
Abstract
Introduction
C
Hydrogen sulfide (H2S) is known to protect endothelial cells against oxidative stress, although the underlying molecular mechanism is only partially understood. The present study demonstrates that H2S protects endothelial cells against oxidative damage by enhancing activator protein 1 binding activity with the sirtuin3 (SIRT3) promoter, thereby upregulating the expression of SIRT3 and its downstream genes, SOD2 and IDH2, as well as improving vascular endothelial function. The present study sheds new light on the molecular mechanism responsible for the cytoprotective effect of H2S through SIRT3 activation in endothelial biology.
The silent information regulator 2 (SIR2) family is functionally important in endothelial cells under oxidative stress (45). Sirtuin3 (SIRT3), one deacetylase belonging to SIR2, increases reactive oxygen species (ROS)-scavenging capacity by enhancing antioxidant enzyme (superoxide dismutase, SOD) activity (6). SIRT3−/− mice showed increased mitochondrial matrix oxidant stress without augmentation of intermembrane space or cytosolic oxidant signaling during sustained hypoxia (43).
Hydrogen sulfide (H2S) is not only a potent antioxidant (19), vasodilator (52), and inhibitor of both vascular smooth muscle proliferation (49) and myocardial apoptosis (8) but also synthesized endogenously in a wide array of cell types either from L-cysteine by cystathionine γ-lyase (CSE) and/or cystathionine β-synthase (CBS) or from cysteine and 3-mercaptopyruvate by cysteine aminotransferase and 3-mercaptopyruvate sulfurtransferase (3-MST) (19). Wen et al. reported that H2S protected endothelial cells against oxidative stress by acting first as an antioxidant and second by maintaining mitochondrial structure and function (44). Several studies suggest that H2S is able to regulate the activity of the sirtuin family, such as upregulation of sirtuin1 (SIRT1) in human PC12 cells (18) and human umbilical vein endothelial cells (HUVECs) (36, 53) and increase of SIRT3 (4) and sirtuin 6 (SIRT6) (12), to exert either physiological or pathophysiological effects. Nevertheless, the precise mechanisms of the antioxidant effect of H2S in endothelial cells remain unclear.
In the present study, we used a slow-releasing H2S donor drug, GYY4137 (17), to examine the antioxidant effect of H2S in endothelial cells and to investigate the downstream signal mechanisms involved. We have identified a completely novel role for SIRT3 in regulating the endothelial response to H2S, thereby raising the possibility that H2S interfering with SIRT3 may be of value in the treatment of cardiovascular diseases, which are underpinned by oxidative stress.
Results
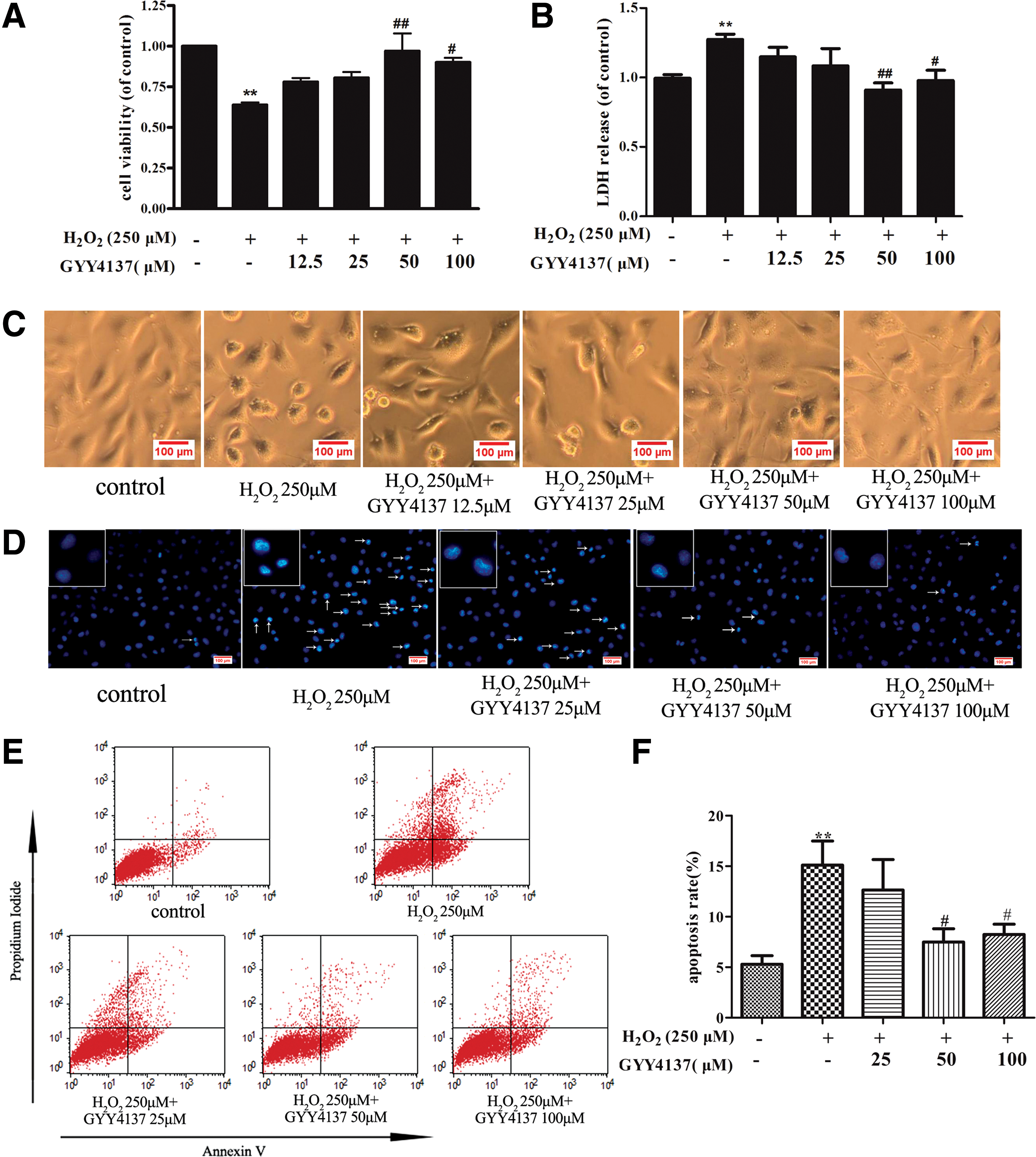
The effect of GYY4137 on H2S concentration, survival, and apoptosis of endothelial cells exposed to H2O2
Assessment of H2S release by amperometry showed that exposure of endothelial cells to H2O2 has no significant influence on H2S concentration in the medium (1.56 ± 0.13 μM vs. 1.37 ± 0.09 μM). Exposure of endothelial cells to GYY4137 (12.5, 25, 50, and 100 μM) before H2O2 treatment enhanced H2S concentration (2.15 ± 0.11, 3.14 ± 0.11, 5.46 ± 2.56, and 6.96 ± 0.38 μM, respectively). Meanwhile, H2O2 caused 36% ± 4% of cell death, and GYY4137 (50 and 100 μM) protected endothelial cells against the toxic effect of H2O2 (Fig. 1A), which were confirmed by lactate dehydrogenase (LDH) assay (Fig. 1B). Similar results were obtained when endothelial cells were visualized microscopically. Exposure to H2O2 caused cells to lose their typical fusiform shape as cobblestones and a rounded appearance, which suggests that a number of cells were dead after H2O2 treatment, while H2S preadministration partially restored cell structure and decreased cell death (Fig. 1C). Apoptosis was also measured by Hoechst 33342 staining and Annexin V/PI-positive staining. H2O2 triggered apoptosis in endothelial cells, as evidenced by the condensation and fragmentation of nuclei, and this apoptotic effect was significantly reduced by H2S (representative photomicrographs are shown in Fig. 1D). Likewise, Annexin V/PI-positive staining, as assessed by flow cytometry, showed greater staining in the presence of H2O2, indicative of apoptosis, which effect was reduced in cells pretreated with GYY4137 (Fig. 1E, F). Collectively, the present results demonstrate that exogenous H2S improves cell viability and decreases apoptosis in H2O2-treated endothelial cells.
The effect of H2S on oxidative stress, mitochondrial function, and mitochondrial permeability potential in endothelial cells exposed to H2O2
Oxidative stress plays an important role in the pathogenesis of cell death. To determine whether the protective role of H2S against apoptosis is related to reduction of ROS, redox status was monitored by dihydroethidium (DHE) oxidative fluorescence microtopography and the dichloro-dihydro-fluorescein diacetate (DCFH-DA) method. Endothelial cells responded to H2O2 with a significant rise in ROS formation and this rise was reduced by pretreatment of cells with GYY4137 (50 and 100 μM) (Fig. 2A, B). In addition, redox status is also related to the activity of antioxidant enzymes because H2O2 also reduced endothelial total SOD and Mn-SOD (SOD2) enzyme activity, while these effects were again reversed by H2S (Fig. 2C, D). In addition, H2S also elevated nitric oxide (NO) content to improve endothelial function after H2O2 treatment (Fig. 2E).

The decreased cellular ROS in H2S-treated cells led us to postulate that mitochondrial function could be impaired by H2O2. To test this hypothesis, we measured the oxygen consumption rate (OCR) in the presence and absence of H2S using the Seahorse XF analyzer. Figure 2F and G shows that cellular respiration and the response to modifiers of mitochondrial function were significantly decreased in H2O2-treated cells. We then performed the MitoStress test to quantify several bioenergetic parameters and we found that H2O2 significantly suppressed the basal respiration and oxygen consumption due to ATP turnover. Maximal respiration and respiratory reserve capacity were also significantly reduced by H2O2, indicating a generally depressed mitochondrial activity. Notably, H2S prevented these effects of H2O2 (Fig. 2F, G). In addition, mitochondrial permeability transition (Δψm) also was determined by JC-1 staining, as depicted in Figure 2H. Treatment of H2O2 resulted in the increase of green fluorescence intensity, but the decrease of red fluorescence intensity, indicating that the Δψm of the cells was significantly decreased. Pretreatment with H2S attenuated H2O2-induced collapse of Δψm in endothelial cells (Fig. 2H). These results suggest that H2S can improve mitochondrial function to limit oxidative stress in H2O2-injured endothelial cells.
The effect of H2S on the MAPK signaling pathway and caspase-3
Mitochondrial ROS are known to activate the mitogen-activated protein kinase (MAPK) pathway, which in turn participates in cell apoptosis. As shown in Figure 3A–C, H2S pretreatment attenuated H2O2-induced phosphorylation of the MAPK family, including p38 MAPK, ERK1/2, and Jun N-terminal kinase 1/2(JNK1/2). Caspases are crucial mediators of apoptosis. Among them, caspase-3 is a frequently activated death protease, catalyzing the specific cleavage of many key cellular proteins (26). As expected, the H2O2-induced expression of cleaved caspase-3 was reversed by H2S (Fig. 3D).

SIRT3 is involved in the protective effect of H2S in endothelial cells
SIR2 is a family of highly conserved NAD-dependent histone deacetylases that act as cellular sensors to detect energy availability and thus modulate metabolic processes, including mitochondrial function (45). SIR2 exists widely in mammals and consists of seven members (SIRT1-SIRT7), which vary in their cellular targeting and location and play a significant role in metabolism, carcinomatosis, cell survival, and other physiological and pathological processes (1). To examine the involvement of SIR2 in the antioxidant effect of H2S in endothelial cells, we examined mRNA expression of SIRT1-7 after exposure to H2O2. The mRNA expression of SIRT1, SIRT3, and SIRT4 was reduced in H2O2-treated cells, while expression of SIRT2, SIRT5, SIRT6, and SIRT7 was unchanged. Expression of SIRT3 (but not SIRT1 or SIRT4) mRNA level was restored to near baseline in cells incubated with GYY4137 (50 μM, 4 h) (Fig. 4A). Moreover, H2S alone did not alter mRNA and protein expression of SIRT3, but it reversed H2O2-induced reduction in SIRT3 expression in endothelial cells exposed to H2O2 (Fig. 4B, C). The positive role of SIRT3 in endothelial cells treated with H2S was further supported by silencing experiments. SIRT3-specific siRNA was transfected into endothelial cells (Fig. 4D) to reduce expression of SIRT3 (37% ±10% vs. 100% ±11%, Fig. 4E, F). SIRT3 silencing abolished the ability of H2S to reverse the H2O2-induced oxidant stress (Fig. 4G, H) and apoptosis in endothelial cells (Fig. 4I–K).

The effect of H2S on SIRT3-regulated signaling in endothelial cells
Two direct mechanisms linking SIRT3 with reduced ROS have been proposed following the identification of isocitrate dehydrogenase 2 (IDH2) and SOD2 as direct targets for SIRT3. SIRT3, as a major mitochondrial NAD+-dependent deacetylase, directly deacetylates and activates mitochondrial IDH2 and leads to increased NADPH levels and thereby an increased ratio of reduced to oxidized glutathione in mitochondria (34). A pivotal mitochondrial ROS-scavenging enzyme, SOD2, can be activated by SIRT3-mediated deacetylation to reduce levels of superoxide anions (6, 28, 38). Therefore, the expression of IDH2 and SOD2 was measured. Exposure of endothelial cells to H2O2 reduced expression of both SOD2 and IDH2, and this effect was reversed by pretreatment with GYY4137 (50 μM, 4 h). H2S failed to reverse this effect of H2O2 in cells transfected with SIRT3 siRNA (Fig. 5A, B). In addition, to test whether the effect of H2S on MAPKs is mediated by SIRT3 in H2O2-injured endothelial cells, we determined the expression of p-JNK, p- p38 MAPK, p-ERK, and total MAPK (p38 MAPK, ERK, JNK) in SIRT3 knockdown cells. As shown in Figure 5C–F, after transfection with SIRT3 siRNA, H2S failed to decrease the phosphorylation of JNK, but it was still able to attenuate the levels of p-p38 MAPK and p-ERK. These results suggest that H2S protects endothelial cells against oxidative damage not only by augmenting SIRT3-mediated IDH2, SOD2, and JNK pathways but also by impacting p38 MAPK and ERK pathways.

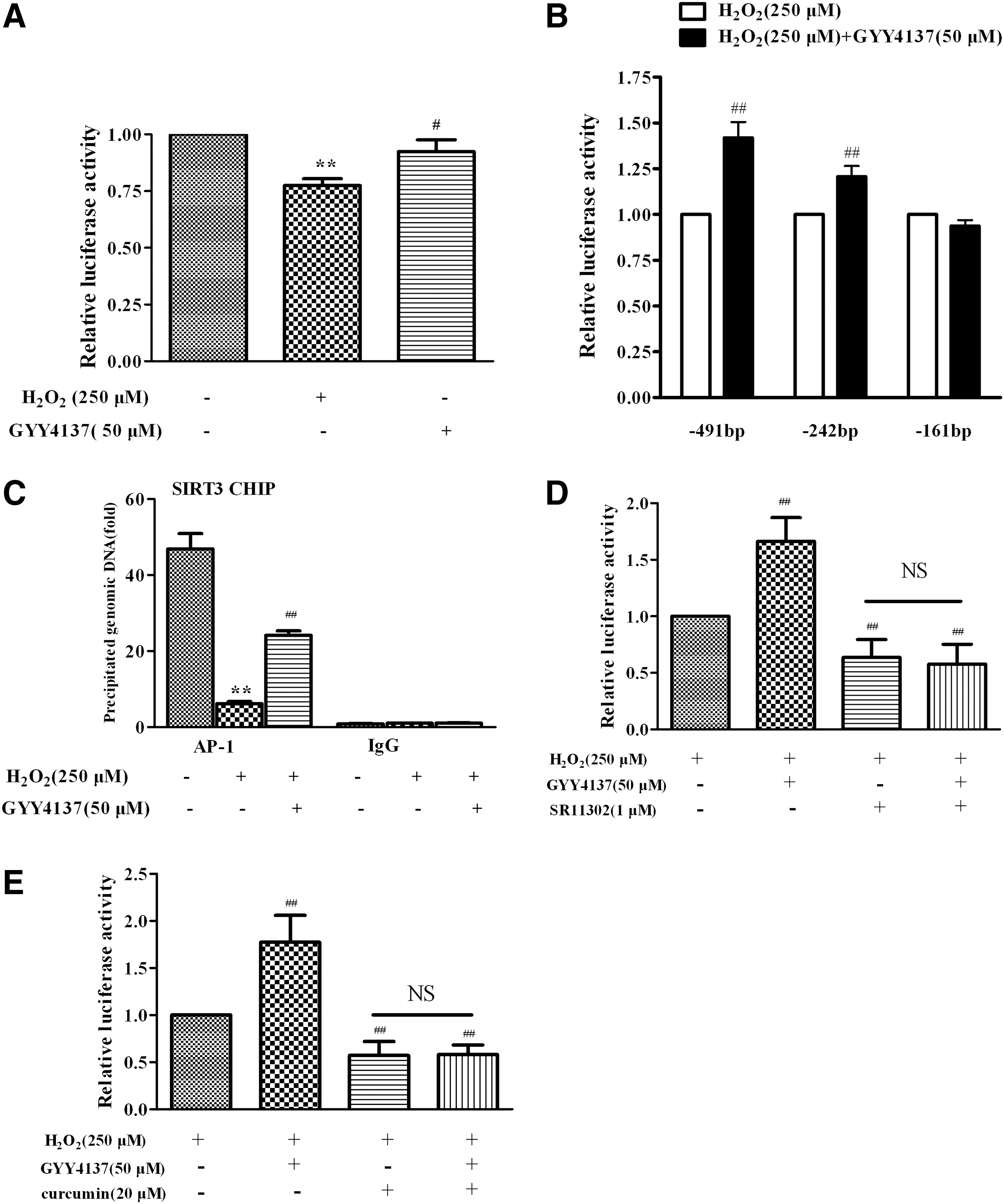
The effect of H2S on SIRT3 gene transcription
To investigate the mechanism by which H2S regulated the transcription of the SIRT3 gene in response to oxidative stress triggered by H2O2, a number of luciferase reporter plasmids containing a series SIRT3 promoter constructs with various lengths were constructed. EA.hy926 endothelial cells were transiently transfected with luciferase reporter plasmids containing the SIRT3 promoter (−491/+146). The reporter assays revealed a diminished SIRT3 promoter activity in endothelial cells exposed to H2O2, which was reversed by H2S (Fig. 6A). With a series of deletion constructs, the stimulatory effects of H2S on SIRT3 promoter activity were observed in −491 Luc and −242 Luc, of which the 5′ ends correspond to 491 bp and 242 bp from the transcription start site, respectively. However, H2S-induced enhancement of SIRT3 promoter activity was abolished in −161 Luc (Fig. 6B), suggesting that the presence of a critical site between 242 bp and 161 bp on the upstream of the SIRT3 promoter was responsible for the effect of H2S on SIRT3 transcription. The putative AP-1 binding site is present in this region of the SIRT3 promoter and the ChIP assay showed that H2S increased AP-1 binding activity with the SIRT3 promoter, which had been decreased in endothelial cells treated with H2O2 (Fig. 6C). The enhanced effect on SIRT3 promoter activity in the presence of H2S was absent when specific AP-1 inhibitor, SR11302 (1 μM) or curcumin (20 μM), was present (Fig. 6D, E). Collectively, these results suggest that H2S upregulated the SIRT3 gene expression via increasing the AP-1 binding activity with the SIRT3 promoter.
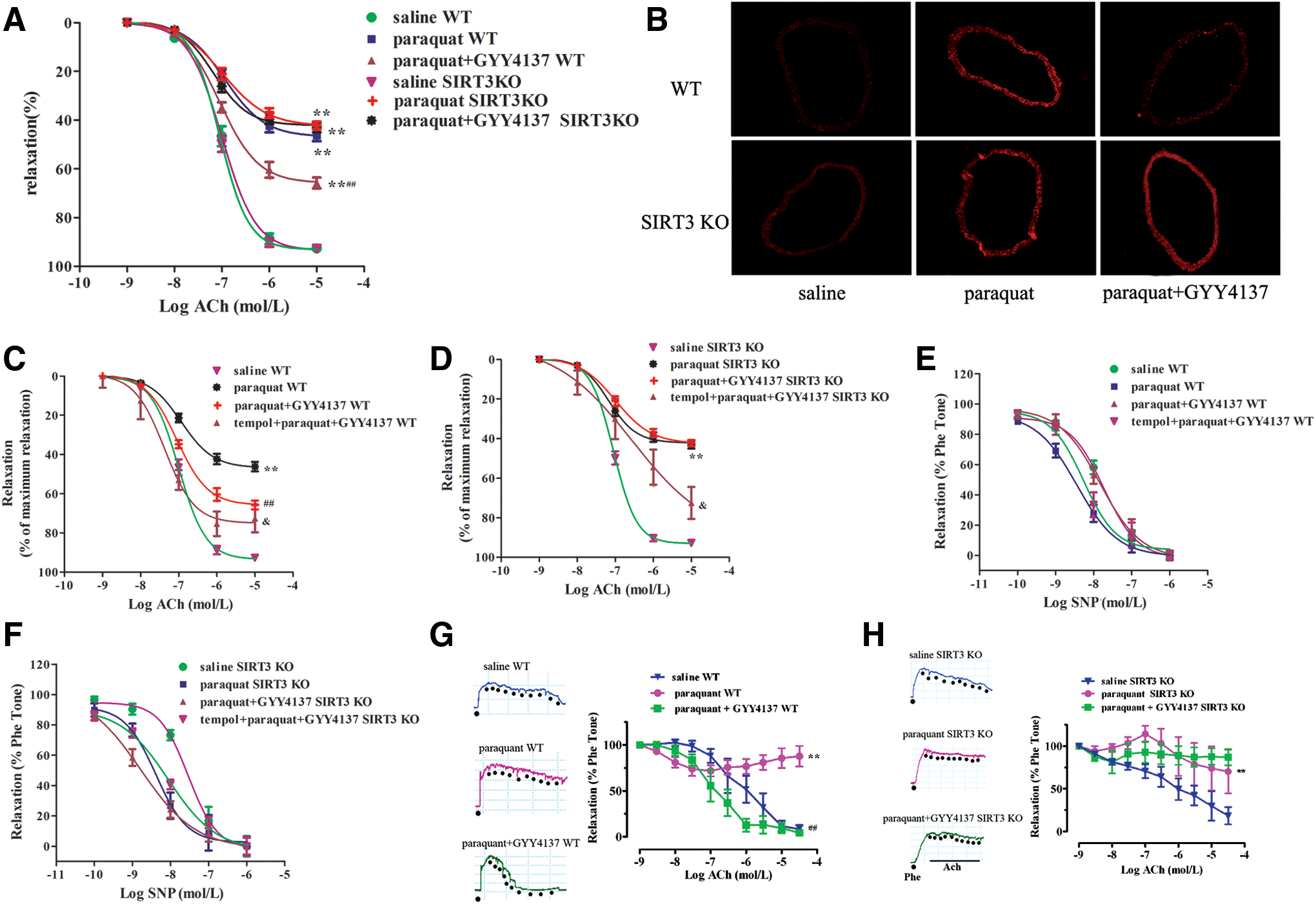
The protective effect of H2S in SIRT3 KO mice
Our results suggest that H2S-regulated SIRT3 is a critical endogenous inhibitor of oxidative damage induced by H2O2 in endothelial cells in vitro. To further explore the pathophysiological significance of H2S-induced SIRT3 expression in vivo, a state of oxidative stress was induced in mice by administration of paraquat (21). Mice treated with paraquat showed impaired endothelium-dependent relaxations and increased oxidative stress in their aortas (Fig. 7A, B). H2S administration improved endothelium-dependent relaxations accompanied by reducing oxidative stress in paraquat-treated mice. More importantly, H2S treatment lost its ability to augment endothelium-dependent relaxations and to lower oxidative stress in the aortas of SIRT3 KO mice (Fig. 7A, B). Although vascular oxidative stress was eliminated by tempol, H2S produced a greater improvement of endothelium-dependent relaxations in tempol preincubated aortas in WT mice, followed by paraquat and GYY4137 administration, than in SIRT3 KO mice (p < 0.05, Fig. 7C, D). In addition, there was no difference in aortic relaxation in response to NO donor (SNP) among all groups, suggesting that the sensitivity of vascular smooth muscle to NO was not altered (Fig. 7E, F). Moreover, H2S treatment augmented endothelium-dependent dilatations in small mesenteric arteries from WT mice, but not SIRT3 KO mice (Fig. 7G, H). These results demonstrate that SIRT3 is most likely required for H2S to inhibit endothelial oxidative damage in vivo, leading to improved endothelial function.
Discussion
Oxidative stress can be broadly defined as an imbalance of ROS production over antioxidant defenses. The mitochondrial respiratory chain is not only a major source of intracellular ROS generation but also an important target for the damaging effects of ROS (31). Endothelial cells are crucial both for vascular homeostasis and for protecting the vasculature against oxidant species. Endothelial cells are replete with CSE and are endogenous sources of H2S. The latter is a powerful antioxidant and it may defend the endothelium against oxidative stressors.
In the present study, oxidative injury was induced by H2O2 and paraquat, both in vitro and in vivo, and the protective effect of H2S was assessed using the donor, GYY4137. Consistent with previous studies (13, 44), the present study shows that H2S protects endothelial cells from cytotoxicity and improves mitochondrial function against H2O2 insult.
The seven mammalian sirtuin orthologs (Sirt1-7) have been studied in diverse disease models, including insulin resistance and diabetes, inflammation, neurodegenerative disease, and cancer, and more recently in cardiovascular pathologies such as cardiac hypertrophy, heart failure, and atherosclerosis (45). mRNA expression of all SIRT1-7 genes was reduced with advanced passages in several kinds of endothelial cells and such reduction was exaggerated in high-glucose-treated cells (22). Our study shows that mRNA expression of SIRT1, SIRT3, and SIRT4 was reduced in H2O2-treated cells, while the expression of SIRT2, SIRT5, SIRT6, and SIRT7 was unaffected. Other studies show that the expression and activity of SIRT1 can be increased by H2S (18, 36, 53). Zheng et al. found that H2S delayed nicotinamide-induced premature senescence of HUVECs via upregulation of SIRT1 (53). H2S also prevented H2O2-induced senescence of HUVECs through SIRT1 activation (36). The present results indicate that SIRT1 mRNA levels decreased in cultured endothelial cells following H2O2 treatment; however, this effect was not restored by H2S cotreatment. As such, our observation contrasted with a previous report that NaHS and GYY4137 were able to reverse H2O2-induced reduction of SIRT1 mRNA in HUVECs and in hyperoxic lungs (36, 41). This discrepancy may be attributed to different regulatory mechanisms after H2O2 exposure in different cell types, the use of different H2S treatment regiments giving rise to different kinetics of H2S release. The observation of increased SIRT3 mRNA by H2S in H2O2-exposed endothelial cells drove us to focus on SIRT3 in the present study.
We found that the effect of H2S was attenuated when SIRT3 was knocked down, suggesting that the protective effect of H2S against oxidative stress and apoptosis depends, at least in part, on SIRT3. SIRT3 plays a role in the maintenance and regulation of normal physiological function of mitochondria (25), which is achieved via its ability to deacetylate acetylated proteins in mitochondria (9, 10, 14). SIRT3 exists as a soluble protein in mitochondria to regulate the acetylation reaction, including those of acetyl-coenzyme A synthase, glutamate dehydrogenase, Ku70, IDH2, a forkhead protein, FOXO3a, and SOD2 (28, 38, 40). Overexpression of SIRT3 can enhance the binding between FoxO3a and the promoter of SOD2 to strengthen the activity of SOD2 transcription (23, 35). SIRT3 may increase mitochondrial ROS-scavenging capacity by enhancing antioxidant enzyme activity, including SOD2 (6). SIRT3 also deacetylates IDH2, which mediates intermediary metabolism and energy production (34, 39). Mitochondrial oxidative respiration is thus enhanced, maintaining mitochondrial energy metabolism and reducing ROS generation. It has been reported that H2S reduces atherosclerotic plaques and endoplasmic reticulum stress by SOD2 activation (7). Likewise, we found that both SOD2 and IDH2, downstream of SIRT3, were activated by H2S in endothelial cells. Therefore, we demonstrated that the SIRT3-SOD2 and SIRT3-IDH2 pathways were also involved in the protective effect of H2S against endothelial dysfunction.
Little is currently known about the molecular mechanism of the regulation of the SIRT3 gene expression. A previous study showed that peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α) activated the mouse SIRT3 promoter, which was mediated by an estrogen-related receptor binding element mapped to the promoter region (15). Our study suggests a potential role of H2S in modulating SIRT3 promoter activity in response to H2O2. This observation agrees with a previous report, indicating that mitochondria, especially during cellular stress or damage, are able to regulate a series of nuclear targeted genes by transcriptional factors acting as mediators of the well-known nucleus–mitochondrion cross talk (3). We also found that H2S could increase AP-1 binding activity with SIRT3 promoter, which was decreased after H2O2 treatment in endothelial cells. The enhanced effect of H2S on SIRT3 promoter activity was absent when specific AP-1 inhibitor, SR11302 or curcumin, was used. This result suggests that H2S can enhance the ability of AP-1 binding into SIRT3 promoter. AP-1 is a multifunctional transcription factor, which regulates gene expression, either positively or negatively, in different types of cells under various physiological and pathological conditions. It is noted that H2S donor inhibits the activation of AP-1, which is bound to the COX-2 gene promoter, in 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion in mouse skin (33). The factors, such as AP-1 and GATA2, have several motif binding sites located within the SIRT3 promoter region (2). Our study is probably the first one to show that H2S increases AP-1 binding activity with the SIRT3 promoter and thus enhances SIRT3 transcription to attenuate endothelial oxidative stress.
The MAPK family is one of the most important downstream signal pathways of oxidative stress. A previous study found that exposure of endothelial cells to H2S increased the phosphorylation of p38 MAPK (24). H2S also activated ERK1/2 to inhibit angiogenic features of human endothelial cells (46). However, others reported that H2S scavenged particulate matter-induced endothelial cell ROS and inhibited the oxidative activation of p38 MAPK (42). Inhibiting ERK decreased the H2S-induced rise in cell migration rate (50). H2S also decreased the expression of caspase-3 to protect human endothelial cells under a hypoxic condition (32). MAPK, as a double-edged sword, plays a vital role in regulating oxidative stress and other pathophysiological processes. Therefore, H2S plays diverse roles in the MAPK signaling pathway in endothelial cells in response to different stimulations. In the present study, prior treatment of endothelial cells with H2S not only conferred protection against death but also reduced activation of intracellular MAPK signaling and apoptosis. Similar results have been reported by other researchers, although using different types of cells (11, 30, 48). In addition, the present results show that the SIRT3 mediates the effect of H2S on JNK, but not p38 MAPK and ERK, in H2O2-treated endothelial cells. Further study is required to establish whether H2S, SIRT3, MAPK, and oxidative stress will form a complicated regulatory circuit. Nevertheless, this study demonstrates a significant role of SIRT3 activation in the protective effects of H2S against H2O2- and paraquat-induced injury of endothelial cells.
In summary, we provide new evidence that SIRT3 plays an important role in the antioxidant effect of H2S in vascular endothelial cells. In addition to inhibition of p38 MAPK and ERK signaling, H2S also enhances AP-1 binding activity with the SIRT3 promoter, which upregulates SIRT3, and subsequent elevation of SOD2 and IDH2 expression and downregulation of JNK activity. We therefore propose that H2S protects endothelial cells against oxidative damage not only by inhibiting p38 MAPK and ERK pathways but also by increasing the expression and activity of SIRT3 as a new mechanism by which H2S protects endothelial cells. Whether H2S donors or other drugs, which can modify SIRT3 signaling, are of value to diminish endothelial dysfunction, a key early event in the development of cardiovascular and metabolic diseases, warrants further investigation.
Materials and Methods
Materials
Unless otherwise stated, all reagents used were obtained from Sigma-Aldrich.
Cell culture and treatment
The EA.hy926 endothelial cell line was purchased from the American Type Culture Collection. Cells were seeded into six-well plates and cultured until they reached about 80% confluence. Dulbecco's modified Eagle's medium (DMEM; Gibco BRL) with 10% (v/v) fetal bovine serum (FBS; Gibco) was replaced immediately before addition of different concentrations of H2S donor, GYY4137 (12.5–100 μM). After 4 h of treatment, cells were incubated in freshly prepared medium containing H2O2 (250 μM; Sigma-Aldrich) for a further 4 h. Microscopic observation of cells to monitor the differentiation status of cultures and to record any cell changes during treatment was conducted using a converted phase-contrast microscope (Olympus).
Measurement of H2S in culture medium
H2S in culture medium was measured using an H2S-specific microelectrode (ISO-H2S-2; World Precision Instruments) connected to a free radical analyzer (TBR4100; World Precision Instruments) as described previously (51). The sensor was set to the 10-nA range and the poise voltage to +150 mV. Before initiation of the experiments, the sensor was polarized and calibrated by adding 4 aliquots of the Na2S stock solution at final concentrations of 0.5, 1, 2, and 4 μM. Concentrations of H2S in the samples were calculated using a standard curve of Na2S.
Cell viability assay
Cell proliferation was measured by Cell Counting Kit-8 (CCK-8; Beyotime) according to the manufacturer's directions. Briefly, EA.hy926 cells (1 × 104) were seeded in a 96-well plate and cultured overnight, then exposed to H2O2 (250 μM, 4 h) after pretreatment with GYY4137 or vehicle (medium, DMEM) for 4 h, followed by addition of 10 μl of the WST-8 mixture to each well. The cells were then incubated for 1 h at 37°C in the incubator. The absorbance was measured in a microplate reader (Biotek Instruments) at a wavelength of 450 nm.
Cell death was evaluated by the quantification of plasma membrane damage, which resulted in the release of LDH. The level of LDH released in the cell culture medium was detected by the LDH cytotoxicity assay detection kit (Beyotime) following the manufacturer's instructions. The optical density was measured spectrophotometrically at 490 nm on a microplate reader (Biotek Instruments).
Quantification of apoptosis
EA.hy926 endothelial cells were seeded in six-well plates and incubated overnight. Cells were then treated with H2O2 (250 μM, 4 h) after pretreatment with GYY4137 (25–100 μM) or vehicle for 4 h. To visualize nuclear morphology, EA.hy926 endothelial cells were fixed in 4% v/v paraformaldehyde and stained with Hoechst 33342 DNA dye (2.5 μg/ml). Uniformly stained nuclei were scored as healthy and viable cells. Condensed or fragmented nuclei were scored as apoptotic. The percentage of cells undergoing apoptosis was also determined by Annexin V/PI staining using the BU-ANNEXIN V-FITC apoptosis detection kit (Biouniquer Technology Co., Ltd.) following the manufacturer's instructions. Annexin V-FITC binding was detected by flow cytometry (BD Biosciences, FL1 filter for Annexin-V-FITC and FL3 filter for PI). The data were analyzed by Cellquest Pro software.
Measurement of ROS in endothelial cells
Cells were exposed to H2O2 (250 μM, 4 h) after pretreatment with GYY4137 or vehicle (DMEM) for 4 h and washed with phosphate-buffered saline (PBS) twice, then switched to serum-free DMEM containing DHE (5 μM; Vigorous Biotechnology Beijing Co., Ltd.) or DCFH-DA (10 μM; Beyotime) as described previously (37, 47). After that cells were washed with PBS again, and the red or green fluorescence was measured with a Zeiss Inverted Microscope (Carl Zeiss). DHE enters the cells and is oxidized by O2 •− to yield ethidium, which binds to DNA to produce bright red fluorescence. DCFH-DA is a lipophilic cell-permeable compound that is deacetylated in the cytoplasm to DCF by cellular esterases. DCF is then oxidized by radicals such as hydroxyl, peroxyl, alkoxyl, nitrate, and carbonate to a fluorescent molecule (excitation 530 nm, emission 485 nm).
Measurement of total SOD and SOD2 activity in endothelial cells
EA.hy926 endothelial cells were treated with H2O2 (250 μM, 4 h) after pretreatment with GYY4137 (25–100 μM) or vehicle (DMEM) for 4 h as described above. Then, cells were washed using PBS and lysed in ice-cold 0.1 M Tris/HCl (pH 7.4) containing 0.5% Triton, 5 mM β-mercaptoethanol, and 0.1 mg/ml phenylmethylsulfonyl fluoride. Lysates were clarified by centrifugation at 14,000 g at 4°C for 5 min and cell debris was discarded. SOD activity was detected using a commercial SOD Assay Kit-WST according to the manufacturer's protocol (Dojindo Molecular Technologies). SOD2 activity was measured in the presence of a CuZnSOD inhibitor (3 mM NaCN) and normalized to total protein content. The highly water-soluble tetrazolium salt, WST-1, which produces a water-soluble formazan dye upon reduction by the superoxide anion, was used to measure SOD2 activity. Absorbance values at 450 nm were measured using a microplate reader (Biotek Instruments).
Measurement of NO
The production of NO in cultured supernatants was measured using an Apollo 1000 single-channel free radical detector employing an amperometric-type NO probe (ISO-NOPF100H; World Precision Instruments), as described previously (27).
Analysis of mitochondrial respiration
EA.hy926 endothelial cells were plated (1.2 × 103 cells/well) in an XF 96-well plate (Seahorse Bioscience) and 18 h thereafter cells were treated with GYY4137 (25–100 μM, 4 h), followed by addition of the medium or H2O2 (250 μM, 4 h) immediately before the medium was changed to unbuffered DMEM containing glucose (10 mM), pyruvate (10 mM), and GlutaMAX (2 mM; Invitrogen). Mitochondrial respiration OCR was measured by a Seahorse Extracellular Flux Analyzer XF96 (Seahorse Bioscience) (5). Briefly, a classical MitoStress test was performed according to the following procedure: (1) basal respiration was measured in unbuffered medium; (2) oligomycin (2 μg/ml final concentration), an inhibitor of ATP synthesis, was injected to determine respiration linked to ATP production; (3) the uncoupler, carbonyl cyanide 4-(trifluoromethoxy) phenylhy-drazone (FCCP, 2 μM), was added to measure maximal respiration; and (4) antimycin A (4 μM) plus rotenone(4 μM) was applied in combination to block respiration due to simultaneous inhibition of complexes III and I, respectively.
Assessment of mitochondrial membrane potential
Mitochondrial membrane potential (Δψm) was measured by a commercial JC-1 kit (Beyotime) following the manufacturer's instructions. Briefly, EA.hy926 were grown on a six-well plate and were treated with GYY4137 (25–100 μM, 4 h), followed by addition of the medium or H2O2 (250 μM, 4 h), then confluent cells were rinsed with PBS and incubated in 1 ml JC-1 staining solution at 37°C for 20 min. Cells were then rinsed twice with JC-1 washing solution and digital pictures for JC monomers (Green fluorescence; 535 nm) and JC aggregates (Red fluorescence; 570 nm) were captured with a Zeiss Inverted Microscope (Carl Zeiss). The ratio of red (J-aggregate)/green (monomeric JC-1) emission is directly proportional to the Δψm.
RNA interference
The following double-stranded RNA oligos specific for SIRT3 (sense, 5′-GCCCAACGTCACTCACTACTT-3′, antisense, 5′-GUAGUGAGUGACGUUGGGCTT-3′) were synthesized by Shanghai GenePharma. Commercially available siRNA to random noncoding sequences were used for control transductions (Shanghai GenePharma). To obtain SIRT3 knockdown cells with transient transfection, cells were transfected with siRNA duplexes at the final concentration of 100 nM using Lipofectamine 2000 reagent (Invitrogen). The cells were used for experiments in 24 h after transfection, and gene silencing was detected by analysis of SIRT3 protein expression with Western blot. Transfected with control siRNA or SIRT3 siRNA for 24 h, EA.hy926 cells were exposed to H2O2 (250 μM, 4 h) after pretreatment with GYY4137 or vehicle (DMEM) for 4 h; ROS and apoptosis were measured as described earlier.
Western blot analysis
Cytoplasmic protein samples were separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred onto a polyvinylidene fluoride membrane (Millipore), and then immunoblotted with primary anti-IDH2 (1:500; Santa Cruz Biotechnology), anti-SIRT3, anti-p38 MAPK, anti-p-p38 MAPK, anti-ERK, anti-p-ERK, anti-JNK, anti-p-JNK, anti-cleaved caspase-3 (1:1000; Cell Signaling Technology), anti-SOD2 (1:500; Abcam), and anti-GAPDH (1:6000; Sigma). Proteins were visualized by enhanced chemiluminescence substrate (Thermo Fisher Scientific, Inc.).
Quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Takara). RNA (500 ng) was added as a template to reverse transcriptase reactions carried out using the PrimeScript™ RT Master Mix Kit (Takara). Quantitative real-time PCR (qRT-PCR) was carried out with the resulting cDNAs in triplicate using SYBR Green remix (Takara) and the ABI 7500 Real-Time PCR System (Applied Biosystems ABI). Experimental Ct values were normalized to 18 s and relative mRNA expression was calculated versus a reference sample. Each sample was run and analyzed in triplicate (Structures of all primers used are listed in Table 1).
Plasmids and luciferase reporter assays
The mSIRT3 promoter (−491 to +146) and pGL3-Basic were provided by Professor Yongsheng Chang of the Chinese Academy of Medical Sciences and Peking Union Medical College. Various fragments of the 5′ flanking promoter region of mSIRT3 were generated by PCR amplification of Luc-491, followed by cloning into pGL3-Basic using the Acc65 I and Xho I sites, as described previously (15).
EA.hy926 endothelial cells were cultured in 24-well plates and cotransfected with luciferase reporter construct (0.2 mg/well) and pRL-TK reporter plasmid (control reporter, 0.006 mg/well) using Lipofectamine 2000 reagent, according to the manufacturer's protocol (Invitrogen). After transfection (24 h), cells were treated with GYY4137 or DMEM as vehicle for 4 h and then exposed to H2O2 (250 μM) for a further 4 h. Cells were harvested in lysis buffer and luciferase activity was measured using the dual-luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to that of control reporter.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed using the Pierce Agarose ChIP Kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's recommendations. In brief, EA.hy926 endothelial cells were treated with H2S (GYY4137 50 μM) for 4 h before H2O2 (250 μM) treatment for 4 h. Protein samples were then precleared with protein A-agarose/salmon sperm DNA (30 min, 4°C), followed by overnight incubation at 4°C with antibodies specific for AP-1 or normal rabbit IgG (as a negative control). The immune complexes were precipitated with protein A-agarose for 1 h. Precipitated genomic DNA was amplified by real-time PCR with primers. Potential AP-1 binding sites on SIRT3 promoter region were amplified with the following primer pairs: 5′-AATCTCCCGGTTTGGCTTCC-3′ (sense) and 5′-CCCGCACGATAACCCGAAGT-3′ (antisense).
Animals and treatment
SIRT3+/− mice were the gift of Professor Hongliang Li (Wuhan University, China). These mice were intercrossed to produce homozygous SIRT3−/− and wild-type 129S1/SvImJ animals. Male SIRT3−/− mice and wild-type (WT) control animals (8–10 weeks) were randomly treated with either GYY4137 (133 μM/kg, i.p.) or vehicle (saline, i.p.) 1 h after paraquat (50 mg/kg, i.p.) or vehicle (saline, i.p.) injection. The doses of both GYY4137 and paraquat used were based upon prior reports in the literature (5, 16). Endothelium-dependent vasorelaxation of mouse aorta and small mesenteric artery was assessed after paraquat injection for 24 h. All animal experiments were conformed to the Guide for the Care and Use of Laboratory Animals published by NIH and also approved by the Committee on Animal Care of Nanjing Medical University (NJMU-ERLAUA-20100112).
Measurement of superoxide formation in mouse aorta
Superoxide production in tissue sections of mouse aorta was detected by fluorescence microtopography using the fluorescent probe, DHE, as previously described (20).
Assessment of endothelium-dependent relaxations
Vasorelaxation of isolated aortic ring segments was determined in organ baths containing oxygenated Krebs' solution. After an equilibration period of 60 min, aortic rings were precontracted with norepinephrine or phenylephrine (Phe, 0.1 μM). Endothelium-dependent or independent relaxation was then assessed in response to cumulative addition of acetylcholine (Ach, 10−9–10−5 M) or sodium nitroprusside (SNP, 10−10–10−6 M) with or without superoxide scavenger tempol (1 mM) preincubation for 60 min. Relaxation at each concentration was measured and expressed as the percentage of force generated in response to norepinephrine.
Statistical analysis
All data are presented as mean ± SEM. Statistical analysis was performed by one-way ANOVA, followed by the Newman–Keuls multiple comparison test as appropriate (Stata13.0 software). p < 0.05 was considered to be statistically significant.
Footnotes
Acknowledgments
This work was supported by grants to Y.J. from the National Basic Research Program of China (973) (2011CB503903), grants to Y.J. (81170083, 81330004, 31371156), to L.X. (81200197), to Y.H. (81200196), and to G.M. (81400203) from the National Natural Science Foundation of China, and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions. Additional support was provided by a grant to M.X. from NIH (R01HL116571), a grant to P.K.M. from the Biomedical Research Council (BMRC) of Singapore, and an operating grant to R.W. from Canadian Institutes of Health Research.
Author Disclosure Statement
No competing financial interests exist.