
Abstract
Introduction
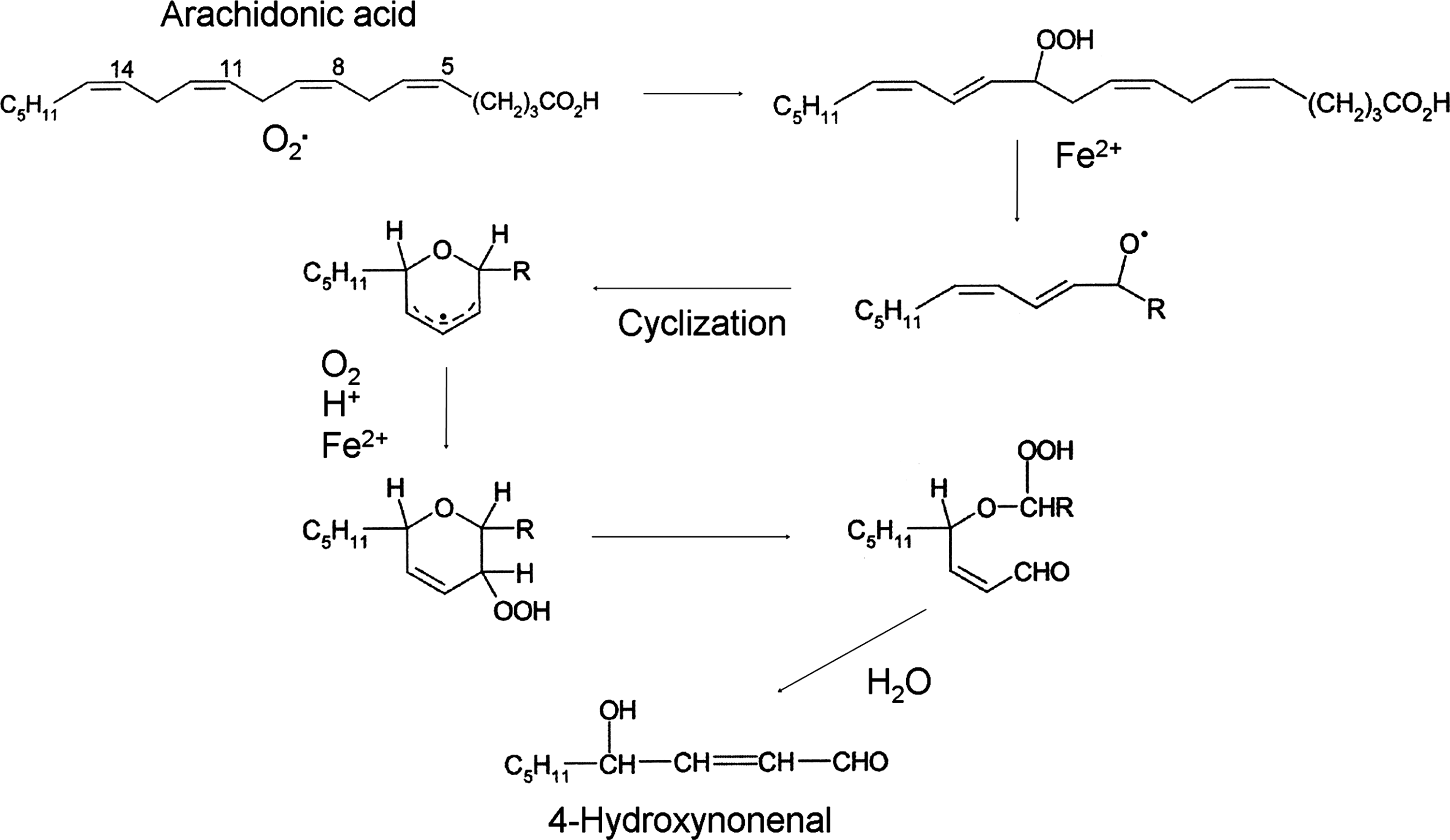
O
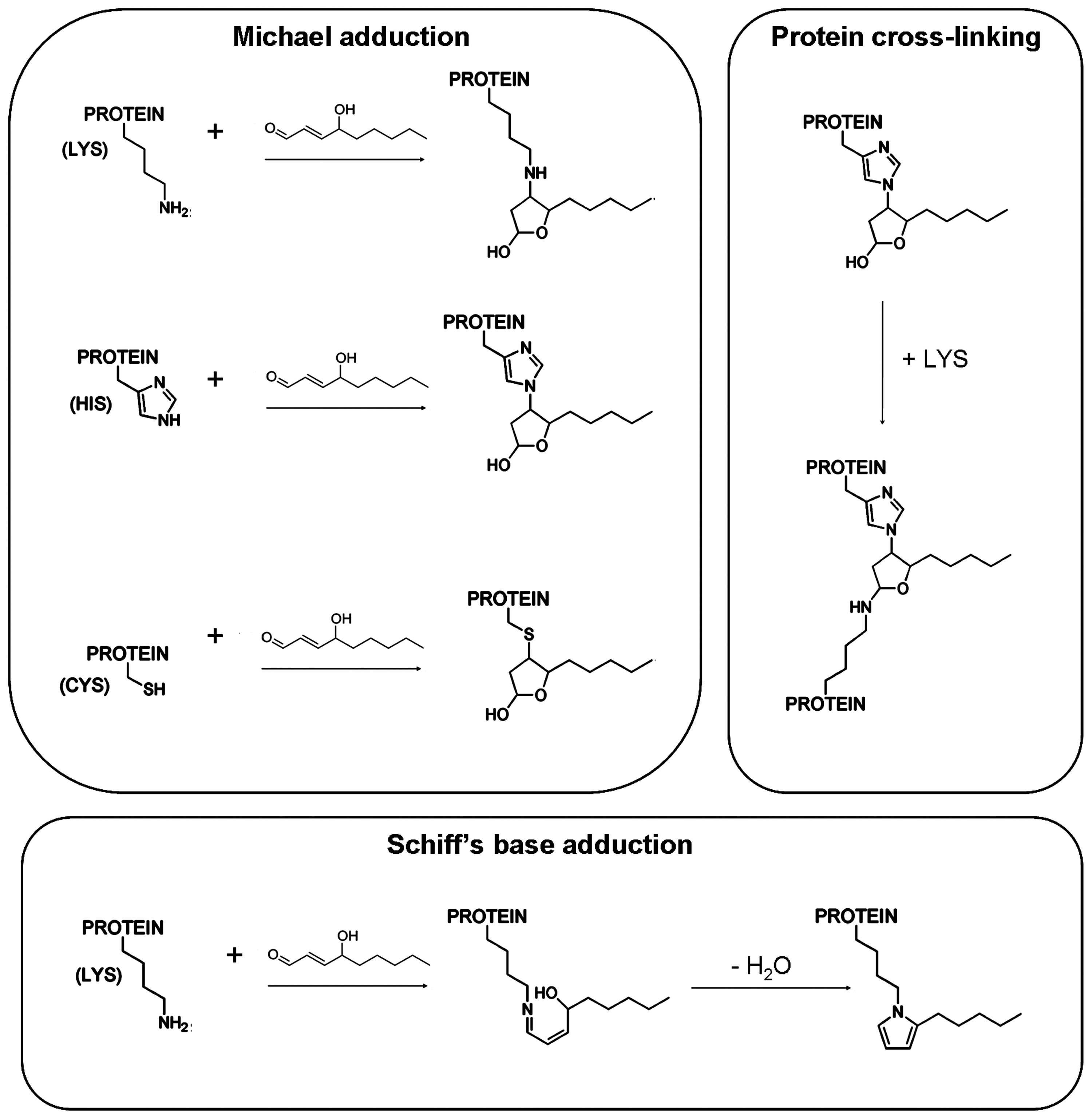
Once formed, HNE is able to affect several signaling processes, as well as gene expression pathways and protein functions. Most of these effects depend on the ability of HNE to bind covalently to functional proteins. Indeed, HNE is a γ-hydroxy-α,β-unsaturated electrophilic compound that preferentially forms 1,4-Michael-type adducts with nucleophiles, such as proteins and DNA. 1,4-Michael addition to 4-HNE occurs readily via the reaction of a nucleophile with C3 of HNE, resulting in the addition of a nucleophile and proton across the HNE carbon–carbon double bond (C=C) (150) (Fig. 2). The addition product subsequently rearranges to a cyclic hemiacetal (lactol) via the reaction of the 4-hydroxyl group with the aldehydic function. Amino acids known to react with HNE via 1,4-addition are Cys, His, and Lys (150). HNE can also react with lysyl residues through Schiff base formation, leading to pyrrole formation. In addition, HNE modification can result in cross-linking of two lysyl residues through reversibly formed Schiff base Michael adducts (134, 221) (Fig. 2).
Due to the high chemical reactivity of aldehydes, mammals have evolved a full set of enzymes converting them to less reactive chemical species and contributing to the control of their steady-state intracellular concentrations, which reflect the equilibria between the rates of formation by LPO and of catabolism into less reactive compounds. The main catabolic reactions are the formation of adducts with GSH, which can occur spontaneously or can be catalyzed by glutathione-S-transferases, the reduction to alcohols by aldo-keto reductases or alcohol dehydrogenases, and the oxidation to acids by aldehyde dehydrogenases (57, 118, 180).
The amphiphilic nature of HNE allows its diffusion across membranes and the covalent modification of cytoplasmic or nuclear compounds far from the site of its origin (135). Similarly, HNE formed outside the cells (i.e., in an inflammatory site or in the plasma) can react with stromal proteins or proteins belonging to adjacent cells, which do not undergo LPO. The targets for HNE are cell-type specific and dependent on both the pattern of proteins expressed by the cell and the aldehyde concentration. Moreover, the modification of specific proteins can have different biological consequences, in relation with the protein function.
In this review, we consider some HNE–protein interactions that have been shown to be involved in the development and evolution of some pathological conditions, such as cancer, neurodegenerative, chronic inflammatory, and autoimmune diseases.
HNE-Protein Adducts in Cancer Cells
Increases of oxidative stress have been demonstrated in the majority of cancer types, while the concentration of LPO products can vary in relation with cell type. The first experiments in this field demonstrated that, in hepatoma cells, the level of LPO products was lower than in normal liver cells (72, 155) and depended on the degree of deviation from the normal phenotype (166). In accordance with these results, Canuto et al. (28) showed that, during rat liver carcinogenesis, the activities of the enzymes metabolizing the toxic aldehydes increased, thus rendering the cancer cells more protected against the cytotoxic effect of aldehydes. Moreover, in hepatoma cells, the majority of HNE was converted to the HNE-GSH conjugate, which was rapidly and efficiently exported from the cell (197). However, the analysis of HNE-protein adducts in different types of tumors by immunoblotting or immunohistochemistry revealed adducts of this kind in renal (138), and colon cancer cells (88), as well as in astrocytic and ependymal glial tumors, in which the incidence of HNE-immunopositive tumor cells increased with increasing grades of malignancy (89).
Oxidative stress and, consequently, the products of LPO were long considered merely involved in carcinogenesis, due to their reactivity with DNA, while other papers demonstrated that oxidative stress and LPO products, such as HNE, also play important roles in the induction of cell cycle arrest, differentiation, and apoptosis in cancer cells (15). Similarly, the presence of HNE-guanosine adducts may not only indicate the mutagenicity of HNE but also indicate its capacity to induce apoptosis in cancer cells. Indeed, the ability to alter DNA is a characteristic of many chemotherapeutic drugs that, through this mechanism, induce apoptosis in actively proliferating cancer cells. Moreover, the concentrations at which HNE can form DNA adducts are rather high and can be achieved only under highly pro-oxidant conditions (216).
In contrast, in several tumor types, the progression of malignancy is accompanied by reductions of oxidative stress, due to the upregulation of antioxidant capacity (199), and the induction of the Nfr2/Keap1 pathway, which negatively regulates the HNE intracellular concentration (151). On the other hand, despite the reduction of intrinsic oxidative stress, the level of HNE-protein adducts in cancer cells may increase, due to the inflammatory response present in tissues surrounding cancer lesions.
In summary, the divergent results regarding the concentration of HNE in tumor tissues of different origins, and the discrepancies between the levels of oxidative stress and the levels of the products of LPO could have diverse causes, including the pattern of HNE-metabolizing enzymes in tumor cells; the lipid composition of the cell membranes, with differing levels of peroxidation-susceptible substrates, such as PUFAs; and the presence of inflammation, which might increase the level of diffusible HNE from neighboring tissues to the tumor cells.
Although the amount of HNE-protein adducts in cancer cells has been often assayed as a means of assessing the level of oxidative stress under diverse experimental conditions, only in some cases the identification and the consequences of HNE-protein adduct formation on cancer cell growth or behavior have been reported. Divergent results obtained in this field document that the formation of HNE adducts can have anti-carcinogenic or pro-carcinogenic effects, depending on the cell type and the specific adduct. In epidermoid carcinoma A431 cells, Liu et al. (119) observed that the signal triggered by the formation and activation of HNE-epidermal growth factor receptor adducts, detected by immunoblot analysis, followed by phosphorylation/activation of Shc adaptor proteins, ERK and JNK, inhibited DNA synthesis and suggested that this HNE-triggered signal transduction cascade selectively worked to suppress cell growth (119).
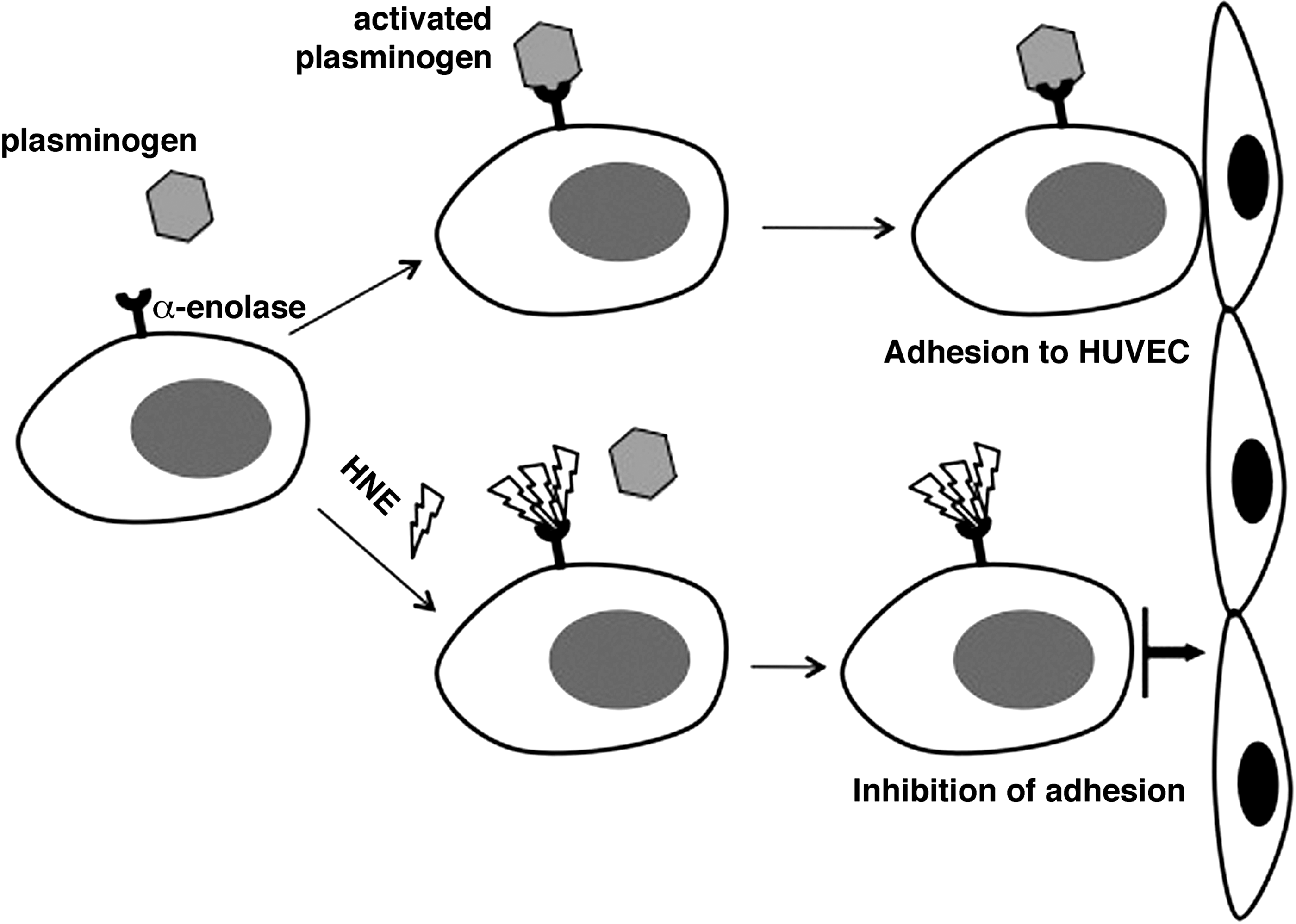
In a previous paper, we analyzed the interaction between HNE and α-enolase in HL-60 human leukemic cells (64), using a combination of two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), immunoblotting, and mass spectrometry. In addition to its enzymatic and transcriptional roles, α-enolase, expressed on the surface of a variety of eukaryotic cells, functions as a strong plasminogen receptor (144). Treatment with HNE strongly inhibited the binding between plasminogen and α-enolase at the surface of HL-60 cells, most probably as a consequence of the formation of HNE adducts with lysyl residues of α-enolase involved in plasminogen binding (7). HL-60 cells, as well as other leukemic cells, display enhanced plasminogen binding, which may contribute to an enhanced fibrinolytic state in leukemic patients (144). The inhibition of plasminogen binding was apparent even at HNE concentrations almost as low as those detected in normal tissues and plasma (1 μM). As a functional consequence, a strong reduction of HL-60 cell adhesion to human venous endothelial cells was produced, which might reduce the invasive and metastatic capacity of HL-60 cells (Fig. 3).
In MDA-MB-231 cells, a triple-negative human breast carcinoma cell line, the analysis of HNE-protein adduct formation revealed that HNE could modify, in a dose-dependent way, the enzyme peptidylprolyl cis/trans-isomerase A1 (Pin1), which catalyzes phosphoserine and phosphothreonine-proline conversions from cis to trans (4). HNE formed Michael adducts with this enzyme, which were detected by matrix-assisted laser desorption ionization/time-of-flight/time-of-flight (MALDI-TOF/TOF) mass spectrometry at the active site residues His157 and Cys113, with Cys113 being the primary site of HNE modification. The molecules that covalently modify critical residues in Pin1 catalytic or binding sites have been shown to induce apoptosis and inhibit cell proliferation, possibly due to their inhibition of Pin1 actions on cell cycle. Thus, it was proposed that some antiproliferative effects observed in cancer cells after exposure to HNE might also depend on the alteration of this enzymatic pathway.
In contrast, in another line of breast cancer cells, MCF-7 cells, and in RKO colon cancer cells, it has been demonstrated that HNE inhibited the AMP-kinase kinase activity of cellular LKB1, a serine/threonine kinase tumor suppressor, which modulates anabolic and catabolic homeostasis, cell proliferation, and organ polarity (209). The authors reasoned that LKB1 would be covalently modified and inactivated by HNE, which might entail increased risks of hypertrophic or neoplastic diseases.
Another HNE effect detected in cancer cells points to an interaction between HNE and peroxisome proliferator-activated receptors (PPARs). PPARs are a superfamily of nuclear receptors, subdivided into three subtypes (α, β/δ and γ), differing for tissue-specific expression, preferential ligand recognition, and biological function (95, 207). In HL-60 cells and in U937 leukemic cells, HNE potentiated the effects of PPARγ ligands, suggesting the existence of mutual interactions between HNE- and PPAR-ligand-related pathways in leukemic cell growth and differentiation (153). In addition, it has been reported that HNE directly binds and activates PPAR β/δ, which, in the liver, exerts a protective action toward chemically induced hepatotoxicity (40). This suggests that HNE, as an endogenous modulator of PPARβ/δ activity, might be involved in the protection from liver disease associated with oxidative damage. In this context, it is of interest that HNE stimulated Glutamate Cysteine Ligase (GCL) activity, through post-transcriptional modification of Cys553 in GCL and Cys35 in the modulatory subunit of GCL in vitro, detected by MALDI-TOF/TOF mass spectrometry. Since GCL catalyzes the first and rate-limiting step in GSH biosynthesis, these results suggest that the stimulation of GCL activity by HNE may concur to a compensatory cytoprotective response, through an increase of intracellular GSH and GSH-dependent detoxifying potential, during periods of oxidative stress (12). The activation of PPAR β/δ by HNE may have anti-carcinogenic effects in breast cancer too (222). Indeed, Yao et al. recently demonstrated that ligand activation of PPAR β/δ in two human breast cancer cell lines inhibited relative breast cancer tumorigenicity and further advanced the development of ligands of PPAR β/δ that are able to specifically inhibit breast carcinogenesis (222).
HNE-Protein Adducts in Neurodegenerative Diseases
HNE-protein adducts have been detected in brain tissues and body fluids in several neurodegenerative diseases, such as Alzheimer's Disease (AD), Huntington's Disease (HD), Parkinson's Disease (PD), amyotrophic lateral sclerosis (ALS), and Down Syndrome (DS) (26, 27, 110, 182, 223). Indeed, the brain is one of the major targets of LPO, as it is highly sensitive to oxidative stress (it consumes about 20–30% of inspired oxygen) and contains high levels of PUFAs.
Among neurodegenerative diseases, the formation of HNE-protein adducts in AD has been extensively documented and a number of comprehensive reviews, describing the proteins involved, have been written by our as well as other research groups (152, 191). The majority of studies in this field adopted proteomic approaches based on the immunochemical detection of HNE-protein adducts with anti-HNE antibodies among cellular proteins separated by 2D-PAGE, followed by Western blotting and identification of immunoreactive spots by mass spectrometry. As seen earlier, while discussing the studies of HNE-protein adducts in cancer cells, very rare studies proceeded to the non-trivial task of actually demonstrating the oxidative modifications of specific proteins by mass spectrometry. These include the adducts of HNE with a regulator of G-protein signaling 4 in PD described next (131). Instead, the pinpointing analyses of ubiquitin carboxyl-terminal hydrolase L1, Cu,Zn-superoxide dismutase (SOD1) and DJ-1 protein in AD and PD conducted by Choi et al. dealt with the oxidation of cysteinyl and methionyl residues (35 –37).
Here, we briefly summarize the most important findings and the most recent insights in this field. Cerebral alterations in AD include synaptic loss (168, 176), neurofibrillary tangles, and amyloid plaques, whose main protein component is the amyloid β (Aβ) peptide, a molecule of 40–42 amino acids, derived from the proteolytic cleavage of integral membrane amyloid precursor protein, by the action of beta- and gamma-secretases (73). It has been demonstrated that Aβ can induce oxidative stress and initiate LPO (27), resulting in the formation of LPO products, including HNE, malondialdehyde (MDA), and others. HNE, in turn, can directly react with the Aβ peptide, through a covalent cross-linking of Aβ peptides, causing an acceleration in Aβ protofibril formations and an inhibition of the production of straight, mature fibrils (184). Another important target for HNE adduction in AD brain tissue is the heme oxygenase protein-1 (HO-1) (191). The activation of this enzyme is one of the earliest events in AD and plays an important role in the response to oxidative stress (158). HO-1 catalyzes the degradation of heme in a multistep, energy-dependent process and represents the rate-limiting enzyme in bilirubin production (123). Its expression is controlled by the Nrf2 transcription factor, as the HO-1 gene contains in its promoter region the antioxidant responsive element (63). In AD brains, the increase of oxidative stress leads to increases of Nrf2 activity and, consequently, increases of HO-1 protein levels. At the same time, oxidative stress induces LPO and HNE formation. The increases of HNE and HO-1 lead to increased formation of HNE adducts of HO-1. While, on the one hand, HNE adduct formation could impair HO-1 function, the loss of HO-1 function was accompanied, on the other hand, by increased phosphorylation of seryl residues of HO-1, leading to HO-1 functional activation. Moreover, the loss of HO-1 function can increase oxidative stress (Fig. 4) (13).

Adducts of HNE with α-enolase have been reported, not only in the HL-60 human leukemic cell line (see the discussion of “HNE-protein adducts in cancer cells”,earlier) but also in the brain tissue of AD patients, where their level correlated with the reduced glucose metabolism and the upregulation of glycolytic enzymes, necessary for counteracting the mounting energy deficit and hypoxic environment (128). In AD brain tissues, the α-enolase level increased to support the increase of glycolytic activity (190). The oxidative modifications of α-enolase lead to a disruption of neuronal energy metabolism and ATP-dependent ion homeostasis. Conceivably, these alterations might compromise the viability of neurons, rendering them more prone to cytotoxicity and apoptosis (191). Reduced glucose utilization and energy production in AD may also be related with the formation of HNE adducts with the neuronal glucose transporter GLUT3 (125) and with the mitochondrial ATP synthase α subunit (149). The latter observation is in agreement with previous results demonstrating a decrease of ATP synthase activity in AD brains (175). In view of the possibly important role of α-enolase as plasminogen receptor at the surface of neurons, Sultana et al. suggested that the formation of HNE adducts with α-enolase might inhibit the conversion of plasminogen to plasmin, involved in the degradation of oligomeric and fibrillar Aβ, thereby preventing the detoxication of Aβ and facilitating neuronal death (191).
Another HNE target in AD brain tissue is represented by collapsin response mediator protein 2 (CRMP2) (149). This protein plays an important role in membrane trafficking, cytoskeletal organization, axonogenesis and neurite outgrowth, and neuronal polarity (161, 162). The formation of adducts of HNE with CRMP2 impairs its activity and might be of pathogenic importance for neurite shortening and the loss of synapses, which are early features of AD (75, 175).
In AD brain, Owen et al. (141) have detected HNE adducts with low-density lipoprotein (LDL) receptor-related protein 1, a membrane receptor involved in Aβ peptide removal. The formation of HNE adducts might lead to protein impairment, which might contribute, in turn, to the extracellular deposition of amyloid substance (141). Moreover, Perluigi et al. demonstrated that SOD1 is HNE modified in the inferior parietal lobule of late-stage AD, which results in the formation of protease-resistant protein aggregates, which are considered highly toxic and can mediate cell death (149). The multiple HNE-protein adducts found in AD point out the relevance of protein modification by HNE in AD initiation and progression.
PD is the most common neurodegenerative motion disorder. Hallmarks of PD are the loss of dopaminergic neurons in the substantia nigra and the presence of cytoplasmic spherical protein inclusions, named Lewy bodies. These inclusions contain various proteins, including α-synuclein (172). Immunohistochemically detectable HNE-protein adducts were significantly increased in nigral neurons of patients with PD (223) and stimulated the aggregation of α-synuclein in vitro (160). Oxidative modification of α-synuclein and adducts of LPO products with this protein have been found in the dopaminergic neurons of the substantia nigra from PD patients (181). Qin et al. (160) demonstrated that incubation of HNE with α-synuclein resulted in the covalent modification of the protein, with approximately six HNE molecules per protein molecule incorporated as Michael addition products. The formation of these adducts prevented fibrillation but might result in the formation of toxic oligomers, which might contribute to the demise of neurons that are subjected to oxidative damage.
The HNE involvement in the pathogenesis of PD has been supported by other observations, indicating a pleiotropic role for HNE-protein adducts. HNE-modified glycolytic enzymes (aldolase A, α-enolase, and glyceraldehyde-3-phosphate-dehydrogenase) have been found by a proteomic approach in the frontal cortex of incidental PD, and dementia with Lewy bodies (67), and this has been suggested to be related with the decreases of enzyme activity and the impairment of glucose metabolism and neurological function in the frontal lobe of PD patients. Moreover, HNE protein adduction can affect G-protein-dependent signaling in PD, whose regulation has been implicated as an important pathogenic factor in PD, as well as in other neurodegenerative diseases. HNE was able to impair this signaling pathway by directly modifying Gαq/11, a subunit of the heterotrimeric G-protein-coupled receptor, as shown by immunoprecipitation and Western blotting (17). HNE could exert similar effects also by modifying and inactivating the regulator of G-protein signaling 4 (RGS4), which increases the GTPase activity of the Gα subunit, as recently demonstrated in a study by Monroy et al., in which the identification of HNE-RGS4 adducts by immunoprecipitation, Western blot, and mass spectrometry was followed by a more refined mass spectrometric analysis, which permitted to detect HNE-modified Cys71, Cys148, and Cys183 (131).
ALS is a motor neuron degenerative disease that occurs both sporadically and as a familial disorder (familial amyotrophic lateral sclerosis [fALS]). Although multiple mechanisms likely contribute to the pathogenesis of motor neuron injury in ALS, it has been suggested that oxidative stress may play a significant role in the pathogenesis and amplification of the disease. The levels of HNE and immunochemically detectable HNE-modified proteins were increased in spinal cord motor neurons of ALS patients, indicating that these modifications were associated with motor neuron degeneration in ALS (146). Using proteomic analysis, Perluigi et al. (148) detected three proteins significantly modified by HNE in the spinal cord of an animal model of fALS, the G93A-SOD1 transgenic mice: (i) dihydropyrimidinase-related protein 2; (ii) Heat-shock protein 70; and (iii) α-enolase. It was suggested that oxidative stress is a major contributing mechanism in the pathogenesis of ALS and that the structural alterations and the losses of functional activity of proteins can contribute to the neurodegenerative process (147).
High levels of oxidized proteins have been found in both HD (reviewed in ref. 24) and DS (48). HD is a dominantly inherited neurodegenerative disorder, caused by the expansion of a CAG repeat in the gene encoding the protein huntingtin (70). It has been suggested that functional defects of mitochondria, which are both important sources of reactive oxygen species (ROS) and targets of ROS-mediated damage, are involved in HD pathogenesis (87). The increase of ROS and the oxidative damage of functional proteins have been associated with pathological neuronal loss in HD. Moreover, a marked increase of HNE adducts has been found by immunohistochemistry in the nucleus caudatus and putamen of HD brains and in the corpus striatum of HD mice, which suggested the therapeutic use of antioxidants to inhibit LPO and protect neurons from oxidative stress-induced cell death, by improving ATP generation and mitochondrial morphology and function (110).
DS is one of the most frequent chromosomal aberrations, resulting from the partial or complete triplication of chromosome 21, characterized by several abnormalities, including premature development of AD neuropathology and by increased oxidative stress, conceivably involved in neurodegeneration (147). Quite recently, Di Domenico et al., by using a redox proteomic approach, have identified various protein targets of HNE in the frontal cortex from DS cases, with and without AD pathology (48). The HNE-modified protein targets identified embraced proteins involved in several biological functions, such as neuronal integrity, axonal transport, cytoskeleton organization, degradative systems, energy metabolism, and antioxidant response. The dysfunction determined by the formation of HNE adducts with these proteins might contribute to the progression from DS to AD. Similar repertoires of aldehyde-modified protein targets had been reported in relation with the other neurodegenerative diseases as well (reviewed in refs. 126, 164, 191).
In recent years, the role of autophagy has emerged as an essential antioxidant pathway in neurodegenerative diseases, because, by permitting the removal of damaged mitochondria and proteins, it can provide an effective antioxidant strategy, independent of the initiating mechanism (66). It has been proved that the accumulation of toxic oxidation products, such as HNE, is a prevalent feature of neurodegenerative diseases and can promote organelle and protein damage, leading to the induction of autophagy (51). Stimulation of autophagy by HNE has been demonstrated also in rat aortic smooth muscle cells (77). The data obtained in these model cells suggested that the autophagic response to HNE could be attributed, in part, to endoplasmic reticulum stress, being a component of the cell survival strategy in response to oxidative stress (71). HNE emerges from the sum of the data reported as an important contributor to the pathogenesis of neurodegeneration, whose build-up in the course of disease modifies functionally important proteins, while promoting the autophagic process as a survival-oriented defense mechanism.
HNE-Protein Adducts in Chronic Inflammatory Diseases
One of the first demonstrations that HNE plays a role in the inflammatory process came from the studies on the effects of HNE on chemotactic-oriented migration of neutrophils. When measured in a Boyden chamber, the latter was stimulated by HNE, even at concentrations of 0.1 μM or less (44). In the next few years, it became evident that HNE is one of the major biologically active aldehydes produced by membrane LPO, in the course of inflammation and oxidative stress, which can accumulate in certain tissues till concentrations of 10 μM or more (49, 201). Experimental ischemia or ischemia/reperfusion was shown to induce early generation of HNE and HNE-dependent protein modifications in the lung (43) or in the isolated rat heart (54). High doses of HNE (50 μM) infused into rat lungs caused perivascular edema with vascular compression and early endothelial cell disruption (76). Moreover, in lung inflammatory disorders, HNE induced lung injury and apoptosis (43).
The hyperproduction of HNE in the adipose tissue of obese patients was shown to contribute also to adipose tissue inflammation, by promoting the release of pro-inflammatory cytokines (reviewed in ref. 39). In C57BL/6 mice fed a high-fat diet, body weight gain and epididymal fat expansion were associated with increases of 4-HNE-protein adducts in adipose tissue detected by Western blotting (211). Excess generation of HNE, acting both as a covalent modifier of cell proteins involved in signal transduction, cytoskeletal organization or cell adhesion, and as a cell signal messenger, has been strongly implicated also in endothelial barrier dysfunction and atherosclerosis (112, 205).
HNE Adduct in Atherosclerosis
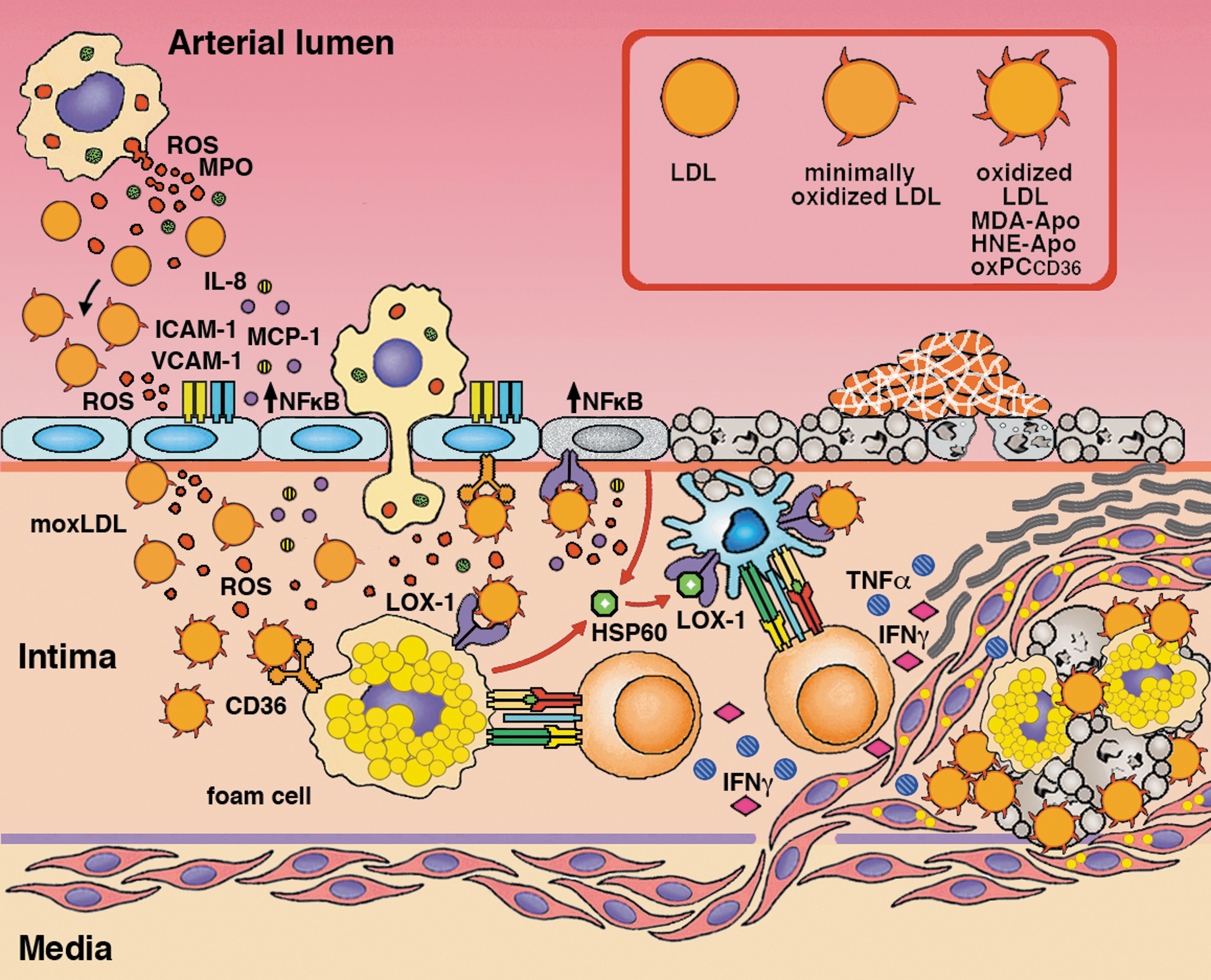
Evidence for the involvement of LPO-derived aldehydes in the alteration of LDL-receptor (LDLr) binding and in the promotion of atheroma formation came from several immunohistochemical analyses of atherosclerotic lesions from human aorta, using antibodies against adducts such as HNE-histidine (202), N ɛ-MDA-lysine (204), and N ɛ-acrolein-lysine [N ɛ-(3-formyl-3,4-dehydropiperidino) lysine] (203), in which intense positivities were associated with cells, primarily macrophages. The role of reactive aldehydes in the pathogenesis of atherosclerosis was also suggested by their increases in plasma, in association with extensive aortic atherosclerosis (142, 169, 170). About 30–40% of the uptake and degradation of oxidized low-density lipoprotein (oxLDL) by mouse peritoneal macrophages is mediated by scavenger receptor SR-AI/II, with CD36 accounting for a further 35% (102, 120). LDL modification by aldehydes enhanced their recognition and uptake by macrophages (79, 80). The formation of aldehyde adducts with lysyl residues of apolipoprotein B (ApoB) in LDL altered the affinity of the latter for the ApoB/E receptor, expressed in most cell types except macrophages, and converted LDL to an atherogenic form that was uptaken by scavenger receptor-bearing cells (macrophages and smooth muscle cells), leading to the formation of foam cells (33, 187, 188, 189). Moreover, modification of human recombinant ApoE with acrolein severely compromised its functional integrity, as for heparin, lipid and LDL receptor binding (196). Acrolein-LDL also induced foam cell formation from macrophages (212).
Phosphatidylcholine (PC) γ-hydroxyalkenal, that is, the γ-hydroxy-α,β-unsaturated core aldehydes still esterified at the sn-2 position of PC, also strongly contribute to the binding of oxLDL by scavenger receptors and to the pathogenesis of atherosclerosis (81, 169). Antibody-based studies revealed the presence of carboxyheptylpyrroles (CHPs) and carboxypropylpyrroles in oxLDL (93), reflecting the presence of protein lysyl adducts in the core aldehydes 9-hydroxy-12-oxo-10-dodecenoyl- acid ester of phosphocholine (HODA-PC), produced by oxidation of 1-palmitoyl-2-linoleoyl-glycero-3-phosphocholine or linoleoyl-2-arachidonoyl-glycero-3-phosphocholine, and 5-hydroxy-8-oxo-6-octenoyl- acid ester of phosphocholine (HOOA-PC), produced by oxidation of 1-palmitoyl-2-arachidonoyl-glycero-3-phosphocholine. The CHP immunoreactivity was also significantly higher in the plasma of patients with atherosclerosis and end-stage renal disease than in healthy controls (93). Chemically synthesized HOOA-PC exhibited properties of a chemical mediator of chronic inflammation. It activated, in a dose-dependent manner, human aortic endothelial cells (HAECs) to bind monocytes and to secrete increased levels of monocyte chemotactic protein-1 (MCP-1) and interleukin-8 (IL-8), which promoted monocyte entry into chronic lesions. HOOA-PC was found unbound and in pyrrole adducts in lipid extracts of oxLDL and human atheromas (80, 156). The binding of oxLDL to CD36 was also partly mediated by the head group of oxidized, but not native PC, in oxidized phospholipids such as 1-palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (18). The scenario emerging from these studies delineated atherogenesis as a result of myeloperoxidase-initiated, free radical-induced production of oxPC, which promoted subendothelial monocyte infiltration and endocytosis of oxLDL by macrophages, accompanied by conversion into foam cells and atheroma formation (169). Thereafter, it was shown that scavenger receptor CD36, another mediator of oxLDL uptake (as well as of recognition and phagocytosis of apoptotic cells) by macrophages, bound oxidized PC derivatives in oxLDL, including HODA-PC and HOOA-PC. These γ-hydroxy-α,β-unsaturated aldehydes, collectively referred to as specific oxidized phospholipids acting via CD36 (oxPCCD36), were potent activators of the CD36-mediated endocytosis of oxLDL by macrophages, promoting the cytotoxic effects of the adducts of oxidized derivatives of phospholipids and cholesterol with proteins (157, 193). oxLDL and individual oxPCCD36 also interfered with the binding of high-density lipoproteins (HDL) to scavenger SR-B1 receptors of hepatocytes, thus inhibiting the HDL-mediated delivery of cholesteryl esters to the liver (10).
The complexity of this process was illustrated by the report that HNE-histidine adducts bound to lectin-like oxidized LDL receptor-1 (LOX-1), a class-E scavenger, multiligand receptor also implicated in atherosclerotic plaque formation (100, 121). Cloned as the main receptor for the binding, internalization, and degradation of oxLDL by endothelial cells (EC) (174), LOX-1 was found to be expressed also in vascular smooth muscle cells, macrophages, fibroblasts, and platelets (8, 34, 132, 220). Its C-type, lectin-like ligand-binding domain was capable of binding diverse ligands such as oxLDL, acetylated LDL (AcLDL), phosphatidylserine, apoptotic bodies, activated platelets, leukocytes, and bacteria (90, 133, 140, 183). LOX-1 has been indicated as a pro-inflammatory factor, with a role in atherosclerosis initiation and progression (34, 53, 92, 208). In endothelial cells, LOX-1 was upregulated on exposure to oxLDL (116). oxLDL binding to LOX-1 induced a decrease in nitric oxide release (42) and the expression of adhesion molecules (114) and MCP-1 (115), while promoting ROS production, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation (41, 42, 127), and apoptosis (116). In macrophages, LOX-1-mediated oxLDL binding stimulated the formation of lipid-laden cells resembling foam cells of atherosclerotic plaques (121, 185) (Fig. 5). Since the upregulation of LOX-1 and downregulation of SR-AI/II and CD36 are induced by cytokines, such as tumor necrosis factor (TNF)-α (101) and transforming growth factor (TGF)-β (52), it is conceivable that LOX-1 play a major role in oxLDL uptake in inflamed atherosclerotic plaques.
In CHO cells stably expressing LOX-1, bovine serum albumin (BSA) modified with HNE, 4-oxo-2-nonenal (ONE), or non-hydroxylated alkenals 2-nonenal and 2-hexenal strongly inhibited the uptake of AcLDL (used as an alternative to oxLDL, in order to bypass the variations in the extent of LDL oxidation), with HNE-BSA showing the strongest inhibitory activity. AcLDL uptake was completely inhibited by anti-LOX-1 antibodies and significantly inhibited also by HNE-LDL, but not by native LDL. BSA modified with these aldehydes, unlike native BSA, was taken up by CHO cells transiently expressing LOX-1, in proportion with the level of LOX-1 expression, highlighting LOX-1 as the receptor responsible for the uptake of aldehyde-modified BSA (100). HNE-LDL uptake was inhibited by the substitutions of critical amino-acid residues of LOX-1, which had been shown to be crucial for oxLDL binding (139), indicating a shared binding site for oxLDL and HNE-LDL on LOX-1. The binding of oxLDL, HNE-LDL, and histidine-LDL to LOX-1 was confirmed with CLTD14, the ligand recognition domain of LOX-1. Moreover, in HAECs, the binding to LOX-1 of HNE-histidine adducts (HNE-LDL, HNE-Nα -acetylhistidine), as well as of oxLDL, but not of LDL and histidine, stimulated ROS formation, an effect that could be inhibited by anti-LOX-1 antibodies. oxLDL, HNE-LDL, and HNE-histidine adducts triggered a redox-sensitive signaling cascade, entailing the phosphorylation of ERK 1/2 and NF-κB (100), which resulted in the expression of genes related to endothelial dysfunction and injury (114, 116).
The ability of oxLDL to function as endothelial cell stressors was largely determined by the extent of their oxidative modification. Minimally oxidized LDL retained their affinity for LDL receptor, activated antiapoptotic signaling, and induced inflammatory changes in macrophages and endothelial cells, resulting in the recruitment of inflammatory cells and the secretion of cytokines and chemokines that promoted further oxidation (1). Further, LDL, LPO, and apolipoprotein modification by reactive aldehydes determined the loss of recognition by the LDL receptor, with a shift to recognition by scavenger receptors, leading to foam cell formation from anti-inflammatory M2 macrophages, which were activated and shifted to a pro-inflammatory phenotype (206, 224). Scavenger receptors expressed on dendritic cells (DCs) (e.g., LOX-1) also mediate oxLDL uptake and the induction of the pro-inflammatory cytokine profile and of differentiation into the mature DC phenotype (136). Vascular-associated DCs (VADCs) thus contribute to the initiation of atherosclerosis (145). Mice receiving DCs pulsed with MDA-LDL exhibited more extensive atherosclerotic lesions, with increased inflammatory signs and antigen-specific immune responses (192) (Fig. 5).
Adaptive immune responses contribute to plaque formation and to the maintenance of the atherosclerotic process. HSP-60, which is involved in the delivery of antigens into the MHC-I presentation pathway (218) and the maturation of DCs (59), is a main target of autoimmune cell-mediated responses in atherosclerosis (25, 69, 97, 98, 124, 167). Infiltration of atherosclerotic lesions with HSP60-specific T cells even appeared to precede the formation of foam cells (96, 129, 219). Intriguingly, HSP-60 is secreted by monocytes (61) and endothelial cells in response to oxLDL (5, 68) and shares with them the LOX-1 receptor (218) (Fig. 5).
HNE-Adducts in Inflammation-Related Diseases
Other inflammation-related diseases associated with the presence of HNE-protein adducts are alcoholic liver disorders (113) and chronic alcoholic pancreatitis, in which the increased formation of HNE-protein adducts was evidenced in acinar cells adjacent to interlobular connective tissue (30). In chronic liver injury, it was demonstrated that HNE was involved in the transdifferentiation of hepatic stellate cells into a myofibroblastic phenotype characterized by proliferation and extracellular matrix deposition, leading to fibrosis (225). The exposure of isolated stellate cells to 1–10 μM HNE led to the detection of HNE adducts with Jun terminal kinase. The translocation of protein adducts determined an increased level of c-Jun mRNA, suggesting that HNE was an activating signal for oxidative stress responses.
An anti-inflammatory role for HNE has been demonstrated by studying NF-κB cell signaling. The latter is the major transcription factor associated with inflammation and oxidative stress (111). Inactive NF-κB is localized in the cytosol, bound to its inhibitory protein, IκB. On activation, NF-κB dissociates from IκB, after which translocation to the nucleus enables DNA binding and transactivation (91). This process is triggered by sequential phosphorylation and ubiquitination of IκB subunit α, followed by proteasomal digestion. The enzyme that catalyzes the ubiquitination of phosphorylated IκB, IκB kinase (IKK), is constitutively active and, in most cases, represents the key regulator of NF-κB activation (23). Ji et al. found covalent adducts of HNE to IKK, by using antibodies against IKK or HNE-protein conjugates in the human colorectal carcinoma cell line RKO and the human lung carcinoma cell line H1299, and demonstrated that HNE binding prevented IκBα degradation and, consequently, inhibited NF-κB activation (86). These authors concluded that, since NF-κB stimulates transcription in response to oxidative stress, its modification by HNE may limit the magnitude of such a transcriptional response.
In another inflammation-related metabolic condition, diabetes mellitus, the increase of oxidative stress and the formation of HNE adducts has been widely reported (45, 200). HNE has been demonstrated to form adducts with some of the proteins involved in the etiopathogenesis of diabetes. Indeed, HNE affected insulin signaling by binding to insulin receptor substrate (IRS)-1/-2 proteins in 3T3-L1 adipocytes, as shown by immunoprecipitation and immunoblotting (47). IRSs are recruited after insulin binding to its receptor and transmit the insulin signal by activating two major pathways: the phosphatidylinositol 3-kinase cascade for glucose, lipid, and protein metabolism and the mitogen-activated protein kinase cascade for cell proliferation and differentiation (171, 214). HNE-IRS adducts likely impair the function of IRSs and favor their degradation, indicating that this aldehyde plays an important role in insulin resistance development and, therefore, could foster the progression to type 2 diabetes (47).
Moreover, HNE seems to be involved in the etiopathogenesis of diabetic cardiomyopathy. Using immunoblotting with anti-HNE antibodies, Lashin et al. demonstrated the presence of HNE adducts with succinyl dehydrogenase in the heart of diabetic rats, which contributed to the functional inhibition of mitochondrial complex-II, amplifying the organelle dysfunction and markedly decreasing oxygen consumption in heart mitochondria. (109). In keeping with these results, Mali et al., using immunoprecipitation, showed that 4-HNE formed adducts with myocardial aldehyde dehydrogenase 2 in mice exhibiting metabolic syndrome/type-2 diabetes mellitus, whose formation was associated with a reduction of the enzyme activity, which might contribute to cardiac hypertrophy and dysfunction (122).
HNE-Protein Adducts in Autoimmunity: Sjögren's Syndrome and Systemic Lupus Erythematosus
HNE-protein adducts have been involved in both innate and adaptive autoimmune responses. Several oxidation-specific epitopes (OSEs) are recognized as endogenous damage-associated molecular patterns (DAMPs) by innate soluble and cell-associated pattern recognition receptors (PRRs). Such OSEs include the oxidation products of membrane phospholipids and PUFAs in LDLs and their adducts, as seen in atherosclerosis (107). PRRs involved include Toll-like receptors, scavenger receptors CD36 and SR-B1, C-reactive protein, complement factor H, and natural IgM antibodies (213), such as those recognizing the adducts of MDA and HNE with LDL, detected in the sera of immunodeficient rag1−/− mice after reconstitution with B-1 cells (38).
A number of interesting observations were also collected, regarding the adducts of HNE with some autoantigenic targets of antinuclear autoantibodies (ANA) characteristically detected in Sjögren syndrome (SS), systemic lupus erythematosus (SLE), and other autoimmune diseases (106). Typical ANA targets in SS include the SS-A/Ro and SS-B/La antigens. The SS-A/Ro antigens comprise a 52-kDa form (SS-A1/Ro52; TRIM21), found in both cytoplasm and nucleus and characterized by a tripartite motif with RING (E3 ubiquitine ligase), B-box and Coiled Coil domains, and a 60-kDa form (SS-A2/Ro60; TROVE2), found mainly in cytoplasm and involved in cell survival to UV damage. Both are components of Ro ribonucleoprotein (RNP) particles, in which they are non-covalently associated with short, non-coding, human cytoplasmic RNAs (hY-RNAs), as in spliceosomal RNPs, and small cytoplasmic RNAs, such as the 5S rRNA precursors of the 60S ribosomal subunit. The 48-kDa SS-B/La antigen is a transcription termination factor for RNA Polymerase III, transiently associated with hY-RNAs in RNPs involved in tRNA processing and histonic mRNA stabilization. Autoantibodies to SS-A2/Ro60 occur in more than 60% of SS patients and 25–40% of SLE patients, as well as in other autoimmune diseases. SS-Ro and SS-La antigens become exposed in apoptotic bodies and blebs of a variable size at the surface of apoptotic cells (29). Apoptotic cardiocytes from fetuses spontaneously aborted, due to the congenital heart block of neonatal lupus, opsonized by maternal anti-Ro and anti-La antibodies, induced the antibody-dependent cell-mediated cytotoxicity (ADCC) of co-cultured macrophages (130). Anti-SS-A/Ro antibodies were involved also in the ADCC in damage of keratinocytes in UV-sensitive SLE (62). It was proposed that, in SLE and SS, both an increased susceptibility of leukocytes to apoptosis (55, 65, 165, 227), possibly related with the overexpression of the E3 ubiquitin ligase SS-A1/Ro52 (56), and an impaired clearance of apoptotic cells by macrophages (117, 165) may be triggers of autoimmunity (173). More interestingly from the standpoint of this review, it was proposed that the breaking of tolerance to self antigens at the surface of apoptotic cells might be promoted by oxidative modifications occurring as a consequence of the oxidative stress that characterizes apoptosis (29, 78).
The role of self-antigen modification by the formation of HNE adducts in the breaking of immunological tolerance was first documented in an early report (217), in which murine serum albumin (MSA), modified in vitro with several unsaturated (MDA, HNE, and heptadienal) and saturated aldehydes (butanal, nonanal), induced strong T-cell-dependent antibody responses. Various T-cell hybridomas, established from immunized mice, recognized MDA- and HNE-modified MSA, but not native MSA, in an MHC-restricted manner. All aldehyde-modified MSA preparations induced strong specific antibody responses, while native MSA did not. Of the former, only HNE-MSA and nonanal-MSA induced crossed antibody responses to unmodified MSA, almost as intense as against aldehyde-modified MSA, indicating that the sensitization of T cells to HNE-MSA adducts favored the intramolecular spreading of the immune response to formerly tolerated epitopes of the native self antigen (Fig. 6) (217). Scofield et al. hypothesized that modification of SS-A2/Ro60 with HNE might facilitate the breaking of tolerance to the native antigen in SS. After immunizing rabbits with either the HNE-modified or the unmodified SS-A2/Ro60, they observed that autoimmunity was established faster and more strongly in the animals immunized with HNE-modified SS-A2/Ro60 (104, 177). Later work provided formal proof that the breaking of tolerance to self antigens in the context of apoptosis required the generation of neoepitopes (143). The immunization of A/J mice with late apoptotic thymocytes, expressing the transgenic hSS-B/La antigen of human origin, was followed by the production of anti-SS-B/La antibodies. Immunization with non-apoptotic cells, expressing the transgenic antigen, had similar, although smaller and slower effects. Instead, no responses ensued either the immunization of transgenic mice with syngeneic thymocytes expressing the transgenic antigen or of wild-type mice with thymocytes expressing autologous SS-B/La (143).
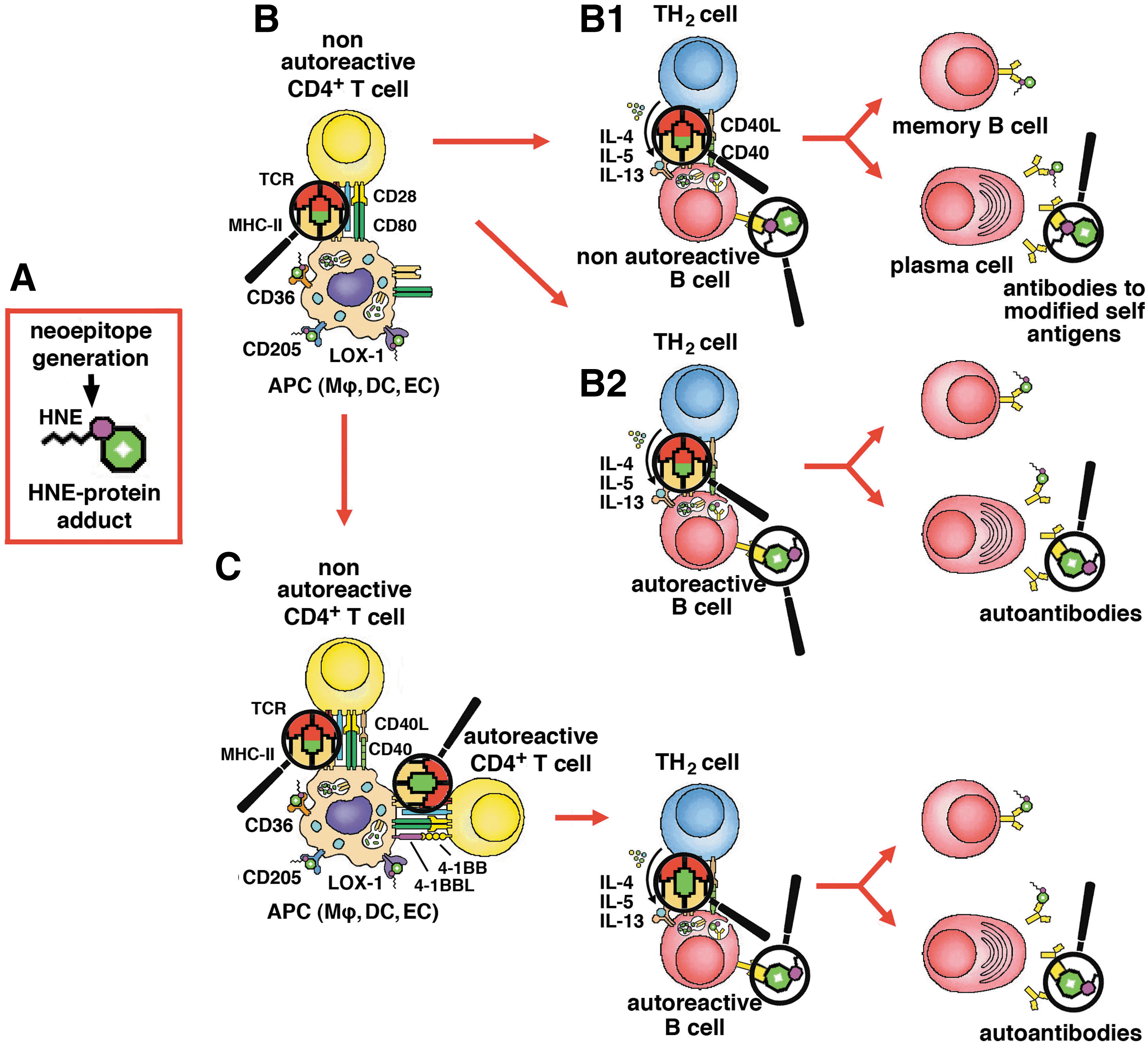
In an extension of this model, an SS-like condition, with anti-SS-A2/Ro60 antibodies, could be induced in BALB/c mice by immunization with a peptide of SS-A2/Ro60 (105). The production of anti-SS-A2/Ro60 and anti-SS-B/La autoantibodies ensued immunization with SS-A2/Ro60, both as such and modified with increasing concentrations of HNE (0.4, 2 or 10 mM). However, antibody production was faster after immunization with low- and, especially, medium-level HNE-modified antigen. The antibodies produced by mice immunized with HNE-modified, but not with unmodified SS-A2/Ro60, included added subpopulations that recognized HNE or HNE-SS-A2/Ro60, but not the unmodified antigen, as well as double-strand DNA (dsDNA), which induced the authors to imply an SLE-like disease, although they did not provide pathological evidence of it. The occurrence of anti-dsDNA and anti-SS-B/La antibodies, after immunization with SS-A2/Ro60, represents an example of intermolecular epitope spreading. The ability of HNE to form adducts with a large number of biological macromolecules might be of help in understanding the broad range of autoantibody responses in SLE and SS. Moreover, immunization with high-level HNE-modified SS-A2/Ro60 was associated with protein aggregation, lower-level antibody responses to unmodified SS-A2/Ro60 and SS-B/La, and a Sjögren-like condition, with reduced salivary flow and lymphocytic infiltration of salivary glands. These results were interpreted as being due to increased bifunctional cross-linking of SS-A2/Ro60 molecules (105), but a different interpretation could be that large, particulate immunocomplexes of aggregated HNE-SS-A2/Ro60 and autoantibodies stimulated the antigen-presenting activity of DCs and macrophages, which skewed the autoimmune response toward a cytotoxic cell-mediated mechanism. The same authors localized the targets of HNE modification within the sequence of Ro60, by using a collection of multiple antigenic peptides, chemically synthesized on the base of the sequences of Ro60 targeted by autoantibodies in SLE (82, 178) and anchored in multiple copies to a heptalysine core. Covalent adduct formation, on exposure to HNE in vitro, mostly occurred in amino-acid sequences participating in the solvent-exposed tertiary structure of Ro60, such as 126–137, 166–172, and 401–195 (103). Quantitative correlations of diagnostic and prognostic interest between markers of LPO, immunological reactivity to lipid-derived reactive aldehydes, and disease activity of SLE were reported. The prevalences and serum titers of MDA- and HNE-specific antibodies were significantly higher in SLE patients than in healthy controls, being also in correlation with the SLE Disease Activity Index (SLEDAI). Analogous correlations were observed between serum levels of MDA and HNE protein adducts and between both SLEDAI scores and antibody levels. Such results underscored the pathogenic role of LPO in SLE and the potential usefulness of anti-MDA and anti-HNE antibodies in predicting its progression (210).
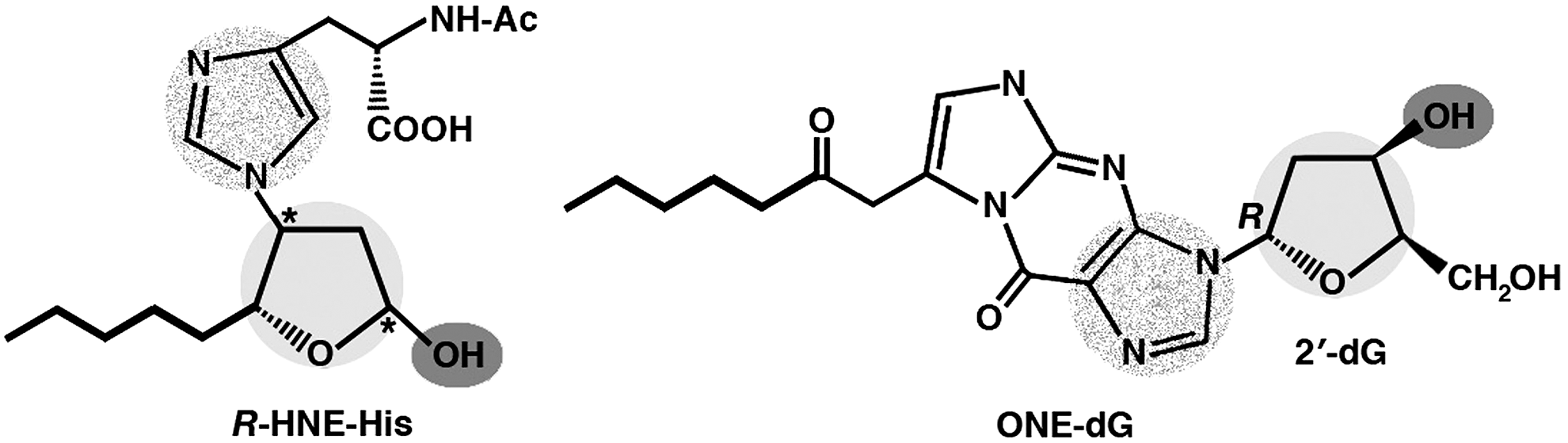
The molecular mimicry between the adducts of HNE and its analogs with proteins, on the one hand, and DNA, in native or modified form, on the other hand, as a mechanism for the production of anti-DNA autoantibodies in response to aldehyde-modified self protein antigens was investigated by Uchida and coworkers. After raising an anti-HNE monoclonal antibody (anti-R mAb 310), which selectively recognized the R enantiomer of HNE-histidine Michael adducts (74), these authors found that the sequences of such anti-HNE mAb strictly resembled those of various clonally related anti-DNA antibodies. Despite this structural similarity, the cross-reactivity of mAb R310 with native dsDNA was limited, but strongly enhanced by the treatment of DNA with ONE, an HNE analog. ONE-2′-deoxynucleoside adducts were identified as alternative epitopes of mAb R310 in ONE-modified DNA. The constituent chemical groups of a common epitope, possibly responsible for the molecular mimicry between the R-HNE-histidine configurational isomers and the 1,N2 -etheno-type ONE-2′-deoxyguanosine adducts, and required for the recognition by bispecific antibodies, were highlighted (Fig. 7). On this basis, it was proposed that endogenous electrophilic molecular species, including HNE, may be immunological triggers of autoimmune disease (2). The same authors further investigated the possible role of HNE-modified proteins as the endogenous prompt for the production of anti-DNA antibodies. Having established a murine hybridoma with the splenocytes of BALB/c mice immunized with HNE-modified keyhole limpet hemocyanin (KLH), they found HNE-specific epitopes in the epidermis and dermis of patients with SLE, pemphigus vulgaris, and contact dermatitis, as well as antibodies against HNE-modified BSA both in the sera of patients affected with SLE, SS, rheumatoid arthritis, systemic sclerosis, and idiopathic inflammatory myopathies, and in the sera of diseased, lupus-prone MRL/lpr mice. On repeated immunization with HNE-modified KLH, mice also developed a distinct population of B cell clones, recognizing native DNA, but not HNE-BSA. In accordance with the work previously cited, the reactivity of anti-HNE B cell clones toward DNA was greatly enhanced by DNA modification with ONE. On the other hand, anti-DNA mAbs cross-reacted with ONE-modified BSA. The data suggested that HNE-specific epitopes formed on HNE generation in cells might serve as sensitizing antigenic determinants for the production of bispecific antibodies against native DNA and ONE-modified proteins (198). Further results in experimental animals and in patients with SLE confirmed that the modification of human serum albumin (HSA) with HNE resulted in the generation of neoepitopes in HSA, which, in turn, was instrumental for the breaking of tolerance to HSA and was accompanied by cross-reactive responses to similarly modified DNA (58). Moreover, anti-ds DNA antibodies from 27 out of 40 patients affected by SLE preferentially bound to HNE-modified HSA, with regard to DNA and native HSA. Analogous results were reported, showing that the immunoglobulin G (IgG) antibodies raised in rabbits against HNE-modified HSA recognized HSA from SLE patients and cross-reacted with native and oxidized goat liver chromatin, while the anti-native/oxidized chromatin antibodies from 41 out of 74 SLE patients also specifically recognized HNE-HSA (3). These findings strongly supported the pathogenetic role of LPO products in autoimmune disease.
HNE-Protein Adducts In Red Blood Cell Aging and Autoimmune Hemolytic Anemia
In autoimmune hemolytic anemia (AIHA), red blood cells (RBCs) coated with autoantibodies on their surface are destroyed at an accelerated rate by splenic macrophages. Mice of the New Zealand Black strain spontaneously develop AIHA with increasing age and serve as an animal model of the disease. Major membrane proteins of RBCs were identified as autoantigenic targets in NZB mice. Autoantibodies eluted from RBC surfaces and mAbs produced by hybridomas established from NZB mice recognized band 3 protein, the major RBC membrane glycoprotein (32, 46). The breakage of tolerance to band 3 protein apparently resulted from the proteolytic removal of its surface domain or other modifications, exposing its membrane-embedded portion (60). More recent studies have provided evidence for the involvement of oxidative modifications of RBC self antigens in the formation of neoepitopes, the loss of tolerance, and the triggering of autoimmunity to RBCs (83). A similar phenotype as in NZB mice, that is, increased production of anti-RBC autoantibodies and accelerated intravascular hemolysis and phagocytic removal of RBCs by Kuppfer cells, along with high levels of ROS in RBCs, was observed in sod1-knockout mice (84, 186). Autoantibodies were directed against HNE, acrolein, and carbonic anhydrase II. Both autoimmune responses and hemolytic anemia were rescued by transgenic expression of human SOD1 in erythroid cells (85). Moreover, immunoblotting and mass spectrometric analyses revealed that exposure of intact human RBCs to HNE resulted in selective HNE-β-spectrin adduct formation and cross-linking of HNE-modified spectrin. Spectrin is the main component of the submembranous cytoskeleton of RBCs and plays a critical role in the stability and strength of RBC plasma membrane. Apparently, local spectrin aggregation might lead to membrane surface area extrusion and loss, by freeing the lipid bilayer from the underlying cytoskeleton (9). As a whole, the observations described earlier are of relevance both for the physiological destruction of RBCs, in view of the reported accumulation of HNE in aging erythrocytes (6), and for their immune-mediated hemolysis, in conditions of enhanced LPO.
Protein-HNE Adducts in Autoimmune Liver Disease and Ferritin-Induced Liver Cytotoxicity
Primary biliary cirrhosis (PBC) is a progressive, nonsuppurative, autoimmune cholangiopathy entailing the selective, cell-mediated destruction of small- and medium-sized (<100 μm in diameter) intrahepatic bile ducts. The immunochemical detection of HNE-modified proteins in liver biopsies revealed HNE-protein adducts in the cytoplasm of biliary cells of small bile ducts in all of 20 patients with PBC. In 30% of patients, HNE-protein adducts were detected also in periportal hepatocytes, in association with higher serum bilirubin levels and histological stage (stage 3, septal fibrosis), in comparison with patients lacking intrahepatocytic HNE-protein adducts. Thus, hepatic LPO may be an early event in bile duct destruction and may contribute to hepatocyte injury and fibrosis during cholestasis in PBC (94).
Non-alcoholic fatty liver disease (NAFLD) covers a pathological spectrum of disease, from relatively benign lipid accumulation (simple steatosis, fatty liver), which is devoid of long-term adverse effects, to progressive nonalcoholic steatohepatitis (NASH), which is associated with necrosis, chronic inflammation, and fibrosis, leading to liver cirrhosis. Adaptive immunity seems to be involved in the progression of NAFLD from steatosis to NASH, as hepatic oxidative stress markers, such as HNE and 8-hydroxydeoxyguanosine, correlated with the severity of hepatic necrosis, inflammation, and fibrosis (31, 179); antibody responses to MDA-modified antigens were associated with increased severity of lobular inflammation or fibrosis (108, 137). In the methionine-choline-deficient (MCD) murine model of NASH, autoimmune responses toward aldehyde-modified self antigens contributed to hepatic inflammation, by promoting T
A major role of HNE and other reactive aldehydes was implicated also in cell death induced by secreted acidic ferritins (20, 21). These appeared to act as soluble mediators of oxidative stress (19), in spite of the reported ability of human H chain ferritin to serve as a cellular antioxidant and apoptosis inhibitor (16, 228). Pathophysiological interest for these observations comes from the reported increases of serum ferritin levels in various pathological conditions, including acute and chronic inflammation and autoimmunity (163, 226). The cytotoxicity of an acidic, H-chain-rich isoferritin (FER-CM) secreted by rat primary hepatocytes in vitro followed a dose-response relationship, marked by the transition from apoptosis to necrosis at concentrations above 100 ng/ml (22). Pro-apoptotic activity was accompanied by modification of cell proteins with HNE, as revealed by cytosolic accumulation of immunocytochemically detectable HNE-histidine protein (HNE-His-P) adducts, especially in the perinuclear area, and DNA damage, as revealed by the formation of micronuclei. FER-CM-induced apoptosis and HNE-His-P immunoreactivity were partially inhibited by the free radical scavenger 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (trolox) and, more completely, by the lysosomotropic iron chelator desferrioxamine (DFO), as well as by proliferative stimulation of rat hepatocytes with epidermal growth factor (EGF) and insulin, whose mitogenic efficacy was reduced, in turn, in the presence of acidic isoferritins (19). It was suggested that these might act as oxidative stress mediators by promoting ferrous iron loading in lysosomes, ROS production, and lysosomal membrane permeabilization. The latter, in turn, might foster cell damage via the release of ferrous ions, ROS and cathepsins, cytosolic amplification of LPO, aldehyde-mediated protein/DNA modification, and mitochondrial outer membrane permeabilization, leading to Fas- and p53-mediated apoptosis or necrosis, depending on the severity of oxidative stress (19). HNE itself was able to trigger p53 and Fas-dependent apoptosis (11). The conclusions drawn in the study cited (19) had some limitations, in that (i) HNE-His-P immunoreactivity varied markedly between different cells treated with FER-CM at the same dosage; (ii) the protection from FER-CM-induced apoptosis and necrosis provided by trolox was only partial, compared with that afforded by DFO and EGF/insulin, as though HNE hyperproduction were not entirely and directly responsible for the observed effects of FER-CM on cell viability. Ways of addressing these aspects might be the use of (i) a selective inhibitor, such as nordihydroguaiaretic acid (215), of reticulocyte 15-lipoxygenase, the enzyme responsible for the conversion of arachidonic acid to 15-hydroperoxy-5,8,11,13-eicosatetraenoic acid, from which HNE is produced by a series of non-enzymatic peroxidation reactions; (ii) cell transfection/transduction and overexpression of the fatty aldehyde dehydrogenase gene (ALDH3A2, FALDH), whose product detoxifies HNE by converting it to 4-hydroxynonenoic acid (39), as already done in 3T3-L1 adipocytes (47).
Conclusions
Results obtained in recent years have shown that the generation of HNE-protein adducts can play important pathogenic roles in several diseases characterized by increases in oxidative stress and, consequently, of LPO and production of reactive aldehydes. However, cancer is peculiar in this regard, as the increases in oxidative stress do not always correlate with increases of LPO, due to differences in membrane lipid composition of cancer cells. Moreover, in cancer cells, the generation of HNE-protein adducts, by leading to apoptosis or to losses of dysregulated functions, can represent a contrast to the progression of disease.
HNE-protein and HNE-DNA adducts can incite autoimmune responses by combined effects on both innate and adaptive immunity. On the one hand, they can act as damage-associated molecular patterns recognized by soluble and cell-associated PRRs, which may favor the uptake and presentation of self antigens by antigen-presenting cells in the context of enhanced levels of costimulation. Furthermore, HNE cross-linking with self antigens can lead to the formation of neoepitopes, which initiate autoimmunity by recruiting T and B cells outside the repertoires of autoreactive T and B cells. Moreover, it has been repeatedly observed, both in the experimental and in the clinical setting, that the breaking of tolerance to a modified self antigen also affected its native counterpart. This effect, which entails the intramolecular spreading of sensitization to other epitopes, reflects both the hapten-carrier relationship linking HNE with its macromolecular targets and the multivalent character of the latter as immunogens. Intermolecular epitope spreading between HNE-modified protein antigens and other proteins or DNA, either in native form or modified with the HNE analog ONE, has been also reported as a reflection of the molecular mimicry and cross-reaction between structurally related epitopes, as well as of the pleiotropic effects of HNE.
Interestingly, in certain chronic inflammatory and neurodegenerative diseases, the presence of HNE adducts can promote adaptive cell responses, by stimulating intracellular GSH synthesis (12), inhibiting the NF-κB activity (86), inducing HO-1 (13), or stimulating autophagy (71). These studies underline the fact that HNE can be considered not only a toxic product of LPO but also a regulatory molecule that is involved in several biochemical pathways.
We believe that, in the next few years, the refinement both of proteomical and of tissue and cell sampling techniques, allowing the individuation of novel cellular targets of HNE, will lead to a better understanding of the mechanisms of HNE action in human diseases.
Footnotes
Acknowledgments
The authors thank the University of Turin and the Compagnia di San Paolo.