
Abstract
Introduction
T
A longer time course of disease progression also potentiates more severe complications as the number of risk factors is also increased. For instance, the risk of a diabetic patient developing cardiovascular disease increases as risk factors accumulate; aging, high blood pressure, cholesterol, and stress contribute to this. Furthermore, diabetes affects multiple organ systems, including the heart, nervous system, and kidneys. Prevention or effective treatment of complications is of paramount importance for improving patient care and quality of life.
The process of removing organelles and long-lived proteins through vesicular sequestration and lysosomal degradation is known as macroautophagy, hereafter called autophagy. In the context of acute myocardial injury, autophagy has been shown to be a critical cardioprotective process (55, 56, 105). Autophagy is the primary mechanism involved in removing whole mitochondria and is, therefore, essential for mitochondrial quality control. Damaged mitochondria that result from cardiac injury can produce reactive oxygen species (ROS) and release death-inducing factors, augmenting cardiac damage (87). Removal of these mitochondria by “mitophagy” prevents additional cell damage and limits tissue injury. However, prolonged autophagy can be detrimental and leads to cardiac atrophy (137). As diabetes is a disease of altered metabolism with slow, sometimes lifelong progression, cardiac function is at risk of deteriorating as a result of long-term dysregulation of autophagy and mitophagy.
Here, we present a discussion of the various factors involved in the progression of cardiovascular complications in diabetes. Cardiomyopathy affects a significant percentage of diabetic patients and greatly contributes to mortality. We discuss the progression of diabetic cardiomyopathy and the contribution of mitochondria to the pathogenesis. In addition, we examine the state of removal of mitochondria via autophagy in the diabetic heart, and draw conclusions about the feasibility of targeting autophagy for the treatment of diabetic cardiomyopathy.
Mechanisms of Autophagy
Autophagy is the primary process by which the cell degrades long-lived proteins and whole organelles, including mitochondria. Removal of mitochondria by mitophagy is critical to the well-being of cardiomyocytes during acute injury such as ischemia/reperfusion (I/R) (55, 56). By quickly degrading dysfunctional mitochondria, the cell can prevent excessive oxidative damage and streamline ATP production.
Autophagy begins with an initiation step that involves activation of the BECLIN1-VPS34-VPS15 complex by ULK1/2 (Fig. 1) (77). This is followed by elongation of the isolation membrane by the Autophagy-related (ATG) proteins ATG12 and ATG5, and conjugation of cytosolic LC3I to phosphatidylethanolamine, resulting in membrane-bound LC3II and expansion of the autophagosome membrane to surround the target material (75, 110). The fully formed autophagosome then fuses with a lysosome, resulting in autolysosome formation and cargo degradation, thus allowing for recycling of the subcomponents.
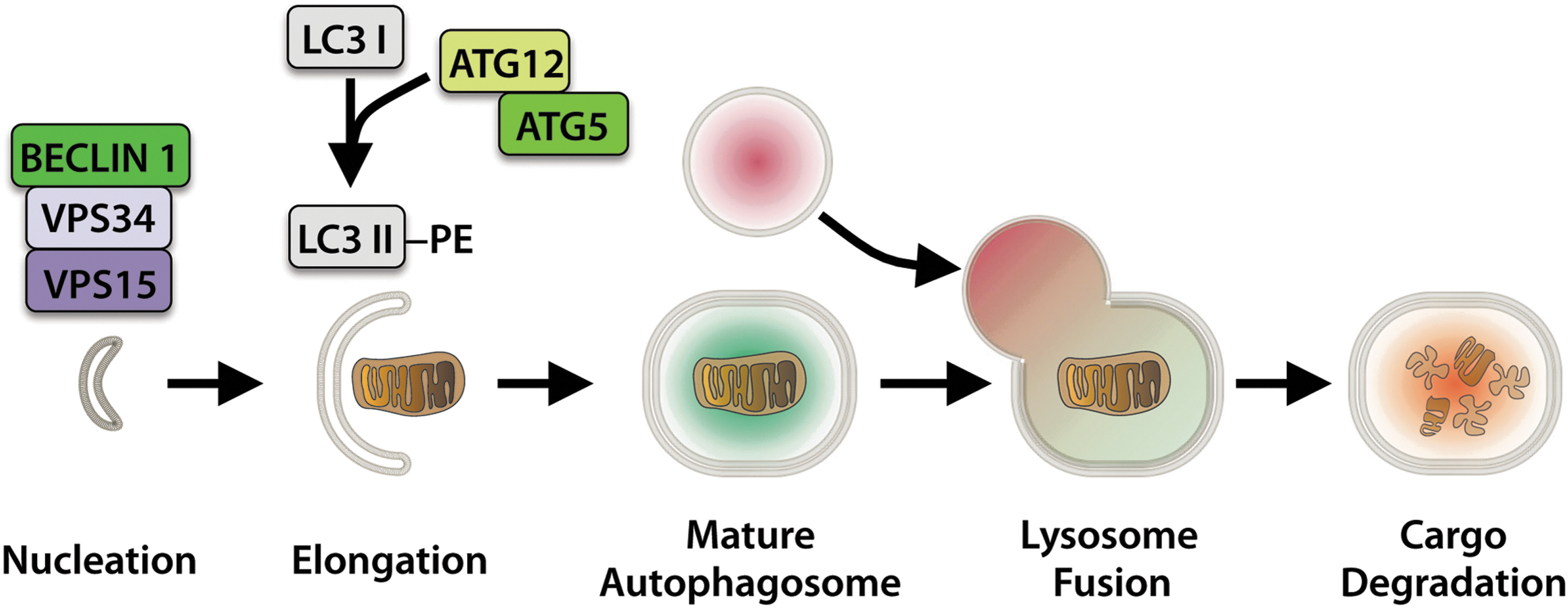
In the past, it was believed that lysosomal degradation through autophagy was of low specificity. However, in recent years, it has become clear that sequestration of cargo by an autophagosome is highly specific where individual dysfunctional mitochondria can be sequestered and degraded by targeted mitophagy to ensure a healthy population of mitochondria in the cell.
Mitochondrial Autophagy
Dysfunctional mitochondria can be detrimental to the health of a cardiomyocyte, necessitating removal of the damaged organelles for myocyte survival (87). There are several pathways that can regulate mitophagy in cells. The first pathway involves labeling of damaged mitochondria for autophagic degradation by PINK1 and PARKIN (Fig. 2). PINK1 is a serine/threonine kinase that is imported and rapidly degraded by mitochondrial proteases in healthy mitochondria (49). Upon loss of mitochondrial membrane potential, PINK1 accumulates on the surface of the mitochondria (73, 116). PINK1-mediated phosphorylation of outer membrane proteins, including MFN2, promotes PARKIN translocation from the cytosol to the mitochondria (22, 104, 116). PARKIN, an E3 ubiquitin ligase, ubiquitinates mitochondrial outer membrane proteins to promote recruitment of the autophagosome (42, 43, 121).
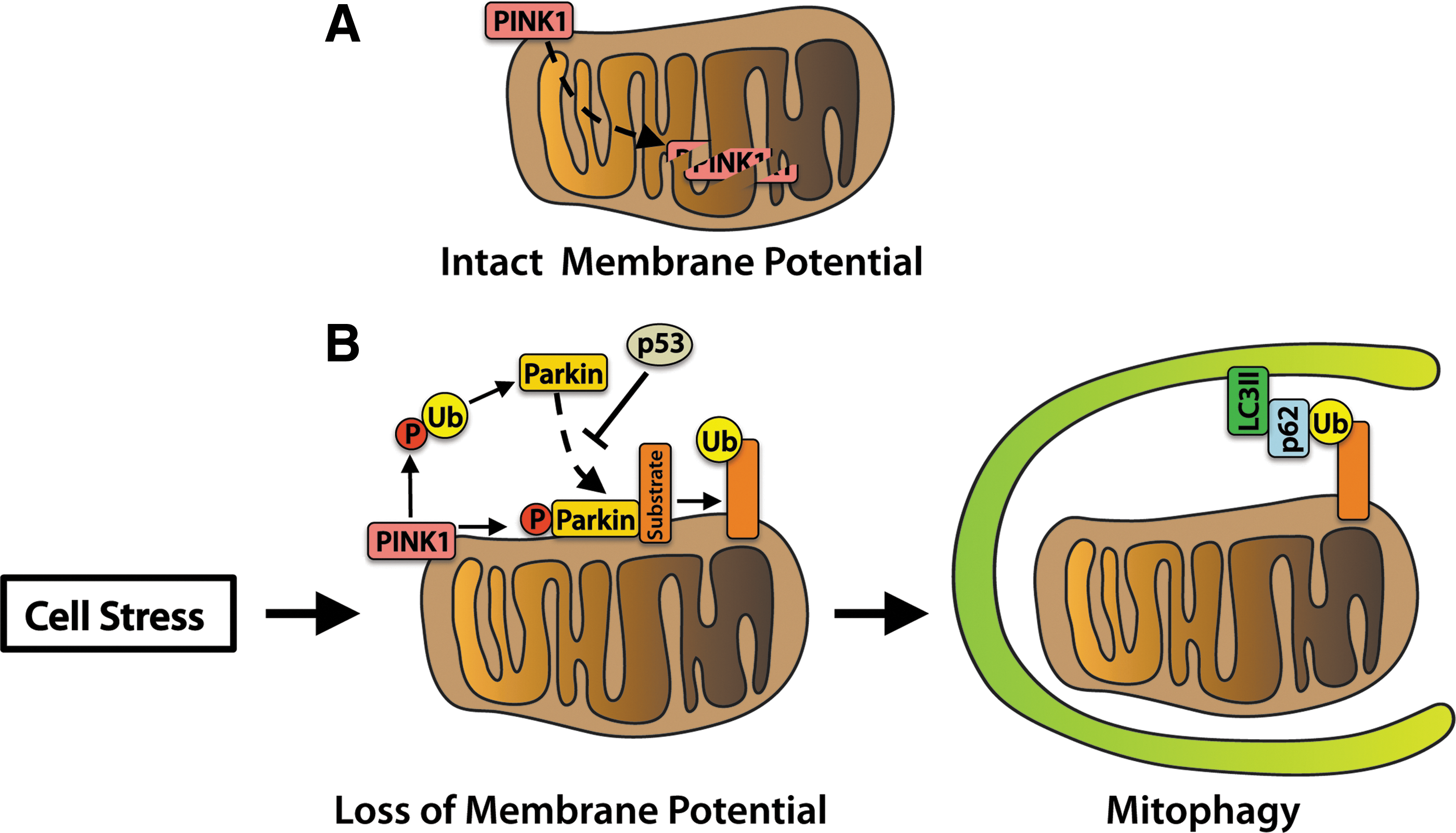
Linkage of the ubiquitin-binding protein p62 to ubiquitin on the mitochondrion and lipidated LC3II on the autophagosome provides a physical attachment point (120). Studies in human subjects found that PINK1 transcript levels are suppressed in skeletal muscle tissue of obese or T2D patients, suggesting that mitochondrial quality control via the PINK1/PARKIN pathway is reduced (136). In addition, impaired PARKIN function may contribute to loss of pancreatic β-cells and the development of insulin deficiency. At the onset of diabetes, the tumor suppressor protein p53 accumulates in the cytosol of mouse β-cells and inhibits PARKIN-mediated mitophagy. Mice deficient in p53 have restored mitophagy and are resistant to β-cell loss induced by streptozotocin (STZ) (63). The PINK1/PARKIN pathway plays an important role in mitochondrial quality control in the heart (16, 68, 90). However, its role in diabetic cardiomyopathy is currently unknown.
Another important mechanism of mitophagy involves a direct interaction between autophagy receptors on mitochondria and the autophagosome (Fig. 3). The BCL-2 family protein BNIP3 and its homologue NIX are known to participate in mitophagy by providing a direct association between mitochondria and LC3II or gamma-aminobutyric acid receptor-associated protein (GABARAP) on autophagosomes. BNIP3 and NIX play important roles in mitochondrial turnover in the myocardium (31). NIX has also been shown to play a critical role in the removal of mitochondria during erythrocyte maturation (138), and BNIP3 overexpression has been shown to activate mitophagy in myocytes (123). BNIP3 and NIX contain LC3-interacting region (LIR) motifs that bind to LC3 on the autophagosome, thereby allowing for a direct interaction without the need for ubiquitin and p62 (57).
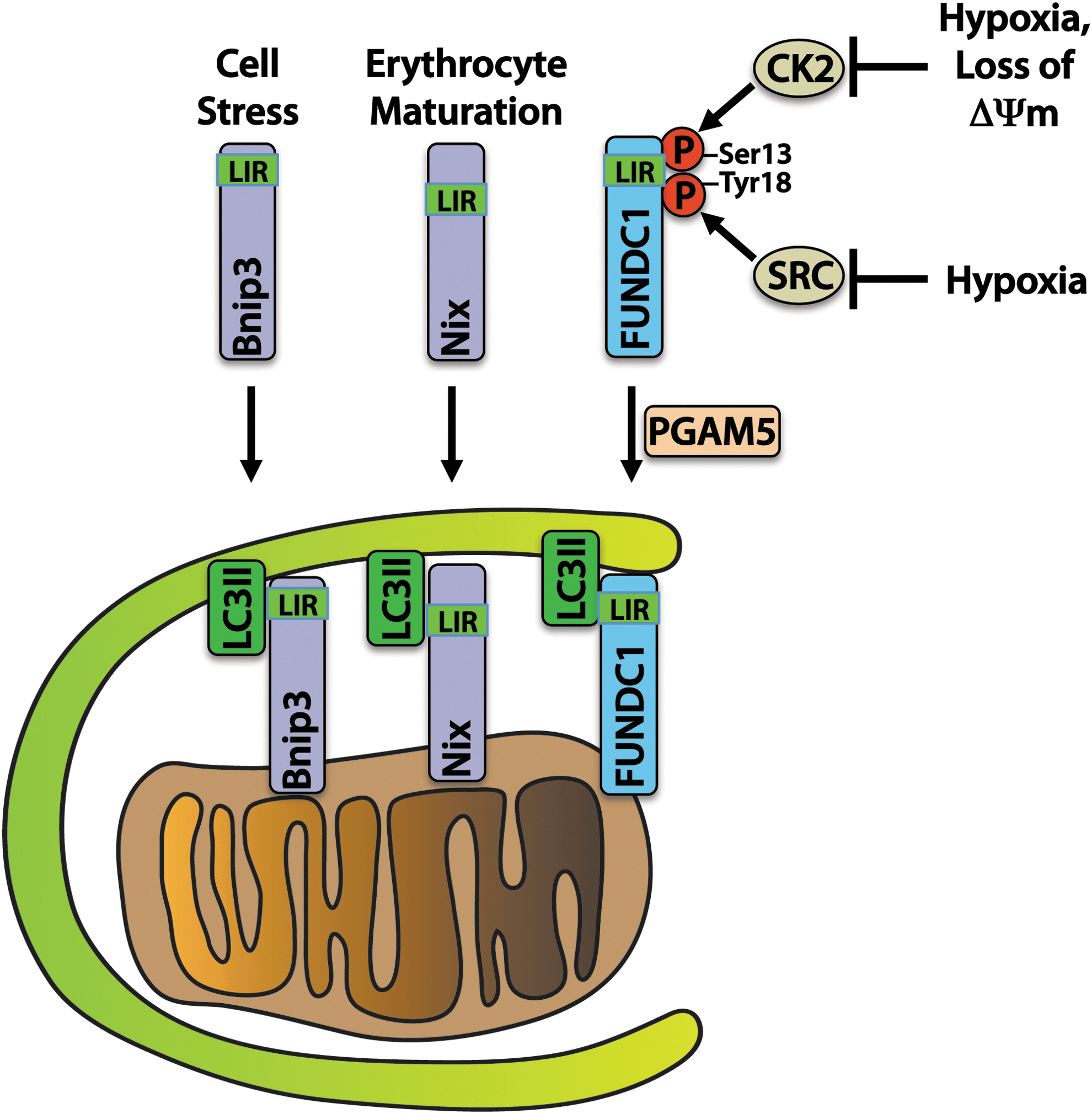
BNIP3 may also be involved in quality control of mitochondrial proteins without complete degradation of the mitochondrion. Overexpression of BNIP3 results in the selective reduction of components of the electron transport chain, possibly through activation of mitochondrial proteases (127). BNIP3 and NIX can also directly activate autophagy by displacing BECLIN1 from its inhibitory interaction with BCL-2 (12). Understanding the key roles that these two related proteins play in regulating autophagy and implementing mitophagy in the diabetic myocardium is therefore of great importance.
FUNDC1 is an outer mitochondrial membrane protein that, similar to BNIP3 and NIX, interacts directly with LC3II via an N-terminal LIR. It is maintained in an inactive state through phosphorylation on Tyr 18 and Ser 13 residues by SRC kinase and CK2, respectively. SRC kinase phosphorylation of FUNDC1 decreases during hypoxia, which triggers binding of LC3II on the autophagosome and stimulates mitophagy in HeLa cells (98). Ser 13 is dephosphorylated in response to hypoxia or FCCP treatment by PGAM5 phosphatase, enhancing LC3 binding (20).
Although the function of FUNDC1 has not yet been investigated in the heart, it likely participates in mitophagy in response to stimuli such as ischemia, given that high transcript levels of FUNDC1 are found in the mouse heart (98). Overall, these studies demonstrate that alternative and redundant mechanisms of mitophagy are in place to ensure the removal of dysfunctional mitochondria in cells. Conversely, the degree to which these three mechanisms of mitophagy (PINK1/PARKIN, BNIP3/NIX, and FUNDC1) are involved in the clearance of mitochondria in the diabetic heart has not been determined (84).
An Alternative Autophagy Pathway
Recently, an alternative pathway of autophagy has been identified in cells. Activation of this pathway has been noted in many different tissues, including the heart, and is capable of removing mitochondria in cells (117). Unlike traditional autophagy, the formation of autophagosomes in this pathway is independent of the ATG proteins. Instead, autophagosome expansion is dependent on the small GTPase RAB9, although the identification of other proteins involved in the elongation of the membrane in this pathway remains elusive. In addition, the canonical phagophore membrane has several potential origination sources, including the endoplasm reticulum, mitochondria, and the plasma membrane (5, 53, 125), whereas membranes involved in alternative autophagy are generated from the trans-Golgi network (117). The two autophagy pathways overlap insofar as both traditional and alternative autophagy require ULK-1 and BECLIN-1 for initiation. Currently, very little is known about the proteins involved in regulating alternative autophagy, and additional studies are required to understand the functional role of this pathway in cells.
Upstream Regulators of Autophagy
Autophagy is a highly regulated process that is controlled by several nutrient and energy-sensing pathways. Two main kinases are involved in sensing energy and nutrient status of the cell. These kinases, mTOR and AMPK, dictate whether the cell will activate energy- and amino-acid-consuming anabolic processes such as cellular growth, or energy- and amino-acid-generating catabolic processes such as autophagy, and convey these messages to ULK1/2. In addition, the Forkhead Box O transcription factors and Sirtuin deacetylases also participate in the regulation of autophagy and are important in controlling responses to stress in cardiomyocytes.
Mechanistic target of rapamycin
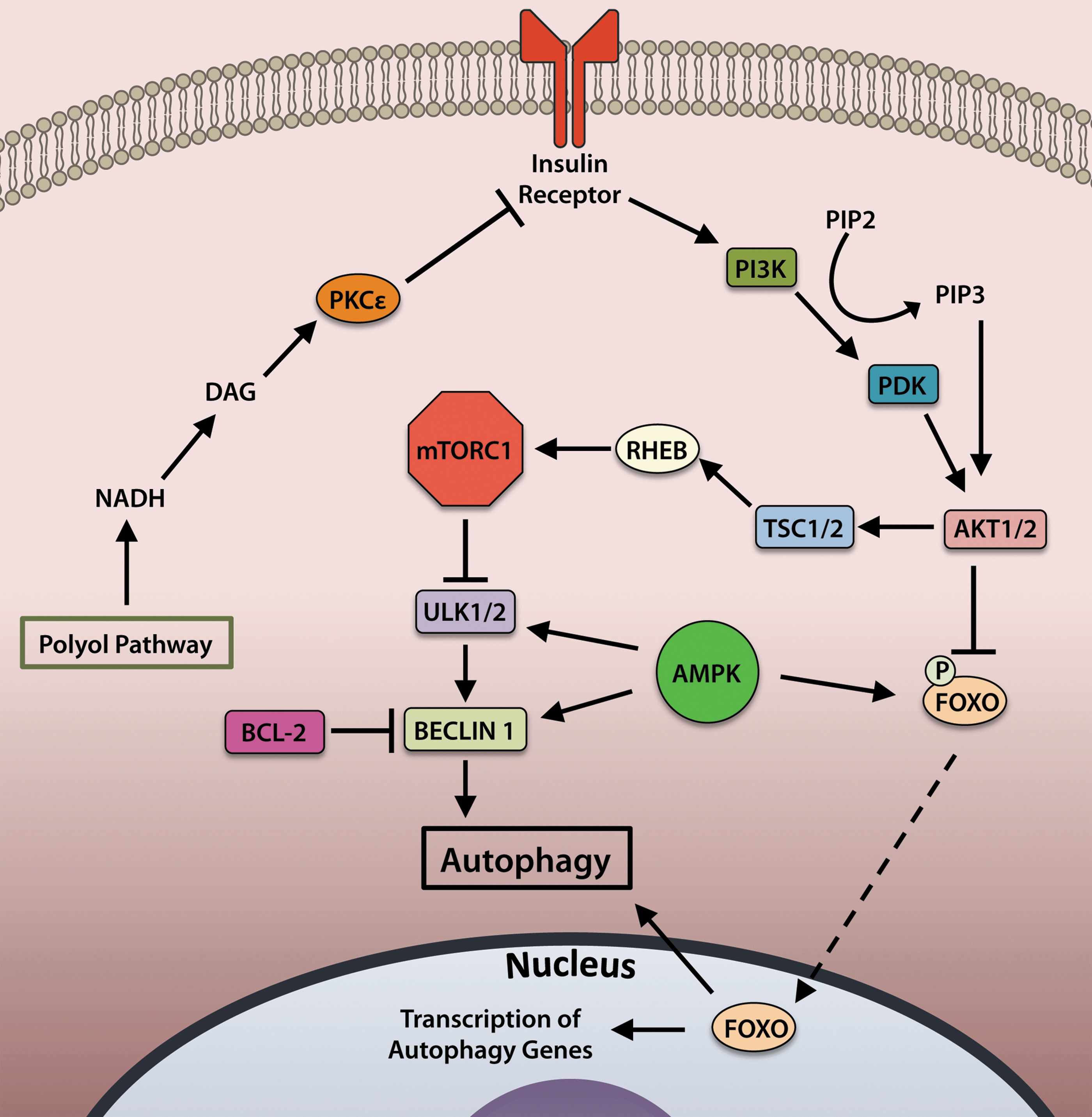
Changes in nutrient or growth factor levels that signal through various cell surface receptors or alterations in amino-acid levels culminate in changes to the activity of Mechanistic Target of Rapamycin (mTOR). mTOR participates in cell survival pathways as a part of mTOR complex 2 (mTORC2), whereas mTOR activity as a part of mTORC1 functions to inhibit autophagy. In addition to mTOR, mTORC1 comprises Regulatory-Associated Protein of mTOR (RAPTOR), Proline-Rich AKT Substrate of 40 kDa (PRAS40), Mammalian Lethal with SEC13 Protein 8 (MLST8), and DEP Domain-Containing mTOR-Interacting Protein (DEPTOR). The complex functions to suppress autophagy and stimulate protein synthesis during times of nutrient sufficiency, and inhibition of mTORC1 via nutrient deprivation shifts the cell away from protein synthesis and toward catabolic autophagy (88).
The activity of mTORC1 is regulated by insulin and growth factors (Fig. 4). Binding of insulin to the insulin receptor stimulates activation of phosphoinositide 3-kinase (PI3K) to mediate conversion of the phospholipid phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which binds to and activates AKT. Active AKT then inhibits activity of tuberous sclerosis complex (TSC)1/2, a negative regulator of the small GTPase Ras homolog enriched in brain (RHEB) (70). RHEB activates mTOR by binding to its catalytic domain. Activated mTOR inhibits autophagy by phosphorylating and deactivating the serine/threonine kinase ULK1/2 to halt initiation of autophagy (65, 74).
mTORC1 also responds to changes in amino-acid levels, which can be altered in the diabetic condition. Amino-acid level sensing and transduction of the status occurs primarily at the lysosome and involves three essential components. A coordinated effort between vacuolar- H+-ATPase (v-ATPase) (164), the Ragulator complex (7), and RAS-related GTP-binding (RAG) GTPases is necessary for mTOR activation (131, 132). mTOR is localized to lysosomes during times of amino-acid sufficiency, presumably to sense nutrient levels. This localization is mediated by active (GTP-bound) RAG complexes (131), which are, in turn, anchored to the lysosome via the scaffolding Ragulator complex (7). v-ATPase responds to accumulating levels of amino acids within the lysosome to activate Ragulator and RAG GTPases, resulting in mTOR activation and localization to the lysosome (164). In the absence of amino acids, v-ATPase associations with Ragulator and RAG proteins are weakened, and mTOR localization is predominantly cytosolic.
AMPK
The energy-sensing kinase AMPK responds to changes in intracellular energy status by activating or deactivating pathways that demand energy input. AMPK is activated during times of cardiomyocyte stress such as ischemia or pressure overload. During states of low energy availability or increased energy demand, such as during exercise (59), glucose deprivation (105), or other conditions in which the AMP/ATP ratio is elevated, AMPK augments autophagy through multiple mechanisms (Fig. 4). AMPK-driven autophagy can occur directly by activation of ULK1/2 or BECLIN 1 (33, 60, 80, 81) or indirectly by acting upstream of mTOR inhibition (71). In addition, AMPK can also promote autophagy via activation of the transcription factors FOXO1 and FOXO3a (discussed in the next section) in cardiomyocytes (103, 141).
FOXO transcription factors
The Forkhead Box O (FOXO) family of transcription factors participates in regulating expression of various factors involved in cell growth, proliferation, and survival. The subfamily members FOXO1 and FOXO3a promote the expression of a number of autophagy genes, including Atg12, Map1lc3A, and Gabarapl1 (Fig. 5) (141). The FOXOs also regulate transcription of BNIP3 and PINK1, which are important regulators of mitophagy (103, 106). Regulation of FOXO activity is controlled by upstream sensors of cellular energy and nutrient levels, as well as by hormones such as insulin.
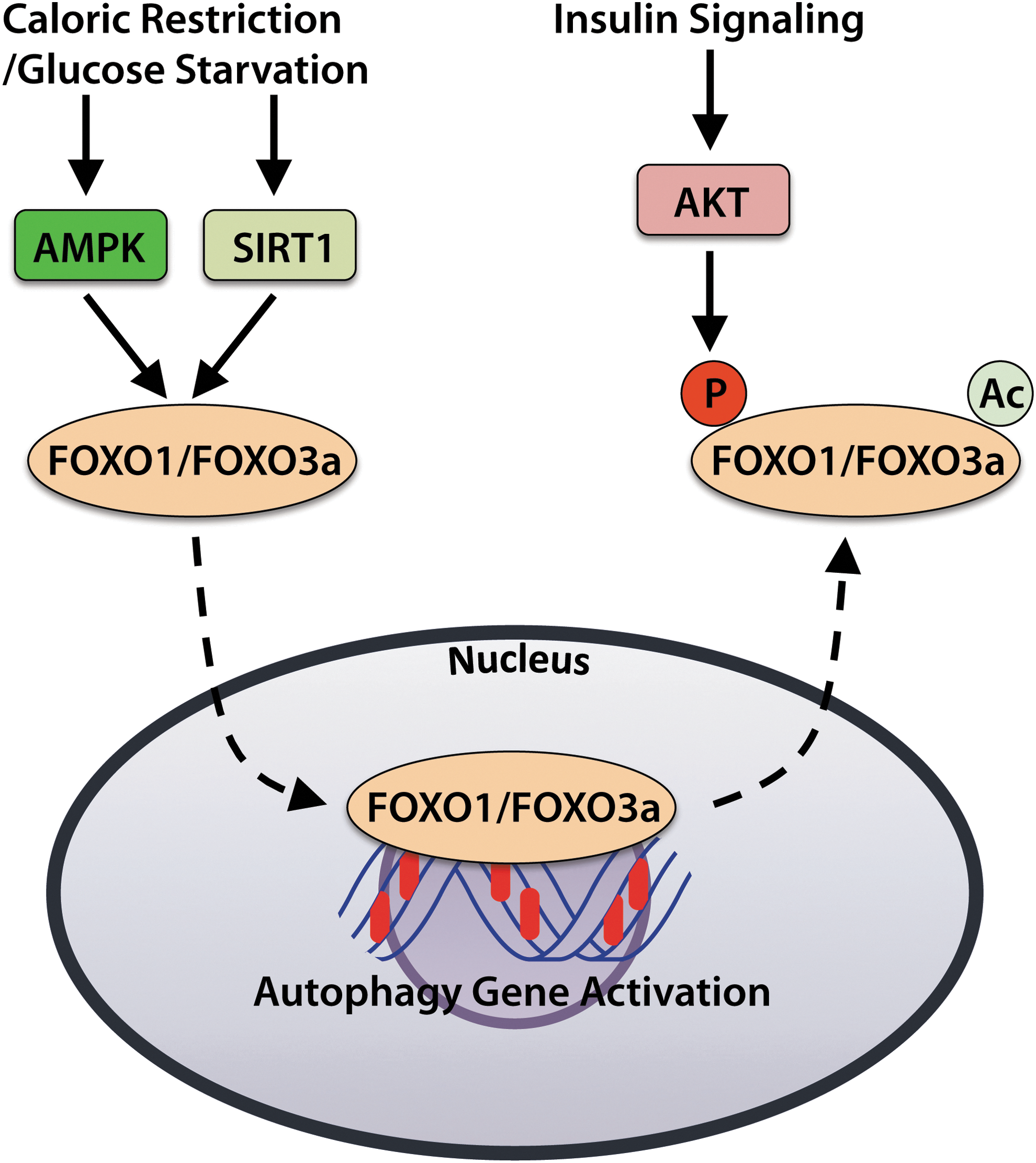
AMPK activates the FOXO proteins when energy levels are low, whereas AKT inhibits FOXOs when growth factors are abundant (18, 50). Phosphorylation of FOXOs by AKT results in deactivation and their export from the nucleus, thereby inhibiting transcription of the genes they control. For instance, AKT-mediated phosphorylation of Foxo3a leads to deactivation and translocation of Foxo3a from the nucleus to the cytosol (18). In contrast, glucose starvation induces FOXO1 and FOXO3 accumulation in the nuclei of neonatal rat cardiomyocytes, which corresponds with elevated autophagy (137, 141).
Sirtuins
The sirtuin (SIRT) family of deacetylases are involved in the regulation of a number of cellular stress responses and metabolic functions. In particular, SIRT1 activates autophagy in response to stimuli such as nutrient deprivation or oxidative stress. In response to caloric restriction (starvation), SIRT1 contributes to the induction of autophagy by deacetylating FOXO transcription factors, activating AMPK, and inhibiting mTOR activity (67, 91). In addition, SIRT1 expression increases during caloric restriction, leading to reduced FOXO3a acetylation and enhanced expression of the autophagy-promoting protein BNIP3 in the kidney (91).
BNIP3 is known to strongly promote mitophagy in cardiomyocytes (123), and evidence of elevated mitophagy was indeed found in kidneys of aged mice subjected to caloric restriction (91). Caloric restriction in the T2D Wistar Fatty (fa/fa) rat model also increases SIRT1 activity and mitophagy in the kidney, and it prevents the development of nephropathy (83). Further, induction of autophagy in response to glucose deprivation is dependent on both SIRT1 and FOXO1 in myocytes (58).
Reactive oxygen species
ROS can be generated from several sites in the cell, including the mitochondria and by NADPH oxidase (NOX) enzymes, and are now recognized as important activators of autophagy (151). In fact, superoxide generated during starvation is believed to be critical for the induction of autophagy in cells (21), and antioxidant treatment can suppress autophagy (149). ROS generated by mitochondria can indirectly activate autophagy by causing oxidative damage to the organelle, which would then require removal via mitophagy (95).
ROS can also promote opening of the mitochondrial permeability transition pore (MPTP) (134), which results in activation of autophagy and mitophagy (35). ROS can also induce autophagy via activation of HIF-1α-mediated expression of BNIP3, resulting in the release of BECLIN1 from BCL-2 and autophagy initiation (161). BNIP3 itself is also activated by oxidative stress (89). ROS has also been implicated in coordinating with NIX in priming mitochondria for mitophagy, which leads to PARKIN translocation (30).
Etiology and Pathogenesis of Diabetic Cardiomyopathy
Diabetes affects organ systems throughout the body and can lead to nephropathy, neuropathy, retinopathy, and cardiomyopathy (45). Diabetic cardiomyopathy is defined by declining contractility and the development of heart failure independent of confounding factors such as hypertension, obesity, or coronary artery disease (78). In its most severe state, diabetic cardiomyopathy is manifested as concentric hypertrophy, ventricular dilatation, and decreased cardiac output. Systolic dysfunction is not apparent until late in the disease pathogenesis, but recent evidence suggests that diastolic dysfunction may be detectable at earlier time points (see Schilling in this Forum). This dysfunction may be attributed to increased interstitial fibrosis that leads to ventricular wall stiffening (4).
Early detection of diabetic cardiomyopathy remains a challenge, yet it may provide a key to improving outcomes and longevity of diabetic patients. The net effects of various factors that influence autophagy in the heart should be carefully evaluated on a case-by-case basis and are confounded by differing effects on tissue type and individual patients' predispositions. The following analysis of the expected impacts of various factors on autophagy in the diabetic myocardium may help summarize the current understanding (Summarized in Fig. 6).

The microenvironment enveloping cardiac myocytes in T1D is distinct from that of T2D. This microenvironment can directly impact the state of autophagy in the heart. The distinguishing condition of T1D is insulin insufficiency caused by the death of insulin-producing pancreatic β-cells. This is in contrast with T2D, which is characterized not by a lack of insulin production but instead by systemic insulin resistance. The net result in both disease states is hyperglycemia, and increased circulating glucose contributes to disease pathogenesis via several pathways that will be discussed later in detail. Thus, impaired insulin signaling disrupts tissue homeostasis both directly and indirectly.
A feature distinguishing T1D from T2D is obesity. T2D is frequently associated with obesity, which carries with it a number of risk factors including elevated circulating low-density lipoprotein and triglyceride levels, hypertension, and atherosclerosis/coronary artery disease (79). In addition, elevated circulating amino-acid levels may influence signaling pathways involving key regulators such as AMPK and mTORC1. Still, some risk factors overlap between T1D and T2D. For instance, postprandial reuptake of amino acids is delayed in patients with T1D (36), thereby contributing to similar systemic effects in the absence of obesity. Likewise, circulating lipid levels are also elevated in T1D animal models and patients, and they can be atherogenic (29, 92).
High glucose negatively affects cardiomyocytes in several ways. First, hyperglycemia leads to increased osmotic stress that can lead to myocyte damage and death. Osmotic stress considered on its own, stimulated by high extracellular sorbitol in the absence of hyperglycemia, can induce cardiomyocyte apoptosis (34, 40). Hyperglycemia can lead to osmotic stress through the formation of sugar alcohols, or “polyols” via a pathway involving aldose reductase and sorbitol dehydrogenase. Aldose reductase is a component of the polyol pathway that is responsible for the reduction of monosaccharides. In particular, the reduction of glucose to sorbitol by aldose reductase has been implicated in the development of diabetic complications (45) (Fig. 7). Sorbitol is usually further metabolized by sorbitol dehydrogenase to produce fructose.
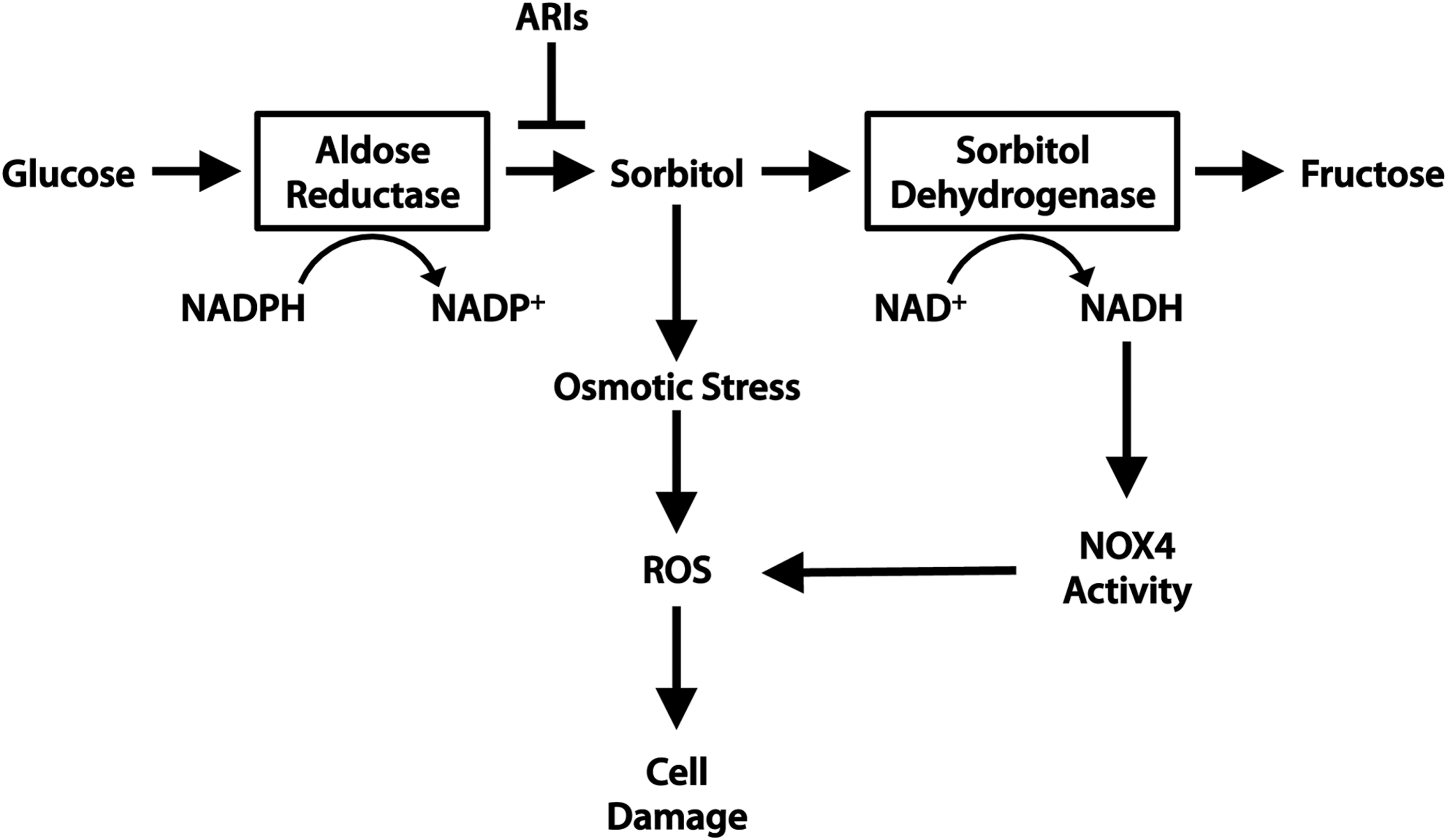
In hyperglycemia, sorbitol accumulates within cells and contributes to osmotic stress. Excessive flux through the polyol pathway has also been shown to promote mitochondrial ROS generation. Inhibition of the polyol pathway by aldose reductase or sorbitol dehydrogenase inhibitors (ARIs or SDIs) protects against simulated I/R injury and oxidative modification of SERCA and Ryanodine receptor (RyR) in rat hearts (147), thereby protecting cardiac function. However, further investigation is required to determine whether hyperglycemia leading to polyol pathway flux affects patients with T2D in the same manner as those with T1D (see Aon et al. in this Forum).
High extracellular glucose is also known to induce cardiomyocyte hypertrophy via several pathways. Activation of the PI3K-AKT signaling pathway by high glucose has been shown to increase protein synthesis in neonatal rat cardiomyocytes (160). However, other studies in neonatal rat myocytes found that high glucose alone is insufficient to cause hypertrophy, and protein synthesis is only increased in the presence of aldosterone (135). This suggests that hyperglycemia sensitizes cardiomyocytes to a hypertrophic response, and that addition of a further strain such as osmotic pressure or oxidative stress is needed to activate the hypertrophic response. Consistent with this, cardiomyocytes cultured in high glucose are more sensitive to acute stress such as I/R injury (99). This is likely due to a combination of factors, including mitochondrial sensitization, osmotic stress, oxidation of cellular components by ROS, and decreased autophagy.
Several factors contribute to the formation of ROS in the diabetic heart. Excessive flux through the polyol pathway can result in a surplus of NADH and FADH2 that donate electrons to the mitochondrial electron transport chain. This can overwhelm the electron transport chain componentry and the resident antioxidant superoxide dismutase 2 (SOD2/MnSOD), thereby leading to excessive superoxide production (45). ROS can also be generated by NOX enzymes. NOX4 is of particular interest, because it has been shown to both positively and negatively affect cardiomyocyte health. An increased availability of NADH from polyol pathway flux may stimulate NOX4 activity. Activation of NOX4 by glucose deprivation of cardiomyocytes or fasting in mice induces cardioprotective autophagy (140).
NOX4-generated ROS is known to contribute to oxidative stress during heart failure and in aging hearts (93), which suggests that chronic activation of NOX4 may be detrimental. Mice deficient in NOX4 have improved cardiac function and reduced myocyte apoptosis in response to pressure overload compared with WT mice (93). Moreover, NOX4 expression and activity increase in STZ (T1D) diabetic rat hearts, contributing to oxidative damage and impaired contractility, and in vivo knockdown of NOX4 prevent this functional decline (100). In addition, enhanced NOX4 activity may be linked directly to hyperglycemia, as cardiomyocytes cultured in high glucose have increased NOX4 expression and activity (100). Therefore, while short-term activation of NOX4 may promote beneficial ROS signaling and autophagy, chronic NOX4 activity appears be detrimental.
Mitochondria in Diabetic Cardiomyopathy
Mitochondria play a critical role in the heart by providing the contracting myocytes with ATP through oxidative phosphorylation. The heart is rich in mitochondria due to the high demand for energy. Therefore, it is particularly susceptible to cellular damage caused by dysfunctional mitochondria. Unfortunately, dysfunctional mitochondria can become excessive producers of ROS and release pro-death proteins that can activate cell death pathways (6). Mitochondrial dysfunction and increased production of ROS play key roles in the development of diabetic cardiomyopathy (see Aon et al. in this Forum).
Mitochondrial respiratory chain complexes I and III are primarily responsible for ROS production in the form of superoxide in response to stress such as I/R (86). Superoxide is both a precursor of other ROS and has the potential to directly modify lipids through peroxidation. In addition, the release of ROS by mitochondria can be potentiated by the phenomenon of “ROS-induced-ROS release,” which can result in opening of the MPTP and release of death-promoting factors (165). Electron transport chain dysfunction is known to occur in diabetic hearts.
Studies have found that many of the respiratory chain components are decreased in STZ diabetic rat hearts, but Complex I subunits are the most affected (8, 9). Overexpression of the mitochondria-targeted antioxidant enzyme phospholipid hydroperoxide glutathione peroxidase (mPHGPx) prevents the loss of these proteins and improves contractile function of diabetic rat hearts, suggesting that oxidation of mitochondrial components is responsible (8).
Deficiencies in the electron transport chain are not only limited to the heart but have also been reported in skeletal muscle biopsies of human patients with T2D (128). Under normal conditions, these dysfunctional mitochondria are degraded by mitophagy. However, if the number of dysfunctional mitochondria exceeds the capability of mitophagy or if the process is impaired, then these mitochondria begin to accumulate. How mitophagy is affected in the diabetic myocardium is currently unknown.
Dysfunctional mitochondria can activate the intrinsic pathway of apoptosis that is regulated by the BCL-2 family proteins (52). The BCL-2 family comprises pro- and anti-apoptotic proteins that are maintained in balance in healthy cells. Anti-apoptotic members contain four BCL-2 homology (BH) domains and include BCL-2, BCL-XL, and MCL-1. The pro-apoptotic members include effector proteins BAX and BAK, which form oligomeric pores to permeabilize the outer mitochondrial membrane, and BH3-only proteins such as BID, BIM, and BAD, which directly interact with anti-apoptotic proteins and the effector proteins to maintain an equilibrium (23).
Increased apoptosis of cardiac myocytes has been observed both in human diabetic patients (39) and in animal models of diabetes (19, 60). In addition, a high-fat diet (HFD) alters the BAX:BCL-2 ratio and increases apoptosis in rat myocardium (44). Hyperglycemia can also directly induce release of cytochrome c through mitochondrial permeabilization in cell culture, resulting in apoptosis (19).
Cell death can also be induced via opening of the MPTP, and studies have found that cardiac mitochondria in diabetic animals are more likely to undergo MPTP opening. For instance, mitochondria from STZ diabetic rat hearts have impaired calcium uptake and are more susceptible to calcium-induced MPTP opening (118, 146). The treatment of mitochondria with a reducing agent (dithiothreitol) or an antioxidant peptide (MTP-131) restores the normal threshold for calcium-induced MPTP opening in mitochondria from STZ rat hearts, suggesting that the increased sensitivity to calcium might be due to oxidation of critical mitochondrial proteins (146).
In addition, opening of the MPTP contributes to loss of cells in myocardial I/R injury, and studies have found that mitochondria in the diabetic heart are more susceptible to MPTP opening. Inhibition of the MPTP in T2D rats attenuates tissue injury during ischemia (15). Collectively, these studies suggest that diabetic cardiac mitochondria are unstable and are more likely to activate both apoptotic and necrotic cell death in response to stress.
Mitochondrial Dynamics
Mitochondrial morphology plays an important role regulating bioenergetic efficiency and mitophagy in cells (97). Mitochondrial fusion is mediated by Mitofusin 1 and 2 in the outer mitochondrial membrane and OPA1 in the inner mitochondrial membrane (54). Mitochondrial fission is regulated by fission 1 protein (Fis1), mitochondrial fission factor (Mff), and dynamin-related protein 1 (DRP1). Fis1 and Mff are localized to the outer mitochondrial membrane, whereas cytosolic Drp1 is recruited to mitochondria during fission (54). Studies have found that mitochondrial morphology plays an important role in regulating bioenergetics (see Galloway and Yoon in this Forum).
Fusion of mitochondria during nutrient deprivation causes an increase in ATP synthesis that helps sustain ATP levels during brief periods of starvation (48). In contrast, excess glucose and fatty acids levels induce mitochondrial fragmentation in β-cells and are associated with increased uncoupled respiration (97). Hyperglycemia has also been reported to cause excessive Drp1-mediated mitochondrial fission in neurons, and reducing fission leads to increased neuronal survival (32). Thus, chronic exposure to excess nutrients leads to excessive fission that is detrimental to cells. Mitochondrial morphology is also altered in diabetic hearts. Type 2 diabetic patients displayed cardiac mitochondrial network fragmentation and a significantly decreased expression of MFN1 (111). However, the functional consequences of altered mitochondrial dynamics in the diabetic myocardium require further investigation (see Galloway and Yoon in this Forum).
Paradoxically, mitochondrial fission is also a prerequisite for mitophagy. Asymmetrical fission of mitochondria generates two bioenergetically different fragments leading to selective sequestration of the mitochondrion with lower mitochondrial membrane potential by mitophagy (148). Recent studies utilizing cardiac specific DRP1 knockout mice have confirmed the importance of mitochondrial fission in mitochondrial clearance (69, 76). These studies demonstrated that mitophagy was abrogated in myocytes lacking DRP1, and this leads to accumulation of dysfunctional mitochondria and development of heart cardiac dysfunction. Conversely, fused mitochondria are protected against mitophagy, possibly because elongated mitochondria are too large to be engulfed by autophagosomes (48).
Since studies indicate that fission is increased in response to an excess nutrient environment, an increase in mitophagy would be anticipated. However, there is an abundance of dysfunctional mitochondria in the diabetic heart (17, 84, 144), suggesting that mitophagy is incapable of clearing these mitochondria. It is likely that chronic exposure to excess nutrients in diabetes will lead to adaptations that interfere with many cellular processes, including autophagy and mitophagy. This adaptation might contribute to the accumulation of dysfunctional mitochondria in myocytes, which can lead to loss of myocytes and the development of diabetic cardiomyopathy.
Autophagy and Mitophagy in the Diabetic Heart
Myocardial autophagy in T1D
Because insulin activates the PI3K/AKT/mTORC1 pathway, it was initially believed that autophagy would be increased in both T1D (insulin deficiency) and T2D (insulin resistance). Consistent with this hypothesis, insulin receptor substrate (IRS) null mice have enhanced autophagic activity in their hearts (126). However, studies have revealed that autophagy is actually suppressed in the hearts of T1D animals (60, 84, 156, 159, 163). For instance, OVE26 mice, a genetic model of T1D, have lower levels of autophagy in their hearts at 5–6 months of age (156, 159). Xu et al. also reported suppressed autophagy after 9 weeks of diabetes in the hearts of STZ mice (159). This study reported that STZ-diabetic mice have lower LC3II/GAPDH ratios than control mice, and experiments with Bafilomycin A1, an inhibitor of lysosomal maturation, indicate impaired formation of autophagosomes in STZ mouse hearts and, consequently, impaired autophagic flux. In addition, the authors report reduced levels of Atg12 and the Atg12-Atg5 complex in the hearts of STZ mice, a potential reason for decreased autophagosome formation (159). The suppression of autophagy in the T1D heart is due to increased mTORC1 activity (159) and/or reduced AMPK activity (156, 159).
In addition, hyperglycemia inhibits autophagy in H9c2 cardiac cells by increasing the interaction between BCL-2 and BECLIN 1 (60). Myocyte apoptosis is greatly elevated in the hearts of diabetic patients: till 85-fold over control patient biopsy samples (39). Because the BCL-2 proteins govern both apoptosis and autophagy, a disruption in their regulation in the diabetic myocardium appears culpable. Evidence of increased binding of BCL-2 to the autophagy inducer BECLIN1 has been observed in animal models of T1D (60), and STZ-diabetic mice show increased cardiomyocyte apoptosis, very much similar to human patients.
He et al. reported that the elevated levels of apoptotic markers in the hearts of STZ-diabetic mice could be ameliorated by treatment with the antidiabetic drug metformin. Metformin treatment was shown to reduce BCL-2 binding to BECLIN1, which has the dual effect of both upregulating autophagy and liberating BCL-2 for apoptosis suppression (60). However, this study only evaluated levels of LC3II in STZ-diabetic mice, and it did not investigate autophagic flux in animals. While the study reports that metformin restores flux in H9c2 cells subjected to high glucose, an in vivo model of chronic diabetes is substantially different from acute glucose treatment of cells. Still, the study raises the possibility that elevated apoptosis and impaired autophagy found in T1D hearts may be related to altered BCL-2 family protein interactions. Overall, these studies indicate that the autophagy pathway is affected at multiple steps in the diabetic myocardium.
The functional role of autophagy in type 1 diabetic hearts
Several studies have investigated whether altered autophagy correlates with development of diabetic cardiomyopathy. Currently, there is a consensus that cardiac autophagy is reduced in type 1 diabetes. However, the functional consequence of this reduction in autophagy remains unclear and controversial. Xie et al. reported that OVE26 and STZ-induced type 1 diabetic mice have reduced autophagy in their hearts, and restoration of autophagic activity with metformin ameliorates diabetic cardiac injury (156). Similarly, He et al. found that activation of AMPK in diabetic mice restores cardiac autophagy and protects against cardiac cell apoptosis, ultimately leading to improvements in cardiac structure and function (60). These results suggest that impaired autophagy contributes to cardiac damage due to reduced clearance of dysfunctional organelles and protein aggregates, and that enhancing autophagy can limit damage in the type 1 diabetic heart.
In contrast, Xu et al. proposed that the reduced cardiac autophagy in type 1 diabetic mice is an adaptive response to prevent excessive autophagic degradation of cellular components. This study found that cardiac damage in STZ and OVE26 type 1 diabetic mice is attenuated in mice deficient in the autophagy proteins BECLIN 1 and ATG16. These mice have significantly improved cardiac function and lower levels of oxidative stress, interstitial fibrosis, and myocyte apoptosis (159). In contrast, cardiac-specific BECLIN 1 transgenic mice have enhanced autophagic activity, resulting in exacerbated diabetes-induced cardiac apoptosis and fibrosis (159). Although type 1 diabetes is associated with mitochondrial dysfunction, this study found that mitophagy proteins PINK1 and PARKIN are significantly reduced in the diabetic hearts, while the mitophagy receptor BNIP3 is unchanged. This suggests that mitophagy is reduced in these hearts despite extensive mitochondrial dysfunction, leading to the accumulation of dysfunctional mitochondria and activation of cell death in myocytes.
Myocardial autophagy in T2D animal models
In contrast to T1D, results from studies on T2D are less consistent and report that cardiac autophagy can be unchanged (94), reduced (27, 51, 139, 158), or even increased (109, 130). However, major inconsistencies in the methods used to assess autophagy between studies likely account for a large portion of the discrepancies in results. For instance, some of these studies report LC3II/I ratios that are unchanged or lower in the hearts of mice with metabolic syndrome. However, inspection of the reported results shows no decrease in LC3II, but instead an increase in LC3I (27, 94), which is not indicative of autophagy suppression but may be due to increased transcription of the LC3 gene. Further, while the ratio of LC3II/LC3I may be informative in cases where only very short-term changes in autophagy are of interest, this ratio alone is insufficient to describe the state of autophagic flux in chronic conditions such as diabetes where transcriptional changes in LC3 may also be occurring.
Some studies speculate on the state of autophagic flux by analyzing degradation of p62, the scaffolding protein that anchors cargo to the LC3II-decorated autophagosome. Guo et al. show that levels of LC3II are decreased while LC3I remains unchanged in the hearts of HFD-fed mice, indicating a lower abundance of autophagosomes (51). They also report that p62 levels are elevated in the HFD group, which is suggestive of decreased autophagic flux. Similarly, Mellor et al. report that the hearts of insulin-resistant fructose-fed mice have an increase in LC3II with no change in LC3I, as well as elevated p62 levels (109), again suggesting impaired autophagic flux. However, because autophagic flux is dynamic, merely evaluating the levels of proteins involved yields only a snapshot of the process. A blockage at the pathway terminus via lysosomal inhibition allows one to evaluate both autophagosome generation and degradation.
Sciarretta et al. found that HFD activates RHEB in the heart, leading to mTORC1 activation and consequent suppression of autophagy (139). Specifically, the authors found reduced levels of LC3II and elevated p62 in HFD-fed mice. Moreover, they found that the number of GFP-LC3-positive autophagosomes in left ventricle cross-sections did not increase in the hearts of HFD-fed mice after administration of the lysosomal-fusion inhibitor chloroquine, suggesting a failure to form autophagosomes rather than increased autophagosome removal.
In contrast, when Xu et al. investigated autophagic flux in the context of insulin resistance and metabolic syndrome, they found that mice fed HFD develop cardiac functional deficits and accumulate LC3II in their hearts, suggesting an increased number of autophagosomes. They also noted the accumulation of p62, indicating impairment in autophagosome clearance. Transmission electron microscopy confirmed the accumulation of autophagosomes and a lack of autolysosomes in the hearts of HFD-fed mice. In addition, HFD-fed mice treated with chloroquine did not accumulate additional LC3II whereas control diet-fed mice did so, confirming that HFD results in impaired clearance of autophagosomes in cardiac tissue (158). Thus, while Sciarretta et al. (139) suggest autophagosome formation is impaired in the hearts of mice fed HFD, Xu et al. (158) suggest impaired autophagosome degradation. Taken together, these studies indicate that the hearts of T2D animals have impaired autophagic flux, although substantial disagreement persists.
While studying autophagy in the hearts of obese animals with insulin resistance is informative because it recapitulates the human disease condition, studies that investigate conditions of insulin resistance in the absence of obesity are equally informative. Mice fed a high fructose diet develop systemic insulin resistance and cardiac dysfunction in the absence of obesity. These mice have markers of impaired clearance of autophagosomes in cardiac tissue, including elevated LC3II/I ratios and accumulated p62 (109). An analysis of flux using pathway inhibition in this model would be of great value to the field, because autophagic flux has not been evaluated in insulin resistance without obesity.
In another model of impaired insulin signaling, mice with cardiac-specific deletions of Irs1 and Irs2 genes have unrestrained autophagic flux, leading to impaired fractional shortening by 4 weeks of age, heart failure, and early mortality. This also demonstrates the importance of endogenous insulin signaling in the suppression of autophagic flux. Notably, these mice show mitochondrial dysfunction that precedes the cardiac functional decline (126). The accumulation of dysfunctional mitochondria suggests a failure of mitophagy despite having enhanced autophagy.
The functional role of autophagy in type 2 diabetic hearts
In contrast to type 1 diabetes, enhancing autophagy in HFD-induced type 2 diabetes appears to be a protective response that reduces cardiac damage. For instance, sustained activation of RHEB and mTORC1 in the hearts of mice on an HFD leads to inhibition of autophagy and exacerbates ischemic injury after a myocardial infarction (139). Many other studies have linked mTORC1 activity to the development of diabetic cardiomyopathy. Völkers et al. found that PRAS40-mediated mTORC1 inhibition prevents the development of HFD-induced diabetic cardiomyopathy (152). Rapamycin, a potent inhibitor of mTOR and inducer of autophagy, improves cardiac function in db/db type 2 diabetic mice (28). In addition, AKT2-deficient mice on HFD have reduced mTORC1 activation and increased autophagy that correlates with reduced hypertrophy and improved cardiac function (158). Of note, knockdown of AKT2 in cancer cells specifically activates mitophagy and can cause cell death from excessive removal of mitochondria (133).
Thus, the cardioprotective effects of AKT2 knockout observed by Xu et al. may be partially explained by an enhancement of mitophagy that expedites removal of dysfunctional mitochondria in the hearts of mice with insulin resistance and metabolic disorder. However, AKT2 knockout mice are known to develop severe diabetes and insulin resistance (24, 41), making systemic inhibition of AKT2 dangerous for patients with T2D. Collectively, these studies demonstrate that enhancing autophagy is cardioprotective in HFD-induced type 2 diabetes. Together, these results highlight the complex role of autophagy in diabetes and the need for additional studies to determine under what conditions autophagy is beneficial versus detrimental in diabetes.
Mitophagy and p53 in diabetes
The tumor suppressor p53 is well known for its role in apoptosis, and loss of p53 is associated with the development of certain cancers (14). Recent studies have also implicated p53 in the development of diabetes. For instance, Hoshino et al. reported that p53-deficient mice were protected against the development of diabetes in models of both STZ-induced type 1 and db/db type 2 diabetes (63). In addition, it is known that hyperglycemia activates p53-mediated cell death in myocytes (37). p53 also promotes cardiac dysfunction in diabetic mice (115). However, p53 has a broad functional role in cells and regulates other processes, including autophagy.
The regulation of autophagy by p53 is very complex, and p53 can both activate and inhibit autophagy in a context-dependent manner (101). Both the intracellular location and levels of p53 can influence its effect on autophagy. For instance, p53 exerts many of its functions by acting as a transcription factor, and nuclear p53 regulates transcription of several genes involved in autophagy, including DRAM (damage-regulated autophagy modulator) and SESTRIN2 (26, 102). Although expression of these genes correlates with increased LC3II levels in cells, the implications of the autophagosome accumulation are not clear. Expression of the p53 target gene DRAM results in increased levels of autophagosomes, as determined by electron microscopy and LC3II levels. However, it is not clear whether DRAM-mediated autophagosome accumulation is the result of increased formation or impaired clearance, as no flux experiments have been completed (26). In addition, DRAM participates in p53-mediated apoptosis, further confounding its role in autophagy (26).
In contrast, cytosolic p53 can inhibit autophagy (101), and it can directly inhibit mitophagy in the heart by interacting with PARKIN, thereby preventing it from translocating to dysfunctional mitochondria. As a result, the removal of damaged mitochondria is abrogated and dysfunctional mitochondria accumulate, leading to cardiac functional decline (64). Inhibition of PARKIN-mediated mitophagy by p53 has also been linked to impaired function of pancreatic β-cells in T1D (63). However, whether cytosolic p53 contributes to the development of diabetic cardiomypathy by inhibiting PARKIN-mediated mitophagy in myocytes remains to be determined.
The study by Hoshino et al. found that pharmacological inhibition of p53 with pifithrin-alpha restores mitophagy and ameliorates accumulation of dysfunctional mitochondria in β-cells in both T1D and T2D (63). Although T1D and T2D have distinct mechanisms, mitochondrial dysfunction in myocytes and other cells is a common characteristic for both types. Thus, it is possible that activation of p53 in the hearts of diabetic mice contributes to loss of myocytes through both activation of apoptosis and inhibition of autophagy. Additional studies are needed to determine whether preserving or enhancing PARKIN-mediated mitophagy represents a potential therapeutic target for both T1D and T2D.
Autophagy in Cell Types Beyond the Cardiomyocyte
Metabolic changes associated with diabetes will impact many other cell types and tissues that can influence cardiac function. For instance, obesity is the result of excess fat storage in the form of white adipose tissue. Because adipocytes regulate fat storage and can release pro-inflammatory cytokines (13), they can have profound effects on cardiac function. Recent studies have found that autophagy and mitophagy play important roles in adipogenesis, and inhibition of autophagy reduces adipose tissue mass (47, 145, 162). Adipocyte-specific Atg7 knockout mice are lean due to reduced white adipose tissue (145, 162), and these mice are also resistant to HFD-induced obesity (162). Abrogation of autophagy in adipocytes results in altered cell morphology and a large number of mitochondria compared with normal white adipocytes (47), which indicates that mitophagy is important for adipocyte maturation. Thus, these studies suggest that activation of autophagy and mitophagy in adipocytes can indirectly contribute to the development of T2D by promoting obesity.
Cardiac fibrosis is a major feature of diabetic cardiomyopathy, where excessive production and deposition of extracellular matrix (ECM) proteins leads to increased myocardial stiffness and subsequent cardiac dysfunction (4). Fibroblasts are responsible for producing the ECM in the heart, and recent evidence suggests that autophagy is an important modulator of fibrosis. Autophagy is involved in the intracellular degradation of type I collagen for the prevention of excess ECM deposition (3, 72, 82). There is also evidence that impaired autophagy promotes differentiation of fibroblasts to myofibroblasts, the profibrotic phenotype of fibroblasts (2, 96). Although studies have reported increased activation of cardiac fibroblasts and fibrosis in diabetic hearts, no studies have examined whether autophagy plays a role in this response. However, considering that autophagy is reduced in T1D hearts (60, 156, 159, 163), it is possible that autophagy plays a role in preventing fibrosis. Hyperglycemia may also participate in the enhanced fibrosis in diabetic hearts, as it both causes activation of cultured fibroblasts (142) and reduces autophagy in cardiac myocytes (85). Thus, several factors may contribute to fibrosis in diabetic cardiomyopathy.
Diabetic patients are also at an increased risk of developing vascular diseases due to defects in endothelial and vascular smooth muscle cell (SMC) function (25). The role of autophagy in atherosclerosis is still poorly understood, and few studies have focused on the role of autophagy in vascular cells in the setting of diabetes. Studies indicate a protective role of autophagy against atherosclerosis in SMCs. Autophagy protects SMCs in vitro against oxidized lipids and excess free cholesterol, while inhibition of autophagy triggers cell death (62, 157).
In addition, sustained hyperglycemia results in the generation of advanced glycation end products (AGEs) that can damage endothelial cells (46). Hou et al. reported that AGEs increase both the number of autophagosomes in endothelial cells and cell death (66). Inhibition of autophagy, however, enhanced AGE-mediated cell death in endothelial cells (155), suggesting that autophagy is functioning to protect against AGE-mediated cellular damage. Although these in vitro studies suggest that enhanced autophagy protects against stress in vascular cells and could potentially influence the stability of atherosclerotic plaques, additional studies are needed to characterize how diabetes affects autophagy and mitophagy in these cells.
Therapies for Diabetes and Diabetic Cardiomyopathy
Many treatment options are available for diabetes and diabetic cardiomyopathy, and it is possible that several of these therapies could affect the status of autophagy. Although no treatments exist that specifically target mitophagy, the effects of available therapies on autophagy and mitophagy have not yet been adequately researched. Several established and experimental treatments may alter autophagy, either positively or negatively, including angiotensin receptor blockers (ARBs), the AMP analog AICAR, ARIs, FOXO1 inhibitors, and resveratrol.
ARBs are first-line therapies for hypertension and heart failure that function to modulate the renin-angiotensin-aldosterone system via inhibition of angiotensin II (AngII) type 1 (AT1) receptors. Recently, a link between AT1 receptors and autophagy has been made. Stimulation of neonatal rat cardiomyocytes overexpressing AT1 with AngII results in hypertrophy and elevated markers of autophagy, both of which are completely abrogated by the ARB candesartan (122). Moreover, AngII has been shown to contribute to insulin resistance in skeletal muscle and SMCs (38, 153, 154). AngII not only has been shown to activate AMPK in vascular SMCs to inhibit proliferation but also has been found to inhibit AMPK in H9c2 rat cardiomyoblast cells to induce hypertrophy (61, 114). However, the effects of AngII and ARBs on mitophagy remain to be determined, and whether ARBs can be utilized to modulate autophagy remains unexplored.
The AMP analog AICAR is not only known to be cardioprotective during I/R injury though inhibition of adenosine kinase and adenosine deaminase, but it has also been shown to induce translocation of the GLUT-4 glucose transporter to the surface of rat ventricular papillary myocytes in an AMPK-dependent manner via a pathway that is independent of insulin signaling and PI3K activation (129). This is an exciting prospect for patients with T2D who may have impaired cardiac glucose uptake. However, the effects of AICAR-mediated AMPK activation on autophagy and mitophagy in the myocardium are not fully understood. Although AICAR potently activates AMPK in rat hepatocytes, autophagy is actually inhibited (108). Radioactive pulse-chase assays were used to measure proteolytic degradation of long-lived proteins by autophagy in the rat hepatocytes. Despite robust activation of AMPK, proteolysis was actually found to be inhibited by AICAR to the same extent as the autophagy inhibitor 3-MA (108).
One possible explanation is that ATP was depleted to such an extent that activation of the highly-ATP dependent process of autophagy became unfeasible despite activation of AMPK (107). Thus, the use of AICAR for the treatment of diabetic cardiomyopathy should be investigated carefully to determine under what circumstances AMPK activation is beneficial versus detrimental.
ARIs aim at inhibiting the polyol pathway and reducing oxidative and osmotic stress in a variety of cell types. Animal studies using ARIs for the treatment or prevention of diabetic neuropathy have shown great promise, but clinical trials conducted since the mid 1980s and ongoing have found ARIs to be not efficacious and potentially toxic to human patients. Nevertheless, ARIs have been shown to be cardioprotective to both diabetic and nondiabetic rats during simulated I/R injury (124), and the activity of aldose reductase may affect autophagy levels in the heart through the accumulation of diacylglycerol (DAG) caused by excess NADH production from polyol pathway flux. DAG-mediated activation of protein kinase C results in inhibition of the insulin receptor signaling pathway and decreased AKT activity, culminating in activation of FOXO transcription factors and increased transcription of autophagy genes (1). Thus, aldose reductase inhibition could help suppress aberrantly activated autophagy and prevent ATP depletion, although no studies have been conducted to determine the effects of aldose reductase inhibition on cardiac autophagy.
Recently, developed inhibitors of FOXO1 aim at lowering circulating glucose levels by reducing hepatic FOXO1-driven gluconeogenesis. In this respect, these inhibitors have shown efficacy in the T2D db/db mouse model (113). Battiprolu et al. also found that FOXO1 and FOXO3 are aberrantly active in hearts of high-fat diet-induced and genetic (db/db) models of T2D. Excessive FOXO1 and FOXO3 activity in T2D may contribute to the development of contractile dysfunction, as cardiomyocyte-specific FOXO1 knockout mice are resistant to HFD-induced cardiac hypertrophy and have improved heart function after prolonged HFD feeding (10). Pharmacologic inhibition of FOXOs may prove to be a viable means of modulating autophagy in the diabetic heart in the future, but the effects of FOXO1 inhibitors on cardiac function or autophagy have not yet been investigated.
Resveratrol is a plant-derived polyphenol that is purported to have health benefits, including improving cardiovascular health, reducing the risk of cancer, reducing severity of diabetic complications, and promoting longevity (Sung et al. in this Forum). Although several of the earlier studies on the health benefits of resveratrol may have been overstated, the compound has the potential to promote autophagy via SIRT1 activation (112). Other studies indicate that resveratrol does not directly activate SIRT1, but instead may act on intermediates such as AMPK to promote activation (11, 119). Therefore, while resveratrol may have beneficial effects in some circumstances, the precise mechanism of action remains unclear.
Concluding Remarks
Inconsistencies in animal models and assays used to evaluate autophagy and mitophagy in diabetes have yielded convoluted and often conflicting results. However, as evidence accumulates, the distinction between Type 1 and Type 2 diabetes grows more apparent. A concerted effort to standardize animal models for future studies should be made to clarify results and refine conclusions. Part of this involves understanding the strengths and limitations of animal models that do not precisely recapitulate the human condition. Findings from studies in animal models of type 1 diabetes tend to be more in accord than findings from type 2 diabetic animals. This may be due to a greater degree of consistency in type 1 animal models, and it also underscores the complexity of the disease state in type 2 diabetes. More refined models of type 2 diabetes would help enhance our understanding of the state of autophagy and mitophagy in the heart, as well as the contributions of autophagy to the pathogenesis of cardiomyopathy.
One hypothesis regarding the state of autophagy in the diabetic myocardium is that T1D may have impaired autophagosome formation, while T2D may have impaired autophagosome clearance. Although this appears to be an undesirable feature of these conditions, it allows for more individualized therapeutic approaches. In the correct context, the ability to selectively upregulate autophagosome formation or enhance autophagosome clearance may prove to be greatly beneficial. The discrepancies between T1D and T2D and the status of autophagy in each may also account for why so many treatments that appear promising in animal models fail to show efficacy in clinical trials. It is therefore of critical importance that future studies on the status of autophagy in diabetes focus on clarifying the state of autophagic flux.
A lack of sufficient studies on mitophagy in the heart and other tissues leaves many uncertainties about the contribution of improper mitochondrial clearance in diabetes. However, a growing interest in the field of mitophagy in diabetes promises to shed more light on the significance of mitophagy in the development of diabetic cardiomyopathy. Targeting mitophagy to combat diabetic cardiomyopathy by enriching the heart's population of mitochondria with healthier organelles is a novel approach and should be investigated more thoroughly with the prospect of decreasing patient mortality and improving quality of life.