
Abstract
Introduction and Mitochondria-Associated Membranes Composition
T
Among these preparations, MAMs (Figs. 1, 2) are the best characterized. The first reports of the direct association of mitochondria and the ER date from the late 1950s (31), and this type of preparation has been subsequently characterized by numerous groups [for recent reviews, see Refs. (94, 114, 125)]. An isolated MAMs fraction is composed of membrane fragments from both the ER and the outer mitochondrial membrane (OMM) that had been in close contact at the time of cellular subfractionation. More recently, the ER portion of the MAMs fraction has been regarded as a detergent-resistant lipid raft (5, 52).
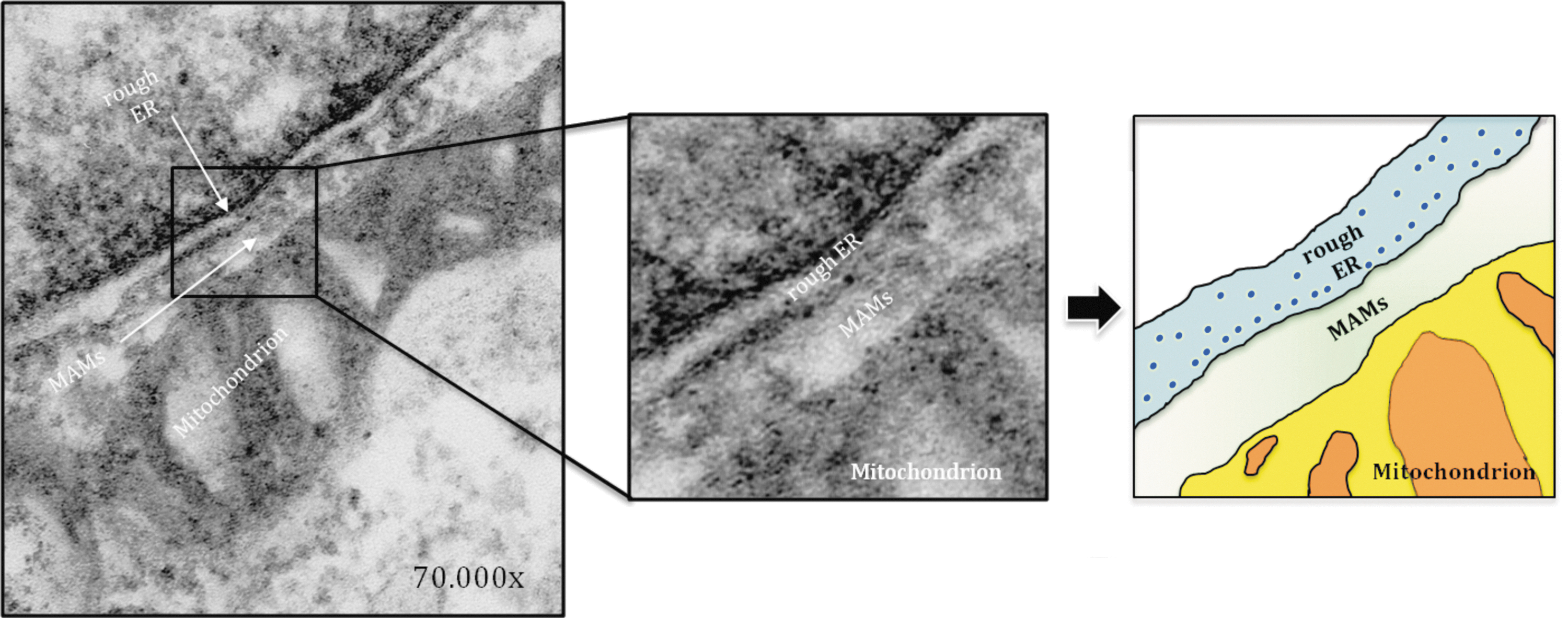

The contact sites between mitochondria and the ER are dynamic structures that are sensitive to the physiological conditions of the cell. This dynamic nature results in a transient and highly variable MAMs composition. The variety of roles played by the MAMs fraction that have been described is related to their unique lipid and protein composition. The studies performed in the past decade that have identified the molecular components of the MAMs fraction have demonstrated that this fraction may contain numerous proteins [more than 75 according to Raturi and Simmen (149), more than 1000 according to Poston et al. (147)]. From this protein composition, it was deduced that MAMs are crucial for numerous cellular processes, including protein sorting [as indicated by the presence of phosphofurin acidic cluster sorting protein 2 (PACS-2)], inflammation [as indicated by the presence of the following inflammasome components: NACHT, LRR and PYD domains-containing protein 3 (NALP3), adaptor ASC and thioredoxin-interacting protein (TXNIP)], ER stress [e.g.,75-kDa glucose-regulated protein (GRP75) and ER resident protein 44 (ERp44)], Ca2+ handling [e.g., the inositol 1,4,5-trisphosphate receptor (IP3R), ryanodine receptor, sigma-1 receptor (Sig1R), p53, and promyelocytic leukemia protein (PML)], mitochondrial contact sites [e.g., the voltage-dependent anion channel (VDAC) and adenine nucleotide translocase (ANT)], lipid synthesis and trafficking [e.g., PSS-1 and PSS-2, serine active site containing 1 (SERAC1), FACL1 and FACL4, acyl-CoA desaturase, fatty acid transport protein 4 (FATP4), phosphatidylethanolamine N-methyltransferase 2 (PEMT2), and several other proteins; for additional details, (158)], protein folding [e.g., calnexin (CNX)], apoptosis [e.g., Bcl-2 and HCLS1-binding protein 3], and modulation of mitochondrial morphology [mitochondria-shaping proteins and chaperone proteins (mitofusins 1 and 2;MFN1 and MFN2, respectively)]. Moreover, MAMs are the predominant subcellular location for γ-secretase activity and for presenilin-1 (PS1) and presenilin-2 (PS2), two proteins that, when mutated, cause familial Alzheimer's disease (FAD) (4, 5) as discussed later in detail. The ER-mitochondrial cross-talk can also be affected by several viral proteins such as human cytomegalovirus vMIA and the p7 and NS5B proteins of hepatitis C virus, which are also targeted to MAMs. Moreover, it has been found that the tumor suppressors PML and p53 can modulate the ER–mitochondria Ca2+ cross-talk by its presence in MAMs (63, 144). In addition, p66Shc, which is a cytosolic adaptor protein that is involved in the cellular response to oxidative stress when phosphorylated at Ser36, has also been identified at the sites of mitochondria-ER association (61, 101). Taking into account that the MAMs fraction contains numerous crucial proteins, as previously mentioned, the involvement of MAMs-mediated disturbances in the pathogenesis of a variety of diseases is not surprising (149). However, the localization of some proteins in the MAMs fraction and the extent of their enrichment remain under debate, and at times, the relationship of these proteins to the MAMs fraction is unclear; see the review by Raturi and Simmen (149).
Relationship Between MAMs and Reactive Oxygen Species
An interesting example of a MAMs-resident reactive oxygen species (ROS)-generating protein is the p66Shc protein. Under physiological conditions, this growth factor adaptor protein (which, in addition to p52Shc and p46Shc, belongs to the ShcA family) is involved in signal transduction via the RAS protein. However, under oxidative stress (exogenous and intracellular), p66Shc can participate in the signaling pathway leading to apoptosis. This dual nature of p66Shc arises from the different (tyrosine and serine) phosphorylation sites within this protein. All ShcA proteins possess a similar domain structure; however, p66Shc (which is a product of an alternatively spliced transcript from the SHC1 gene) contains an additional N-terminal proline-rich collagen-homology domain (CH2) containing a serine phosphorylation site (Ser36) that is important for the aforementioned “proapoptotic” properties (40, 170) as well as a functional region (CCB) that is responsible for its interaction with cytochrome c (64). As observed for p52Shc and p46Shc, when phosphorylated, p66Shc has been found to bind the Grb2/SOS complex and, in this way, competes with p52Shc for binding to Grb2. This finding suggests that p66Shc can function as a dominant negative regulator of the RAS-mediated signaling pathway (122, 131). Under oxidative stress caused by UV radiation or H2O2 treatment, p66Shc can be phosphorylated at Ser36 by one of the serine-threonine kinases [protein kinase Cβ (PKCβ) (59), c-Jun N-terminal kinase (JNK), or extracellular signal-regulated kinase (ERK)] (79, 100, 146). Moreover, phosphorylation of p66Shc at Ser36 can be mediated by apoptosis signal-regulating kinase 1 (ASK1)-JNK phosphorylation (110). This phosphorylation initiates the following cascade of events: (i) isomerization of Ser36-phosphorylated p66Shc by the prolyl isomerase Pin1, (ii) dephosphorylation of Ser36-P-p66Shc by phosphatase A2 (PP2A), (10) and (iii) translocation of p66Shc to the mitochondria and/or MAMs fraction, where it participates in ROS production (64, 146) (Fig. 3). Additional details on the “nature” of p66Shc can be found in the comprehensive review by Migliaccio et al. (121). It should also be noted that the ROS-sensitive fluorescent probes used in studies describing the ability of MAMs-resident proteins to produce different ROS unfortunately cannot precisely detect ROS. For example, the CM-H2DCF probe used for the measurement of hydrogen peroxide can also detect other ROS, due to the fact that the oxidation of H2DCF to DCF is a two-step process: First, the DCF radical is formed, and it is then (second step) oxidized to DCF in a reaction with molecular oxygen (163, 176). Unfortunately, the first step of H2DCF oxidation can be mediated by various radical species, such as hydroxyl radicals, carbonate radicals, and nitrogen dioxide, as well as by thiyl radicals resulting from thiol oxidation (42, 71, 88, 181, 182). Moreover, alteration of the fluorescent probe signal can be caused by superoxide radicals (formed in the second step of H2DCF oxidation), which can be dismutated to hydrogen peroxide and cause self-amplification of the signal (67, 128). Similarly, dihydroethidium (DHE), and MitoSOX, which are used for the measurement of superoxide, are also not completely specific. For example, DHE and MitoSOX can undergo unspecific oxidation by ONOO− or •OH into ethidium or mito-ethidium, respectively (88). For this reason, even if the exact form of ROS (e.g., superoxide or H2O2) is stated in the text of the original paper commented on in our review, other ROS should also be taken into consideration due to the constraints imposed by probe chemistry.

The first reports addressing the cytosolic localization of p66Shc indicated that it could also be localized in the mitochondrial matrix. Moreover, Orsini et al. found that p66Shc can interact with mitochondrial Hsp70 and regulate Ψm (mitochondrial membrane potential) (133). Subsequent studies have revealed that mitochondrial p66Shc is present in the mitochondrial intermembrane space (IMS), where it interacts with cytochrome c and, as a redox enzyme, produces H2O2 (64). However, the determination that nearly 35% of the mitochondrial p66Shc localizes within the IMS (64) appears to be an overestimation (101, 179). Regarding this model, Gertz et al. have proposed that the N-terminus of p66Shc forms a redox module and serves as a thiol-based redox sensor. Under oxidizing conditions, p66Shc can be “activated” due to the formation of two disulfide bonds, resulting in the reversible tetramerization of p66Shc. In contrast, under normal conditions, glutathione and thioredoxins protect against p66Shc tetramerization by reducing potentially formed disulfide bonds and, in this way, inactivating the proapoptotic properties of p66Shc (56).
Direct involvement of p66Shc in mitochondrial ROS production has been repeatedly described by several groups (37, 64, 102, 103, 146). However, it remains unclear whether p66Shc can translocate across the OMM and resides in the IMS, where it can interact with cytochrome c (64), or whether it binds to the OMM from the cytosolic side (101). In the model in which p66Shc is localized in the IMS, p66Shc has been proposed to interact with cytochrome c (due to the CCB domain present in p66Shc) and transfers electrons from cytochrome c to molecular oxygen, which results in H2O2 formation. p66Shc present in the MAMs fraction can interact with an as yet unidentified OMM protein and, thus, can participate in ROS production (Fig. 3). Our studies have shown that p66Shc can be found not only in the cytosol and MAMs (179) but also in the PAM fraction (101). Interestingly, the level of p66Shc in the MAMs fraction increases in an age-dependent manner, corresponding well to mitochondrial H2O2 production, which has been found to increase with age (101). Lebiedzinska et al. performed studies on fibroblasts from patients with diagnosed mitochondrial disorders and demonstrated that p66Shc can also be phosphorylated at Ser36 in the event of intracellular oxidative stress caused by mitochondrial dysfunction (102, 103). In the case of these studies, it is very difficult to distinguish whether the “activated” p66Shc pathway in fibroblasts from patients participates in superoxide (O2 −•) or H2O2 production. Admittedly, the administration of hispidin (an inhibitor of PKCβ phosphorylation of p66Shc) decreases cytosolic and mitochondrial O2 −• levels in these cells, indicating that p66Shc may be involved in the production of this type of ROS. However, hispidin also causes an increase in the levels of cytosolic and mitochondrial dismutases (SOD1 and SOD2), which are enzymes catalyzing the reduction of superoxide anions to H2O2. Therefore, a decrease in the O2 −• level after hispidin treatment not only can be caused by the inhibition of p66Shc Ser36 phosphorylation but also may be SOD1 and SOD2 specific. ROS production by p66Shc appears to be a specialized function whereby electrons are removed from the ETC to catalyze the partial reduction of molecular oxygen (64). The redox activity of p66Shc accounts for the decrease in ROS levels observed in p66Shc knockout cells (121) and is also responsible for an altered mitochondrial metabolism under basal conditions that is characterized by lower oxygen consumption (129).
Interestingly, the connection between p66Shc and ROS seems to have significant physiological relevance in the case of hypertension. It appears that p66shc mediates hypertension-associated, cyclic stretch-dependent, endothelial damage and that in stressed cells, activation of integrin α5β1 and c-Jun N-terminal kinase enhances the phosphorylation p66shc at Ser36 and, thus, ROS production (166). The results of these experiments can be extrapolated to an organismal level as well. It has been demonstrated that in mice lacking p66shc, age-related and hyperglycemia-induced endothelial dysfunctions (24, 32, 65, 93) as well as the extent of atherosclerosis (126) are diminished.
Additional examples of proteins that can produce ROS and that are localized in the MAMs fraction include Ero1-Lα (which is present in several tissues and most cell types) and Ero1-Lβ (which is abundant in cells with a high secretory capacity) (2, 47, 149). Although Ero1-Lα is a luminal ER oxidoreductase, it presumably binds to the ER membranes in regions involved in MAMs formation, resulting in greater than 75% of Ero1-Lα localization in the MAMs fraction (58). Ero1-Lα, in addition to protein disulfide-isomerase (PDI), is responsible for the formation of disulfide bonds and, hence, plays an essential role in protein folding (21, 45). Enyedi et al. have shown that the activity of Ero1-Lα results in significant H2O2 formation in the ER. Briefly, oxidative protein folding consists of two major steps: (i) The FAD-bound Ero1 protein oxidizes the PDI (e.g., ERp44 or ERp57) and (ii) PDI subsequently catalyzes the formation of disulfide bonds within newly synthesized/folding proteins (3). Ero1-Lα uses oxygen, which is finally reduced to H2O2 as the electron acceptor on the oxidation of PDI (74). Studies performed on yeast have indicated that greater than 25% of the ROS produced during protein synthesis/folding is related to yeast homolog Ero1p activity (172). The interplay between Ero1-Lα and ERp44, which is an ER luminal chaperone protein (with thioredoxin activity) that is also present in the MAMs fraction, is responsible for the regulation of Ca2+ release from the ER via IP3R1. Ero1-Lα in the ER-mitochondrial hot spots interacts with IP3R1, oxidizing it to potentiate the release of Ca2+ during ER stress (2). Interestingly, hypoxic conditions result in the complete relocation of Ero1-Lα from MAMs, indicating that its intracellular localization depends on oxidizing conditions (58). Moreover, the ability of Ero1-Lα to modulate ER-mitochondria Ca2+ communication may substantially affect the induction of apoptosis (40, 169). In contrast, ERp44 interactions with IP3R1 under reducing ER conditions inhibit Ca2+ transfer to the mitochondria (76); however, the oxidation of IP3R1 by Ero1-Lα is accompanied by ERp44 dissociation from the complex with IP3R1, which enables its full activation (2, 109) (Fig. 3).
Additional details on these topics can be found in the following reviews (149, 165).
MAMs in Cell Death Pathways
Several studies have highlighted and broadened the functional roles of MAMs in a variety of cellular processes, from lipid synthesis/transport, Ca2+-signaling, and ER stress to mitochondrial morphology.
In addition to these roles, MAMs play a key role in the initiation and amplification of cell death. In the next sections, we will illustrate how these contact sites serve as signaling platforms that are capable of determining cellular life and death decisions through the regulation of programmed cell death events.
Apoptosis
In the mitochondrial pathway of apoptosis, stress signals induce mitochondrial outer membrane permeabilization (MMP), which results in permeabilization of the OMM. MMP then facilitates the release of several proteins that usually reside in the mitochondrial intermembrane and intracristal space. These proteins include cytochrome C (cyt-C), apoptosis-inducing factor (AIF), and Smac/DIABLO. Finally, these proteins trigger or facilitate the formation of a caspase-activating complex, the apoptosome, which results in the activation of effector caspases (66). It is therefore clear that one of the key steps of mitochondrial apoptosis is the permeabilization of the OMM. This event is highly regulated by members of the BCL-2 family, particularly through the activity of BAX, BAK, and BID. Under normal conditions, the proapototic protein BAK is situated in the cytosolic compartment. On apoptosis induction, BAX inserts into the OMM, where it associates with BAK or BID. Such complexes stimulate MMP and the release of cyt-C by forming pores in the OMM. Nevertheless, how this molecular opening induced by BAX/BAK/BID occurs is still controversial. A first putative mechanism suggested the cooperation of BAX with other proteins to form and open a pore in the inner membrane (the so-called mitochondrial permeability transition pore [mPTP], permeability transition pore complex) allowing water and proteins till ∼1.5 kDa to pass through (98).
The mPTP is a multiprotein complex that forms at the junctions between the inner and outer mitochondrial membranes. The current mPTP model is built around the F1/Fo ATP-synthase and is composed of several elements, including ANT, VDAC, BAX, and BAK (12, 14, 15).
Opening the pore results in matrix swelling and OMM disruption, which, in turn, promote the release of proteins from the IMS.
An alternative model of MMP was proposed, which did not indicate a key role for the PTP. This mechanism is regulated by BCL-2 proteins, which act directly on the OMM. For example, it was found that anti-apoptotic BCL-2 family members work to block the MMP, while proapoptotic members can act to activate BAK/BAX/BID or interfere with anti-apoptotic BCL-2 family members (48, 99).
Other studies suggest a critical role of VDAC in the MMP and apoptosis (162). Initially, the overexpression of VDAC leads to apoptosis in a variety of cell types (186). In addition, several works address the molecular pathway involving VDAC. In fact, under physiological conditions, antiapoptotic BCL-2 members interact with VDAC and regulate its function, which is to shuttle ATP from the mitochondrial matrix to the cytoplasm (73). However, the scenario is not actually quite so simple: Recent evidence shows that the contribution of VDAC to cell death can be isoform and stimulus dependent. Accordingly, it has been found that VDAC1 silencing promotes apoptosis, whereas silencing of VDAC2 has the opposite effect (38).
MMP is not the only crucial event in apoptosis. There is also a major change in the membrane potential (in the plasma membrane potential [Ψpm] and the mitochondrial transmembrane potential [Ψm]) accompanying MMP. Under normal conditions, the K+ concentration is much higher in the cytosol than in the extracellular fluid. A continuous, low K+ efflux, via K+-channels, is essential for the maintenance of Ψpm, which is vital for ion and volume homeostasis. It has been reported that during mitochondrial swelling and MMP, Ψpm collapses, resulting in dissipation of intracellular [K+], cell shrinkage, DNA fragmentation, and loss of membrane asymmetry, with aberrant exposure of phosphatidylserine residues on the plasma membrane surface (35). Thus, a gross perturbation of Ψpm appears to occur in the postmitochondrial stage of apoptosis.
Ψm also contributes to mitochondrial apoptosis. A prominent dissipation of Ψm takes place in the early stage of the apoptotic process after the opening of the PTP. Nevertheless, studies suggest that loss of Ψm could be a consequence of the apoptotic-signaling pathway. For example, during the etoposide-induced apoptosis of L929 fibroblasts, loss of Ψm occurs as a late event after nuclear alterations and BAX translocation to the mitochondria (91). Moreover, it has been suggested that loss of Ψm is not required for cyt-C release, but only for the release of AIF (112). Considering these remarks, it is clear that dissipation of Ψm is a classic feature of apoptosis, but its effective role in this pathway remains to be addressed.
Cardiolipin
To play crucial roles in cellular bioenergetics and cell survival, mitochondria require the correct import of a large number of proteins from the cytosol. Lipids play a key role in this “mitochondrial protein sorting.” Of the mitochondrial lipids, the dimeric phospholipid CL is characteristic of this organelle, and it is responsible for the stability of several IMM protein complexes (33). Reduced levels of CL induce a collapse of Ψm with consequent blocking of the Ψm-dependent protein translocation into mitochondria. Moreover, during apoptosis, CL is redistributed between the IMM and the OMM, and its oxidation induces the release of proapoptotic factors (53, 86). Although the molecular mechanism of CL translocation remains elusive, MAMs have been proposed to play a key role in CL movement. In demonstration of this theory, the state and efficiency of MAMs are prime determinants for CL transfer and recruitment to the OMM (78, 152) (Fig. 4).
Mitochondrial network
The shape of the mitochondrial network is also important to the functioning and health of the cell. During apoptosis, the mitochondrial network undergoes dramatic rearrangements, and it is generally accepted that it collapses into small spherical structures in response to apoptotic stimuli.
Furthermore, as discussed earlier, mitochondrial dynamics and morphology are modulated by a correct assemblage of ER-mitochondria contacts. In wild-type cells, during apoptosis, mitochondrial fission facilitates mitochondrial fragmentation, the collapse of Ψm, and the release of IMS-stored apoptogenic factors. For example, inhibition of fission through DRP1 RNA interference retards the release of cyt-C from the IMS (107). Consistent with these data, DRP1-KO-derived MEFs display a delay in cyt-C release and retardation of caspase activation (81), and RNA interference targeting MFF induces mitochondrial elongation, with a consequent delay in cyt-C release and blocking of the apoptotic program.
Taken together, these data suggest an important role of fission-related proteins during the progression of the apoptotic program. After the induction of apoptosis, the fission protein DRP1 translocates to the OMM, with consequent augmentation of mitochondrial fragmentation. Indeed, it has been reported that during apoptosis cytosolic BAX/BAK proteins travel to the OMM and interact with DRP1 and MFN2. As a result, these BCL-2 member proteins promote SUMO modifications of DRP1, which stably associates with the OMM (18, 177). In addition, BAX/BAK induce tBID activation, which is correlated with blocking of mitochondrial fusion, most likely through inhibition of the mitofusin protein MFN-2 (90). The activation of fusion proteins (such as MFN-1/-2 and OPA1) that are indispensable for the maintenance of normal mitochondrial morphology and calcium uptake by mitochondria antagonizes apoptosis progression (51).
Accordingly, inhibition of OPA1-mediated fusion through RNA interference leads to fragmentation of the mitochondrial network, concomitant with the dissipation of the mitochondrial membrane potential. As a consequence of these events, cyt-C is released, and executioner caspases are activated (28, 132).
The apoptotic program is also promoted by depletion of PACS2. Indeed, the absence of PACS-2 induces the caspase-dependent cleavage of BAP31 to yield the pro-apoptotic fragment p20, causing mitochondria to fragment and uncouple from the ER (164).
Overall, these findings indicate that disruption of the apposition of mitochondria with the ER and the consequent increase in mitochondrial fragmentation are functionally linked to apoptosis induction.
Calcium (Ca2+)
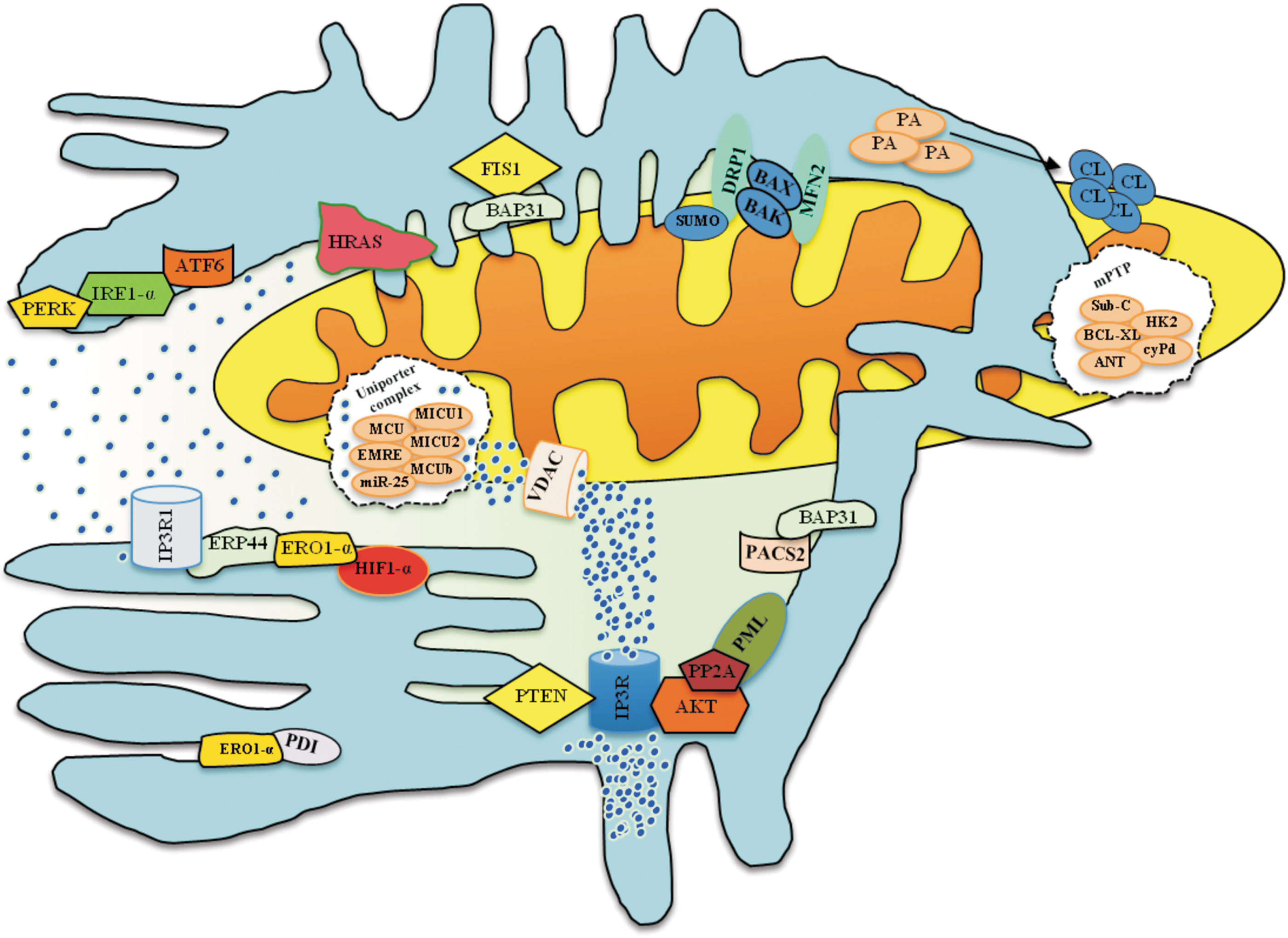
Ca2+ homeostasis is fundamental to numerous cellular mechanisms, including cell death. Elevation of the intracellular Ca2+ concentration is dependent on either Ca2+ influx from the extracellular space through the plasma membrane or Ca2+ release from intracellular Ca2+ stores, such as those in the ER. The mitochondria are equally important for Ca2+ signaling. Different works have demonstrated that Ca2+ release from the ER results in cytosolic Ca2+ increases that are paralleled by similar or even increased cycles of mitochondrial Ca2+ uptake (11, 116).
This unique characteristic of the mitochondria is principally due to the large electrochemical gradient Ψm and due to the existence of MAMs. As describe earlier, the close apposition of the mitochondria and ER creates Ca2+ hotspots, which have been found to play pivotal roles in several cellular functions, including the highly efficient transmission of Ca2+ from the ER to the adjacent mitochondrial network to stimulate oxidative metabolism. Apoptosis is also intimately connected to the regulation of Ca2+ handling promoted by MAMs. An excess of Ca2+ flows out of the ER into mitochondria via IP3 receptors (which present at high levels in the MAMs compartment) and promotes the apoptotic program (142). For example, several studies have correlated MAMs with increased Ca2+ transfer in sensitization to apoptosis. For example, the Ca2+ fluxes into mitochondria induce the oligomerization and activation of BAX, which promotes the permeabilization of the OMM and, ultimately, the release of pro-apoptotic factors into the cytosol. Other proteins that link the ER to mitochondria have been found to exert control over pro-apoptotic Ca2+ fluxes in stressed cells.
A growing body of research highlights the key role of the oncogene H-RAS in the maintenance of tumor survival and proliferation. Although the clear molecular mechanisms underlying these processes are not well established, a recent study illustrated a direct link between Ca2+ regulation and HRAS-driven transformation. In this work, Rimessi et al. identified HRAS localized to the MAMs compartment, suggesting the possibility that this localization may serve as a strategic point to regulate the transmission of Ca2+ from the ER to mitochondria. Indeed, after the induction of oncogenic HRAS, global intracellular Ca2+ perturbation accompanied by a dysfunction of mitochondrial physiology has been observed (153).
PTEN (protein phosphatase and tensin homolog deleted on chromosome 10), a tumor suppressor that downregulates AKT and localizes to the MAMs compartment, was recently shown to be able to interact with the AKT/IP3R complex (11), leading to a reduction of its phosphorylation and an increase in Ca2+ release. The tumor suppressor PML also modulates the ER-mitochondria Ca2+ signaling platform. This protein was found to localize to the ER and MAMs, where it interacts with IP3R3, regulating the ER-mitochondria Ca2+ flux and apoptosis. Furthermore, using PML-KO-derived MEFs and a PML chimera that exclusively localizes to the outer surface of the ER (erPML), our laboratory demonstrated that PML also acts as a suppressor of other oncogenic pathways. Indeed, loss of PML promotes a reduction of PP2A activity at the ER and an increase of AKT activity. Consequently, the hyper-phosphorylation of IP3Rs mediated by AKT inhibits ER Ca2+ release and causes the cell to be less sensitive to Ca2+-mediated apoptotic stimulation (62).
Other central players in the ER-mitochondria Ca2+ flux include a series of chaperones and oxidoreductase, which also localize to the ER/MAMs compartment. ERp44, an ER luminal protein of the thioredoxin family, was found to directly interact with IP3R1, inhibiting its channel activity. Specifically, Higo et al. demonstrated that ERp44 directly inhibits the channel activity of IP3R1 in a pH-, redox state-, and [Ca2+]ER-dependent manner (76). Furthermore, in this study, it was demonstrated that ERp44 also reinforces the ERO1alpha/oxidoreductase system. ERO1-α is an enzyme that modulates the activity of PDI. ERO1-α localizes to the MAMs and ER, where it can interact with and control the activity of HIF1-α, modulating the hypoxic response to disulfide bond formation (120). Based on these observations, the reported intimate relationship between ERp44 and ERO1-α could play a key role in the modulation of IP3R–dependent calcium signaling and, thus, in the regulation of the apoptotic program.
It is clear that the release of Ca2+ from ER stores is an essential component of cell survival processes, particularly apoptosis. On release from the ER, Ca2+ is taken up by mitochondria through the mitochondrial calcium channel uniporter (MCU), which together with its regulators (MICU1, MICU2, MCUb, EMRE, MCUR1, and miR-25), it constitutes the MCU complex (113, 115). Next, the Ca2+ flux promotes apoptosis induction through the activation of the mPTP, which, in turn, promotes cyt-C release and caspase activation. As described earlier, the mPTP is composed of several components, including proteins localized at MAMs (12). Based on this observation and considering the key role of the mPTP in cell survival, it is possible to predict a direct and novel role of ER-mitochondria apposition in mPTP regulation and, thus, in the onset of apoptosis. Several findings suggest the existence of this intimate relationship. mPTP opening is an important event in cardiomyocyte cell death during ischemia-reperfusion (I/R). In addition, peroxidation of CL by ROS in the presence of Ca2+ induces mitochondrial permeability transition and cyt-C release in rat heart mitochondria. Thus, increased levels of peroxidized CL and Ca2+ might lead to the opening of the mPTP. Furthermore, these effects were not observed for nonoxidized CL and were inhibited by cyclosporin A and bongkrekic acid (138, 143). Considering that MAMs are prime determinants of CL homeostasis and Ca2+ handling, it is possible that these membranes play an active role in the regulation of mPTP opening during I/R.
Other indications of the existence of this relationship might be found by considering p66Shc. As described earlier, p66Shc is a MAMs protein that is strongly regulated by ROS and is involved in Ca2+ handling. Once activated, p66Shc oxides reduced cyt-C and catalyzes the reduction of O2 to H2O2 using electrons from the respiratory chain. The generated H2O2 leads to mPTP opening and consequent caspase activation and apoptosis (64).
The ER not only is one of the main sites of intracellular Ca2+ storage in the cell but also is considered the primary site for the synthesis and folding of proteins for the entire cell. When the capacity of the ER machinery to fold proteins becomes inadequate, the cell enters into a dangerous state, known as “ER stress.” When ER stress becomes too severe and the dedicated signaling pathways (such as UPR) cannot alleviate this condition, a lethal signal may be turned on, giving rise to cell death, usually in form of apoptosis.
On ER stress three sensor proteins of UPR-system (ATF6, PERK, and IRE1-α) generally stimulate an increase of ER and a gain of function of chaperone activity. Consequently, the ER morphology considerably undergoes changes, and the ER-mitochondria contact sites become stronger and closer. This increased coupling results in major oxygen consumption, a stronger reductive capacity, and augmented Ca2+ transfer. This increase in mitochondrial activity is observed only where the reticular and mitochondrial networks are redistributed. Overall, these findings suggest that the increased juxtaposition between the ER and mitochondria observed during ER stress could contribute favorably to cellular adaption to stressful conditions (17).
Nevertheless, these dependent ER stress sensor proteins can modulate the transmission of signals between the ER and mitochondria in a UPR-independent manner. For example, ablation of the protein PERK, which was recently discovered to be enriched in the MAMs fraction, in addition to the ER compartment, promotes perturbations of ER-mitochondria contact sites and reduces apoptotic activity in response to agents inducing ROS production, Ca2+ transfer, and ER stress. Collectively, these data reveal that a conserved MAMs structure is indispensable for transmitting Ca2+ as well as ROS-mediated signals to the mitochondria after ROS-based ER stress (174). Accordingly, cells lacking MFN2 exposed to ER stress display a weaker ER-mitochondria interaction and a reduction of apoptosis. Furthermore, other recent studies have highlighted the key role of the ER-mitochondria juxtaposition in propagating apoptosis in the presence of pro-oxidant inducers of ER stress (25, 62, 137).
Autophagy
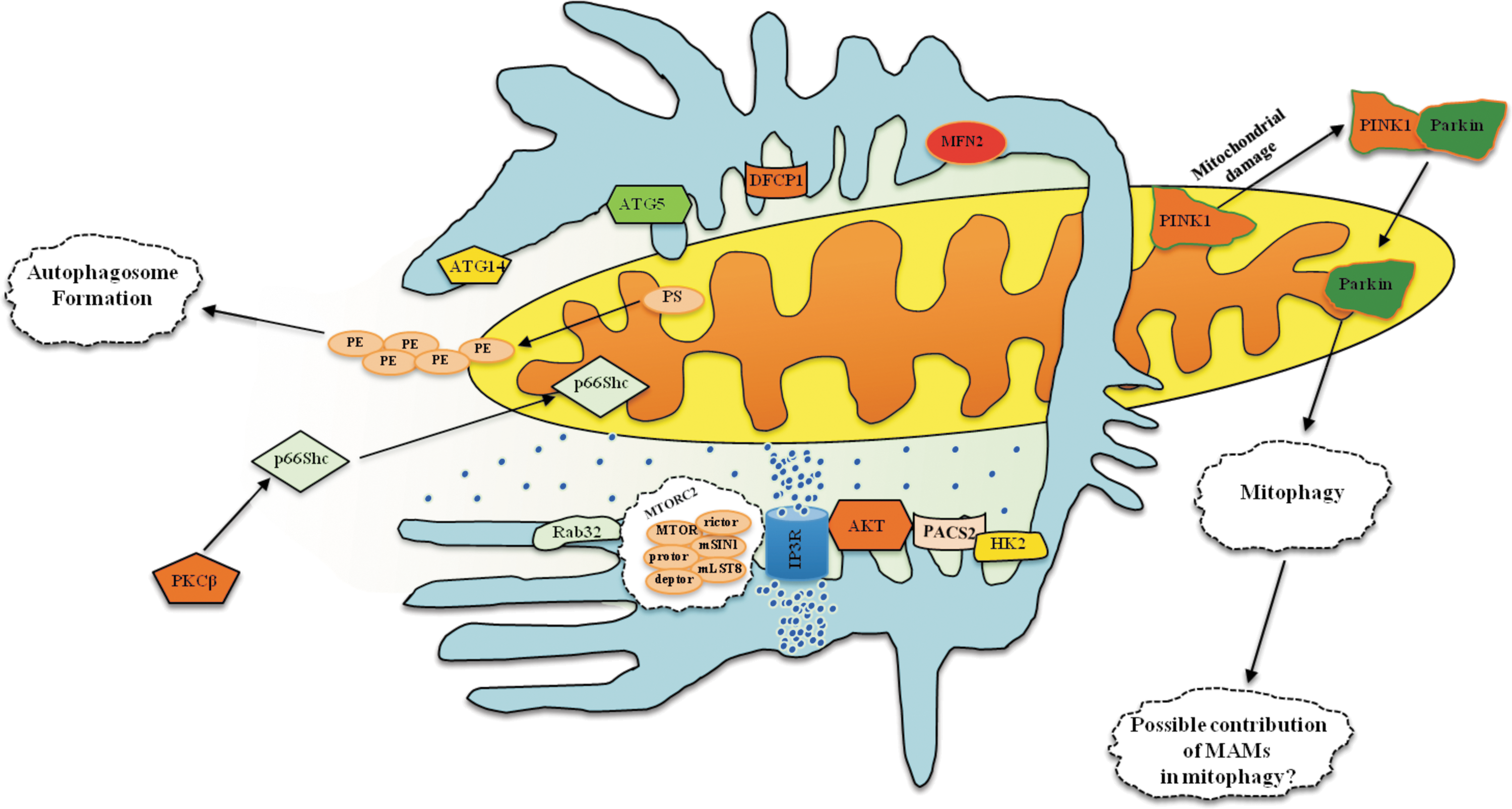
MAMs are not only important for the coordination of mitochondrial integrity, Ca2+ handling, and apoptotic activity but also a primary element for the initiation and execution of the autophagic machinery. The mitochondria and ER are involved in autophagosome biogenesis. Initially, several ER proteins were found to localize to the autophagosome membrane. Then, electron and fluorescent microscopy analyses demonstrated that the ER and the initial isolation membrane of autophagosomes (the phagophore) are juxtaposed, and on starvation-induced autophagy the cell forms the so-called omegasome, a phosphatidylinositol 3–phosphate compartment connected to the ER that is fundamental for autophagosome formation. Alternatively, the autophagosome might be derived from mitochondria. It has been demonstrated that during starvation, the OMM “gifts” mitochondrial-derived membranes that will be used for autophagosome biogenesis. Furthermore, a crucial role of the ER-mitochondria connection in autophagy induction has been demonstrated. Indeed, MFN2 KO-derived cells are not able to undergo autophagy, supporting the possibility of a contribution of lipids from the ER to mitochondria (70). In addition, these studies provided a further indication of the intimate relationship between MAMs and the autophagosome. However, the importance of ER-mitochondria contact sites for autophagosome formation remained controversial.
This question was answered in 2013 by an elegant study published in Nature, where it was demonstrated that autophagosome formation starts at MAMs. Immunofluorescence and electron microscopy, in addition to subcellular fractionation, showed that specific preautophagosome/autophagosome markers (ATG14 and ATG5) localize to MAMs under starved conditions. Consistent with these findings, on starvation, DCFP1 (double FYVE domain-containing protein 1) translocates to the omegasome. In contrast, in PACS2 KO and MFN2 KO cells, both the accumulation of autophagic markers and the translocation of ER-related proteins are significantly reduced, indicating a stable role of MAMs during the completion of autophagosomes (72). In spite of these findings, the role of MAMs in the molecular mechanism of autophagy remains to be elucidated.
It is well known that the main regulator of autophagy is the serine/threonine kinase mTOR (mechanistic target of rapamycin) (95). This kinase exists in two protein complexes: mTOR complex-1 (mTORC1) and -2 (mTORC2). Interestingly, it has been found that mTORC2 localizes to MAMs, where it activates AKT. Once activated, AKT appears to control MAMs integrity and mitochondrial physiology through PACS and hexokinase (HK2) phosphorylation. Furthermore, mTORC2-AKT regulates IP3R3 phosphorylation and Ca2+ release at MAMs (8). Despite this, the involvement of mTORC-2 in autophagy remains debatable. Nevertheless, it has been found that Rab32 (described earlier to be an MAMs protein essential for autophagosome formation) is critical for the regulation of mTORC2 activity (29). Overall, these findings may suggest an active role for this complex in autophagosome formation and the regulation of autophagic activity.
Other MAMs proteins also appear to regulate autophagy. One example is p66Shc, a protein that plays an important role in controlling the mammalian life span (146). It has recently been found that PKCβ and p66Shc overexpression leads to a reduction of autophagic activity and their roles in the modulation of autophagy appear to be interrelated. In support of this hypothesis, overexpression of PKCβ drives a strong increment of p66Shc phosphorylation and an augmentation of the transfer of p66Shc to the mitochondrial compartment. Consistent with this finding, MEFs derived from PKCβ KO mice display a strong and significant reduction of both the phosphorylation and mitochondrial localization of p66Shc (141).
As has been well reviewed, different forms of specialized autophagy have been discovered in recent years (75). One of the most important is mitophagy, which is responsible for the selective removal of damaged and exhausted mitochondria. Several studies have demonstrated that two genes, PINK1 and PARK2, are involved in the maintenance of a healthy population of mitochondria.
Parkin, encoded by the PARK2 gene is a cytosolic E3 ubiquitin ligase that mediates the ubiquitylation of a number of target proteins. Parkin has been found to be cytoprotective under various conditions, and it has been reported to play a role in mitochondria under stress conditions (127). Parkin ubiquitinates several OMM proteins, including VDAC, MFN, DRP1, BCL2, and BAX (54, 55). When severe damage impacts a population of mitochondria, the PINK1-parkin axis recognizes defective mitochondria, rapidly isolates them from the mitochondrial network, and, finally, degrades them through the ubiquitin-proteasome and autophagic pathways (151). Recently, it has been demonstrated that mitochondria supply membrane material not only during serum starvation but also during drug-induced autophagy, introducing a novel mechanism of parkin-associated mitophagy.
Cook et al. demonstrated that stress and starvation conditions and treatment with drugs that are capable of promoting autophagy increased both PINK1 and parkin localization to the mitochondrial compartment. Furthermore, using confocal and electron microscopy, these authors showed that mitochondria labeled with parkin are not only engulfed by the forming autophagosome but also used to form new a autophagosome. Accordingly, inhibition of the mitophagic process through PINK1 knockdown restores normal functional mitophagy in cells (30). In conclusion, this work could open new avenues for the discovery of novel roles of the ER-mitochondria contact sites in the regulation of the molecular pathways of general and selective autophagy (Fig. 5).
MAMs and Inflammation
In addition to their established role as a signaling hub for Ca2+ and lipid transfer between the ER and mitochondria, MAMs have recently been shown to play a central role in the modulation of various key processes, including inflammasome signaling. A link between inflammation and the ER-mitochondria interface was established for the first time in 2011 in a study by Zhou et al., who demonstrated a new role for mitochondria in NLRP3 inflammasome activation (190).
The NOD-like receptors (NLRs) are composed of 22 human genes that are characterized by the presence of a central nucleotide-binding oligomerization (NACHT) domain, C-terminal leucine-rich repeats (LRRs), and an N-terminal effector domain. On activation, select NLR family members form multiprotein complexes (termed inflammasomes) that serve as platforms for caspase-1 activation and the subsequent proteolytic maturation of the potent proinflammatory cytokine IL-1 β. (171). Due to its association with numerous inflammatory diseases, the NLRP3 inflammasome is currently the most fully characterized, well-studied inflammasome. The key components of a functional NLRP3 inflammasome include NLRP3, the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and caspase-1 (159). On sensing a wide variety of danger signals, NLRP3 oligomerization and recruitment of ASC and procaspase-1 trigger the autoactivation of caspase-1 and the maturation and secretion of proinflammatory cytokines, such as IL-1 β.
Resting NLRP3 localizes to the cytosol and ER structures, whereas on inflammasome activation using nigericin (an antibiotic) or monosodium urate, both NLRP3 and its adaptor ASC colocalize to the MAMs fraction. Thus, by virtue of its ER-mitochondria localization on activation, the NLRP3 inflammasome is strategically located to receive signals emanating from mitochondria.
Previous studies have indicated that ROS represent a common integrator across several stimuli that activate the NLRP3 inflammasome. In addition to the ER and peroxisomes (19), mitochondria are the primary source of ROS. However, the source of NLRP3-activating ROS and the associated underlying mechanisms remain unclear.
Zhou and colleagues observed a correlation between mitochondrial ROS activity and the presence of IL-1 β in the supernatant of the human THP1 macrophage cell line. To avoid cellular damage, ROS-generating mitochondria are constantly removed by mitophagy; these authors found that inhibition of the autophagic machinery affects IL-1 β production, resulting in the accumulation of defective and ROS-producing mitochondria, which potentiates NLRP3-dependent inflammasome activation.
Although these results indicated that the prolonged presence of damaged, ROS-producing mitochondria is implicated in inflammasome activation, this evidence remained indirect. However, this finding suggests that MAMs may become locally enriched in ROS or ROS-derived signaling molecules and recruit ROS-sensing proteins. Consistent with this hypothesis, these authors inhibited the activity of the OMM channel VDAC and demonstrated that the knockdown of VDAC1/2 selectively abrogates NLRP3 inflammasome formation. In contrast, VDAC, and thus mitochondria, are not essential for the activation of AIM2 or IPAF inflammasomes. VDAC activity is regulated by hexokinase and Bcl-2 family members; the overexpression of Bcl-2 leads to partial VDAC closure and a concomitant decrease of mitochondrial Ca2+ levels and ROS production. Stimulated macrophages isolated from Bcl-2-overexpressing transgenic mice exhibit decreased levels of IL-1 β compared with cells from wild-type mice (Fig. 6).

Additional observations provide support for a pivotal role of mitochondria in NLRP3 inflammasome activation. ROS production can be specifically induced in mitochondria by inhibiting key enzymes of the electron transport chain. Addition of the complex I inhibitor rotenone results in a partial loss of Ψm and strong ROS production, as observed for the complex III inhibitor antimycin A; indeed, both drugs lead to NLRP3 inflammasome activation (111, 190).
Furthermore, antioxidants specifically targeting mitochondria appear to block inflammasome activation and inflammation in general (20, 124).
Bulua et al. identified mitochondrial ROS as a driver of inflammation in tumor necrosis factor receptor-associated periodic syndrome (TRAPS), which is an autoinflammatory disorder caused by missense mutations in the type-1 TNF receptor (TNFR1), and potentially in other autoinflammatory diseases (20). When mitochondrial respiration or ROS production is inhibited, inflammatory cytokine production in response to LPS is blunted. These results suggest a more general role for mitochondrial ROS in the induction of inflammatory cytokines upstream of inflammasome activation. In TRAPS, increased mitochondrial ROS result from enhanced oxidative phosphorylation; blockade of ROS production by the mitochondria provides a new therapeutic strategy for reducing the symptoms of TRAPS and other inflammatory states.
Taken together, these and other data suggest a model in which a wide range of danger signals converge to cause increased generation of mitochondrial ROS. A rise in mitochondrial oxidant production could therefore represent the common currency of all of these divergent stress signals, with subsequent activation of the NLRP3 inflammasome. For instance, although there is compelling evidence that mitochondrial oxidants regulate the inflammasome, the mechanisms through which inflammatory signals can regulate mitochondrial function and the precise mechanism by which the release of ROS can trigger NLRP3 activation have yet to be defined. The precise molecular target of mitochondrial ROS is poorly understood, and there is little insight regarding the specific role of oxidants in inflammatory circumstances.
Another NLRP3 binding partner, TXNIP, redistributes to MAMs/mitochondria in response to oxidative stress (157) or NLRP3 inflammasome activation (189). In resting cells, TXNIP interacts with TRX and is therefore unavailable for NLRP3 interaction.
Inflammasome activators, such as uric acid crystals, induce the dissociation of TXNIP from thioredoxin in an ROS-sensitive manner and allow it to bind NLRP3 and translocate to MAMs/mitochondria (189), raising the possibility that TXNIP is involved in IL-1 β production through NLRP3 under ER stress conditions. TXNIP is a critical signaling node that links ER stress and inflammation. TXNIP is induced by ER stress through the PERK and IRE1 pathways, induces IL-1 β mRNA transcription, activates IL-1 β production by the NLRP3 inflammasome, and mediates ER stress-mediated β-cell death (134) (Fig. 6).
MAMs play a central role not only in the communication between the ER and the mitochondria but also in maintaining various cellular processes such as the antiviral response.
Recently, a new role has been identified for the adaptor MAVS (mitochondrial antiviral signaling protein) as the mitochondrial anchor for NRLP3 inflammasome formation (167). MAVS is a well-known mitochondrial protein that plays a crucial role in RIG-like receptor (RLR) signaling pathways leading to type I IFN induction and NF-κB activation (160). Specifically, MAVS contains an N-terminal CARD-like domain and a C-terminal transmembrane domain that targets the protein to the mitochondrial membrane. The mitochondrial localization of MAVS represents the first example of a mitochondrial protein that plays a pivotal role in innate immunity. Viral RNAs are recognized in the cytosol by the helicases RIG-1 or MDA5 (melanoma differentiation-associated gene 5). The N-termini of RIG1 and MDA5 contain two CARD domains that interact with the CARD domain of the mitochondrial adaptor MAVS. After the recruitment of transactivators, MAVS induces phosphorylation of IRF3 and IRF7 and activation of NF-κB, leading to the production of type I IFNs and proinflammatory cytokines, respectively. It has recently been proposed that during RNA infection, RIG-1 is recruited to the MAMs to bind MAVS (77). Dynamic MAMs tethering to mitochondria and peroxisomes subsequently coordinates MAVS localization to form a signaling synapse between membranes. Importantly, the hepatitis C virus NS3/4A protease, which cleaves MAVS to support persistent infection, targets this synapse for MAVS proteolysis from the MAMs, but not from the mitochondria, to ablate RIG-1 signaling of immune responses. These results identify an innate immune signaling synapse in which the MAMs serves as the central scaffold that coordinates MAVS-dependent signaling of the RIG-1 pathway between mitochondria and peroxisomes (Fig. 6). Collectively with the role of the MAMs in NLRP3 inflammasome signaling (190), these findings indicate that the MAMs plays a central role in initiating both the innate immune and the inflammatory responses to infection.
Recently, Jacobs et al. demonstrated that gp78, which is an E3 ubiquitin ligase active in the ER-associated degradation (ERAD) pathway and localizes to the ER-mitochondria interface, is a novel regulator of RLR signaling (83). In addition to the enteroviruses coxsackie virus B and poliovirus, the depletion of gp78 results in a robust decrease of vesicular stomatitis virus infection and a corresponding enhancement of type I IFN signaling. Mechanistically, gp78 modulates type I IFN induction, altering both the expression and signaling of MAVS. These studies indicate an unexpected role for MAM-localized gp78 E3 ubiquitin ligase in the negative regulation of MAVS signaling. These results suggest two parallel pathways by which gp78 regulates MAVS expression and signaling: One pathway requires its E3 ubiquitin ligase and ERAD activity, whereas the second pathway requires the gp78 C-terminus and occurs via the association between this region and the N- and C-terminal domains of MAVS.
In addition to RIG-1, several other proteins that function in the area surrounding the mitochondria fine-tune the activities and functions of MAVS. Among these proteins, STING (STimulator of INterferon Genes) is particularly interesting, as it is enriched at MAMs; this protein interacts with RIG-1 and binds MAVS to activate a TBK-1- and IRF3-dependent cascade that ultimately induces the expression of type I IFN (82).
These results suggest that MAMs delineate a signaling synapse (involving both mitochondrial and ER components) that is essential for optimal antiviral responses.
Recently, it has been demonstrated that MAVS is required for optimal NLRP3 activity, mediating recruitment of NLRP3 to mitochondria, as well as promoting the production of IL-1 β and the pathophysiological activity of the NLRP3 inflammasome (167). The recruitment, which depends on a short N-terminal sequence in NLRP3, promotes ASC “speckle” formation and the downstream biochemical events associated with the activity of the inflammasome. Contrary to these results, which support a role for MAVS in the activation of the NLRP3 inflammasome via nonviral stimuli such as LPS plus nigericin or LPS plus ATP, Park et al. demonstrated that MAVS regulates NLRP3 activation primarily in response to stimuli that directly engage MAVS, such as infection with Sendai virus (140). Activation of MAVS signaling by Sendai virus infection promotes NLRP3-dependent caspase-1 activation, whereas knockdown of MAVS expression clearly attenuated the activation of NLRP3 inflammasomes in THP-1 and mouse macrophages (Fig. 6). These results suggest that MAVS facilitates the recruitment of NLRP3 to the mitochondria and may enhance its oligomerization and activation by bringing it in close proximity to mitochondrial ROS.
Given the important roles of the MAMs in different cellular processes, it is not surprising that numerous viral proteins target this structure. One well-characterized example is the human cytomegalovirus glycoprotein UL37 exon 1 (16) that traffics into the MAMs during permissive infection and induces alteration of the MAMs protein composition. This glycoprotein targets MAMs with two mitochondrial targeting signals (150) and is able to reduce ER Ca2+ contents, possibly by modulating the amount of Ca2+-regulating chaperones and oxidoreductases such as BiP present on MAMs (188) or by increasing the targeting of GRP75 to the VDAC/IP3R/GRP75 ternary complex (16).
More recently, human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) has been suggested to be present on the ER, MAMs, and OMM, possibly via the integration of its C-terminal transmembrane domain (80). Vpr injures OMM and causes loss of Ψm by post-transcriptionally reducing the expression of MFN2 and increasing mitochondrial deformation. Vpr also markedly decreases cytoplasmic levels of DRP1 and increases bulging in MAMs. Collectively, these results suggest that Vpr-mediated cellular damage may occur via an alternative protein transport pathway from the ER through the MAMs to the mitochondria, which is modulated by MFN2 and DRP1 (Fig. 6).
As we have outlined thus far, MAMs play a central role in the formation and regulation of the NLRP3 inflammasome. In addition to the two aforementioned nigericin and crystalline activators, several stimuli can trigger inducible activation of the NLRP3 complex, including extracellular ATP released by dying cells (117), cholesterol crystals (44), RNA and Ca2+ (85).
Murakami et al. elucidated a critical role for Ca2+ mobilization in the activation of the NLRP3 inflammasome by multiple stimuli (123). These authors demonstrated that blocking Ca2+ mobilization inhibits the assembly and activation of the NLRP3 inflammasome complex and that during ATP stimulation, Ca2+ signaling is pivotal in promoting mitochondrial damage. C/EBP homologous protein, which is a transcription factor that can modulate Ca2+ release from the ER, amplifies NLRP3 inflammasome activation, thus linking ER stress to the activation of the NLRP3 inflammasome. Ca2+ signaling is not sufficient for NLRP3 inflammasome activation, as indicated by the inability of the Ca2+ ionophore ionomycin to induce IL-1β production; however, only stimuli that mobilize Ca2+ in a manner leading to mitochondrial damage would activate the NLRP3 inflammasome.
Lee et al. recently confirmed and updated the role of Ca2+ in inflammation; these authors found that the murine Ca2+-sensing receptor (CaSR) strongly activates the NLRP3 inflammasome, which is mediated by increased intracellular Ca2+ and decreased cellular cyclic AMP (105). CaSR activates the NLRP3 inflammasome through phospholipase C, which catalyzes inositol-1,4,5-trisphosphate production, thereby inducing release of Ca2+ from ER stores; the increased cytoplasmic Ca2+ promotes the assembly of inflammasome components. Moreover, G-protein-coupled receptors can activate the inflammasome, indicating that increased extracellular Ca2+ functions as a specific amplifier of inflammation (155). Activation is mediated by signaling through the Ca2+-signaling receptor and GPRC6A via the phosphatidyl inositol/Ca2+ pathway.
Recently, Nakahira et al. demonstrated that treatment with LPS and ATP release mtDNA into the cytosolic compartment and that this requires activation of the NLRP3 inflammasome, directly contributing to downstream activation of caspase-1 (124) (Fig. 6).
Recently, Oelze et al. demonstrated that glutathione peroxidase-1 (GPx-1) ablation in aging animals has a substantial impact on the burden of oxidative stress and injury (130). They observed that the age-dependent increase in the infiltration of cardiovascular tissue with leukocytes is more pronounced in GPx-1-deficient mice, suggesting that GPx-1 deficiency may lead to an inflammatory phenotype of the vasculature, which is a condition that has been reported to contribute to increased oxidative stress and vascular/endothelial dysfunction (178).
These findings give rise to further speculations. For example, MAMs may be a key player in aging-related, low-grade inflammation, as aging is associated with increased mitochondrial ROS formation, and MAMs contain several redox-sensitive components of the NLRP3 inflammasome.
Collectively, these findings highlight the central role of MAMs in the coordination of inflammasome formation and antiviral immunity. Future studies are necessary to fully explore the biological significance of MAMs during these processes.
MAMs Deregulation in the Pathogenesis of Neurological Disorders
Alterations in mitochondrial and ER homeostasis and the link between MAMs homeostasis and cellular derangement are common features of several neuronal diseases in which genetic models and environmental factors permitted the identification of common traits in the pathogenic routes. Indeed, with the increasing amount of information on MAMs, considerable evidence indicates that MAMs play an important role in neuronal disease. Analysis of the proteins present at MAMs through a mass spectrometry-based proteomic characterization from mouse brain tissue was conducted in association with a quantitative validation method to distinguish true MAMs proteins from contaminating proteins. Poston et al. (147) identified several proteins related to different neuronal-based diseases, such as movement disorders (chorea and Parkinson's disease), genetic disorders (Huntington's disease), and neurodegenerative diseases (schizophrenia, dementia, and seizures). This evidence suggests that several neuronal disorders share an alteration of MAMs homeostasis, where two crucial organelles (the mitochondrion and the ER) for neuronal cells are in close contact with a sustained cross-talk. We can therefore speculate that neuronal diseases are MAMs-related disorders.
Here, we briefly discuss alterations in MAMs functions that appear to be important in the pathogenesis of selected human neuronal pathologies. We will summarize only the possible link between MAMs and these pathologies, referring readers to the literature for details of the individual pathologies.
Ca2+ homeostasis represents a link between MAMs and neuronal diseases, particularly through the alterations in Ca2+ cross-talk between the ER and mitochondria at MAMs, which are a hotspot of Ca2+ signaling domains (142).
Various experimental observations have suggested that an alteration of intracellular Ca2+ homeostasis contributes to the development of FAD and, more generally, to the pathogenesis of AD. Indeed, mutations in PS have been shown to alter ER Ca2+ release, affecting the mitochondria in a number of cell models (69, 106, 108). Interestingly, PSs are highly enriched in MAMs (158).
Different, and, in some cases, contrasting, hypotheses have been proposed. Some authors argue that ER Ca2+ overload is because wild-type PSs, but not the FAD mutants, can form Ca2+-permeable leak channels in the ER (173), thus providing a clear case for enhanced Ca2+ release in their pathological model. Subsequent studies from other groups have presented results that are not entirely consistent with this hypothesis (26, 49, 57, 92, 187). This experimental discrepancy has been explained in part by the observation that PS1 and PS2 play distinct roles: PS2, but not PS1, modulates the ER-mitochondria tethering by increasing the number and/or the extent of their contact sites and, in turn, the Ca2+ cross-talk between the ER and mitochondria. FAD-linked PS2 mutations lead to a larger increase in ER-mitochondria interactions and, consequently, to an altered (and deleterious) Ca2+ transfer from the ER to mitochondria (Fig. 7).
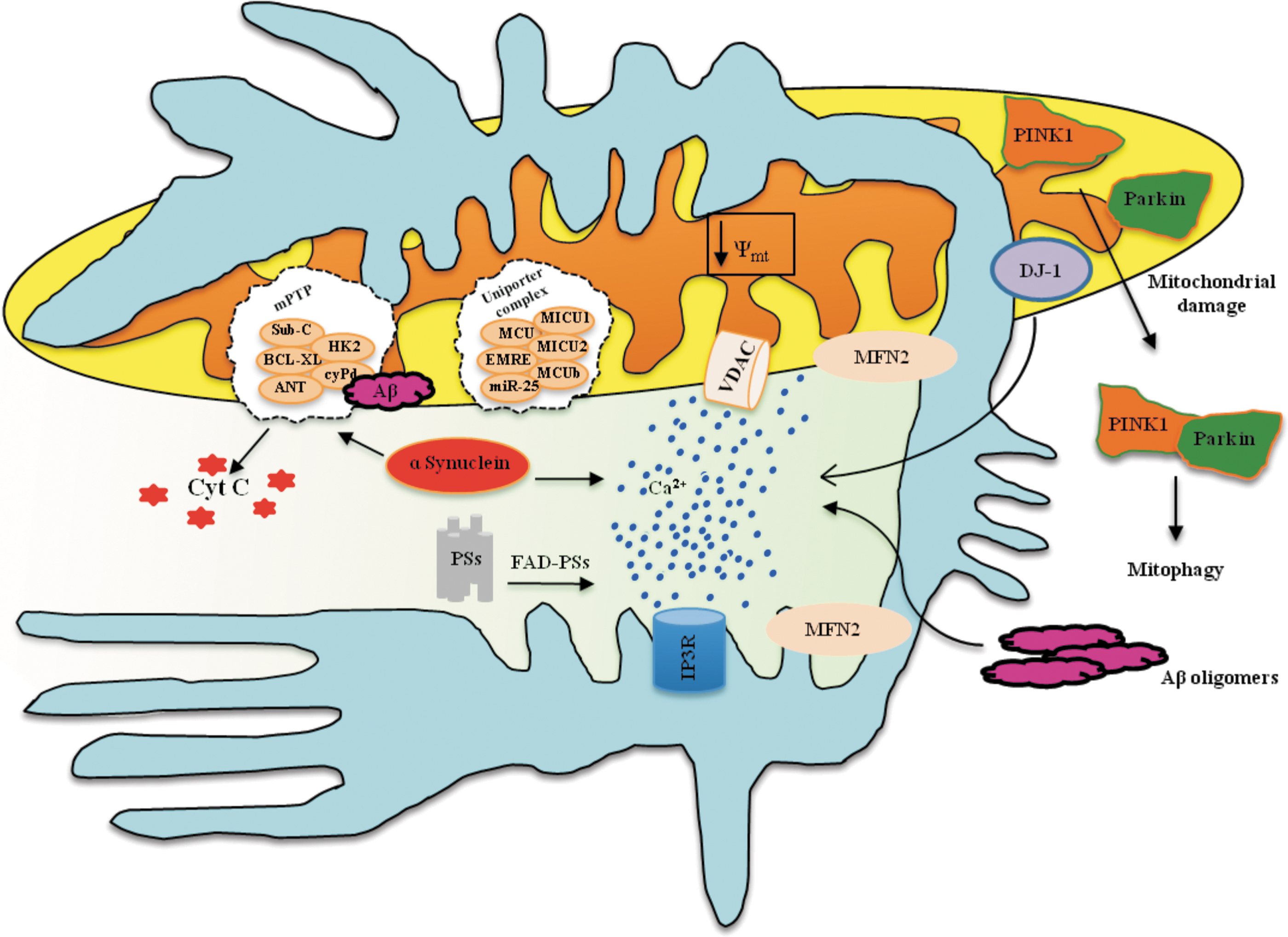
There is currently a consensus that an increased association between MAMs and mitochondria is linked to the pathogenesis of AD; indeed, in different models of AD, the area of close apposition between mitochondria and the ER (i.e., the MAMs) and the processes that occur at MAMs are enhanced compared with wild-type cells (158). Consistent with these observations, γ-secretase and PS activity is lower in MFN2-deficient cells that contain very few MAMs than in wild-type cells (5). Interestingly, this occurs not only in FAD but also in sporadic AD. However, these results were obtained in only fibroblasts from patients, not in neurons.
Several studies support the theory that the neuronal demise is potentiated by vascular alterations in the early stages of the disease. Recently, it has been demonstrated that amyloid-β (A β) induces ER stress in brain endothelial cells and triggers a mitochondria-mediated apoptotic cell death pathway involving ER-to-mitochondria Ca2+ transfer, decrease of Ψm, and release of proapoptotic factors (50), thus suggesting the participation of MAMs in this pathogenetic route (Fig. 7).
Regardless of how the synergistic “Ca2+ hit” occurs, mitochondrial dysfunction appears to be an obligatory downstream step in the pathogenesis of AD. Electron microscopy analysis of mitochondria in various regions of AD-affected brains revealed significant morphological organelle alterations (6).
A β peptides increased neuronal ROS production, activated the mitochondrial fission proteins DRP1 and FIS1, and caused mitochondrial fragmentation (7).
Moreover, A β peptides modulate the mPTP (Fig. 7). A β peptides have been shown to inhibit mitochondrial respiration (34) and facilitate the opening of the mPTP. However, knockout mouse models for cyclophilin D, which is an essential component of mPTP activity, exhibit improved cognitive abilities and a minor A β-mediated reduction of long-term potentiation (43).
MAMs also play a key role in Parkinson's disease (PD) (Fig. 7). This hypothesis is supported by the identification of a cohort of proteins involved in the familial forms of PD that appear to share intracellular localization of MAMs and possibly indicate a signaling role for Ca2+. Specifically, mutations were reported in genes encoding for α-synuclein, DJ-1, PINK1, and parkin.
α-Synuclein is localized to mitochondria-associated ER membranes (68). The different levels of α-synuclein oligomerization have been linked to cell death. In particular, a heterogeneous mixture of small oligomers of α-synuclein can lead to Ca2+ dysregulation to the point of mPTP activation and commitment to neuronal cell death (36). Studies have shown that α-synuclein positively affects Ca2+ transfer from the ER to the mitochondria (22, 89, 119, 161). This effect is correlated with an increase in the number of MAMs through the C-terminal of the α-synuclein domain (22). Moreover, it has been proposed that the accumulation of α-synuclein in the cells causes the redistribution of α-synuclein to localized foci and reduces the ability of mitochondria to accumulate Ca2+, resulting in augmented autophagy with the risk of altering normal mitochondrial homeostasis (22).
DJ-1 is largely cytoplasmic, except for a pool localized in mitochondria, most likely within the IMS and in the matrix; whereas little, if any, DJ-1 is associated with the outer and inner mitochondrial membranes (41). Numerous studies support the role of DJ-1 in mitigating oxidative stress and in the maintenance of mitochondrial homeostasis (41). A recent study proposed that DJ-1 modulates ER-mitochondrial Ca2+ cross-talk, favoring the tethering between the two organelles and, thus, MAMs formation (135).
Parkin is a ubiquitin-protein ligase that localizes to the mitochondrial matrix, where it enhances mitochondrial gene transcription and biogenesis in proliferating cells (84). Parkin has been proposed to play a neuroprotective role, promoting the clearance of damaged mitochondria through the mitophagic process (127). In Drosophila, parkin null mutants exhibit defects in mitochondrial function and increased oxidative stress. In contrast, overexpression of parkin in cultured cells prevents mitochondrial swelling and stress-induced apoptosis (84). Parkin overexpression stimulates MAMs formation and, in turn, physically and functionally enhances ER–mitochondria coupling, thus favoring Ca2+ transfer from the ER to the mitochondria and energy metabolism (23).
As previously described, parkin associates with PINK1 in mitochondrial quality control pathways, promoting the selective degradation of damaged mitochondria through mitophagy (175, 185).
PINK1 is unambiguously localized to mitochondrial membranes, and PINK1 overexpression protects cells from mitochondrial depolarization and apoptosis (183). Loss-of-function mutations are responsible for dopaminergic neuronal degeneration in Drosophila (139). Overexpression of parkin has been shown to rescue the mitochondrial dysfunction caused by PINK1 deficiency (184).
Finally, a mutant form of PINK1 has been shown to exacerbate mitochondrial alterations (disturbing mitochondrial Ca2+ fluxes) promoted by an α-synuclein mutant, thus suggesting cooperative roles for these two proteins (118).
GM1-gangliosidosis represents an additional example of a MAMs-related pathology. This neurodegenerative disease is characterized by GM1-ganglioside (GM1) accumulation within MAMs in brain tissue (156).
In addition, series of independent observations highlighted a possible role for MAMs in Huntington's disease. Mutant, but not wild-type huntingtin (Htt; the protein that, when mutated, is responsible for the disease), localizes to the mitochondrial membranes in neurons (136). Htt forms a ternary complex with Htt-associated protein-1A (HAP-1A) and IP3R (a protein enriched at MAMs). In this complex, mutant Htt, but not wild-type Htt, facilitates Ca2+ release from the ER and renders neurons more sensitive to Ca2+-mediated cellular dysfunction via mitochondrial Ca2+ overload (9) and mPTP opening (Fig. 8) (27).
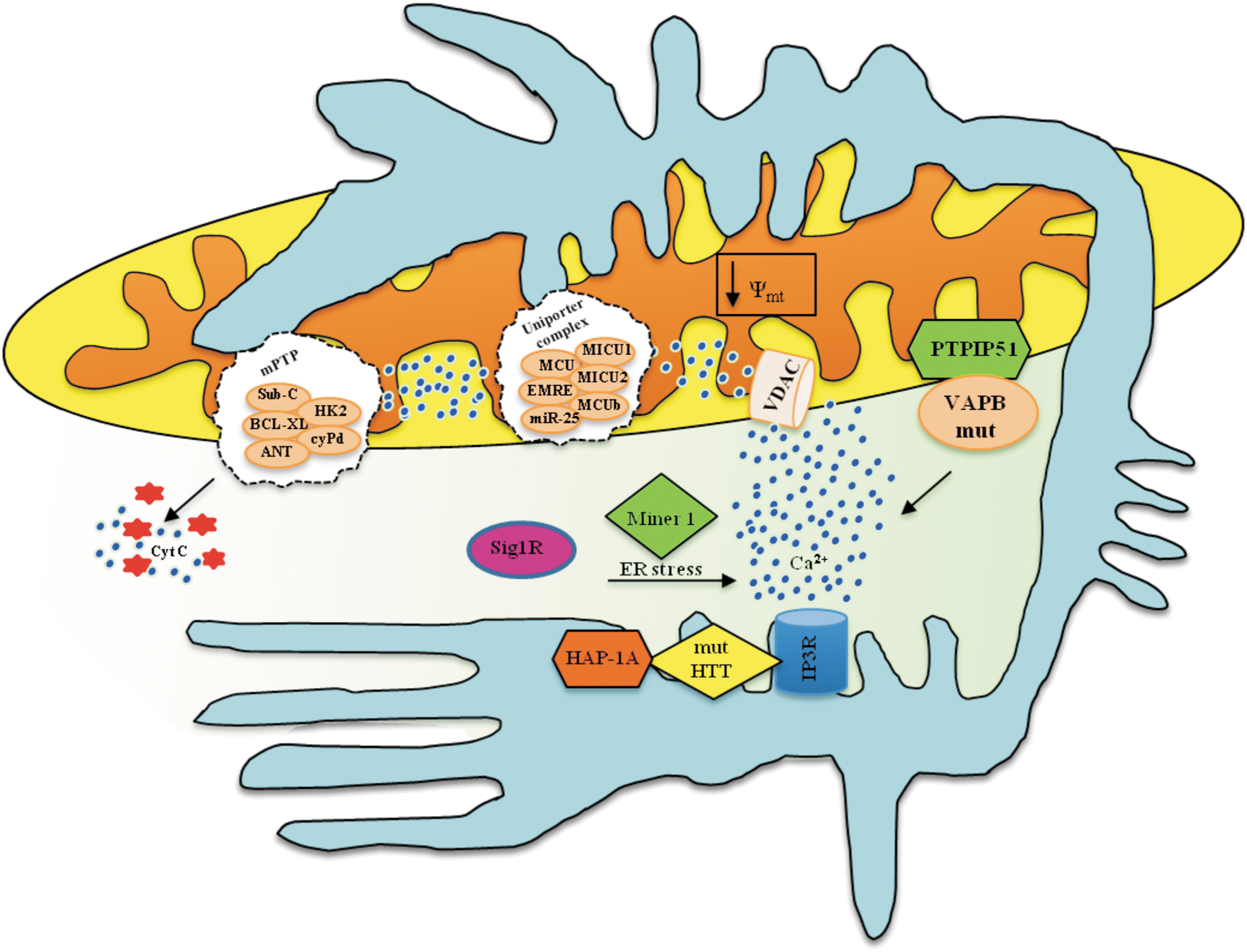
MAMs involvement appears to also be important in amyotrophic lateral sclerosis (ALS). Vesicle-associated membrane protein-associated protein B (VAPB) has been proposed to play a role in the pathogenesis of this neurological disease, and recent studies localize this protein at MAMs, where it interacts with protein tyrosine phosphatase interacting protein 51 (PTPIP51), which is an outer mitochondrial membrane protein, and regulates ER-mitochondrial Ca2+ cross-talk. The VAPBP mutant associated with ALS has been demonstrated to alter its binding to PTPIP51 and exacerbate ER-mitochondrial Ca2+ transfer (39) (Fig. 8).
Sig1R, which is another important protein that is particularly enriched at MAMs and is involved in Ca2+ homeostasis, has been proposed to be involved in the pathogenesis of ALS (148) (Fig. 8).
Interestingly, in several studies, mitochondrial dysfunction has been reported to be frequently associated with demyelination, whereas proper mitochondrial function is required for correct oligodendrocyte differentiation and myelination. Bonora and colleagues reported that correct mitochondrial Ca2+ signaling and, thus, MAMs activity are impaired in conditions mimicking the proinflammatory environment to which the oligodendrocytes are exposed in multiple sclerosis patients. These abnormalities result in inefficient oligodendrocyte differentiation (13).
Finally, MAMs are also emerging as critical intracellular domains in the pathogenesis of the incurable disease Wolfram syndrome. This concept is demonstrated by the observation that Miner1, which is a protein that, if mutated, causes Wolfram syndrome, is enriched at MAMs, and is involved in ER stress and Ca2+ signaling around MAMs, as previously described for other proteins involved in mitochondrial structure and physiology (180).
These results highlight MAMs involvement in the pathogenesis of several neurological disorders. Studies on this topic are ongoing in several groups, including ours, and we predict that in the future, new findings will consolidate this concept for the disorders presented here and for numerous others.
Concluding Remarks
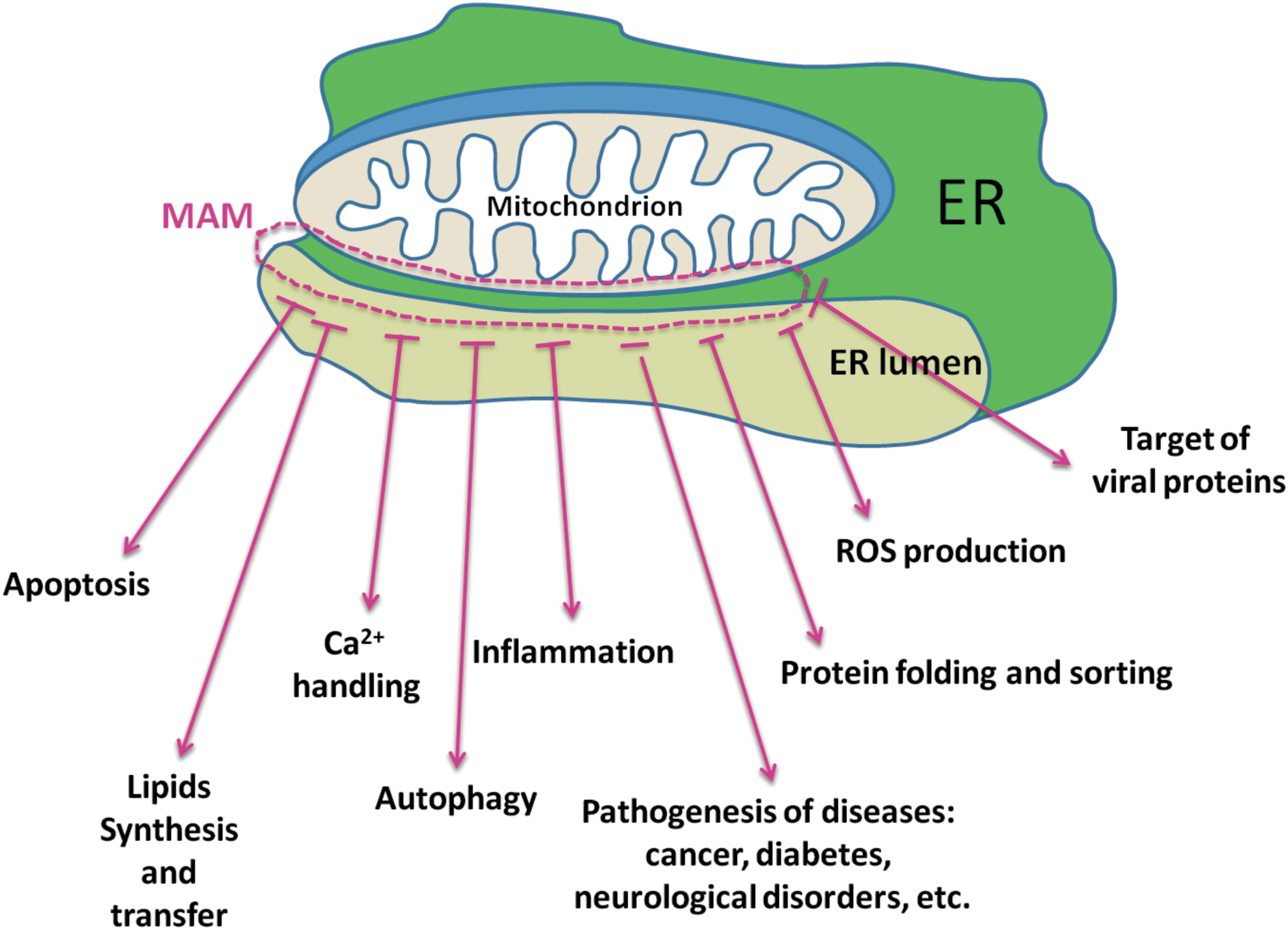
Overall, the picture emerging from the study of MAMs appears to be extremely complex (Fig. 9), with several uncertainties to be resolved. Nevertheless, the common role of MAMs in important physiopathological pathways is clear, providing a leading theme for future studies. However, whether alterations in MAMs represent a response to the disease pathogenesis or directly contribute to the disease has not yet been unequivocally established. In any case, MAMs represent a promising pharmacological target for several important human diseases. Finally, the following important questions remain open: (i) What are the entire proteomes of the MAMs in the different cell and tissue types under normal conditions and under stress or pathological conditions? (ii) Are the MAMs identical within the cell, or is any cell endowed with several different types of MAMs with respect to protein composition? (iii) What are the precise molecular mechanisms of MAMs formation? and (iv) How are the proteins and lipids targeted to MAMs?
Footnotes
Acknowledgments
This study was supported by the Italian Association for Cancer Research (AIRC) and local funding from the University of Ferrara to Paolo Pinton and Carlotta Giorgi; and from Telethon (GGP11139B), the Italian Cystic Fibrosis Foundation (Grant 19/2014), the Italian Ministry of Health, the Italian Ministry of Education, and the University and Research (COFIN, FIRB, and Futuro in Ricerca) to Paolo Pinton. Mariusz R. Wieckowski and Jerzy Duszynski were supported by the Polish National Science Center (UMO-2011/11/M/NZ3/02128), the Polish Ministry of Science and Higher Education grant W100/HFSC/2011 and Grant HFSP RGP0027/2011. Simone Patergnani was supported by a research fellowship from FISM (Fondazione Italiana Sclerosi Multipla), cod. 2012/B/11.