
Abstract
Introduction
T
Myocardial Energy Substrate Metabolism Under Physiological Conditions
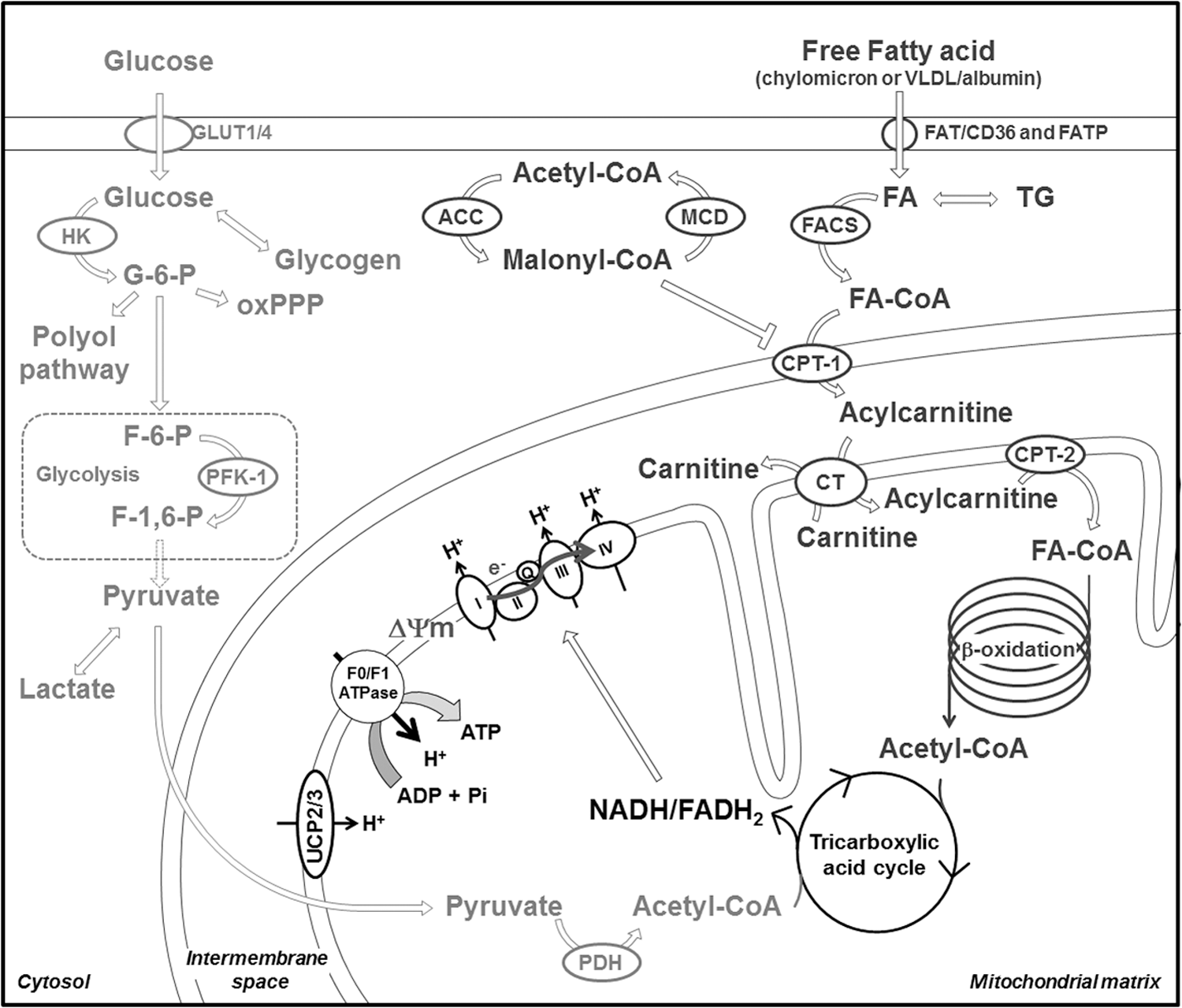
Under resting conditions and in inter-prandial phases, the healthy heart obtains 70%–80% of the energy from the oxidation of free fatty acids (FFA) and the remainder from carbohydrates. These metabolites are defined as cardiac energy substrates. The reader is referred to extensive reviews on this topic (53, 82 –84); however, a synthetic description of cardiac substrate metabolism is necessary to better understand how its derangement can favor oxidative stress in dCM and HF.
Fatty acid metabolism
Circulating FFA enter cardiomyocytes either by passive diffusion or by facilitated transport due to cell membrane free fatty acid translocase (FAT), also known as “cluster of differentiation 36” (CD36). Deletion (45) or inhibition (55) of FAT/CD36 causes a 50%–60% decrease in cardiac FFA uptake. In the cytosolic compartment, long-chain FFA are converted into long-chain fatty acyl-coenzyme-A (CoA) esters by fatty acyl-CoA synthase and successively conjugated with carnitine by carnitine palmitoyltransferase-1 (CPT-1) to form long-chain fatty acylcarnitine (99) (Fig. 1). CPT-1 is the key-limiting step determining the rate of FFA entry in mitochondria, and its activity is regulated allosterically by the inhibitor malonyl-CoA (42, 53, 58, 68). The concentration of malonyl-CoA depends on the balance between its synthesis catalyzed by acetyl-CoA carboxylase (ACC) (52, 74), and its degradation is catalyzed by malonyl-CoA decarboxylase (MCD) (21, 75) (Fig. 1), the activity of which is regulated by the so-called “fuel sensor” AMP-activated protein kinase (AMPK) (22, 33). In response to low levels of chemical energy, AMPK is activated and exerts an inhibitory action on ACC with consequent reduction of malonyl-CoA levels and an increase in CPT-1 activity. In mitochondria, long-chain fatty acyl-CoA undergo complete degradation in the β-oxidation cycle, leading to the production of acetyl-CoA, which is then catabolized in the tricarboxylic acid cycle (TCA) to generate FADH2 and NADH, that is, the reducing equivalents that feed the electron transport chain (ETC) (Fig. 1).
Carbohydrate metabolism
As mentioned earlier, while the heart relies mainly on FFA, ∼10%–40% of the acetyl-CoA used for ATP production is derived from the oxidation of pyruvate, the final product of lactate dehydrogenation and glycolysis (29, 82, 98) (Fig. 1).
Glucose transport into cardiomyocytes is facilitated by the glucose transporters (GLUTs) of sarcolemma membrane. The main cardiac GLUT isoforms are GLUT1 and GLUT4 (38), with the first constitutively active, while the second translocates to the sarcolemma in response to insulin, increased workload, and ischemia (61, 102). AMPK activation is also involved in this process (62). Once in the cardiomyocyte, glucose is phosphorylated by hexokinases (20). Glucose-6-phosphate derived from exogenous glucose and intracellular glycogen pool (14) undergoes progressive breakdown through the glycolytic pathway or, to a lesser extent, the oxidative pentose phosphate pathway (oxPPP) and the hexosamine biosynthetic pathway (43) (Fig. 1).
Most of the pyruvate generated from glycolysis or lactate dehydrogenation is decarboxylated in mitochondria to form acetyl-CoA (70). This reaction is a key-limiting step for carbohydrate oxidation and is catalyzed by pyruvate dehydrogenase (PDH), located in the mitochondrial matrix. PDH is finely regulated by specific PDH kinase (PDK) and phosphatase and functions as a sensor of fuel availability. For instance, high rates of FFA β-oxidation lead to increased levels of acetyl-CoA/free CoA and NADH/NAD+ ratios and subsequent activation of PDK, which, in turn, inhibits PDH. The mutual inhibition between FFA and carbohydrate oxidation was first described by Randle in the 1960s and generally referred to as the “Randle cycle” (64, 71).
TCA and ETC
TCA is responsible for the oxidation of acetyl-CoA generated from energy substrate breakdown. The chemical energy is accumulated in reducing equivalents and utilized for the synthesis of ATP by the ETC. Each cycle of the TCA generates three NADH and one FADH2, in addition to one molecule of GTP.
The oxidation by the ETC of all reducing equivalents available to mitochondria leads to an electrochemical gradient that, while essential for ATP synthesis, can also promote ROS production under pathological conditions such as diabetes.
Transcriptional control of cardiac metabolism
Enzymes involved in FFA and carbohydrate metabolisms are finely regulated at transcriptional level. Important transcription factors for FFA metabolism are the three isoforms of peroxisome proliferator-activated receptors (PPARs): PPARα, PPARβ/δ, and PPARγ (35, 100). They form heterodimers with the retinoid X receptor-α (RXRα) and the PPARγ coactivator-1 (PGC-1) and bind to the PPAR response element located in the promoter regions of many genes encoding for proteins involved in metabolism. PPARα is highly expressed in the heart, activated by long-chain FFA, and regulates the expression of numerous enzymes involved in different steps of FFA metabolism, from uptake (FAT/CD36, FATP1) to mitochondrial entry (CPT-1, MCD) and β-oxidation (acyl-CoA dehydrogenase, 3-ketoacyl-CoA thiolase) (35, 100) (Fig. 2).

Metabolic Alterations Promoting Cardiac Oxidative Stress in dCM
Type I and II diabetes are characterized by hyperglycemia and hyperlipidemia. This excess of circulating substrate concentration, along with the lack of insulin or insulin resistance in type I and II diabetes, respectively, leads to changes in myocardial metabolism characterized by enhanced FFA oxidation and markedly impaired glucose oxidation (11, 95). This cardiac metabolic phenotype favors oxidative stress, that is, the imbalance between mitochondrial and/or cytosolic ROS generation and antioxidant defenses. Most of the studies on cardiac metabolism in dCM have been performed in experimental models and only a few have been performed in patients. Oxidative stress was documented in atrial tissue from patients with type II diabetes (2). The authors of this study found an increased mitochondrial hydrogen peroxide (H2O2) generation and high levels of hydroxynonenal-modified and 3-nitrotyrosine-modified proteins, two hallmarks of oxidative stress. Interestingly, H2O2 emission, reflecting the net balance between H2O2 generation in the ETC and scavenging by the matrix antioxidant system, was increased regardless of the type of oxidized substrate or the respiratory state. Moreover, the atrial tissue displayed impaired mitochondrial capacity to oxidize palmitoyl-carnitine and glutamate. A strength of this study was the recruitment of an appropriate control group, in that both nondiabetic and diabetic patients had a body mass index of ∼32 kg/m2. However, the contribution of obesity in the development of mitochondrial alterations in atria of diabetic patients could not be determined. A more recent study in a similar model of atrial tissue from type II diabetic patients elucidated this particular aspect (60). Interestingly, the authors showed that both diabetes and obesity impair atrial muscle contractility, but only diabetes also causes mitochondrial dysfunction and oxidative stress. They concluded that hyperglycemia is the major driver of oxidative stress in the diabetic heart. In the next few paragraphs, we will try to separate the relative contribution of glucose and FFA from the pathogenesis of cardiac oxidative stress in dCM.
Glucose metabolism and ROS generation
Nonmitochondrial ROS generation
Hyperglycemia triggers nonmitochondrial ROS generation through several interconnected pathways, forming a complex network (Fig. 3). A recently identified mechanism is of particular interest, since it involves the uptake of glucose by cardiomyocytes through a less known transporter, namely the sodium/glucose co-transporter (SGLT1) (6). Rat cardiomyocytes exposed to a high glucose concentration displayed the activation of the small GTPase Ras-related C3 botulinum toxin substrate 1, followed by activation of the NADPH oxidase Nox2 and therefore superoxide production. Interestingly, Nox2 can also be activated by 3-O-methyl-
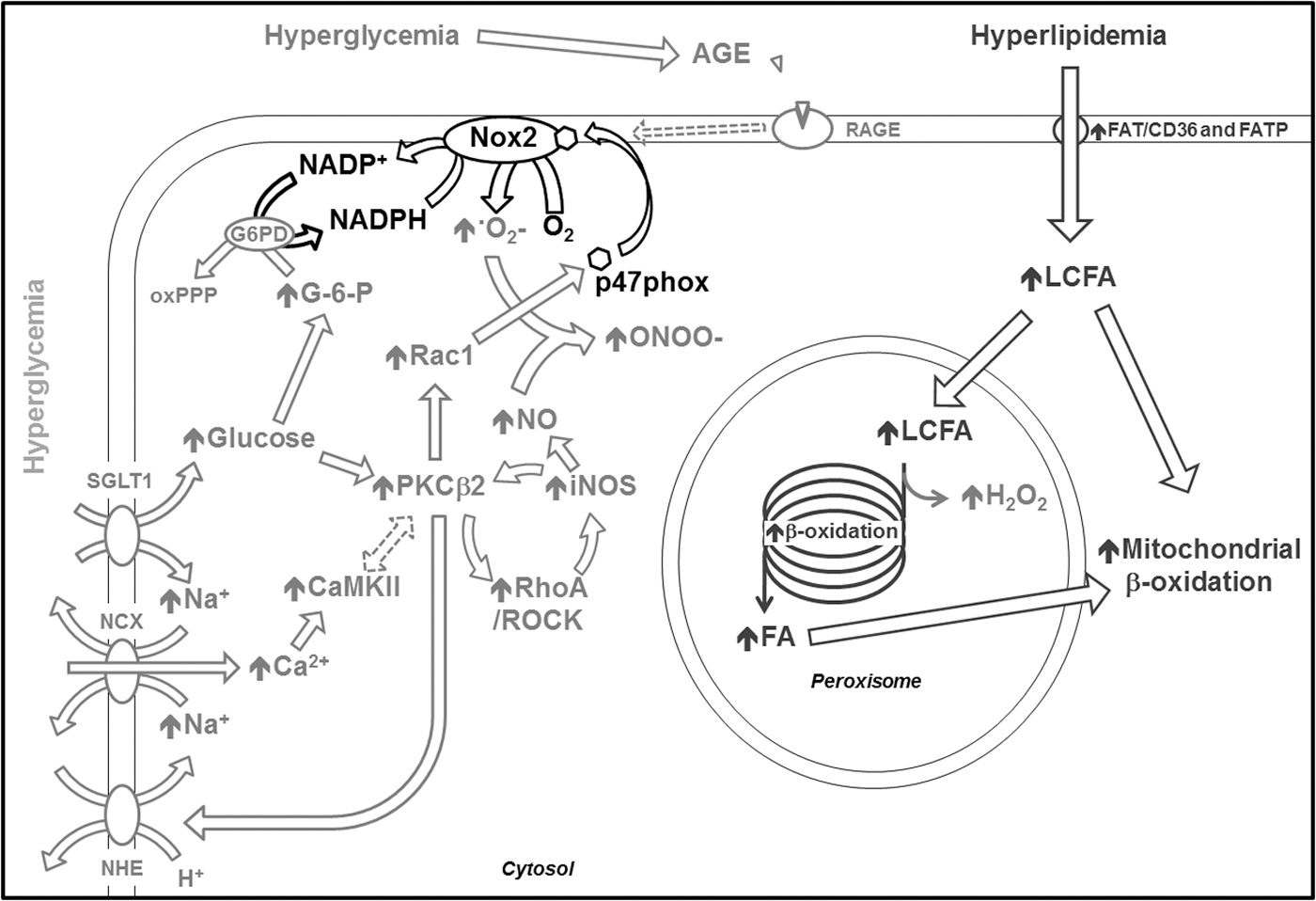
The studies mentioned earlier either demonstrated (76) or suggested (6) the involvement of PKC in the activation of Nox2 in response to high glucose concentrations, in accordance with other reports. In fact, it has been shown that Nox2 activation in cardiomyocytes exposed for 12 h to high glucose concentration was prevented by the PCKβ2 inhibitor LY333531 (96). Another study in cardiomyocytes exposed for 24 h to high glucose concentrations or isolated from type I diabetes rats found that PCKβ2 induction by hyperglycemia is involved in the production of ROS through a positive feedback loop involving the RhoA/Rho kinase pathway and the overexpression of the inducible nitric oxide synthase (78) (Fig. 3). These findings have been further supported by a more recent report (48). Of note, inducible nitric oxide synthase upregulation is known to be associated with nitrosative stress caused by peroxynitrite, the product of NO and superoxide reaction (36).
Another pathway linking hyperglycemia to ROS generation via NADPH oxidase involves the calcium/calmodulin-dependent kinase II (CaMKII) (66) (Fig. 3). The authors of this study found hyper-phosphorylation of CaMKII caused by elevated intracellular Ca2+ concentration in a rat model of type I diabetes, as well as in cultured neonatal rat cardiomyocytes exposed to high glucose concentrations. The proposed mechanism is the following: High glucose concentrations activate the Na+-H+ exchanger, leading the Na+-Ca2+ exchanger to work in a reverse mode to compensate for the increased intracellular concentration of Na+. The consequent cellular influx of Ca2+ causes CaMKII activation. Interestingly, Na+-H+ exchanger upregulation driven by PKC in response to high glucose has been shown in cultured vascular smooth muscle cells (97). Moreover, a link between PKC and CaMKII was already found in the hippocampus of type I diabetic rats (50).
Hyperglycemia also contributes to the generation of advanced glycation end-products (AGE) that, through their receptor RAGE, can induce oxidative stress (Fig. 3). While only a few groups explored the involvement of AGE and RAGE in cardiac oxidative stress, more solid evidence has been found in other cell types such as cardiac endothelial cells or pancreatic β-cells [for review, see Daffu et al. (19)]. Moreover, AGE can increase cellular oxidative stress through the inhibition of antioxidant defenses.
Mitochondrial ROS generation
Two hypotheses attempt to link mitochondrial respiration and ROS level: the “mild uncoupling” (9, 44) and the more recent “redox-optimized ROS balance” (4, 18). Mild uncoupling is related to the mitochondrial membrane potential, while redox-optimized ROS balance is related to the redox environment as the intermediate between mitochondrial respiration and ROS level. In mitochondria, superoxide is produced through complexes I and III of the ETC (37). High levels of ROS in cardiac mitochondria have been found in diabetic humans (2) and experimental animals (76). Transgenic mice overexpressing manganese superoxide dismutase (SOD), that is, the mitochondrial isoform of this enzyme, are protected against oxidative damage caused by type I diabetes (77). Moreover, increased catalase expression or inhibition ETC complexes I and II with rotenone and thenoyltrifluoroacetone, respectively, attenuate ROS generation in cardiomyocytes from animals with type I and II diabetes, pointing to mitochondria as the primary source of ROS in these models (101) (see Aon et al. in this Forum). Other interesting results linking hyperglycemia to mitochondrial ROS generation came from a study demonstrating, in bovine aortic endothelial cells, a blockade of three pathways involved in hyperglycemia-induced alterations by the normalization of mitochondrial ROS (65). Unfortunately, this study did not investigate the signaling pathway linking hyperglycemia to mitochondrial ROS generation.
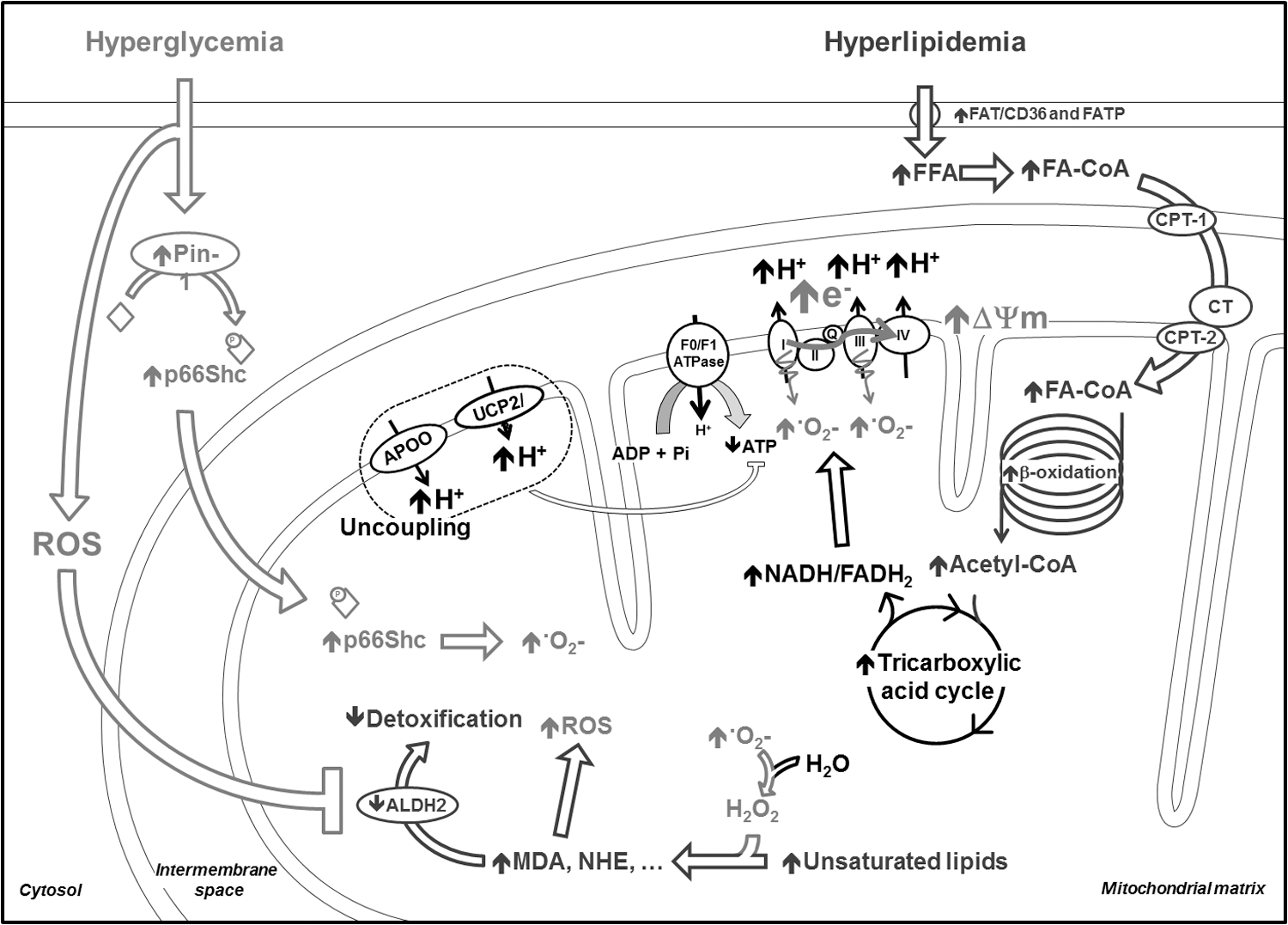
Regarding the potential pathways involved in hyperglycemia-induced ROS generation, an elegant study recently described the increased expression and activity of the peptidyl-prolyl cis-trans isomerase (Pin1) and of the Ser-36 phosphorylation by the growth factor adapter protein (p66Shc), associated with an increased mitochondrial superoxide concentration in human aortic endothelial cells exposed to high glucose concentrations (67) (Fig. 4). Interestingly, the mitochondrial superoxide level was normalized when the cells exposed to high glucose concentrations were treated with an anti-Pin1 siRNA. Moreover, the same authors also showed Pin1 overexpression in aorta of type I diabetic mice. Using Pin1−/− transgenic mice, the authors confirmed the involvement of this enzyme in the hyperglycemia-induced mitochondrial superoxide overproduction. Finally, an increased Pin1 expression and activity was also found in the blood of type II diabetic patients and correlates with HbA1c and fasting plasma glucose concentrations (67). Pin1 activity also correlates with the concentration of 8-iso-prostaglandin F2α, a marker of oxidative stress. This study was focused on vascular dysfunction; however, it may be considered of interest in the cardiology field, since other studies demonstrated a beneficial effect of p66Shc deletion on cardiac function and oxidative state in models of diabetes (56, 59). For instance, isolated rat cardiomyocytes transduced with a dominant-negative p66Shc exhibit attenuated high glucose-induced ROS production and more stable mitochondrial energetics (56). Moreover, mice with type I diabetes display an increased phosphorylation and translocation of p66Shc in mitochondria (Fig. 4).
Other potential mitochondrial sources of ROS are monoamine oxidases (MAO) A and B. Although no available data support the involvement of MAO in dCM, a possible pathogenic role of these constitutive ROS generators has been recently found in mice with cardiac overload (39, 40) and it is plausible that these enzymes are involved in hyperglycemia-induced cardiac ROS generation in the setting of dCM. Interestingly, increased levels of MAO-A have been found in the heart of the streptozotocin-induced diabetic rat, which were normalized by multiple antioxidant administration (46).
Finally, the end products of lipid peroxidation (oxidation of unsaturated lipid by H2O2) such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE) (1) are known to be involved in mitochondrial ROS generation (47). Interestingly, a recent study in type I diabetic rats found that hyperglycemia-induced ROS inhibits aldehyde dehydrogenase 2 (ALDH2), an enzyme involved in the oxidation and detoxification of aromatic and aliphatic aldehydes such as HNE (94). ALDH2 decreased activity is also correlated to the plasmatic concentration of glycosylated hemoglobin and cardiac alterations in another study using type I diabetic rats after an intraperitoneal injection of streptozotocin (93). Such findings revealed interactions between lipid- and glucose-induced ROS generation and between nonmitochondrial and mitochondrial ROS generation, thus adding a further level of complexity to the ROS generation network.
Lipid metabolism and ROS generation
As described earlier, cardiac FFA uptake and oxidation is enhanced in patients (69) and in animal models of type I and II diabetes (11, 13, 95). During the early stages of the disease, insulin resistance mainly causes this metabolic alteration. In more advanced stages, high FFA levels activate PPARα, which, in turn, upregulates the enzymes involved in FFA uptake and β-oxidation (26). Large amounts of the reducing equivalents NADH and FADH2 are produced due to enhanced FFA β-oxidation and acetyl-CoA oxidation in the TCA. They cause an excessive flux of protons across the ETC with consequent hyperpolarization of the inner mitochondrial membrane (Fig. 4). The final effect is the inhibition of complex III in the ETC and the excessive superoxide generation by complexes I and III (10, 80, 86). An increase in reverse electron flow has also been suggested as a potential mechanism (10). Other sources of superoxide have been identified in mitochondria (10), for instance, the electron transfer flavoprotein:Q oxidoreductase associated to FFA β-oxidation (80).
Interestingly, it has been recently shown that tumor suppressor p53 and synthesis of cytochrome c oxidase 2 are enhanced by hyperglycemia-induced ROS in both type I and II diabetic mice. This phenomenon leads to increased ETC complex IV activity and consequent enhancement of FFA oxidation and ROS production by the ETC (63), suggesting another possible interaction between hyperglycemia and hyperlipidemia-induced ROS. Of note, ROS generation by ETC is counteracted by uncoupling proteins (UCP), which allow the flux of protons back in the mitochondrial matrix, hence favoring oxygen consumption uncoupled from ATP production. In fact, the UCP isoform 3 has been found to be upregulated in type I diabetic hearts (13). Unfortunately, based on the mild uncoupling hypothesis, this potentially protective mechanism necessarily lowers cardiac efficiency (15). Another protein recently identified as potentially involved in ETC uncoupling is the apolipoprotein O (88). On the one hand, this protein prevents ETC-derived ROS generation, while on the other hand it depletes mitochondrial-reducing equivalents, thus favoring the accumulation of FFA intermediate metabolites in cardiomyocytes. Among these, ceramides are known to directly interact with the ETC and induce ROS generation (28).
Finally, the FFA β-oxidation occurring in peroxisomes, although minor, can become a source of ROS in the diabetic heart, as recently shown in a mouse model of a high-fat diet (73) (Fig. 3).
Despite abundant data supporting the detrimental role of enhanced FFA oxidation in diabetic hearts, some experimental evidence suggests an apparently paradoxical acute beneficial effect of palmitate on redox homeostasis and cardiac function in type II diabetes mouse models (24, 85). When challenged with high glucose and the beta-adrenergic receptor agonist isoproterenol, cardiomyocytes isolated from db/db mice, a model of type II diabetes, display mitochondrial ROS overproduction and contractile dysfunction (85). These alterations could be prevented not only by the anti-oxidant molecule glutathione but also by exogenous palmitic acid. The mechanisms underlying this interesting phenomenon are not yet clear. The beneficial effects of palmitate on redox balance and cardiac contractility have been recently confirmed in the Zucker Diabetic fatty rat (8), a model of type II diabetes. The authors of this study demonstrate that palmitate is able to rescue contractile function of heart trabeculae harvested from diabetic (but not from normal) hearts exposed to high glucose. These beneficial effects are associated with an increased scavenging capacity of the antioxidant system (glutathione and thioredoxin) and reduced oxidative stress (8). In this regard, Aon et al. recently reviewed the cellular management of lipid excess focusing on the involvement of lipid droplets and hypothesized a “metabolic channeling of lipid utilization from close contacts between mitochondria and lipid droplets” (3).
Metabolic alterations and antioxidant defenses
Under physiological conditions, antioxidant defenses neutralize ROS to prevent oxidative stress. They include SOD, gluthathione peroxidase, catalase, thioredoxin, vitamins C and E, and coenzyme Q10 (35, 36, 57, 81, 100). Diabetes is characterized by an impairment of antioxidant defenses, which further amplifies oxidative stress due to increased ROS production. These biochemical alterations have been well characterized in the heart and recently reviewed (36). However, very little is known about the specific role played by hyperglycemia and hyperlipidemia. It has been recently shown that hyperlipidemia induces catalase overexpression in mice subjected to a high-fat diet (73). In this model, the increased concentration of H2O2 induces the activation of c-Jun N-terminal kinase, which promotes the phosphorylation of the transcription factor family FOXO, leading to catalase overexpression (73). The capacity of rat hearts to counteract the oxidative stress caused by a chronic high-fat/high-sugar diet by upregulating thioredoxin reductase-2 has been recently shown, although the signaling involved in this mechanism was not investigated (27). Conversely, other authors found an increased expression of the thioredoxin endogenous inhibitor named thioredoxin interacting protein, and therefore reduced anti-oxidant defenses, after hyperglycemia in patients with diabetes and in rodents with type I and II diabetes (16). Lower pools of reduced glutathione and thioredoxin reductase-2 have been found in energetically/redox stressed mitochondria isolated from hearts of type II diabetic db/db mice (85). The authors of the study attribute a direct causative role to these alterations in determining cardiomyocyte contractile dysfunction.
Although no evidence is yet available in hearts, it should be mentioned that AGE have also been involved in the impairment of antioxidant defenses in human embryonic kidney cells in culture (15). Cells treated with two different AGE types presented an increase phosphorylation of FKHRL1, which subsequently leads to a 70% downregulation of the MnSOD in mitochondria (15). Finally, the polyol pathway was found to be hyperactivated in the heart of BBZ rats, which develop spontaneous type II diabetes: The concentration of both sorbitol and fructose, products of this pathway, was increased in diabetic rat hearts at 12 and 48 weeks of age (49). Although this study did not investigate the involvement of this polyol pathway upregulation in oxidative stress, others hypothesized its causative role in the decreased availability of NADPH necessary for glutathione reductase (91).
Metabolic Alterations Promoting Cardiac Oxidative Stress in HF
Role of altered glucose metabolism
Very little is known about the involvement of metabolic alterations in the generation of ROS in HF. Moreover, some discrepancies are found among the few studies that investigated this phenomenon, due to complexity of the HF syndrome and its evolution. One of the first studies proposing a link between metabolic alterations and oxidative stress in HF was performed in dogs with pacing-induced HF (31). The authors provided evidence that NAD(P)H oxidase, fueled by NADPH deriving from an upregulated oxPPP, is an important source of superoxide in failing hearts. NADPH might also feed another superoxide generator, the uncoupled NO synthase. The inhibitor of NAD(P)H oxidase gp91ds-tat abolished superoxide production in cardiac tissues homogenates, further confirming the involvement of this enzyme. While no changes in Nox-2 and Nox-4 expression were observed, the authors found an increased interaction between Nox-2 and its subunit p67phox, leading to its activation. The same group then performed a similar study in left ventricular tissue samples collected from HF patients (30). Not only they confirmed the findings obtained in dogs, but also, by using chelerythrine and PP2, respectively PKC and Src kinase inhibitors, provided evidence that both PKC and Src kinase coordinate G6PD and NAD(P)H oxidase activation. Moreover, using the same HF dog model, other authors demonstrated decreased protein and transcript levels of aldose reductase in the myocardium (79), indicating polyol pathway downregulation. One possible interpretation of these findings in HF dogs and patients is that the co-existence of cardiac enhanced glucose uptake, oxPPP upregulation, and decreased polyol pathway might lead to an excessive flux through NADPH-generating enzymes that, in turn, might feed not only the anti-oxidant system (glutathione peroxidase) but also superoxide generators such as NADPH oxidase and uncoupled NO synthase (Fig. 5). To further explore, in vivo, the link between glucose metabolism and oxidative stress, the effects of raising glycemic levels from ∼80 to ∼170 mg/dL were tested in dogs with pacing-induced HF (92). In response to this acute glycemic peak, cardiac isoprostane output, an index of oxidative stress, increased by approximately twofold, whereas oxPPP inhibition normalized oxidative stress and enhanced cardiac oxygen consumption, glucose oxidation, and stroke work. The clinical implications of these findings are evident. For instance, they suggest that physiological postprandial glycemic peaks might induce a marked cardiac oxidative stress in HF patients. In theory, an attenuation of the oxPPP activity would mitigate this stress. But the balance between the anti-oxidant and the pro-oxidant effects of reducing equivalents provided by the oxPPP is very delicate. In fact, mice with 60% genetic reduction of G6PD activity were not more protected against cardiac remodeling and dysfunction after myocardial infarction or transverse aortic constriction. On the contrary, the outcome in these mice was slightly worse compared with the control (34). In Dahl salt-sensitive rats fed a high-salt diet to induce progressive cardiac dysfunction, the oxPPP was found to be activated during the HF stage (41), consistent with findings in dogs (31). In these animals, administration of dichloroacetate, a PDH stimulator (Fig. 1), enhanced cardiac function and survival of the animals and activated the cardiac oxPPP. Although they could not provide direct evidence for it, the authors of this study attributed the beneficial effects of dichloroacetate to oxPPP activation. However, it is also possible that dichloroacetate redirected glucose from the oxPPP to the oxidative glycolytic pathway, thus subtracting substrate from NADPH-generating enzymes (Fig. 6). Only direct tracking of the entire glucose metabolic fate can help draw the right conclusions.
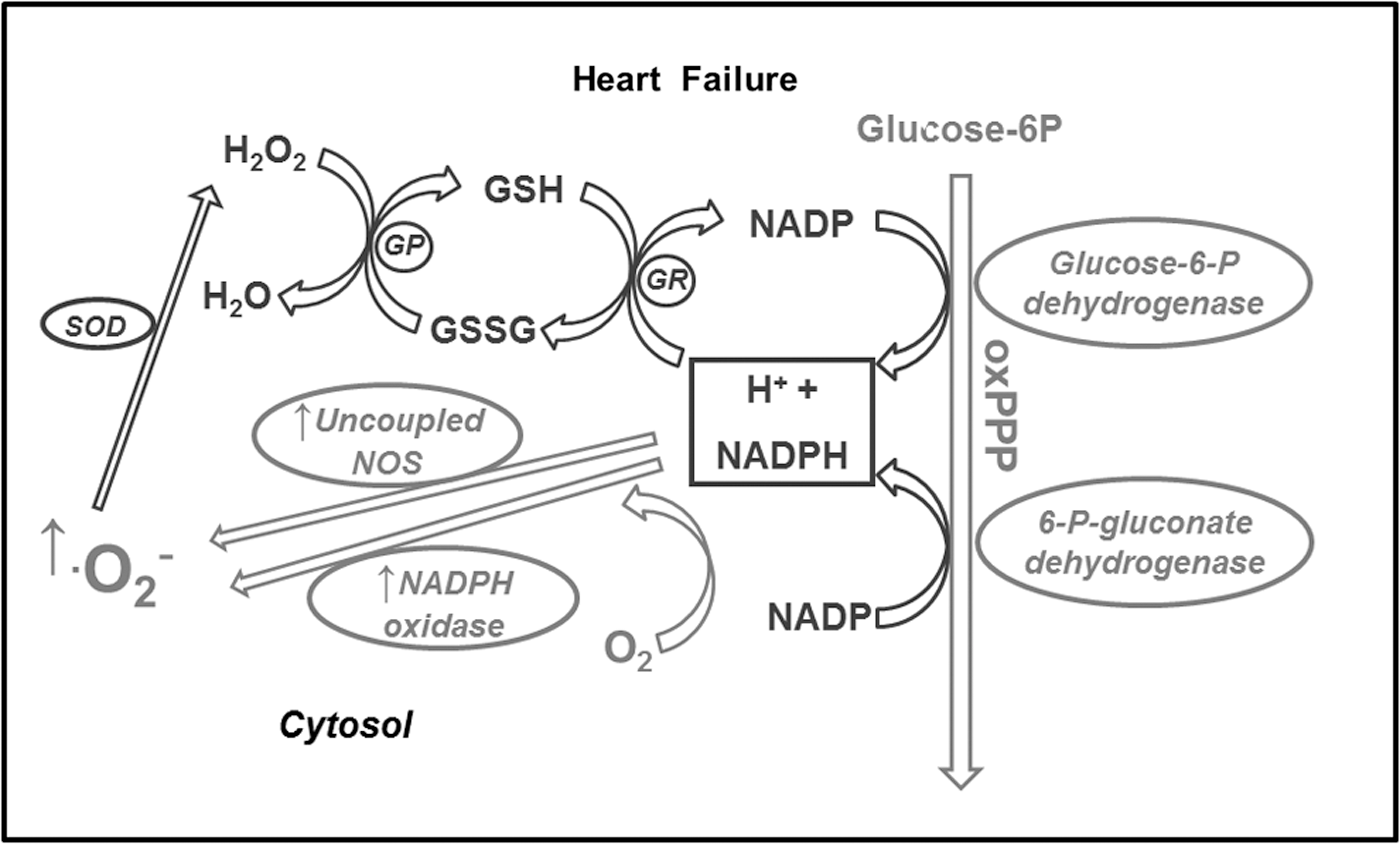
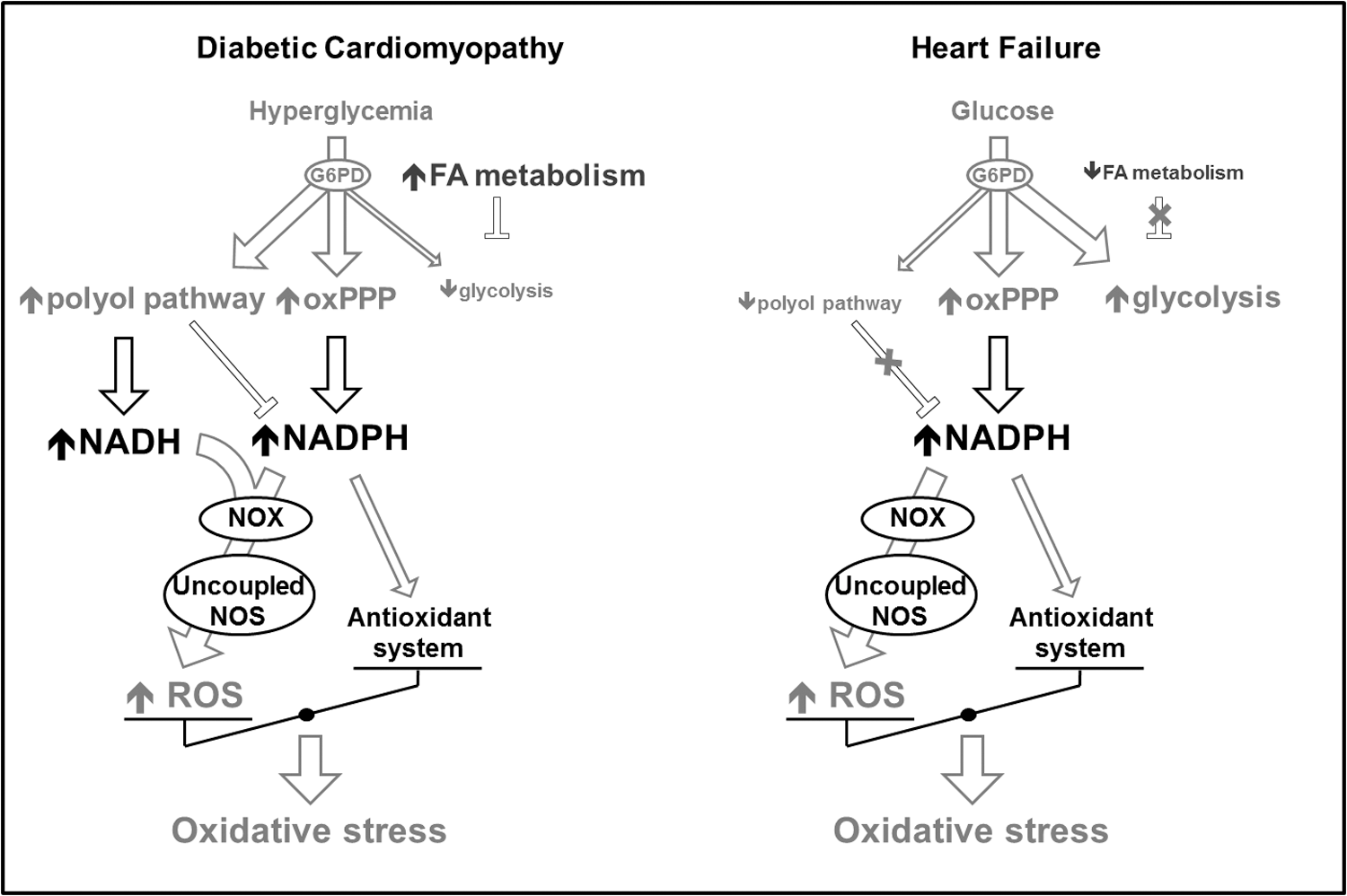
Currently, an alternative tool to explore overall glucose metabolism and therefore elucidate these complex aspects might be provided by computational analysis of the metabolomic profile in myocardial extracts. Using metabolomics data in intact Langendorff-perfused hearts (glucose as the only substrate), Cortassa et al. (17) were able to calculate the control of the glucose fluxome, including glycolysis, oxPPP, glycogenolysis, and polyol pathways. They found that the higher the glycolytic flux, the lower the oxPPP and polyol pathways. Otherwise stated, the structure of control reveals that an overall increase in the glycolytic flux will decrease the flux redirection through oxPPP and polyol pathways. In dCM, due to hyperglycemia, the glycolytic enzymes are impaired by fatty acid metabolism and Randle cycle, probably leading to an overfeeding of oxPPP and polyol pathways and an exaggerate competition for NAD(P)H, limiting its availability for glutathione peroxidase. In the absence of hyperglycemia, the polyol pathway activation probably plays a very minor role in HF (Fig. 6). Glucose must necessarily enter the glycolytic or the oxPPP. In addition, as previously mentioned, the polyol pathway is downregulated in HF (79) while the oxPPP is upregulated (31). Therefore, if further studies confirm it, the different involvement of polyol pathway and oxPPP might be considered a hallmark difference between glucose metabolism-induced oxidative stress in dCM and HF.
Role of altered FFA metabolism
The failing heart metabolic phenotype is characterized by a decreased FFA oxidation, as described in the previous paragraphs. PPARs and PGC-1, transcription factors responsible for the regulation of enzymes involved in FFA metabolism (Fig. 2), are downregulated in HF (5). Interestingly, these same transcription factors might also regulate the expression of enzymes that play a role in the anti-oxidant defense. In PGC-1α knockout mice, the decreased expression of antioxidant proteins such as SOD2 and thioredoxin reductase-2 likely contributed to decreased survival after transverse aortic constriction (54). In fact, myocardial oxidative stress markers 3′-nitrotyrosine and 4-hydroxynonenal were increased in association to more severe signs of HF compared with wild type. Similar results were found in PGC-1β-deficient mice (48, 72).
Conclusions
Despite the opposite metabolic alterations found in dCM and HF, they seem to have at least one common denominator, namely the generation of ROS. However, simply by evaluating the current literature, it is difficult to identify alterations of the energy substrate metabolic pathways, shared by the two pathological conditions, which might be responsible for enhanced oxidative stress. One of these alterations is perhaps the oxPPP over-activation (Fig. 6). In both dCM and HF, an increased glucose channeling in the oxPPP leads to the generation of a large amount of NADPH feeding the NAD(P)H oxidase and, subsequently, increasing ROS generation. While not surprising in the failing heart, which relies mainly on glucose, the over-activation of this pathway is less intuitive in dCM, especially in insulin-resistant patients, and calls for the search of potential therapeutic targets, such as glucose transporter SGLT1 and the PKC, apparently playing a central role in ROS generation associated to carbohydrate metabolism. Cardiac metabolic modulation is still a potentially interesting option for the treatment of dCM and HF and might also prove efficacious to mitigate oxidative stress (51).
Footnotes
Acknowledgments
Supported by National Institutes of Health Grants P01 HL-74237 and R01 HL-108213.