiHOME and 12,13-DiHOME. Linoleic acid levels are high in the Western diet, and recent studies indicate that dietary imbalance between ω-6 and ω-3 PUFAs leads to an adverse cardiovascular and metabolic profile, thereby contributing to the pathogenesis of nonalcoholic fatty liver disease, diabetes, and cardiovascular disease (141). Moreover, at least in rats, linoleic and α-linolenic acid consumption over three generations exerted cumulative effects on the regulation of hepatic expression of genes related to lipid metabolism (25). While such studies can only provide indirect evidence for a detrimental consequence of increase linoleic acid metabolism, there have been clear demonstrations of the toxicity of derivatives of linoleic acid (87, 107, 135).
Unfortunately, it appears that there is a surprisingly high degree of inter-individual variability in certain aspects of lipid metabolism (119), which renders responsiveness to ω-3 fatty acid supplementation probably too variable to ever see clear effects in large population studies. Indeed, variables such as age, the underlying health status of individuals, the dose, duration of exposure, and relative proportion of EPA and DHA in the supplements given (22, 41, 91, 96) can complicate the situation. However, assessing individual responsiveness to ω-3 fatty acids may be a way to increase the effectiveness to PUFA dietary supplementation (119).
In contrast to the ω-6 PUFAs, the ω-3 PUFAs are usually attributed anti-inflammatory effects—particularly when considering the metabolic syndrome, type 2 diabetes, and cardiovascular disease (86). Despite early positive reports of the benefits of supplementing diets with fish oils on cardiovascular outcomes (86), larger and better controlled clinical trials have failed to reveal any clear clinical benefit (38). Genetic variation may underlie some of the inconsistent results, as the available literature hints at gene variants that influence tissue ω-3 PUFA status and response to fish oil supplementation (100). Animal studies have, however, delivered encouraging information, and the progressive substitution of dietary ω-3 PUFA for saturated fatty acids in atherosclerosis-prone LDL-receptor knockout mice led to a decrease in plasma lipid levels, especially cholesterol. ω-3 PUFA supplementation also decreased expression of pro-inflammatory markers in the aorta and was associated with the deceleration of atherosclerosis (26). Clearly, more detailed and exhaustive studies are needed to determine the real role of the different PUFA epoxides and diols in the pathophysiology of cardiovascular disease and the therapeutic potential of sEH inhibition either alone or in combination with dietary PUFA substitution/supplementation.
Footnotes
Acknowledgments
The authors acknowledge the work of the many groups whose work it has not been possible to cite here because of space limitations. Work performed in the author's own laboratory was supported by the Deutsche Forschungsgemeinschaft (SFB 1039/A6 and Exzellenzcluster 147 “Cardio-Pulmonary System”).