
Abstract
Background
Discovery in vitro
N
In 1997, 20 years after discovering the endoperoxide to levulinaldehyde rearrangement, we secured the first evidence that LGs are generated in vivo. Using anti-LGE2 antibodies raised against proteins modified with pure LGE2 obtained by chemical synthesis, we detected disease-related elevations in levels of LGE2 adducts in blood from patients with atherosclerosis (AS) or end-stage renal disease (RD) (79). That LG-protein adducts can be formed via a PGH synthase-dependent pathway (7) has now been demonstrated by activating human platelets with exogenous arachidonic acid or thrombin (8). Formation of these adducts is inhibited by indomethacin, a PGH synthase inhibitor, and is enhanced by an inhibitor of thromboxane synthase, an enzyme that metabolizes PGH2. Thus, enzymatic cyclooxygenation of arachidonate leads to the production LGs in whole cells through a spontaneous rearrangement of PGH2 that competes with its enzymatic conversion to thromboxane A2.
Radical-induced cyclooxygenation produces isoLGE2
There is an alternative mechanism for the formation of LGE2. We had anticipated that a free radical pathway would also produce LGE2 adducts. Nevertheless, we were surprised to find that free radical-induced oxidation of low-density lipoprotein (LDL) in vitro generated LGE2-adduct immunoreactivity (80). Although free polyunsaturated fatty acids, for example, arachidonate released from arachidonyl phospholipids (PLs), are the substrates for cyclooxygenase (COX), we had expected that isoLGE2 formed on oxidation of LDL would be esterified in PLs. Our LGE2-protein antibodies did not detect protein adducts of the LGE2 ester of 2-lyso-phosphatidylcholine (PC) prepared by chemical synthesis. Therefore, phospholipase activity associated with the LDL particle must hydrolyze the protein adducts of isoLGE2-PC exposing the unesterified epitopes. Hydrolytic release of lyso-PC is known to accompany oxidation of LDL (88). Seven years later, in an animal model of oxidative injury, treatment of rats with CCl4 was shown to cause the formation of protein adducts of isoLGs esterified in PLs that eventually undergo phospholipolysis in vivo (12). This is consistent with our earlier finding that protein adducts of an LGE2-PL ester are substrates for snake venom PLA2 (80).
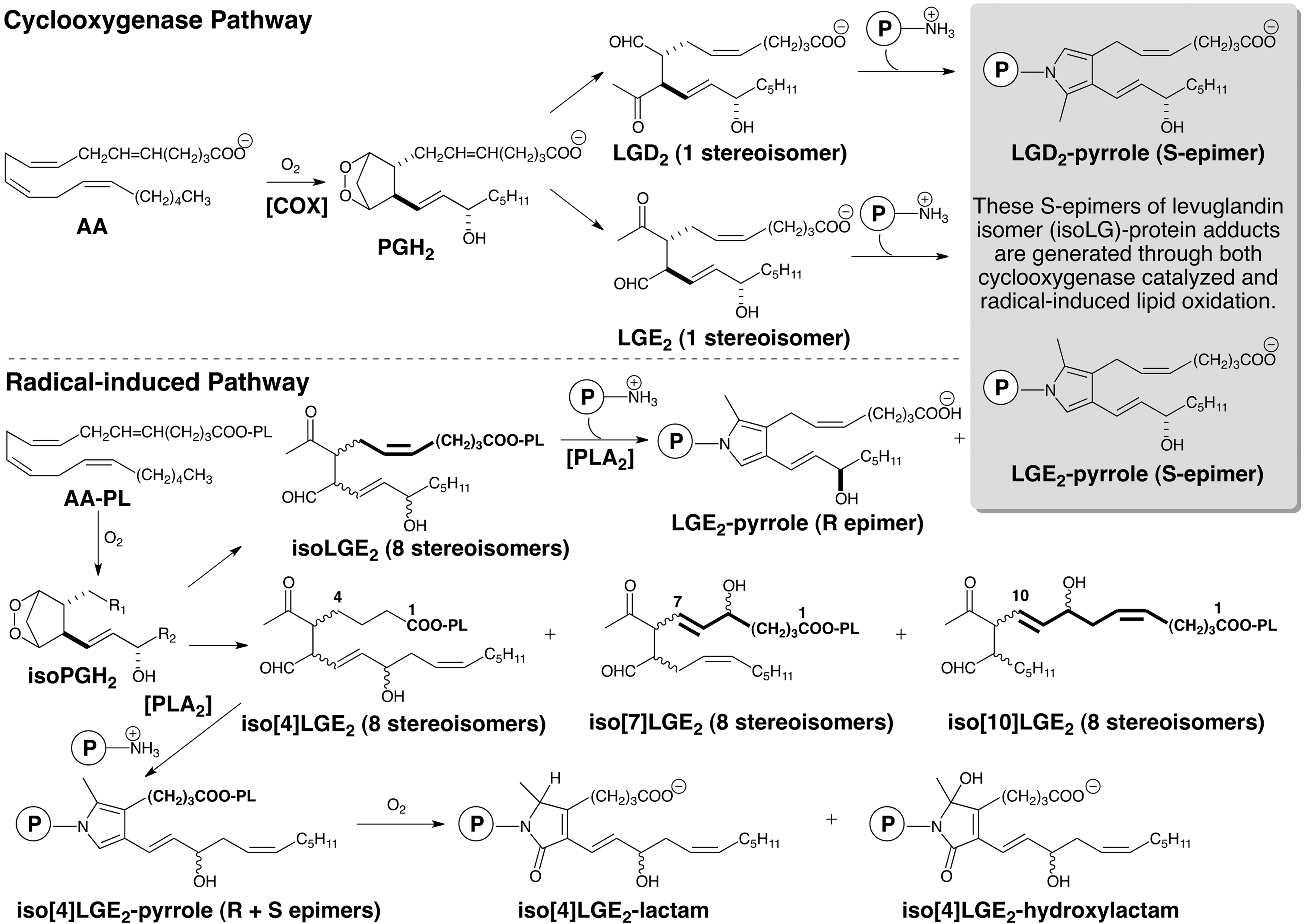
Structural isomers of LGE2: unique products of radical-induced cyclooxygenation
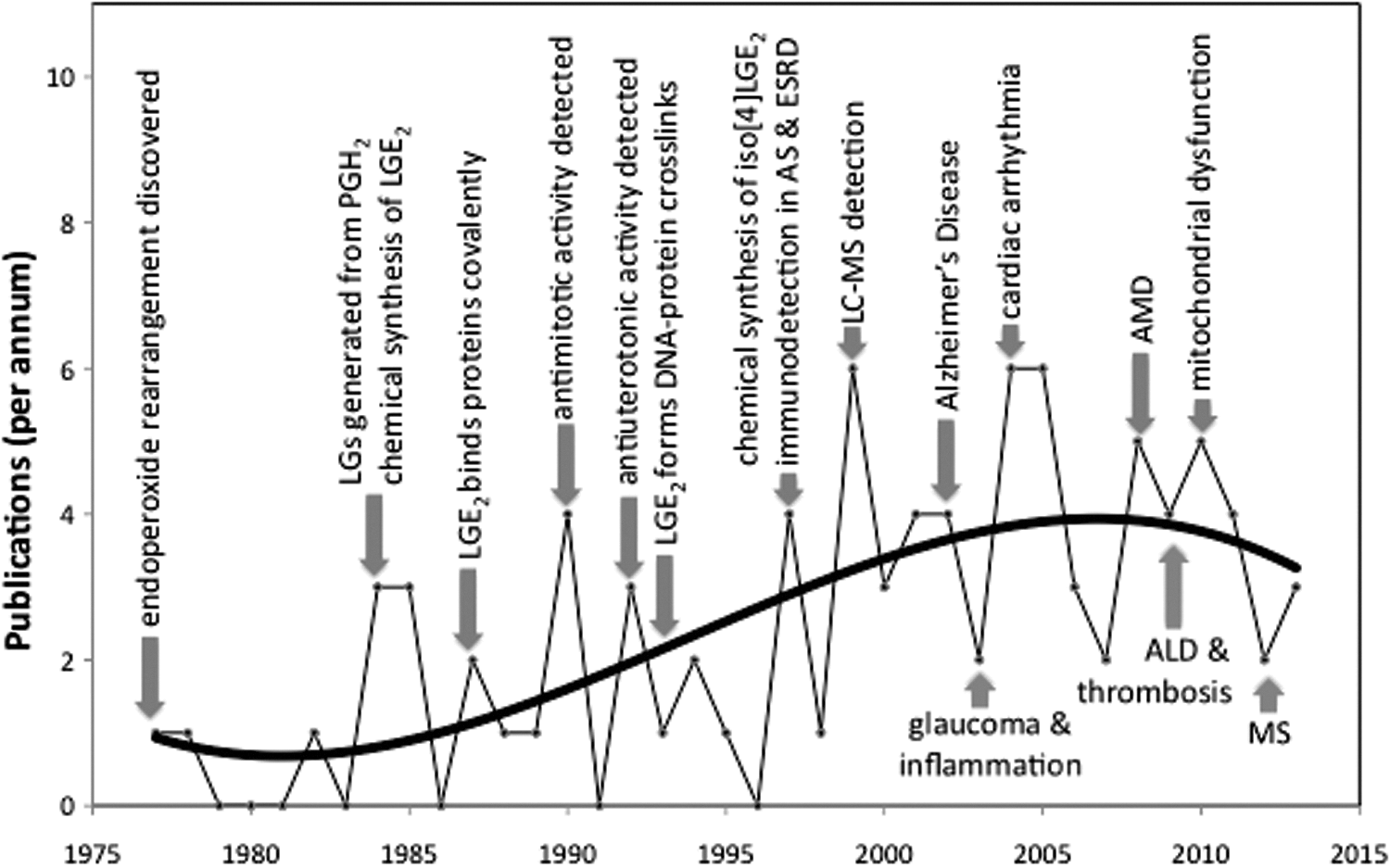
Detection of LGE2 adducts in vivo, for example, the LGE2-pyrrole (Fig. 1), with anti-LGE2 antibodies cannot distinguish between their production through enzymatic cyclooxygenation and radical-induced cyclooxygenation because the exactly same LGE2 isomer can be produced by either pathway. However, while the COX-promoted cyclooxygenation of arachidonate only generates LGE2 and LGD2 with prostanoid side chains, we postulated that 8 LGE2 stereoisomers (referred to collectively as isoLGE2) are only one of the eight structurally isomeric families of isoLGs (each also consisting of eight stereoisomers) that can be generated from AA-PC by nonenzymatic cyclooxygenation (80). The structural isomers of LGE2 and LGD2 are named with a bracketed numeral [n], for example, iso[4]LGE2 (Fig. 1), to indicate the number of carbon atoms in their carboxylic side chains. Most importantly, these structural isomers with non prostanoid side chains and their protein adducts can only arise through free radical-induced cyclooxygenation. Using anti-iso[4]LGE2 antibodies, we secured the first unambiguous evidence that LG isomers are also produced in vivo through free radical-induced cyclooxygenation. Mean levels of both isoLGE2- and iso[4]LGE2-protein adducts in plasma from patients with AS (n=16) or RD (n=8) are about twice those in healthy individuals (n=25) (73, 79). These elevated levels are not related to variations in age, total cholesterol, or apoB. A linear correlation (r=0.79) between plasma isoLGE2- and iso[4]LGE2-protein adduct levels in all 49 individuals is consistent with a common free radical-induced mechanism for the production of both oxidized lipids in vivo. The correlation is even stronger (r=0.86) for patients with AS or RD. Furthermore, isoLG-protein adduct levels are more strongly correlated with disease than are total cholesterol or apoB, suggesting an independent defect that results in an abnormally high level of oxidative injury associated with AS and RD (63). Since then, the association of isoLGs with a growing list of diseases has emerged (Fig. 2).
It is important to understand that immunoreactivity detected with anti-LGE2 or a single-chain (scFv) antibody that binds with isoLGE2 adducts (22) does not provide a tool for unambiguous distinction between the COX and free radical-induced generation of isoLGs (71), because isoLGE2 and the LGE2-pyrrole (S-epimer) are products of both pathways (Fig. 2). The conclusion that immunoreactivity detected with an isoLGE2-specific “D11 ScFv” is indicative of free radical-induced oxidant stress is incorrect, because COX-mediated cyclooxygenation cannot be ruled out (22, 93). We recently generated a mouse monoclonal antibody (mAb) against iso[4]LGE2-protein adducts that is highly selective for adducts of iso[4]LGE2 versus those of isoLGE2. Since iso[4]LGE2 is only produced through free radical-induced cyclooxygenation, this anti-iso[4]LGE2 mAb provides an excellent tool for unambiguously detecting the operation of that pathway, because it does not respond to products of the COX pathway. This antibody may also prove useful for enrichment of isoLG-modified proteins in biological extracts by standard immunoaffinity techniques, and thus facilitate proteomic identification of protein targets of isoLGs in vivo.
Throughout this review, I have eschewed the “isoketal” terminology introduced by others, because it is a misleading alternate nomenclature that refers to the products of the free radical-induced cyclooxygenation as “isoketals” to “distinguish them from the products of the cyclooxygenase pathway” (3). However this is a false dichotomy since the products of the COX pathway are also generated through free radical-induced cyclooxygenation. The claim that “isoketals are…analogous to cyclooxygenase-derived levuglandins” (2) is incorrect, because the levuglandins generated through COX and free radical-induced cyclooxygenation are the exact same molecules and not analogs. Levuglandins are a subclass of isoLGs (Fig. 1).
Identification of specific proteins modified by isoLGs
Identification of the proteins that are modified in vivo by isoLGs was first attempted by Western blot analysis of two-dimensional SDS-PAGE using antibodies against adducts and subsequent proteomic analysis (39). Although the proteins identified co-migrate with immunoreactivity against specific adducts, proteomic evidence establishing the in vivo presence of isoLG-derived modifications of specific aminoacyl residues of proteins in biological samples had never been reported.
An arachidonic acid analogue with a terminal alkyne (al-AA) was used to modify proteins in cultured cells by lipid oxidation products generated through exposure to iron ions and ascorbate or t-butylhydroperoxide (53). Extraction of the resulting covalently modified proteins was then achieved through cycloaddition of the alkyne with a biotin-linked azide. A target protein, that is, the cardiac sodium channel Nav 1.5, in the extract was then identified by Western blot analysis using antibodies specific for the protein. The conclusion that covalent modification of the protein involved the generation and adduction an isoLG was supported by inhibition of protein modification by a scavenger that is particularly reactive toward isoLGs (53).
Recently, using targeted LC-MS/MS analysis of tryptic peptides from protein extracts of human retina, we demonstrated that peptide mapping and sequencing of an in vivo isoLG-modified protein can be used to unambiguously establish that a specific protein, cytochrome P450 (CYP)27A1 modified with an isoLG on a particular lysyl residue is present in the human retina (vide infra) (16).
Isolevuglandin structural diversity and the common core
One factor that complicates characterizing the biological occurrence of isoLGs and their pathological involvements in disease is the generation of a vast array of structural isomers through cyclooxygenation of polyunsaturated fatty acyls with 3, 4, 5, or 6 C=C bonds alternating with CH2 groups, that is, doubly allylic methylenes (Fig. 3). We reported the chemical synthesis of 17-isoLGE4 (84). Subsequently, evidence was obtained confirming the presence of protein adducts of isoLGE4 or its stereo and structural isomers in vivo (3), and elevated levels of these protein adducts were detected in cerebrospinal fluid from multiple sclerosis patients (29).

Although hundreds of different isoLGs may be produced in vivo, the reactions that produce all isoLGs and their biological chemistry is expected to be very similar owing to the localization of their reactive functionality in a “common core” structure (highlighted in Fig. 4) as well as identical carboxyl and alkyl termini in their side chains. The isoLGs derived from various polyunsaturated fatty acyl PLs differ only by having various hydrocarbon chains inserted into their upper and lower side chains. These differences are not expected to influence the biological chemistry of the γ-ketoaldehyde core or the derived pyrroles. These pyrroles are electron rich and prone to oxidation. Our discovery of the free-radical induced formation of isoLG-derived protein adducts was subsequently confirmed by collaborative LC-MS/MS studies that we initiated with Roberts at Vanderbilt University. Those studies demonstrated the formation of LG isomers on free radical-induced in vitro oxidation of arachidonic acid (AA), and revealed that oxidation of the initial isoLG-pyrrole adducts generates lactams and hydroxylactams (13). These structures were identified by exhaustive proteolysis of isoLG-modified protein that delivered the corresponding lysyl lactams and hydroxylactams (Fig. 4). Additional complexity in the products arising from isoLGs is found for some isoLGs that have 1,4-diene moieties in their sidechains. Further oxidation can convert the 1,4-diene moieties to 1-hydroxy-2,4-dienes (5).
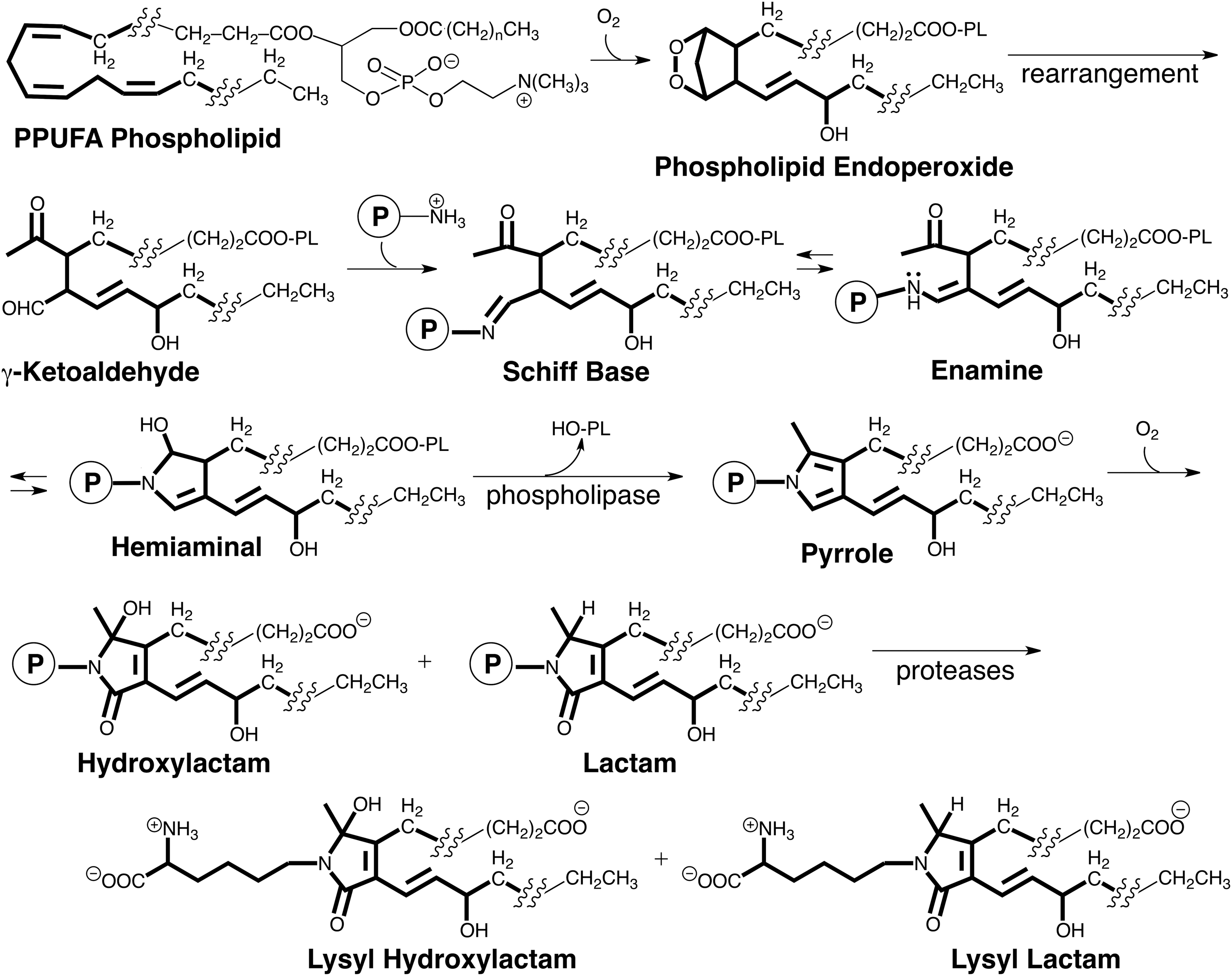
Besides differences in the isomeric composition of isoLGs produced through the COX and free radical-induced pathways, the cyclooxygenation substrates for these two pathways are also distinct. Free polyunsaturated fatty acids, for example, arachidonate released from arachidonyl PLs are the substrates for COX. That PL esters of polyunsaturated fatty acids are the primary targets in vivo for radical-induced cyclooxygenation was demonstrated in a rat model of oxidant injury to the liver that results in free radical-induced oxidation of polyunsaturated PLs (12). After exposure of rats to CCl4, both lactam adducts and Schiff base adducts were detected in the liver. The lactam adducts were present as free acids, but >97% of the Schiff base adducts were apparently esterified to PLs because treatment of the proteins with base caused a 42-fold increase in the level of the Schiff base free acid detected (12). Thus, endoperoxide intermediates and the isoLGs generated through their rearrangement remain esterified in PLs that bind rapidly to proteins to generate Schiff base adducts. These metastable adducts undergo a slow subsequent dehydration to deliver pyrrole intermediates that are ultimately oxidized to produce hydroxylactam and lactam end products that were detected by exhaustive proteolysis that delivers lysyl hydroxylactams and lysyl lactams (Fig. 4).
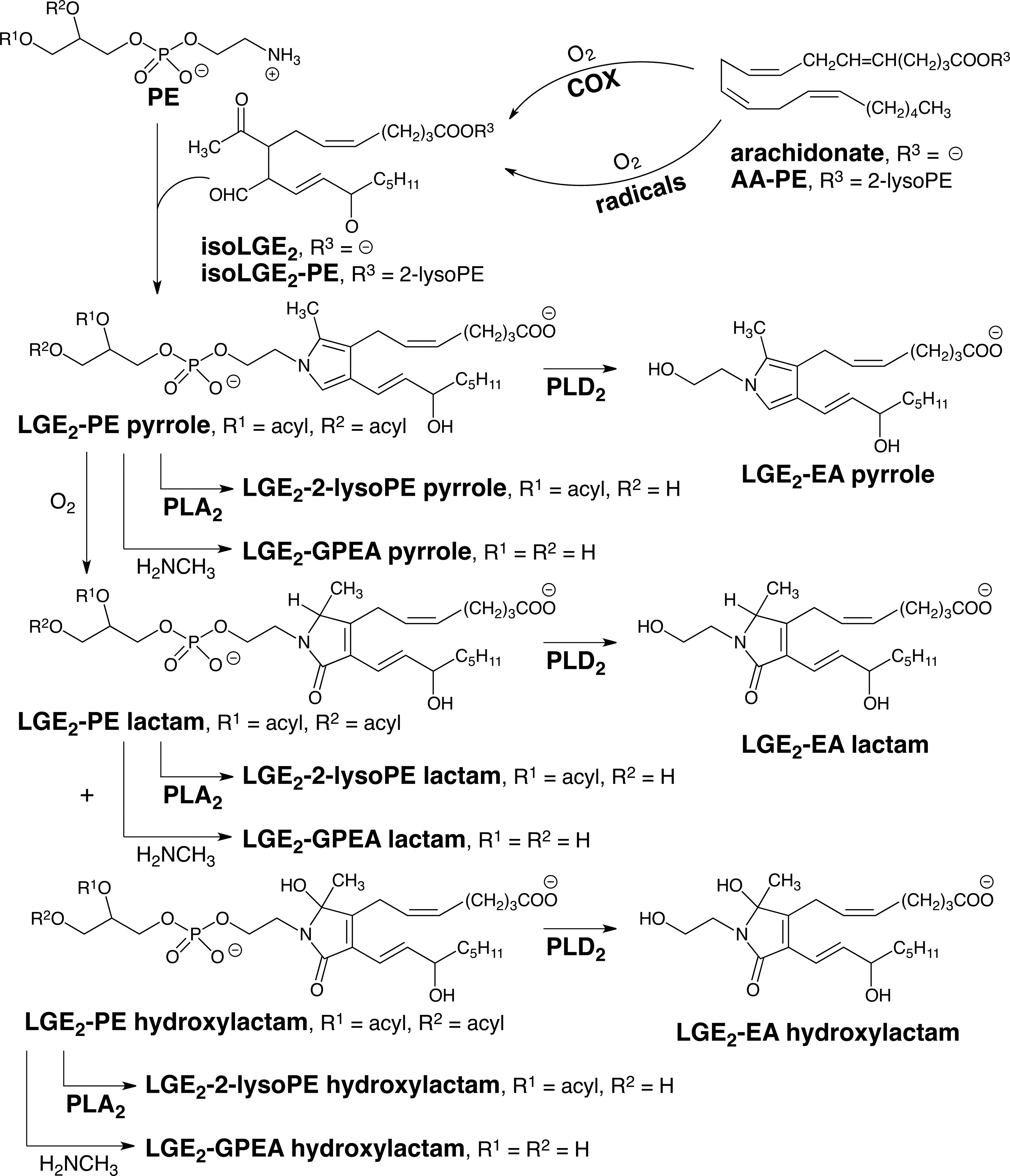
Using LC–MS/MS, we showed that isoLG adducts that incorporate the amino group of phosphatidylethanolamines (PEs) into isoLG-hydroxylactams (Fig. 5) are generated in vivo (47). Quantitative analysis of these hydroxylactams is achieved with samples that are an order of magnitude smaller, and sample processing is much simpler and less time consuming than required for measuring protein-derived isoLG-lysyl lactams. In vivo, PEs incorporate a variety of acyl groups esterified to the 1 and 2-positions. Treatment of heterogeneous mixtures of isoLG-modified PEs with phospholipase A2 generates mainly 1-palmitoyl-2-lysoPE-isoLG adducts (47). Even greater simplification is achieved by treating such mixtures of isoLG-PEs with methyl–amine (33) or sodium hydroxide (34) that converts them into isoLG adducts of glycerophosphate ethanolamine (GPEA, Fig. 5). Alternatively, treatment with PLD2 converts the isoLG-PEs into isoLG adducts of ethanolamine (isoLG-EAs, Fig. 5) (90).
Interestingly, LGs also form covalent adducts with the guanidine group of protein arginyl residues, but the adducts are unstable, readily undergoing hydrolysis to produce ornithine residues (98). Stable adducts of LGs with the deoxycytidyl primary amino group in DNA have also been characterized (14).
Inflammation fosters the production of isoLGs
Using a mouse Candida sepsis model of inflammation, we observed 3.5- and 2.7-fold increases in iso[4]LGE2 and isoLGE2 adducts of plasma proteins after pathogen exposure in wild-type mice (62). Myeloperoxidase (MPO) uses H2O2 together with NO2 − to generate nitrogen dioxide, a free radical capable of initiating lipid peroxidation. That MPO contributes to the formation of isoLG protein adducts in vivo under inflammatory conditions is suggested by a 34% reduction in protein adduct content in plasma proteins from MPO knockout mice (p=0.003) even though an increase might be anticipated because the inflammatory state is more severe (persistent) in the knockout mice.
In a murine model of allergic airway inflammation produced by sensitization to ovalbumin with subsequent daily aerosol challenge, increased isoLGE2-adduct immunoreactivity was found in airway epithelial cells at 24 h after the first aerosol challenge and at 5 days in alveolar macrophages (93). Collagen surrounding airways and blood vessels, and airway and vascular smooth muscle, also exhibited increased immunoreactivity after ovalbumin challenge. High levels of isoLGE2-adducts appeared in epithelial cells within an hour, declining after 24 h and dropping to baseline levels by day 5 in spite of continual daily challenge. In contrast, baseline levels of isoLGE2-adducts were present in alveolar macrophages after an hour, rising modestly by 24 h and reaching a maximum at day 5. These contrasting time courses suggest that isoLGs in macrophages arise from macrophage uptake and accumulation of isoLG adducts shed from epithelial cells or generated in the extracellular matrix. This conclusion is supported the observation that LGE2-modified LDL binds with and is degraded on receptor-mediated endocytosis by macrophage cells (40). Both the COX and free radical-induced pathways could be involved in the production of the isoLGE2 detected with the ScFv antibody used in this study. Stimulation of A549 cultured lung epithelial cells with the cytokine IL-1β followed by treatment with arachidonic acid results in upregulation of COX-2 and elevation of isoLGE2-histone levels in the nucleus, and the formation of these isoLGE2 (presumably the pure LGE2 stereoisomer) adducts is inhibited by pretreatment of the cells with the nonsteroidal anti inflammatory drug indomethacin (15).
In a mouse model of alcoholic liver disease, free radical-induced oxidative damage owing to inflammation is indicated by an accumulation of iso[4]LG adducts in liver tissue. Here, there is no ambiguity as to COX versus free radical-induced generation of the isoLG detected, because iso[4]LGE2 is only produced through free radical-induced cyclooxygenation. More than160% higher mean levels of iso[4]LGE2-PE-hydroxylactam (p<0.001) were detected by LC-MS/MS in liver homogenates from chronic ethanol-fed mice (32.4±6.3 ng/g, n=6) compared with controls (12.1±1.5 ng/g, n=4) (47). Immunohistochemical analysis of liver tissue from these mice found elevated levels of iso[4]LGE2 and isoLGE2 immunoreactivity in the ethanol-fed mice compared with controls (67). That isoLGE2 was produced by free radical-induced lipid oxidation was confirmed by the failure of COX 1 or 2 deficiency to prevent the ethanol-induced generation of LGE2 adducts. Pathological markers of ethanol-induced hepatic injury, including serum alanine aminotransferase, hepatic triglyceride, and cytochrome P450 (CYP)2E1, were elevated in response to ethanol feeding. Tumor necrosis factor (TNF)α can induce mitochondrial reactive oxygen species production in hepatocytes (23). Animals deficient in TNFα receptor 1 (TNFR1) (96) or treated with TNFα neutralizing antibody are protected from ethanol-induced lipid peroxidation and liver injury (41). IsoLG adduct formation was reduced in both TNFR1- and CYP2E1-deficient mice. In summary, ethanol feeding enhanced isoLG–protein adduct production via a TNFR1/CYP2E1-dependent, but COX-independent, mechanism in mouse liver.
Children born prematurely have significantly (p=0.008) elevated mean levels of blood plasma iso[4]LGE2-adduct immunoreactivity, 22.7±5.9 nmol/ml (n=6) compared with 15.3±5.0 nmol/ml (n=16) in children with no significant birth events (24). This suggests that chronic inflammation is a heretofore-unrecognized consequence of premature birth or postnatal treatment protocols that result in a failure to develop fully competent antioxidant defenses.
Pyridoxamine and analogues trap isoLGs inhibiting their adduction to proteins
By screening various primary amines as potential scavengers of γ-ketoaldehydes, a model γ-ketoaldehyde, 4-oxopentanal, was found to react extremely rapidly with pyridoxamine (PM), with a second-order rate constant at physiological pH being approximately 2300 times faster than that of Nα-acetyllysine (1). The extreme reactivity of PM was unique to γ-dicarbonyls owing to a mechanism involving facilitation by the phenolic proton of ring closure to deliver a pyrrole from an unstable initial Schiff base adduct. PM prevented adduction to ovalbumin and inactivation of RNase A and glutathione reductase by isoLGE2, and inhibited the formation of isoLG-protein adducts in platelets activated with arachidonic acid (21). PM and lipophilic PM analogues, such as salicylamine, are noncytotoxic isoLG scavengers that block the formation of LG adducts of proteins in cells without inhibiting the catalytic activity of the COXs and thus without interfering with the formation of prostaglandins (97). They are useful for assessing the biological consequences of isoLG production in vivo (vide infra). The oral administration of salicylamine can be used to assess the contribution of isoLGs in animal models of disease (100). They also have been used in experimental animals in vivo to assess the pathophysiological contribution of levuglandins in diseases associated with COX upregulation, as have other primary amines that compete with protein lysyl ɛ-amino residues. Thus, glucosamine at doses that did not inhibit prostaglandin biosynthesis totally abolished formation of LG-protein adducts after induction in A549 lung cancer cells of COX-2 by IL-1β (11, 42, 89).
Pathological Activities of Isolevuglandins
Inhibition of tubulin polymerization and mitosis
LGE2 inhibits the first synchronous cell division of fertilized sea urchin eggs with an ED100 ∼25 μM (52). Tubulin is the major constituent protein of the microtubule, an important component of the eukaryotic cytoskeleton, and an integral part of the mitotic spindle. Presumably, LGE2 can enter cells and bind to tubulin, thus preventing microtubule assembly and inhibiting mitosis. Indeed, LGE2 inhibits GTP-induced assembly of bovine microtubule protein (∼1.4 mg/ml, ∼85% tubulin, and 15% microtubule-associated protein) with a slightly higher ED100 ∼80 μM (52). The LG did not cause depolymerization of preassembled tubulin. Complete inhibition of assembly occurred on binding of only ∼2 molecules of LG per tubulin dimer, one to each subunit. These observations suggest that a small number of key residues essential for microtubule assembly are modified by LGE2 in the dimer state but are inaccessible to LGE2 in the polymerized state.
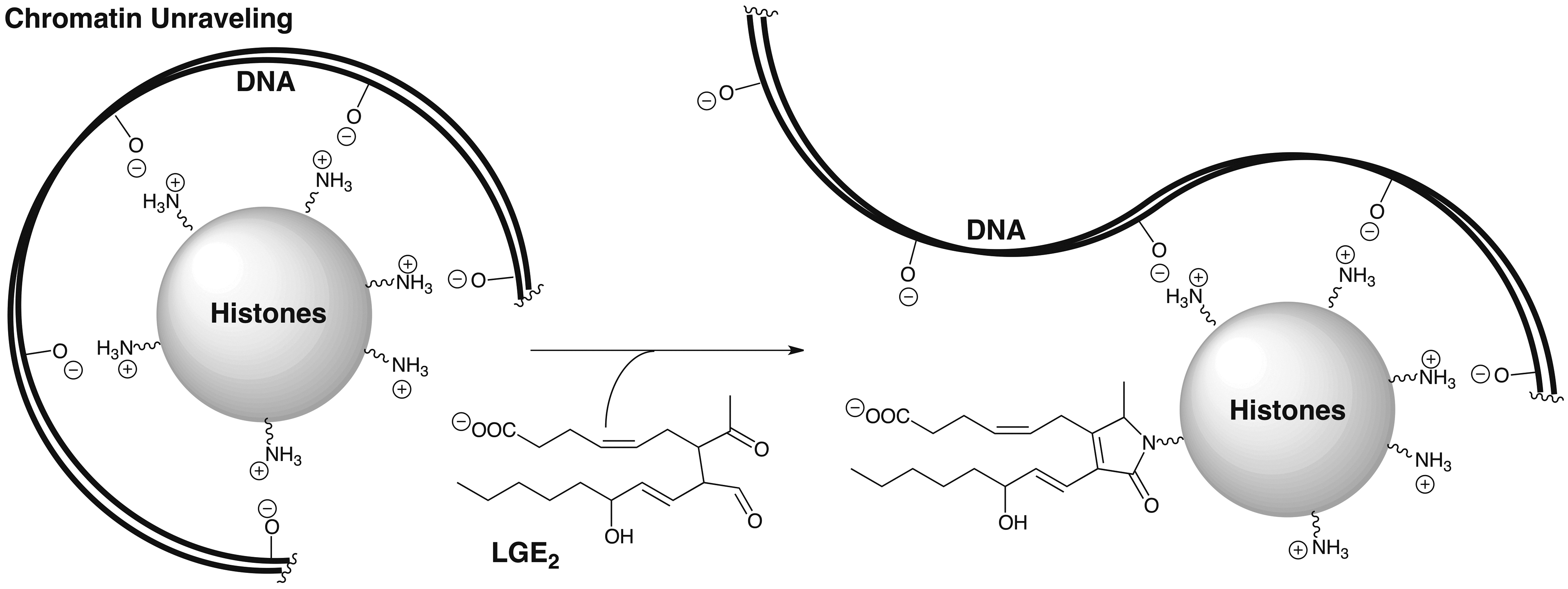
Modification of histones and disruption of their nucleosomal complex with DNA
To stimulate COX-2 expression, cultured lung epithelial cells (A549) were treated with IL-1β or macrophages cells (RAW264.7) were treated with LPS and IFNγ. Subsequent addition of arachidonic acid resulted in the generation of LG-protein adducts of histones (15). Interestingly, a strong preference for modification of histone H4 and, to a lesser extent, of H3/H2B was observed. Changes in lysyl modifications of H4, in particular, are a common hallmark of human cancers. This discovery links inflammation and COX-2 expression that is associated with the development of many cancers, with histone modification by LGs. DNA is “packaged” into nucleosomes by winding the negatively charged phosphodiester-linked polynucleotide around histone octamers that are positively charged owing to protruding histone lysyl residues (Fig. 6). Conversion of the lysyl primary amino group into a pyrrole or lactam, neither of which is protonated at physiological pH, in conjunction with the addition of a negatively charged carboxyl group, could disrupt histone-DNA binding (Fig. 6). It is tempting to speculate that this may increase DNA transcriptional access to previously silent oncogenes and contribute to the development of cancer.
Modification of cytochrome c in mitochondria impairs respiration
Positively charged lysine residues on cytochrome c are responsible for its association with the negatively charged, cardiolipin-rich mitochondrial inner membrane as well as with cytochrome c oxidase and cytochrome c reductase (65). Modification of lysine residues on cytochrome c is expected to weaken these interactions by converting positively charged lysyl amino groups into derivatives that lack a protonated amine, and introducing negatively charged carboxylates. Treatment of mitochondria with isoLGE2 impairs maximal respiration that is restored by the addition of unmodified cytochrome c (87). Apparently, modification of cytochrome c by isoLGs reduces its affinity for the electron transport chain complex, thereby disrupting respiration.
Disruption of the blood–brain barrier
The potent cytotoxicity of isoLGs can damage the blood–brain barrier. Direct injection of 100 nmol of isoLGE2 into the substance of the cerebral hemisphere of rats caused pallor and loss of cellular constituents typical of necrotic cells, and the marginal area became hypercellular because of the infiltration by macrophages (82). A dose-dependent extravasation of Evans Blue revealed loss of blood–brain barrier integrity.
DNA-protein cross-linking
LGE2 causes repair-resistant DNA–protein cross-links and cell death (LD50=230 nM) in V79 Chinese hamster lung fibroblasts (51). After removal of any unadducted free LGE2 and the return of the cultures to normal growth medium, additional DNA-protein cross-links continued to form over the ensuing 6–24 h. The results suggest that LG adducts to DNA or protein are not repaired, but react further at sites on protein or DNA in close proximity to the initial adducts, forming cross-links in a slow phase of the process.
Protein-protein cross-linking
Treatment of proteins with isoLGs generates protein–protein cross-links (43). The consumption of monomeric ovalbumin by reaction with 15 equivalents of LGE2 is almost complete within 2 h. Initially, polyacrylamide gel electrophoresis of the reaction product reveals that the amount of dimer rises rapidly, reaching 30% within 2 min, but then drops, and the formation of a “ladder” of various oligomers is readily apparent after 30 min. Only high-molecular-weight oligomers are present after 4 h. Malondialdehyde or 4-hydroxynon-2-enal caused no discernable oligomerization under the same conditions. Thus, isoLGE2 is orders of magnitude more effective in generating protein–protein cross-links than these other electrophilic products from arachidonate oxidation that bind with and cross-link proteins.
Although many molecules of isoLGE2 bind within 1 min to each molecule of protein, protein–protein crosslinking is completely prevented by adding a large excess of glycine to the reaction mixture at 5 min after combining the protein and isoLG, resulting in protein-glycine crosslinks (44). Thus, isoLGs serve as molecular glue, sticking to a protein and then bonding it through a covalent chemical link with another molecule of protein. Potentially, this can generate large heterogeneous aggregates of diverse proteins. It can also result in covalent linkages between proteins and nucleophilic functionality in small molecules such as glycine and spermine (6), a polyamine that is abundant in both the cytosol and nucleus. It is likely, although as yet unproven, that isoLGs also cross-link proteins with ethanolamine PLs resulting in anchoring proteins to cell membranes.
COX promoted oligomerization of amyloid β1-42: Alzheimer's disease
Accumulation of amyloid β (Aβ) in the brain is associated with the pathogenesis of Alzheimer's disease (AD) (28, 83). Dimers and higher oligomers of Aβ are abundant in regions exhibiting neuronal pathology (50, 66), and soluble oligomers of Aβ1-42 are cytotoxic to neurons (45, 81, 94). Incubation of monomeric Aβ1-42 with PGH2 or LGE2 caused the formation of oligomers, resembling protofibrils in contrast with the slower spontaneous oligomerization of this peptide that produced long fibrils (10). The average amount of LG-protein adducts is 12-fold higher in AD brains than in control brains with the magnitude of the increase correlating with the pathological evidence of severity (99). That the LG-protein adducts isolated from AD brain do not result from free radical-induced lipid oxidation was indicated by LC-MS/MS chromatogram of the isoLG-lysine lactam obtained through exhaustive proteolysis of AD brain proteins that exhibited a single peak (99). In contrast, the isoLG-lysine lactam obtained from the mixture of isolevuglandin isomers generated by radical-catalyzed oxygenation of arachidonic acid exhibited multiple peaks. After 24 h incubation with isoLGE2, Aβ1-42 becomes toxic to primary cultures of mouse cerebral neurons; whereas Aβ1-42 itself incubated for 24 h without the levuglandin exhibited little toxicity. The oligomers formed from a reaction of Aβ1-42 with isoLGE2 exhibit immunochemical similarity with neurotoxic amyloid-derived diffusible ligands.
Most remarkably, the formation of these neurotoxic oligomers occurred at a molar ratio as little as 0.1 LGE2 per Aβ1-42 demonstrating that intermolecular cross-linking cannot be the sole mechanism for oligomerization (9). It is tempting to speculate that modification of Aβ1-42 by LGE2 serves as a seed to accelerate oligomerization. These observations support the view that a 60%–80% reduction in risk of developing AD associated with the use of nonsteroidal antiinflammatory drugs for at least 2 years (42, 89) involves the inhibition of isoLG production through the COX pathway. These observations laid the foundation for a new biochemical paradigm for the participation of the COX enzymes in AD. A recent study on the effect of an isoLG scavenger on dementia in mice added important support for this view. Thus, oral administration of salicylamine to a mouse model of dementia, protected the mice against the development of age-related deficits in spatial working memory (20). This observation suggests that trapping of isoLGs may mitigate the development of cognitive dysfunction in patients with AD.
Inhibition of proteolysis: neurodegeneration and glaucoma
Covalent modification of proteins, for example, ovalbumin (OA) or the amyloid peptide Aβ1-40, with isoLGE2 conferred resistance to processing by the 20S proteasome (19). Furthermore, both isoLGE2-OA and isoLGE2-Aβ1-40 competitively inhibit the chymotrypsin-like activity of the 20S proteasome for processing nonadducted proteins. Similarly, a ubiquinated iso[4]LGE2-modified protein, that is, calpain-1, inhibited 26S proteasome activity, while unmodified calpain-1 had no effect (31). Presumably, the bulky isoLG adduct sterically hinders the processive passage of the protein through the peptide tunnel of the proteosome where its protease activity resides. This competitively inhibits access of nonadducted proteins to the active site (19). Covalent modification of the 20S proteasome with isoLGE2 only causes a minor decrease in its protease activity (19). Presumably, this is because the active site, which is buried within the central cavity of this multienzyme complex, is relatively inaccessible compared with other proteasomal lysine residues.
In vivo, modification of the proteasome by isoLGs occurs in the presence of other proteins. In a model of this environment, isoLGE2 was added to cell lysates. Under these biomimetic conditions, proteosomal activity is strongly inhibited, but the kinetic behavior is not consistent with competitive inhibition (19). When newly generated, a nascent isoLGE2-protein adduct can bind nearly two equivalents of glycine (44). It is possible that this strong inhibition involves cross-linking of protein substrates with the proteasome. The nascent isoLG-protein adduct, for example, a Schiff base, can serve as an “activated monomer” that is capable of sticking to (cross-linking with) the proteasome, resulting in irreversible binding of the isoLG-modified substrate (Fig. 7). Thus, adduction with isoLGE2 might convert proteins into “suicide substrates” that covalently bind to, and consequently irreversibly inhibit, their target enzyme (102). However, direct evidence for this inhibition mechanism, for example, characterization of an enzyme cross-linked with its substrate by an isoLG, has not been reported.
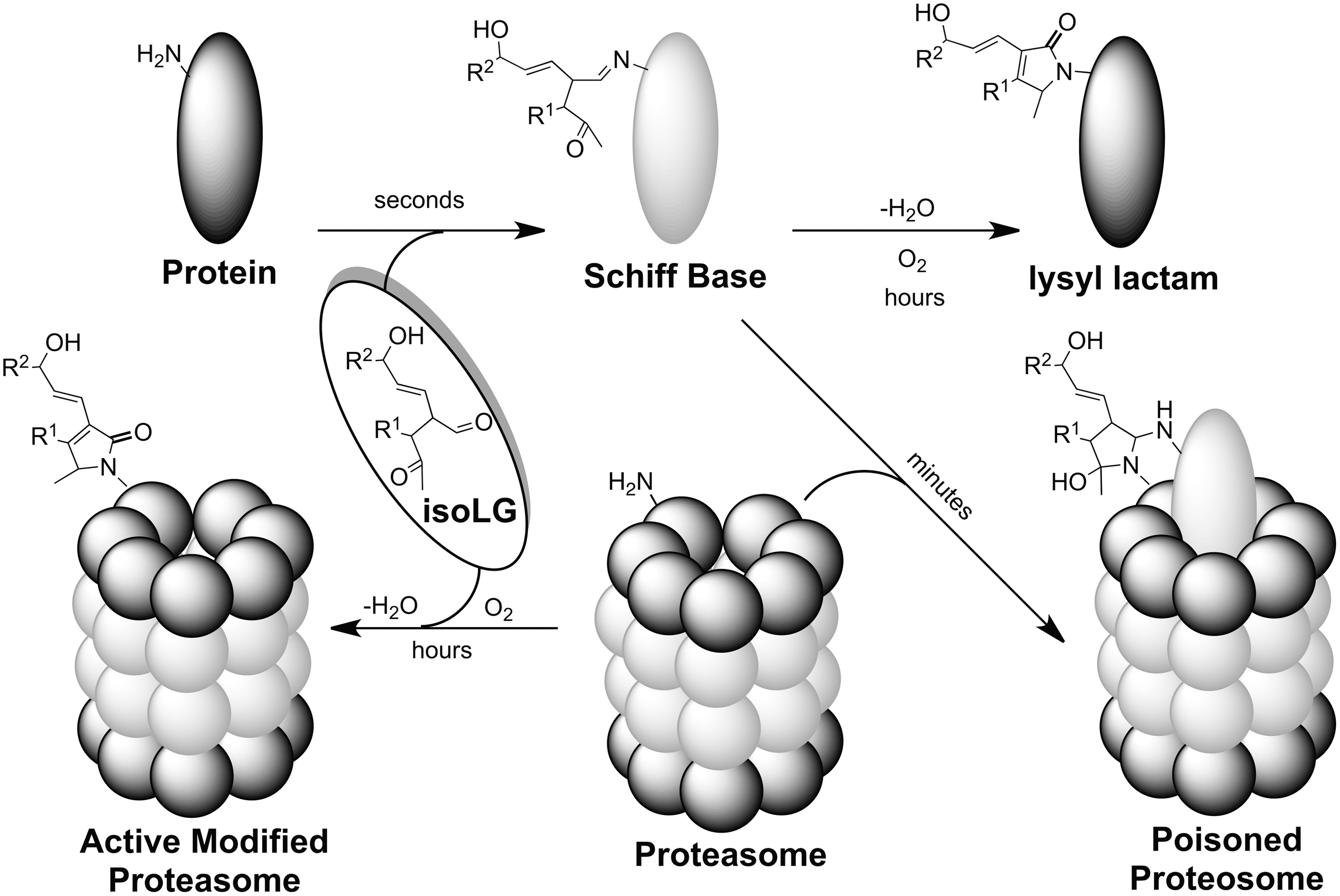
Proteasomal degradation of proteins is an important regulatory mechanism. Inhibition of the proteasome can promote accumulation of ubiquinated proteins (57) and phosphorylated neurofilaments (49), and foster apoptosis (48, 92). For example, isoLG-protein adduction to the 26S proteasome might inhibit the ATP-dependent degradation of ubiquinated proteins and, thus, contribute to the accumulation of ubiquitinated proteins present in the neurofibrillary tangles that are a hallmark of AD. Addition of isoLGE2 to neuroglial cells in vitro dose dependently inhibited proteasomal activity (IC50=330 nM) and induced cell death at higher concentrations (LC50=670 nM). Since this concentration was 100-fold lower than that previously reported for 4-hydroxynon-2-enal, isoLGs are “among the most potent neurotoxic products of lipid oxidation heretofore identified” (19). It is tempting to speculate that a similar process could convert a signaling peptide into an irreversible agonist whose binding with its receptor would result in dysregulation of a signaling pathway, because the receptor would remain in a permanently activated state.
Reactive oxygen species play a pathogenic role in primary open-angle glaucoma by fostering changes that reduce permeability of the trabecular meshwork (TM) tissue and, consequently, impede aqueous humor outflow, resulting in elevated intraocular pressure. We found elevated levels of calpain-1 in glaucomatous TM (31). However, calpain activity in glaucomatous TM is only about 50% of that in controls. This paradox is explicable by the facts that isoLG-protein adducts accumulate in human glaucomatous TM and that modification by isoLGs renders calpain-1 inactive. Thus, treatment of calpain-1 with iso[4]LGE2 in vitro results in covalent modification, inactivation, the formation of high-molecular-weight aggregates, and resistance to proteasomal digestion. Iso[4]LGE2-modified calpain-1 undergoes ubiquitination (32), and its loading impairs the cellular proteasome activity (31). Apparently, interference with proteasomal activity, owing to protein modification by isoLGs, contributes to glaucoma pathophysiology by decreasing the ability of the TM to modulate outflow resistance. Thus, lipid-derived oxidative protein modifications interfere with the housekeeping and/or regulatory functions of proteases, that is, calpain-1 and the proteasome. Upregulation of calpain-1 expression in a futile attempt to ameliorate the problem, exacerbates the pathology owing to efficient inactivation of the newly generated enzyme by adduction of isoLGs. In contrast, higher calpain-1 activity, which appears to be under translational control, was observed in glaucomatous optic nerve compared with control (32). Therapeutic neuroprotection strategies using calpain-1 inhibitors will require consideration of such anatomic differences in its activity and biosynthesis.
Elevated levels of iso[4]LGE2-adducts are present in astrocytes derived from the glaucomatous optic nerve head compared with those from controls. To model the environment in primary open-angle glaucoma, astrocytes isolated from rat brain cortex and human optic nerve were subjected to 0–150 mmHg pressure for 3 h. Levels of iso[4]LGE2-modification increased in direct proportion to a pressure of 50% above that found in control cells (30). Longer exposure resulted in a large increase in levels of iso[4]LGE2-adduct immunoreactivity. These observations suggest that chronic and/or prolonged exposure to pressure can contribute to the elevated iso[4]LGE2-protein adduct immunoreactivity observed in glaucomatous astrocytes. Apparently, reduced aqueous outflow caused by accumulation of isoLG-modified proteins causes increased intraocular pressure that promotes the generation of more isoLGs.
Inhibition of an oxidase: age-related macular degeneration
Patients with age-related macular degeneration (AMD) have mean blood levels of iso[4]LGE2-PEs (5.2±0.4 ng/ml, n=15) that are elevated ∼53% (p<0.0001) compared with those of healthy volunteers (3.4±0.1 ng/ml, n=15) (37). The formation of cholesterol-rich deposits in the retina (drusen) are a hallmark of AMD (46, 68). Mitochondrial cytochrome P450 (CYP)27A1 is a sterol C27-hydroxylase that eliminates cholesterol and likely 7-ketocholesterol from the retina. Recently, we found that modification of this protein with iso[4]LGE2 in vitro diminished enzyme activity (16). Only three Lys residues, Lys134, Lys358, and Lys476, readily interacted with the isoLG in vitro. Analysis of a protein extract from a human retinal sample by proteolysis followed by multiple reaction monitoring demonstrated that the peptide AVLK358 (-C20H26O3)ETLR was present in the retinal protein extract. Such protein modification presumably impairs sterol elimination, contributing, for example, to cholesterol-rich lesions associated with AMD. Subsequently, the specificity of isoLG modification was confirmed, providing direct evidence that isoLG adduction impairs enzyme activity and supporting the hypothesis that isoLG modification in the retina is detrimental to CYP27A1 enzyme activity, potentially disrupting cholesterol homeostasis. Thus, the effect of Lys358 modification on enzyme activity was investigated by characterizing catalytic properties of Lys358 as well as Lys476 CYP27A1 mutants both before and after isoLG treatment (17). The K358R mutant was less susceptible to isoLG-induced loss of catalytic activity than the wild type (WT), whereas the K476R mutant was nearly as vulnerable as the WT.24 Thus, modification of Lys358, a residue involved in redox partner interactions, is the major contributor to isoLG-associated loss of CYP27A1 activity.
To test whether scavenging of isoLGs could prevent modification of retinal proteins, mice were exposed to a bright light that caused retinal isoLG-adduct formation (18). In contrast, pretreatment of mice with PM, an efficient scavenger of γ-ketoaldehydes (vide supra), suppressed the accumulation of retinal isoLG adducts, and morphological changes in photoreceptor mitochondria were not as pronounced as in untreated animals (18). These experiments demonstrate that preventing the damage to biomolecules by lipid peroxidation products, a novel concept in vision research, is a viable strategy to combat oxidative stress in the retina. Whether this strategy can be of clinical utility for the prevention of the pathological sequelae of isoLG generation in human subjects deserves clinical investigation.
Modification of ion channels: cardiac arrythmia
AT-1 cells are an atrial tumor myocyte cell line that expresses a model integral membrane potassium ion (K+) channel that is relevant to oxidative ischemia/reperfusion injury-induced cardiac dysrhythmias (85, 95). IsoLGE2 caused a pronounced dose-dependent inhibition of the rapidly activating delayed rectifier K+ current Ikr (IC50=2.2 μM) (12). Activating and deactivating currents were suppressed equally, suggesting that these effects were neither voltage- nor gating dependent. Moreover, Ikr was not restored with washing, suggesting that the inhibition was not caused by a reversible allosteric effect but rather by an irreversible covalent modification of channel proteins. The inhibition was not manifested immediately, requiring 60 min to achieve full inhibition when tested at a concentration of 10 μM isoLGE2. Because isoLGEs adduct to proteins within seconds, the more prolonged time course for Ikr inhibition may be caused by time-dependent diffusion into the membrane and/or cross-linking of channel proteins.
The cardiac sodium channel NaV1.5 is a key determinant of electrical impulse conduction in cardiac tissue (53). Acute myocardial infarction leads to diminished sodium channel availability, both because of decreased channel expression and because of greater inactivation of channels already present. Myocardial infarction leads to significant increases in reactive oxygen species and lipoxidation products. The effects of reactive oxygen species on NaV1.5 function in whole hearts can be modeled in cultured myocytes, where oxidants shift the availability curve of INa to hyperpolarized potentials, decreasing cardiac sodium current at the normal activation threshold. Both the general oxidant tert-butyl-hydroperoxide and isoLGE2 potentiate inactivation of cardiac Na+ channels in human embryonic kidney (HEK)-293 cells and cultured atrial (HL-1) myocytes (26). Furthermore, isoLGE2were generated in the epicardial border zone of the canine healing infarct, an arrhythmogenic focus where Na+ channels exhibit similar inactivation defects. These observations suggested that Na+ channel dysfunction evoked by lipid peroxidation is a mechanism for ischemia-related conduction abnormalities and arrhythmias. Further studies confirmed that exposure to oxidants induces lipoxidative modification of NaV1.5 by isoLGs and demonstrated that PM and analogues, which act as isoLG scavengers, block voltage-dependent changes in sodium current by the oxidant tert-butylhydroperoxide, both in cells heterologously expressing NaV1.5 and in a mouse cardiac myocyte cell line (HL-1) (53).
IsoLG-modified ethanolaminephospholipids: AS
Elevated levels of LG-protein adduct immunoreactivity in blood from patients with atherosclerosis provided the first evidence for the formation and pathological significance of LG-adducts in vivo (75). The data in Table 1 paradoxically show that cholesterol levels are significantly elevated in older individuals (N[62]) who do not have AS compared with atherosclerosis patents (AS[63]) of a similar average age. This probably reflects the success of therapeutic interventions, including diet or drugs, that reduce total cholesterol levels, but apparently not isoLG levels, for the AS patients. The elevated isoLG-adduct levels, detected by immunoassay with anti-isoLG antibodies, indicate a defect that results in an abnormally high level of oxidative injury that is associated with AS but is independent of the classical risk factors, total cholesterol, or LDL. That defect is almost certainly an elevated susceptibility toward oxidative injury. While LDL and total cholesterol levels may correspond with those of fatty acyl precursors of oxidized lipids, isoLG adduct levels represent a direct measure of oxidative injury that is intimately associated with the formation of foam cells. Because LG-protein and isoLG-protein adducts can accumulate for days or weeks over the lifetimes of proteins (86), which are extended by their resistance to proteasomal degradation (vide supra), they represent a convenient dosimeter that provides a cumulative index of oxidative stress. More recently, we showed that isoLG adducts that incorporate the amino group of PEs into isoLG-hydroxylactams (4) are also potential biomarkers for assessing risk for oxidative stress-stimulated diseases (47).
Radical-induced oxidation of LDL to an oxidized form (oxLDL) is accompanied by the formation of isoLG-adducts, and it fosters massive unregulated endocytosis by a class of macrophage receptors that recognize the lipid-based protein modifications, but not native LDL (25). This uptake, and impaired processing, leads to accumulation of large quantities of partially digested lipoprotein and globules of cholesterol esters in the macrophage cells, giving them the appearance of being filled with foam. The resulting “foam cells” are the progenitors of fatty streaks that evolve into atherosclerotic plaques. We found that LGE2-modified LDL binds with and is degraded by receptor-mediated endocytosis by macrophage cells (40). Both binding and uptake, leading to degradation, are blocked by an excess of oxLDL, suggesting that LGE2-derived modifications generated on oxidation of LDL contribute to binding and uptake of oxLDL by macrophages. It must be noted here that this biological activity is also elicited by other products of lipid oxidation. Thus, we subsequently identified a family of oxidatively truncated PLs whose binding with macrophage CD36 receptors also promotes endocytosis of oxLDL (60, 61).
Platelet hyper reactivity produces a prothrombotic state that plays an important role in the pathogenesis of AS. Incubation of platelets with 1 nM isoLGE2 potentiates platelet aggregation induced by collagen, and is accompanied by the formation of thromboxane B2, and phosphorylation of both cytosolic phospholipase (cPL)A2 and p38 MAPK. Since isoLGE2 can be produced by both COX-mediated and free radical-induced cyclooxygenation, arachidonate oxidation through either or both of these pathways could produce such platelet activation. Some lipid-derived protein modifications, such as carboxyalkypyrrole derivatives of protein lysyl residues, induce a prothrombotic state by receptor-mediated responses, for example, by serving as agonists of the toll-like receptor 9 (59). Alternatively, the activation of cPLA2 through phosphorylation may be a consequence of isoLG-induced modification of platelet plasma membrane aminophospholipids and proteins that could conceivably alter the membrane structure and affect membrane fluidity which could promote activation of PLA2 (2). It is noteworthy that, since activation of platelets by exogenous agonists is accompanied by COX-dependent formation of isoLGE2-protein adducts (8), isoLGE2 can promote its own generation. In other words, isoLGE2 could function as the mediator of an autocatalytic cycle that provides a mechanism for amplifying the effects of isoLGE2 on thrombosis.
PE is a major target of endogenously generated isoLGs. MPO is an oxidative enzyme released by activated neutrophils that associates with circulating high-density lipoprotein (HDL) (38) and is an important generator of isoLG-modified plasma protein in vivo (62) Incubation of HDL with MPO generated at least 10-fold greater amounts of isoLG-PE than isoLG-protein adducts (34).
PE adducts of isoLGE2 (isoLGE2-PE) dose dependently induce cytotoxicity (LC50 2.2 μM) in human umbilical vein endothelial cells (HUVECs) (90). As noted earlier, “inflammation fosters the generation of isoLGs.” Recently, it was reported that the opposite is also true, that is, the generation of isoLGs fosters inflammation. Thus, isoLGE2-PE also fosters the expression of inflammatory cytokines and endoplasmic reticulum (ER) stress signaling in HUVECs (35). It induced expression of adhesion molecules and increased MCP-1 and IL-8 mRNA in HUVECs. Chemical inhibitors of the ER stress response reduce the extent to which isoLGE2-PE activates inflammatory responses (35). This is consistent with a developing paradigm linking oxidized lipids, ER stress, inflammation, and AS (36). ER stress response proteins are markedly increased in atherosclerotic lesions (91). In addition, fluorescently labeled isoLGE2-PE rapidly internalized to the ER (35). IsoLGE2-PE induced C/EBP homologous protein CHOP and BiP expression and p38 MAPK activity, and inhibitors of ER stress reduced isoLGE2-PE -induced C/EBP homologous protein CHOP and BiP expression as well as EC activation, consistent with isoLGE2-PE inducing ER stress responses that have been previously linked to inflammatory chemokine expression. Thus, isoLGE2-PE is a potential mediator of the inflammation induced by lipid peroxidation. Furthermore, isoLGE2-PE induces monocyte adhesion to endothelial cells (35). Endothelial cell activation and monocyte adhesion initiate AS by promoting migration of monocytes into the subendothelial space where they become foam cells, the precursors of atherosclerotic plaques.
Since isoLGE2 is also produced through COX-mediated cyclooxygenation, these biological activities of isoLGE2-PE also link COX activity to ER stress and inflammation. Blocking the contribution of the COX pathway to the generation of isoLGE2-PE adducts is almost certainly an important component of the therapeutic benefit of low-dose aspirin for reducing the risk of AS.
NAPE-PLD prevents accumulation of inflammatory and cytotoxic isoLGE2-PEs
Preventing the accumulation of bioactive proinflammatory isoLGE2-PEs is important for protecting cells against inflammation and oxidant injury. N-acyl phosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) was recently shown to serve as a promiscuous mammalian PLD for N-modified PEs that metabolizes isoLGE2-PEs (37). Blocking the expression of this enzyme with siRNA specific for NAPE-PLD transfected into HEK293 cells abrogated their ability to hydrolyze isoLGE2-PEs to isoLG-modified ethanolamine that is an order of magnitude less inflammatory than isoLGE2-PE. NAPE-PLD is widely expressed, with the highest expression in tissues such as brain, kidney, and testis that are particularly sensitive to oxidative injury (58). In brain, NAPE-PLD localizes to a variety of brain cells, including neurons, astrocytes, cerebral endothelial cells, and microglia (101). As noted earlier, isoLGE2-PE is rapidly trafficked to the ER (35). Consequently, it is especially noteworthy that NAPE-PLD is preferentially localized to the cisternae of smooth ER in hippocampal mossy fibers (56). NAPE-PLD expression is acutely upregulated in response to spinal cord contusion in rats (27) as would be expected if it has a role in ameliorating the pathological consequences of oxidative damage, for example, by promoting clearance of isoLGE2-PEs.
Conclusions and Prospects
When we reported the discovery that the bicyclic peroxide core of the prostaglandin endoperoxide spontaneously rearranges to levulinaldehyde, we noted that “the biological ramifications of such transformations demand scrutiny” (69). It is now apparent that this biological chemistry of lipids is emerging as a rich source of molecular level insights into some pathological consequences of COX activity and oxidative stress. We discovered that a diverse family of lipid-derived levulinaldehydes, isoLGs, is produced through both COX and free radical-induced cyclooxygenation of polyunsaturated fatty acids and their PL esters. IsoLGs have been associated with (i) inhibition of tubulin function, (ii) modification of histones and disruption of their nucleosomal complex with DNA, (iii) cross-linking proteins with DNA, (iv) impairment of mitochondrial function through modification of cytochrome c, (v) disruption of the blood brain barrier, (vi) promotion of Alzheimer's disease through COX-associated isoLG-induced oligomerization of Aβ1-42, (vii) promotion of neurodegeneration and impairment of aqueous outflow in the eye contributing to glaucoma, (viii) inhibition of proteosomal processing of proteins that could impair its regulatory functions, (ix) accumulation of cholesterol that contributes to AMD owing to modification of an oxidase, CYP27A1, (x) cardiac arrythmia owing to disruption of ion channels through modification by isoLGs, and (xi) promotion of AS by converting PEs into proinflammatory isoLGE2-PE agonists and fostering monocyte adhesion to endothelial cells. The activity of these inflammatory mediators is diminished or eliminated through NAPE-PLD-mediated metabolism. Trapping isoLGs with sacrificial primary amines, with PM and analogues being uniquely effective, can be used to prevent the formation of isoLG adducts, for example, with proteins or ethanolamine PLs, in vivo. Nonsteroidal anti-inflammatory drugs can be used to inhibit the COX-promoted generation of the stereoisomerically pure isoLGs, for example, LGE2 or LGD2, which have the same absolute configuration as prostaglandins.
Although isoLGs are structurally diverse, their common core renders their biological chemistry predictable although by no means simple. Besides this structural diversity, additional complexity is engendered by an extraordinary proclivity to form adducts with the enormously diverse universe of proteins, ethanolamine PLs, and nucleic acids, altering their function and cross-linking them into cryptic conglomerations. It is also important to note that the formation of isoLGs through free radical-induced cyclooxygenation is accompanied by the cogeneration of many other lipid oxidation products and derived protein modifications. Deconvoluting the relative biological importance of this plethora of molecules remains a redoubtable challenge.
Some simplifying principles are also emerging. Although proteins can become covalently linked to many molecules of isoLGs, as little as one isoLG molecule can suffice to destroy protein function owing to preferential modification of a specific protein lysyl residue. IsoLG modification of proteins or ethanolamine PLs can also result in gain of function. Nothing is known about the dependence on those new biological activities on the structures of the adducted isoLGs. Just as protein adducts of the structurally isomeric isoLGE2 and iso[4]LGE2 can be readily distinguished by antibodies, it is reasonable to anticipate that receptor recognition of these protein adducts or the corresponding PE adducts can vary with isoLG structure. It seems likely that some of the gain-of-function biological activities are receptor mediated and, therefore, may depend on isoLG structural isomerism. It is premature to categorically conclude that for adducts of all isoLGs the “biological activity of all of the isomers would be expected to be nearly identical” (2). This hypothesis can only be tested by studying the biological responses to adducts of pure isoLG structural isomers that are only readily available by chemical synthesis.
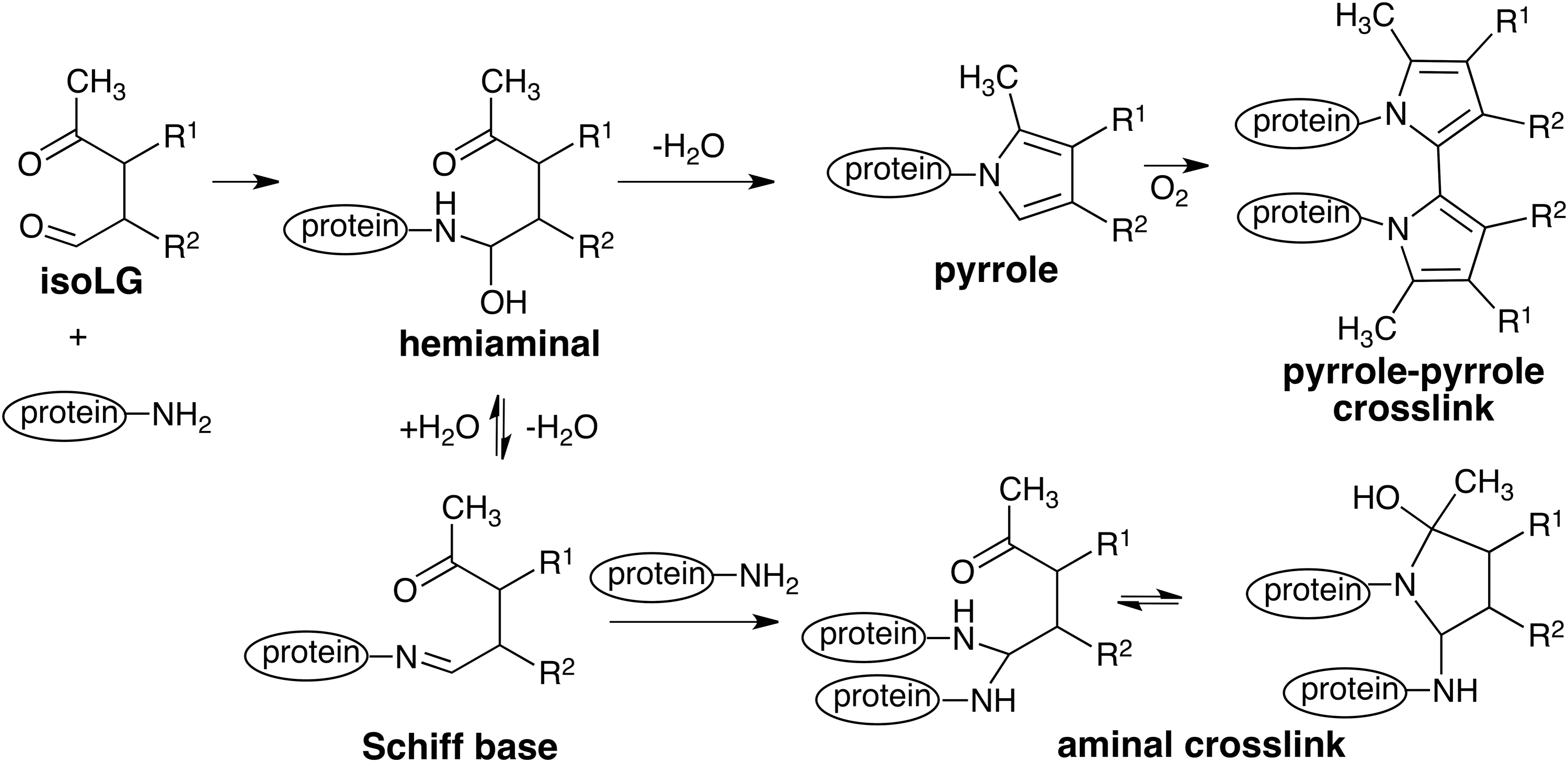
Although protein–protein cross-linking accompanies COX-promoted oligomerization of amyloid peptide as well as a reaction of calpain-1 with isoLGs, non cross-linked adducts are also formed. So the relative pathological importance of cross-linked versus non cross-linked adducts is not yet known. Two different structures have been proposed for protein–protein cross-links caused by isoLGs, that is, a bisaminal and a pyrrole-pyrrole dimmer (Fig. 8). No direct evidence is available to support either of these hypotheses.
Owing to the complexity of the task, gaining a comprehensive understanding of the “biological ramifications” of isoLG formation and adduction with biomolecules demands a rare combination of skills in organic synthesis, mass spectrometry, biochemistry, and cell and animal biology.
Footnotes
Acknowledgments
The portions of this work conducted in the Salomon laboratories were supported by grant (GM021249) from the National Institute of General Medical Sciences of National Institutes of Health. Dr. Salomon thanks his students and collaborators whose names appear in the references for their industrious, meticulous, persistent, and thoughtful contributions to their studies.