
Abstract
Introduction
M
The endosymbiotic origin of mitochondria defines several distinctive hallmarks of these organelles. Mitochondria, whose proteome accounts for 900–1500 polypeptides (146, 165), retained two membranes, the outer mitochondrial membrane (OM) and the inner mitochondrial membrane (IM), respectively. These phospholipid bilayers segregate two mitochondrial compartments—the matrix and the intermembrane space (IMS) (Fig. 1). The large matrix compartment houses multiple metabolic enzymes and the mitochondrion's own small circular genome (mtDNA) and the machineries necessary for its replication and expression. A recent study showed that ∼500 proteins reside in the matrix of mammalian mitochondrion (156). The IMS compartment is smaller and contains fewer (∼60) proteins (82, 183). The large portion of mitochondrial proteins—including the electron transport chain and F1FO ATPase multiprotein complexes (collectively known as the oxidative phosphorylation [OXPHOS] system)—localizes to the IM. The IM proteome is particularly enriched in the specific invaginations of the membrane termed cristae. The OM subproteome is estimated to contain ∼100 polypeptides (198). Another hallmark of the mitochondrial proteome is its bigenomic nature. The vast majority of proteins comprising the mitochondrial proteome are encoded by nuclear genes, synthesized in the cytosol, and subsequently imported into the organelle, while only 13 of the ∼1500 mitochondrial proteins are derived from mtDNA (18, 147, 179), These unique properties of mitochondrial architecture impose several significant challenges to mitochondrial functions and necessitate constant monitoring by MQC (Fig. 2). One challenge stems from the inherent generation of reactive oxygen species (ROS) by the electron transport chain complexes of OXPHOS (2, 9, 61) (Fig. 2A). During this process, some electrons may leak from the electron transport chain and rapidly react with molecular oxygen. Various reports estimate that 0.3%–2% of the mitochondrial O2 consumption may be diverted toward ROS formation (2, 9, 61). Incomplete reduction of O2 by escaping electrons leads to the formation of highly reactive superoxide anion, which can further promote formation of other ROS and reactive nitrogen species (RNS) radicals (161). While free radical-scavenging mechanisms are in place (82), they are not always sufficient to eliminate harmful ROS/RNS that are highly damaging to nucleic acids, proteins, and lipids (61, 161). In addition, many ROS are now recognized as important signaling molecules essential for intracellular communication and stress response (161). Therefore, as exemplified by a number of failed trials aiming to correct mitochondrial damage by antioxidant treatment (30, 29), massive neutralization of ROS may be equally detrimental for cells.

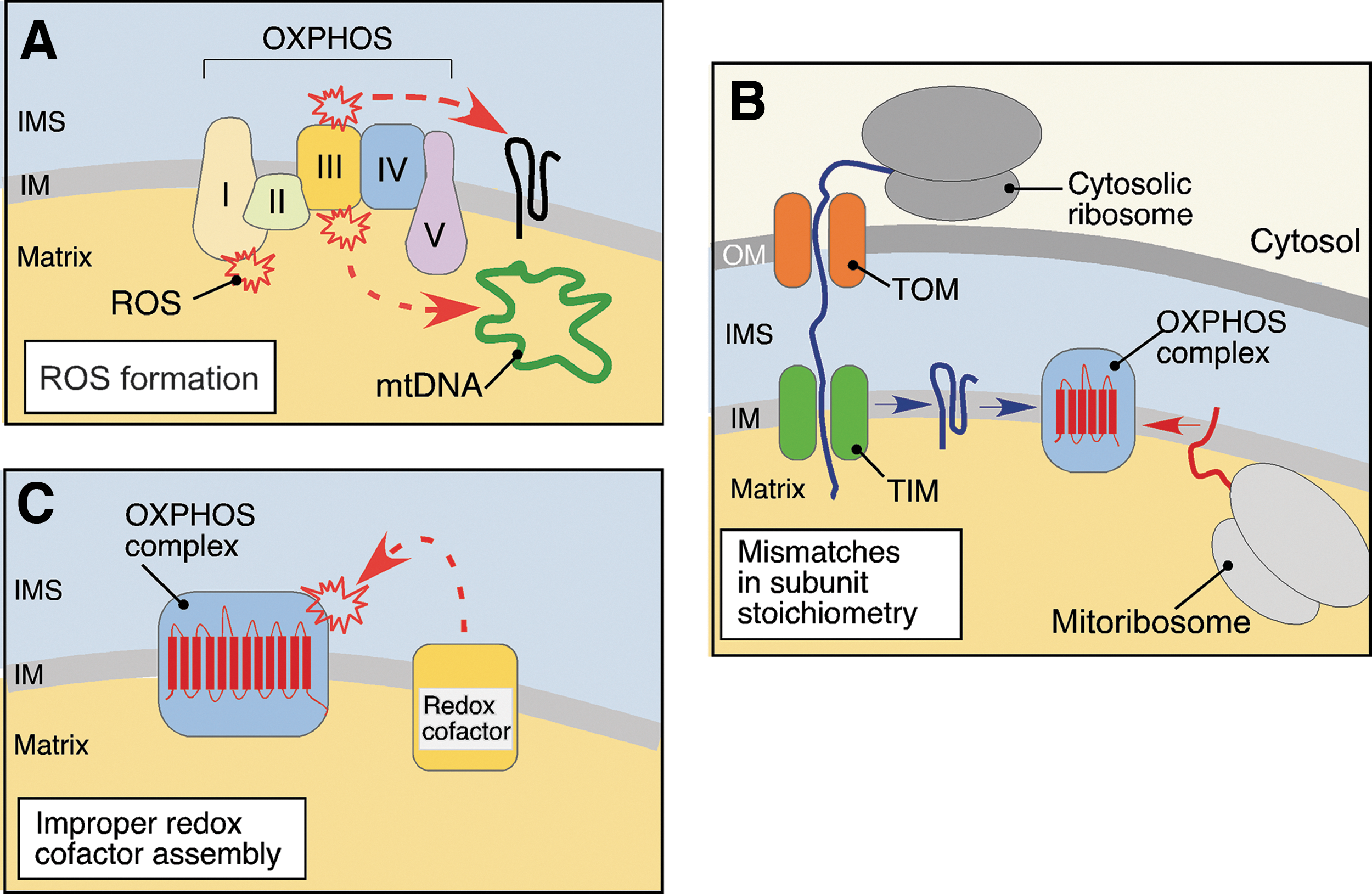
The other challenge arises from the dual genetic origin of several OXPHOS complexes (complexes I, III, IV, and V) (Fig. 2B). Biogenesis of these respiratory units requires tightly coordinated expression, sorting and folding of both mtDNA- and nuclear-coded polypeptides, and their subsequent assembly into stoichiometric complexes within the IM (18, 128). If such coordination fails, unfolded or orphaned subunits that are prone to misfolding and/or aggregation may accumulate. Finally, the electron transport chain complexes contain multiple redox cofactors, which when improperly assembled or stalled in the assembly process, can act as pro-oxidants and further contribute to the challenging biochemical environment in the organelle (31, 99, 100, 132) (Fig. 2C). If unopposed, these challenges can distort mitochondrial protein homeostasis and propel progressive mitochondrial failure. The acute failure at one of the aforementioned risk sites may trigger a so-called vicious cycle—a series of deleterious events leading to the gradual increase of mitochondrial damage. For instance, accumulation of unassembled or misfolded polypeptides can impede biogenesis and/or function of the OXPHOS, which in turn will produce excessive ROS. These can further disrupt the mitochondrial proteome through induction of mutations in mtDNA and additional perturbation of protein folding, leading to more ROS and ROS-induced damage and ultimately, to the demise of the organelles (18, 57).
In this review, we summarize the current knowledge about the MQC mechanisms by which cells cope with biochemical stresses arising from the unique functional and organizational properties of mitochondria, and how these mechanisms sustain normal mitochondrial function and integrity. We will primarily focus on the proteolytic facet of MQC and outline recent advances and current concepts in the field.
Overview of Mitochondrial Protein Quality Control
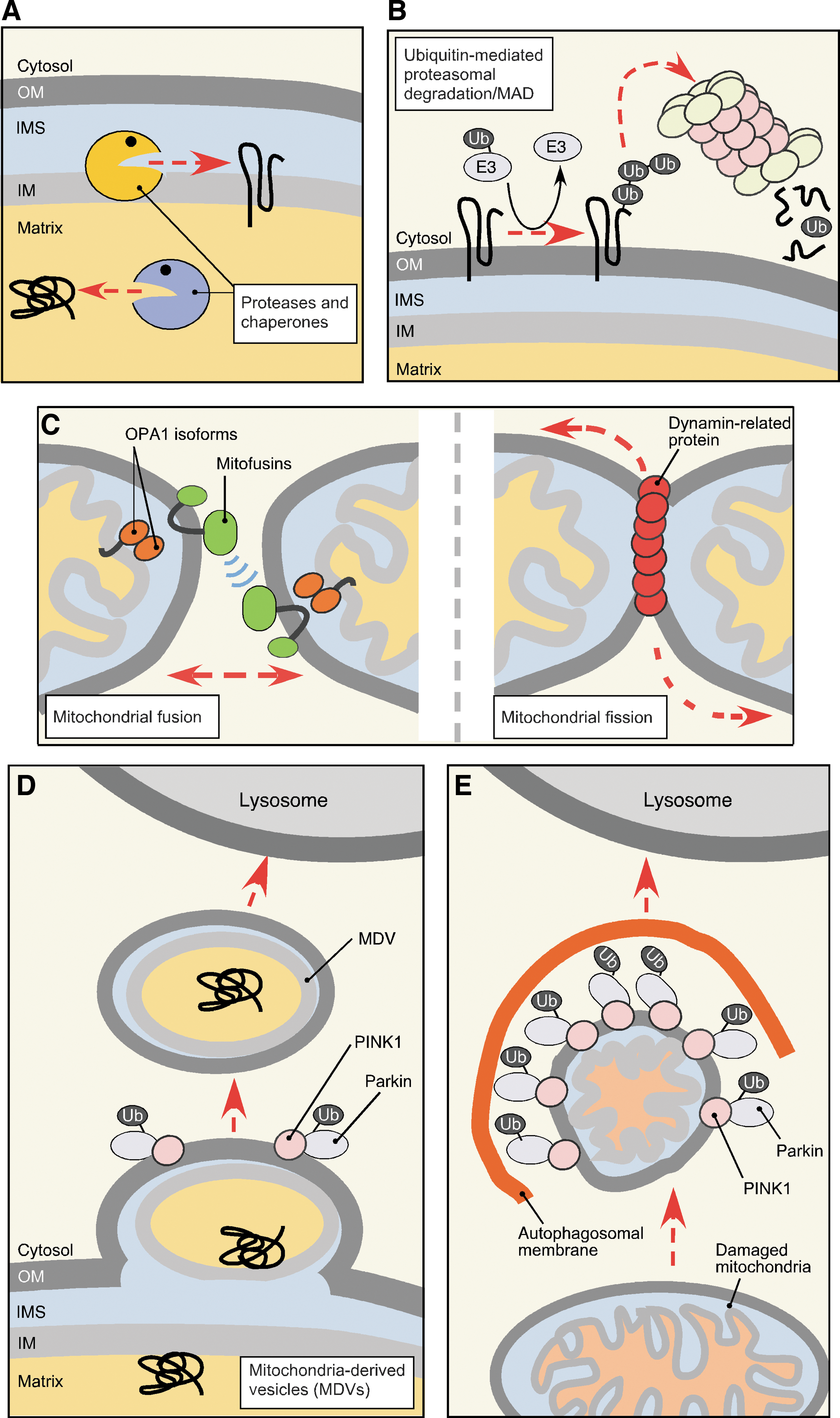
Because of inherent susceptibility of mitochondria to biochemical stresses, elaborate multilayer quality control mechanisms have evolved that survey, repair, or eliminate damaged mitochondria. Depending on the extent of damage, MQC mechanisms can engage at several levels (Fig. 3). The molecular level of MQC (referred hereafter as protein MQC [PMQC]) includes a network of evolutionary conserved mitochondrial proteases and chaperones distributed across mitochondrial compartments (Fig. 3A), as well as cytosolic proteolytic systems like the ubiquitin–proteasome system (UPS), which can associate with the OM (79, 96, 116, 175) (Fig. 3B). Although the number of studies addressing PMQC in mammalian cells is steadily growing, most of our current knowledge stems from studies in bacteria and the yeast model. The key molecules involved in PMQC are summarized in Table 1. One layer of PMQC is represented by ATP-dependent chaperones of mtHsp70 and Hsp60 heat-shock protein families, responsible for the sorting, folding, and disaggregation of proteins in the matrix compartment (136, 187). Similarly, the Hsp70-type and Hsp90-type chaperones operate in the cytosol and prevent aggregation and facilitate transport of unfolded newly synthesized or nascent polypeptides into mitochondria (60, 75, 197). The proteolytic facet of PMQC includes multiple conserved proteases (7, 18, 21, 157). The proteases are distributed across mitochondrial subcompartments and generally can be divided into two groups: (i) ATP-dependent, also known as AAA+ (ATPase associated with diverse cellular activities) proteases and (ii) ATP-independent proteolytic enzymes. The former group includes ClpXP and Lon/Pim1 proteases that reside in the matrix and degrade oxidatively damaged or aggregated polypeptides in the compartment (24, 25, 26, 34, 94, 77, 171, 181). Another two AAA+ proteases localize to the IM. The m-AAA (matrix AAA) protease has its active site exposed to the matrix side of the IM where it performs its quality control functions (11). The active site of i-AAA (intermembrane space AAA) peptidase faces the IMS (113, 190). These proteases appear to be important for removal of dysfunctional or orphaned proteins that are intrinsic to or associated with the IM (11, 105, 112, 113, 190). The AAA+ proteases typically exist as homo-oligomeric (Lon/Pim1, i-AAA) or hetero-oligomeric (m-AAA, ClpXP) complexes, and their proteolytic activity is coupled to ATP hydrolysis (13, 22, 159, 181). Such molecular architecture also permits chaperone-like functions of AAA+ proteases and appears to confer the ability to recognize misfolded or unassembled proteins and refold, and/or extract these polypeptides from phospholipid bilayers (159). The second group of proteases is more heterogeneous and includes the following: (i) processing peptidases (MPP, Oct1, Icp55, Cym1, and IMP) involved in sequential removal and/or degradation of mitochondrial targeting sequences (MTS) and thus proper biogenesis, sorting, and stabilization of matrix-and IM-targeted proteins (3, 73, 92, 131, 133, 176, 185); (ii) soluble peptidases (Prd1/Neurolysin, Atp23, and Ynm3/Omi) that seemingly contribute to MQC in the IMS (92, 143, 145, 180, 199); and (iii) IM-bound proteases (Pcp1/PARL, Oma1) implicated in the quality control of the IM proteome and regulation of mitochondrial dynamics (8, 19, 54, 58, 78, 83, 97, 127).
Protein names in brackets indicate aliases. “/” denotes alternative subcellular localization.
Cyto., cytosol; IM, inner mitochondrial membrane; IMS, intermembrane space; OM, outer mitochondrial membrane; Mult., multiple cellular locations.
Growing evidence indicates that the cytosolic UPS also represents an important facet of PMQC. First, UPS has been shown to participate in quality control and removal of several mitochondria-targeted proteins before or during their import into the organelle (7, 35, 116). Moreover, UPS can access the OM subproteome and mediate retrotranslocation and degradation of OM resident proteins—a process termed mitochondria-associated degradation (MAD) (79, 96, 116). MAD apparently relies on p97/Cdc48 AAA+ protein (79, 193), which is also involved in a well-described extraction of ubiquitylated proteins from the endoplasmic reticulum (ER) (37, 88).
Several MQC mechanisms are available on the organellar level (Fig. 3C–E). Fusion and fission events (Fig. 3C) mediate organellar dynamics and facilitate mitochondrial biogenesis, and even redistribution of mtDNA and proteome throughout the mitochondrial network (39, 41, 64, 196). Such redistribution due to fusion of several mitochondria permits the dilution of damaged molecules and/or replenishment of depleted components in malfunctioning organelles (41, 196). A phenomenon known as stress-induced mitochondrial hyperfusion (164, 177) represents an example of coupling between mitochondrial fusion and cellular stress response and highlights the significance of mitochondrial dynamics in stress management. Upon homeostatic insults like oxidative stress or starvation, mitochondria in the stressed cells form highly interconnected networks thereby increasing content mixing, ATP production, and protecting mitochondria from autophagic removal (68, 138, 155, 164, 177). When transient stress protection via mitochondrial hyperfusion is not sufficient, damaged organelles are removed from the network through fragmentation (fission) events (178). Mitochondrial fission serves to increase the number of mitochondria in the cell before mitochondrial biogenesis or cellular division, as well as to segregate dysfunctional or depolarized mitochondria away from the healthy network (144, 178, 196). Once malfunctioning (e.g., severely depolarized) mitochondria have been segregated, the components of their OM subproteome that are involved in establishing contact/tethering sites with other mitochondria are ubiquitylated and proteolyzed by UPS to prevent their rejoining with healthy mitochondria (56, 66, 172, 189). Then, damaged organelles are removed via another facet of organellar MQC (Fig. 3E)—a mitochondria-specific type of autophagy known as mitophagy (see Refs. 119, 195 for detailed review). An acute overwhelming stress such as treatment with oxidants causes massive fragmentation of the mitochondrial network followed by initiation of apoptosis (194). Conversely, genetic inhibition of mitochondrial fission increases apoptotic resistance and cell survival (109, 154).
Finally, a novel MQC mechanism has recently been described by the McBride laboratory (Fig. 3D). Mitochondria-derived vesicles (MDVs), which carry selected oxidized cargo and deliver this cargo to lysosomes, have been reported to facilitate MQC (137, 170). The MDV route appears to function at both normal and oxidative stress conditions and is independent of mitochondrial dynamics and mitophagy (166, 170). Although the identity of the vesicle cargo has not been fully characterized, this mechanism offers a potential strategy to remove segments of mitochondrial membranes containing damaged, hard to dissociate protein complexes and/or reactive prosthetic groups such as heme, which cannot be catabolized within the mitochondrion.
Mitochondrial Subproteomes and Their Regulation by PMQC
Matrix subproteome
Virtually all proteins of the dense matrix subproteome are synthesized in the cytosol and imported into the compartment as precursors in an unfolded state (136). Proper maturation and folding of these proteins in the matrix are facilitated by several PMQC factors (Fig. 4). First, mtHsp70 and its J-type cochaperones participate in the import of precursor proteins through the TIM23 IM translocase complex and later, in conjunction with the Hsp60-Hsp10 chaperone system, they promote folding of the imported polypeptides (136, 187). Second, proteolytic removal of the N-terminal MTS is mediated through the action of the two-subunit MPP processing metallopeptidase complex (73, 131, 176). Some proteins undergo an additional processing by the mitochondrial intermediate peptidase (MIP/Oct1), which removes additional residues following the MTS (131, 176). Recent studies identified an additional intermediate cleaving aminopeptidase Icp55 that stabilizes multiple matrix proteins via the removal of a single, potentially destabilizing N-terminal amino acid residue following the MTS (131, 133, 185). Interestingly, Oct1 processing also appears to contribute to such stabilization (184). This situation resembles the N-end rule protein stabilization pathway described for the cytoplasm (173); however, the downstream protease(s) degrading destabilized polypeptides remain(s) to be identified.
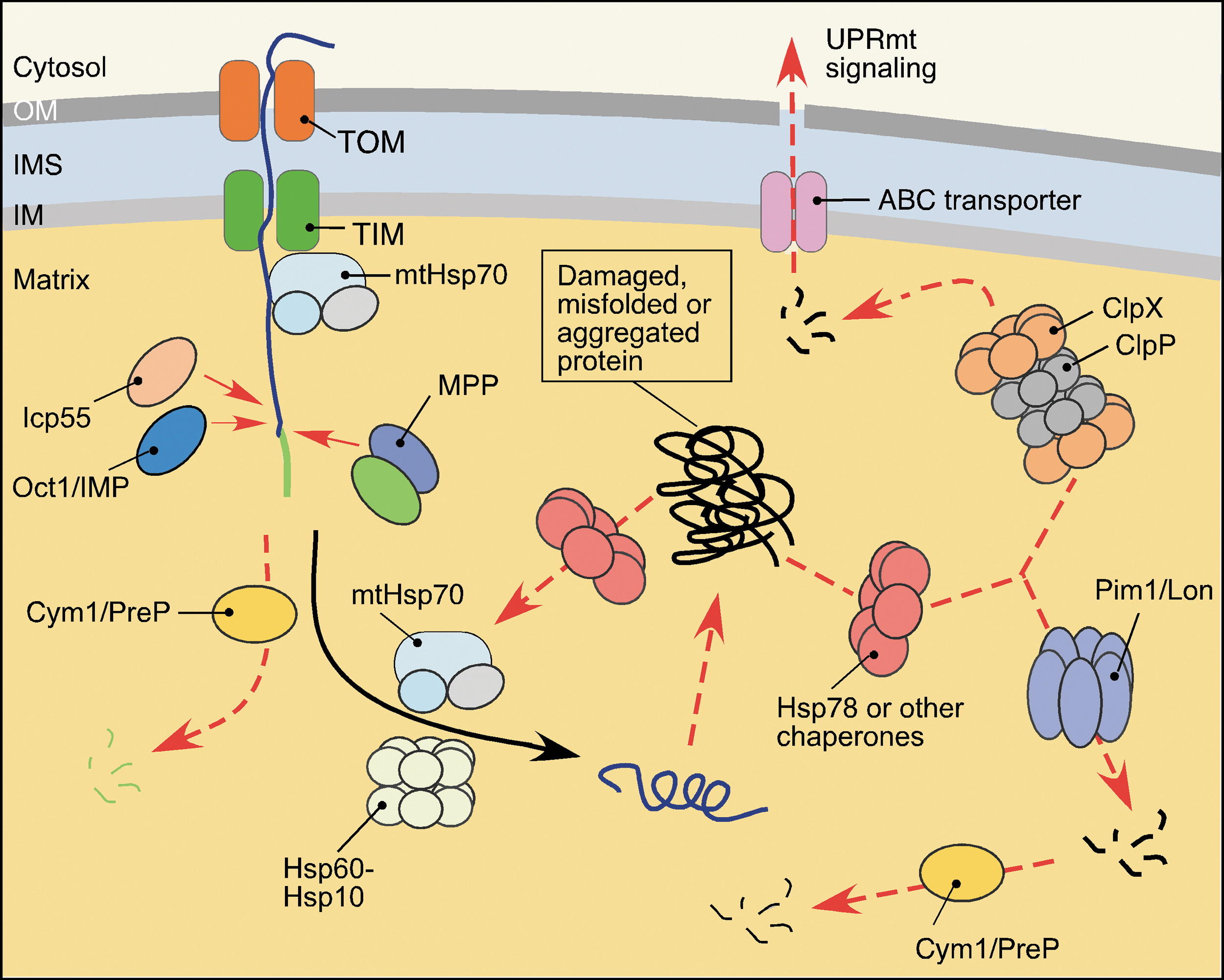
The mitochondrial presequence peptidase, Cym1/PreP, also appears to contribute to the matrix MQC. Originally misidentified as an IMS-localized enzyme, this conserved metallopeptidase has been implicated in the clearance of free targeting peptides generated by MPP and MIP, as well as small (up to 65 amino acid residues), unstructured oligopeptides (92, 176), which upon accumulation may impair mitochondrial integrity (85, 139). Also, studies on mammalian PreP showed that the peptidase is required to prevent mitochondrial accumulation of the amyloid-β (Aβ) peptide, which upon accumulation can cause mitochondrial dysfunction (59).
Finally, two conserved AAA+ serine proteases, Lon/Pim1 and ClpXP, are found in the mitochondrial matrix (94, 181) (Fig. 4). The Lon/Pim1 protease exists as a large homo-oligomeric complex with each subunit containing both ATPase and serine protease motifs (167, 181). It preferentially targets heat-damaged or oxidatively damaged proteins (24, 25, 26, 34). While protein misfolding and/or loss of prosthetic groups appear to contribute to substrate recognition by mitochondrial Lon (72, 121, 186), the exact recognition determinants remain to be identified. Studies in yeast indicate that Lon/Pim1 cooperates with ClpB-type AAA+ chaperone Hsp78 (24, 186) and mtHsp70 (160) to accelerate disaggregation/degradation of aggregated protein. The significance of this cooperation in higher eukaryotes remains unclear, as metazoans lack an apparent Hsp78 ortholog. In addition, Lon plays an important role in regulation of stability and expression of mtDNA via proteolytic control of the abundance of mitochondrial transcription factor TFAM (117, 124).
Unlike Lon, the ClpXP protease is a hetero-oligomeric complex consisting of two stacked ClpP serine protease heptamers topped with two hexameric rings formed by AAA+ ClpX subunits (22, 94). Reportedly, the ClpX component participates in recognition of misfolded polypeptides and channels them into a proteolytic chamber formed by the ClpP subunits (12, 22). Although multiple studies implicate bacterial ClpXP as a protein quality control protease (22), its exact role within the mitochondrion remains elusive. Of particular interest is the recently postulated function of ClpXP in the mitochondrial unfolded protein response (UPRmt) in nematodes (76, 77). Peptides generated via ClpXP-mediated proteolysis of unfolded proteins in the matrix are extruded from mitochondria into the cytosol, whereby they trigger a specific transcriptional response that promotes expression and synthesis of nuclear-borne mitochondrial chaperones and proteases to restore mitochondrial proteostasis (76). This mechanism serves to sense and correct imbalances between the proteins of nuclear and mitochondrial origin, particularly subunits of the OXPHOS complexes. Recently, a Lon-regulated facet of UPRmt was reported in a roundworm model. The bZip transcriptional factor ATFS-1 required for UPRmt signaling is destined to the mitochondrial matrix where Lon degrades it; however, under stress conditions, ATFS-1 is stabilized and trafficked to the nucleus where it initiates the transcriptional response (76, 135). Although UPRmt is conserved among worms, mice, and humans (191), it remains to be determined if stress-sensing and signaling mechanisms in mammals are identical to the ones described for nematodes.
Inner membrane subproteome
The mitochondrial IM is among the most proteinaceous biological membranes and houses a significant portion of mitochondrial proteome, including the OXPHOS complexes. Assembly and function of the reactive respiratory complexes create major challenges for mitochondrial protein homeostasis (Fig. 2). Several mechanisms are in place to assure normal biogenesis and maintenance of the proteins residing in the IM. First, a large number of dedicated chaperones and chaperone-like assembly factors assist and regulate biogenesis and maintenance of the respiratory complexes (63, 125, 128). The second set of mechanisms controlling IM proteostasis involves proteolytic enzymes (Fig. 5). The two-subunit, IM-bound IMP proteolytic complex—similar to Oct1 and Icp55 peptidases—was shown to stabilize its substrate proteins (131). For instance, processing of the Mgr2 subunit of TIM23 translocase by IMP stabilizes Mgr2 and promotes TIM23 assembly (86).

The membrane-bound m-AAA and i-AAA metalloprotease complexes—surveying the matrix and the IMS sides of IM, respectively—are two major factors that provide quality control of the IM subproteome (7, 18, 21, 112, 113). The importance of coordinated actions between the IM AAA proteases is highlighted by the observation that simultaneous loss of these molecules in yeast is lethal (111, 112). The m-AAA protease is a large hetero-hexameric complex typically formed by Yta10/AFG3L2 and Yta12/SPG7/Paraplegin subunits in yeast and humans (11, 103). An additional rodent-specific subunit AFG3L1 that can substitute for AFG3L2 has been described (107). Also, studies on mammalian m-AAAs showed that other forms of the enzyme consisting of AFG3L2 subunits only (in humans) or AFGL32 and AFG3L1 (in rodents) do exist (103). The cryo-EM reconstruction of the yeast m-AAA revealed that the protease complex is ring shaped and contains a central pore, which may be restricted to unfolded polypeptide segments (108). This finding provides insights into the substrate recognition by mitochondrial AAA metallopeptidases, however, the detailed mechanism is yet to be determined. The molecular architecture of the i-AAA protease is similar to one of the m-AAA complex, except that the former proteolytic machine is always a homo-oligomer formed by six copies of the Yme1/YME1L peptidase (69, 70). Known substrates of the AAA metalloproteases include surplus, misassembled, and/or damaged subunits of the OXPHOS complexes that are intrinsic or peripheral to the IM (10, 45, 84, 105, 112, 113, 134, 148, 168, 190). In addition, via its involvement in maturation of the nuclear-borne constituent of the large mitoribosomal subunit MrpL32, the m-AAA is critical for synthesis of mtDNA-coded polypeptides (33, 140). The i-AAA complex is an important regulator of the IM's functional integrity and dynamics via several proteolytic events. One such event is constituent regulatory proteolysis of the PRELI protein family members Ups1 and Ups2 involved in transport, synthesis, and accumulation of mitochondrial phospholipids (46, 152). Second, the i-AAA is involved in biogenesis of the short isoform of the optic atrophy 1 (OPA1) dynamin-related GTPase, which is central to mitochondrial dynamics and mtDNA maintenance (8, 71). The versatility of the IM AAA proteases is impressive and remains to be understood. A possible explanation is provided by cooperation of these proteolytic machines with other proteases and adaptor-like proteins. For instance, the conserved IM-associated peptidase Atp23 involved in maturation of the respiratory complex V (143, 199) reportedly cooperates with the Yme1 proteolytic complex to degrade Ups1 (152). Likewise, two adaptor-like proteins Mgr1 and Mgr3 have been shown to facilitate degradation of model substrates by the i-AAA in yeast (51, 52). The evolutionary conservation of Mgr3 suggests that the YME1L complex may also operate in a similar manner. Similarly, overexpression of the matrix AAA+ protein Afg1/LACE1 (1, 110) has been shown to facilitate degradation of several m-AAA protease substrates in a series of respiratory-deficient yeast mutants (99). However, it remains to be determined if Afg1 is a bona fide adaptor/cooperating partner of the m-AAA complex. In addition, the m-AAA protease exerts overlapping activity with the ATP-independent membrane-bound metallopeptidase Oma1 complex (97, 100). Studies by our group established that Oma1 per se is also an important stress-activated protease required for cell survival (32). In mammalian cells, Oma1 mediates rapid proteolytic processing of the long membrane-anchored form of OPA1 (L-OPA1), thereby promoting IM fragmentation and subsequent cellular MQC actions in response to homeostatic insults (8, 19, 200). Conversely, the mitochondrial network remains intact or even hyperfused in stressed Oma1-deficient mouse embryonic fibroblasts (19, 152). While the exact mechanism of stress sensing by Oma1 only begins to emerge (19, 32, 120, 200), it is clear that Oma1 activation is central to the regulation of IM proteostasis and dynamics upon various stresses and/or pathological states.
Pcp1/PARL is yet another proteolytic module intrinsic to the IM. It belongs to the rhomboid family of serine proteases and mediates intramembrane cleavage of several IM polypeptides in yeast, including cytochrome c peroxidase Ccp1 (processed in conjunction with m-AAA protease) (174) and yeast OPA1 ortholog Mgm1 (81). Interestingly, the latter polypeptide does not appear to be an Oma1 substrate either under normal or under stress conditions (53, 114), thereby suggesting partial evolutionary repurposing of Oma1 function. Reciprocally, PARL does not seem to cleave OPA1 in vivo (53). Instead, recent studies have implicated mammalian PARL in constituent proteolytic removal of the phosphate and tensin homolog-induced putative kinase 1 (PINK1) —a crucial regulator of mitophagic (whole mitochondria) and nonmitophagic (respiratory chain complexes) mitochondrial turnover (48, 89, 182, 189). PINK1 is translated in the cytosol and imported into the mitochondria. Under normal conditions, the protein is sorted to the IM where it is degraded in a PARL-dependent manner (48). This pathway is blocked in severely depolarized or ATP-depleted mitochondria, whereby PINK1 is stabilized and accumulates on the OM subsequently recruiting and activating E3 ubiquitin ligase Parkin/PARK2, which in turn triggers segregation and mitophagic removal of malfunctioning mitochondria (87, 93, 106, 119, 195). Interestingly, a recent study demonstrated the important role of the PINK1-Parkin pathway in the formation of MDVs under mitochondrial stress conditions (126).
IMS subproteome
Relatively little is known about the IMS MQC (Fig. 5). Recent studies identified the i-AAA protease as an important regulator of the IMS subproteome through its involvement in the folding and degradation of unassembled and/or misfolded small TIM proteins (20, 162). Of note, small TIM proteins themselves have been postulated to chaperone several folding reactions in the IMS (82). Similarly to Cym1/PreP, the IMS-localized oligopeptidase Prd1/Neurolysin appears to degrade cleaved presequences and small oligopeptides (92, 131, 176), thereby preventing their accumulation in the IMS. The high-temperature requirement A2 (HtrA2/Omi/Ynm3) serine protease is a highly conserved enzyme, whose functions and mechanism of action are understood primarily via studies on its bacterial orthologs—stress-inducible quality control peptidases DegP and DegS (44). HtrA2/Omi is a homotrimer and appears to be the only mitochondrial protease containing a PDZ-domain required for recognition of exposed hydrophobic stretches of misfolded proteins. The postulated modulation of Htr2A via PARL-assisted processing (40) and PINK1-mediated phosphorylation suggests its involvement in regulation of mitophagy (151). However, while studies in yeast and mammalian models indicated the role of Htr2A in thermotolerance (145), prevention of accumulation of aberrant respiratory chain subunits (123, 129), and even identified several substrates of the protease (91), its precise role in the IMS MQC awaits clarification.
Outer membrane subproteome
Recent findings provide several important insights into the PMQC mechanisms controlling OM proteostasis (Fig. 6). In addition to the aforementioned cytosolic Hsp70 and Hsp90 chaperones assuring proper delivery and likely the removal of nascent and/or newly synthesized unfolded polypeptides to be inserted into the OM (60, 122, 197), additional MQC modules have been characterized recently. The role of UPS in OM proteostasis becomes increasingly evident. Although it has been initially thought that the UPS may only intercept misfolded or damaged proteins en route to the mitochondrion (7, 35), numerous studies have suggested that mitochondria-localized polypeptides can be ubiquitylated and subsequently removed by the UPS in the process known as MAD (96, 116, 175). Consistently, several E3 ubiquitin ligases such as Mdm30, MITOL/MARCH-V, and MULAN (56) were found to associate with the cytosolic side of the OM. Likewise, the ubiquitin ligase Parkin is recruited to depolarized mitochondria (56, 66, 119, 172, 189, 195). Additional mitochondria-associated modifiers like SUMO ligases have also been described (56). Subsequently, multiple OM proteins have been identified as targets of these ligases and/or UPS (56, 66, 158). The discovery of VCP/p97/Cdc48-associated mitochondria stress responsive 1 (Vms1) protein in yeast that translocates from the cytosol to the mitochondrial OM upon stress and recruits the Cdc48-Npl4 complex, provided further mechanistic understanding of MAD (79, 80). The AAA+ protein VCP/p97/Cdc48 complex is involved in multiple cellular processes but plays a key role in the extraction of ubiquitylated ER- and OM-anchored proteins for proteasomal degradation (37, 88, 193). Reciprocally, loss of Vms1 results in accumulation of ubiquitylated OM proteins and mitochondrial damage (79). Vms1 apparently is not the only VCP/p97/Cdc48-Npl4 recruiting factor, as a fraction of the complex can still associate with dysfunctional mitochondria in the absence of Vms1 (55, 79). Therefore, additional UPS recruitment factors are likely to exist and remain to be identified. Another intriguing line of findings suggests the involvement of MAD in retrotranslocation and removal of several IM- and matrix-localized proteins (14, 15, 122). However, more work is needed to corroborate this postulate.

Recent studies identified yet another OM-associated AAA+ protein Msp1/ATAD1 required to prevent accumulation of mislocalized ER-destined tail-anchored proteins in the OM, thereby maintaining proper mitochondrial function and morphology (43, 142). It has been proposed that Msp1 represents a novel PMQC component involved in the extraction of mistargeted tail-anchored polypeptides. It remains to be determined if Msp1 constitutes an additional branch of MAD or is an independent facet of the OM MQC.
PMQC in Disease and Aging
Over time, a combination of increasing oxidative damage, failing protein homeostasis, and mitochondrial and organellar QC capacity can contribute to cellular aging and instability of mtDNA and mitochondrial proteome (27, 41). The resulting decline in mitochondrial health can primarily affect organs and tissues with high energetic demands and contribute to the onset of cardiovascular, neurodegenerative, and complex metabolic diseases like cancer.
Reduced activity/abundance of Lon/Pim1 and ClpP proteases has been demonstrated in cells from a patient with late-onset autosomal dominant hereditary spastic paraplegia (type SPG13) and aged rat hepatocytes (17, 74). In line with these findings, overexpression of Lon in the Podospora anserina fungal model of aging significantly extended the lifespan of the organism (118). Interestingly, the same effect is achieved through the depletion of P. anserina ClpP (62). The prospective role of Lon in cancer stems from its regulation by hypoxia-inducible factor HIF1α and Lon's role in hypoxia-induced remodeling of the respiratory chain (65, 153). Indeed, a recent study identified Lon as a prospective anticancer target (28).
The ability of the oligopeptidase PreP to prevent accumulation of mitochondria-targeted Aβ peptides (59) indicates its protective role against Alzheimer's disease (AD). Consistently, the activity but not abundance of PreP is attenuated in AD patients' brain mitochondria and in AD transgenic mouse models (4). The exact role of PreP in the onset of AD is yet to be determined.
Mutations in genes encoding the subunits of m-AAA protease are known with both familial and sporadic forms of autosomal recessive hereditary spastic paraplegia (Paraplegin) (38, 140), spinocerebellar ataxia type 28 (AFG3L2) (49), and spastic ataxia-neuropathy syndrome (150) in humans. Studies in mice also indicate that the AFG3L2 subunit of m-AAA is important for the survival of Purkinje cells (5) and anterograde mitochondrial transport in murine cortical neurons (102), thereby highlighting the role of failing m-AAA function in late-onset neurodegeneration. In addition, a recent report identified a variant of Paraplegin, which is linked to enhanced ROS generation and several clinical phenotypes, including type 2 diabetes mellitus and coronary artery disease (6).
Depletion of OMA1 in the mouse model reduces energy expenditure and specifically leads to obesity, hepatic steatosis, and altered thermogenesis/cold stress resistance (154). These phenotypes are likely due to defective L-OPA1 processing and inability to initiate fragmentation of the mitochondrial networks in response to metabolic and/or oxidative insults (154, 192). Also, a recent high-throughput sequencing study of patients with familial and sporadic forms of amyotrophic lateral sclerosis (ALS) identified several mutations in the conserved residues of OMA1, thus implicating OMA1 as a prospective ALS-causing gene (47).
The IMS protease HtrA2/Omi plays an important role in protecting neurons from degeneration (90, 91, 123) and has been associated with Parkinson's disease (PD) (98, 169). Likewise, a PD-associated mutation in PARL has been recently identified (163). Although in vitro studies highlight the importance of MAD for prevention of neurodegenerative pathologies such as PD, its physiological role in human health awaits further investigation. Two lines of evidence implicate MAD in mitochondrial disease. First, a murine knockin model of p97 mutations (associated with inclusion body myopathy and Paget's bone disease) presents with mitochondriopathy-like phenotypes (16). Second, the PINK1-Parkin system, which likely represents a facet of the MAD that links molecular and organellar MQC—is clearly associated with neurodegenerative processes. Loss-of-function mutations in Parkin have been described in juvenile PD patients and account for ∼50% of the familial cases of PD (101).
Concluding Remarks and Perspectives
The role of functionally intertwined multilayer MQC mechanisms in assuring mitochondrial health becomes increasingly evident. It is important to note that humans sustain mitochondrial damage not only from age-related decline in mitochondrial function but also from causes that can affect younger populations. For instance, aggressive anticancer or antiviral therapies may enhance the formation of malfunctioning mitochondria in nontarget cells in patients undergoing such treatments (42). Modulation of the activity of certain MQC components can therefore be a promising approach to enhance cellular health and lifespan (62, 118). Reciprocally, proteases like Lon can be prospective targets in anticancer therapies to promote the death of drug-treated malignant cells (28, 67).
A number of important unresolved questions regarding MQC persist. For instance, the mechanisms by which MQC components promptly recognize mitochondrial stress signals remain obscure. Likewise, it remains to be determined how MQC modules in different mitochondrial subcompartments coordinate their actions in response to homeostatic insults. The regulation of nuclear output and physiological responses via function/dysfunction of MQC modules is another exciting question. Answering these questions is likely to yield new insights into MQC in health and disease and may lead to novel or supplementary therapeutic and preventive approaches against mitochondria-related maladies.
Footnotes
Acknowledgments
We apologize to those authors whose work we were unable to cite due to space limitations. This work was supported by NIH grants P30GM103335 (to the Nebraska Redox Biology Center), 1R01 GM108975 (to O.K.), 5P20RR024485-02, and 8 P20 GM103542-02 Redox COBRE subproject award (to S.C.). We thank Drs. Dennis Winge and Rodrigo Franco for critical reading of the article.