
Abstract
Introduction
O
Cisplatin-induced ototoxicity occurs in 30% of patients treated with cisplatin and yet the mechanism of cisplatin ototoxicity is uncertain. From our study, we observed that Cx43 accelerates cisplatin-induced cell death either in a gap junction-dependent or -independent manner in auditory cells. Study results also revealed significant decrease in cisplatin-induced cell death by gap junction reduction such as siRNA-mediated gene inhibition or pharmacological inhibition. In addition, Cx43 siRNA significantly decreased cell death in the nonjunctional region and this finding was not changed by disruption of Cx43 trafficking with brefeldin A. HeLa cells expressing the cytoplasmic C-terminal domain showed enhanced sensitivity. This result/process implies that the prodeath activity of Cx43 protein is independent of the gap junction. These findings may provide a new strategy against cisplatin-induced ototoxic damage, enabling hearing preservation in cancer patients undergoing chemotherapy.
Cisplatin is transferred into hair cells through transporters, including copper transporter 1 and organic cation transporter 2, and induces NADPH oxidase 3 (NOX3) and superoxide-generating enzyme-mediated production of reactive oxygen species (ROS). Excessive ROS production results in redox imbalance and eventually becomes a mediator of cell death (28). Despite several studies on the mechanism of cisplatin-induced cytotoxicity, the ototoxic pathophysiology remains unclear and most studies have focused on the intracellular mechanisms.
Gap junctions are one of the most important channels involved in intercellular communication in the cochlea (31). Gap junctions permit the direct intercellular transfer of molecules <1 kDa. Each gap junction is formed by apposition of two hemichannels from adjacent cells, and each in turn comprises six protein subunits termed connexins (Cxs) (55). The Cx consists of four transmembrane domains, two extracellular loops, a cytoplasmic loop, a cytoplasmic amino-terminal (NT), and a carboxy-terminal (CT) domain (43). To date, the Cx family comprises 21 members in the human and 20 in the mouse, and these are expressed in a tissue-specific manner (28). Among them, Cx43 is the most widely studied in humans given that Cx43 is the most ubiquitous Cx protein found in human tissues, including heart, brain, and kidney (46), and its role in cellular events, such as differentiation, proliferation, and cell death (16, 51).
In the inner ear, five types of Cxs, including Cx43, Cx26, Cx29, Cx30, and Cx31, are expressed in supporting cells that surround hair cells and lateral wall cells in the cochlea. These Cxs play a role in K+ circulation, which is essential for generating the endocochlear potential (26). Previous studies revealed the importance of Cx gap junction networks for hearing functions by reporting that Cx mutations caused hearing loss and were associated with the interruption of K+ flow in the inner ear (31). However, the role of gap junctions and Cxs in cisplatin-induced ototoxicity remains unknown.
Gap junctional intercellular communication (GJIC) plays an important role in various physiological cell functions (42, 54). In particular, under pathological conditions, gap junctions can promote either cell survival or cell death depending on the type of tissue or the extent of injury. The former is referred to as the Good Samaritan effect and the latter the bystander effect (48). So far, hemichannels and Cxs are considered simply as structural components for gap junction formation. However, there is ample evidence suggesting that hemichannels can also be opened or closed under pathological conditions, including changes in redox status, membrane depolarization, and Ca2+ influx, regardless of the absence of gap junctions. The opening of unopposed hemichannels released molecules ≤1 kDa, such as nicotinamide adenine dinucleotide, ATP, glutamate, glutathione, and inositol triphosphate into extracellular fluid, where these molecules play roles as paracrine and/or autocrine mediators between distant cells and, in turn, participate in various cellular functions (9, 36, 39). In addition, several studies involving gene deletion mutants reported that the Cx protein itself could affect cellular function in a junction-independent manner (32). Our previous study described the role of gap junctions as bystanders to cisplatin-induced cell death (28). Then, we questioned the critical roles of the nonjunctional Cxs in cell death, and we hypothesized that Cx43 participates in cisplatin-induced auditory cell death in both gap junction-dependent and -independent pathways.
The purpose of the present study was to investigate the role of junctional and nonjunctional Cxs in ototoxic drug-induced auditory cell death by focusing on Cx43 in the cochlea.
Results
Effects of gap junction inhibition on cisplatin-induced toxicity under junctional and nonjunctional conditions
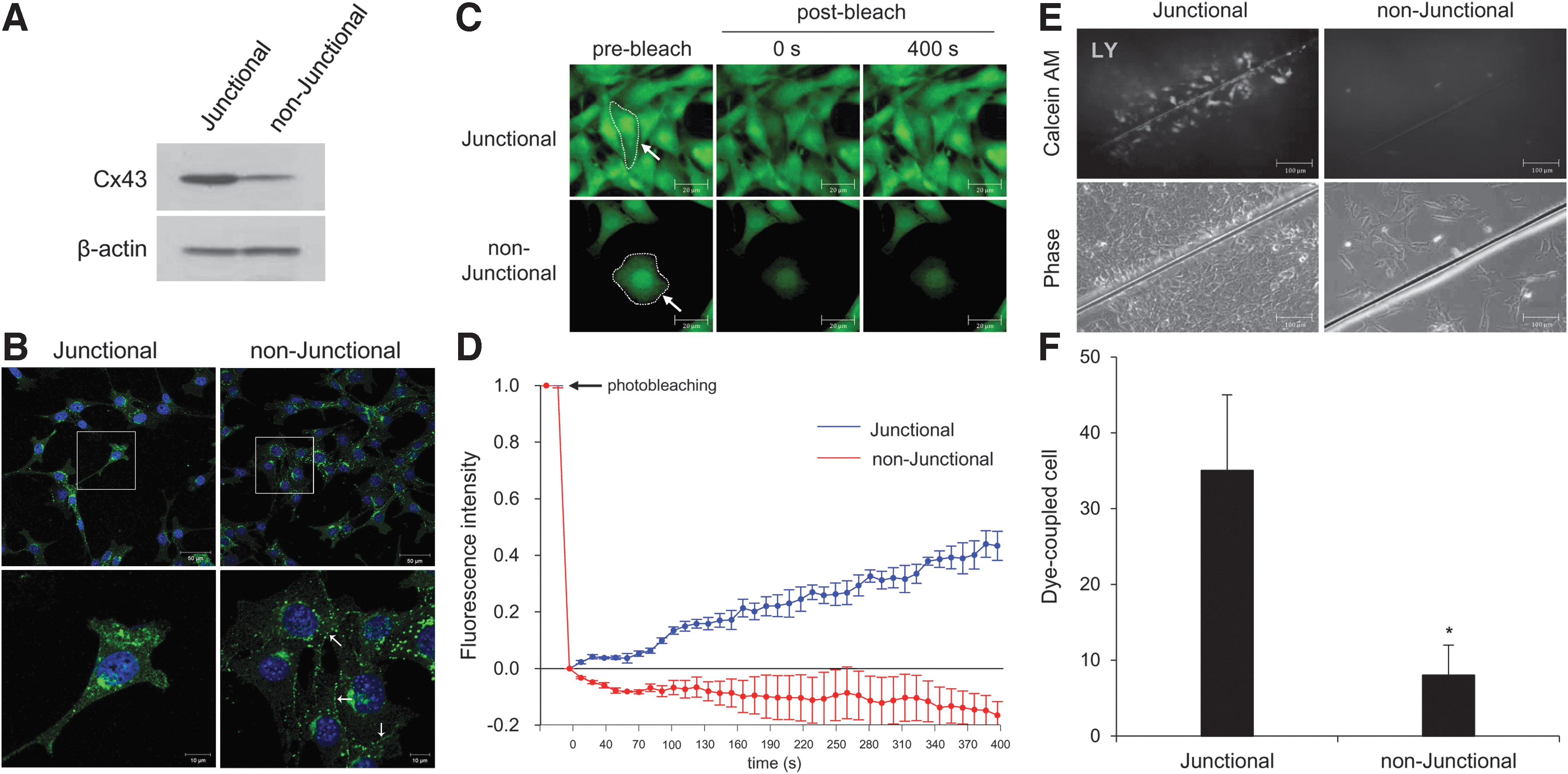
Based on cell density, we developed a gap junctional and nonjunctional model in mouse organ of Corti-derived HEI-OC1 (House ear institute-organ of Corti 1) cells. Since gap junction plaques are formed at cell–cell contact sites, cell density-dependent regulation is a basic factor referred to as either a gap junctional or nonjunctional condition (51). However, cell density control could not rule out the possibility of gap junction formation, therefore we applied more acceptable methods toward nonjunctional conditions. As an initial experiment to check these conditions, patterns of Cx43 expression and GJIC were tested using two techniques: (a) fluorescence recovery after photobleaching (FRAP) and (b) scrape load dye transfer (SLDT) as previously described (28).
As shown in Figure 1A, Cx43 was found to be widely expressed in HEI-OC1 cells. The intercellular gap junction plaques were observed between neighboring cells, while higher levels of Cx43 protein were expressed in high-density cell culture compared with low-density cell culture (Fig. 1B). In the FRAP assay, a region of interest (ROI) in a cell was photobleached by exposure to an argon laser under quenching of the green fluorescence tracer dye, calcein AM (2 μM/L, 0.623 kDa). Cells were monitored every 10 s for 400 s to observe the recovery of fluorescence intensity. As shown in Figure 1C and D, at high density, HEI-OC1 cells recovered about 42.3% ± 5.2% of their prebleach signal within 400 s of photobleaching. However, at low cell density, fluorescence intensity did not recover and decreased by 16.3% ± 4.8%. In line with this approach, we performed the SLDT assay at both high and low cell densities. Lucifer yellow (LY) dyes are capable of penetrating gap junctions and were added to cell culture media. Dye-transferred cells were observed at high cell density, while no dye-transferred cells were observed at low cell density (Fig. 1E, F). Taken together, our results indicate that the nonjunctional conditions in auditory cells were successfully prepared. We next evaluated the effect of Cx43 on cisplatin-induced ototoxicity in junctional and nonjunctional conditions.
To investigate the role of junctional and nonjunctional Cx43 on cisplatin-induced toxicity, we used 18 alpha-glycyrrhetinic acid (18α-GA) or Cx43 small interfering RNA (siRNA). 18α-GA, which is a derivative of licorice roots, can rapidly block GJIC and hemichannels (52). In the FRAP assay, the relative intensity of fluorescence decreased by 40.4% ± 1.0% (p > 0.05) and 60.2% ± 9.0% (p < 0.05) when cells were treated with 18α-GA and Cx43 siRNA, respectively, in comparison with control (Fig. 2B, C and Supplementary Fig. S1; Supplementary Data are available online at
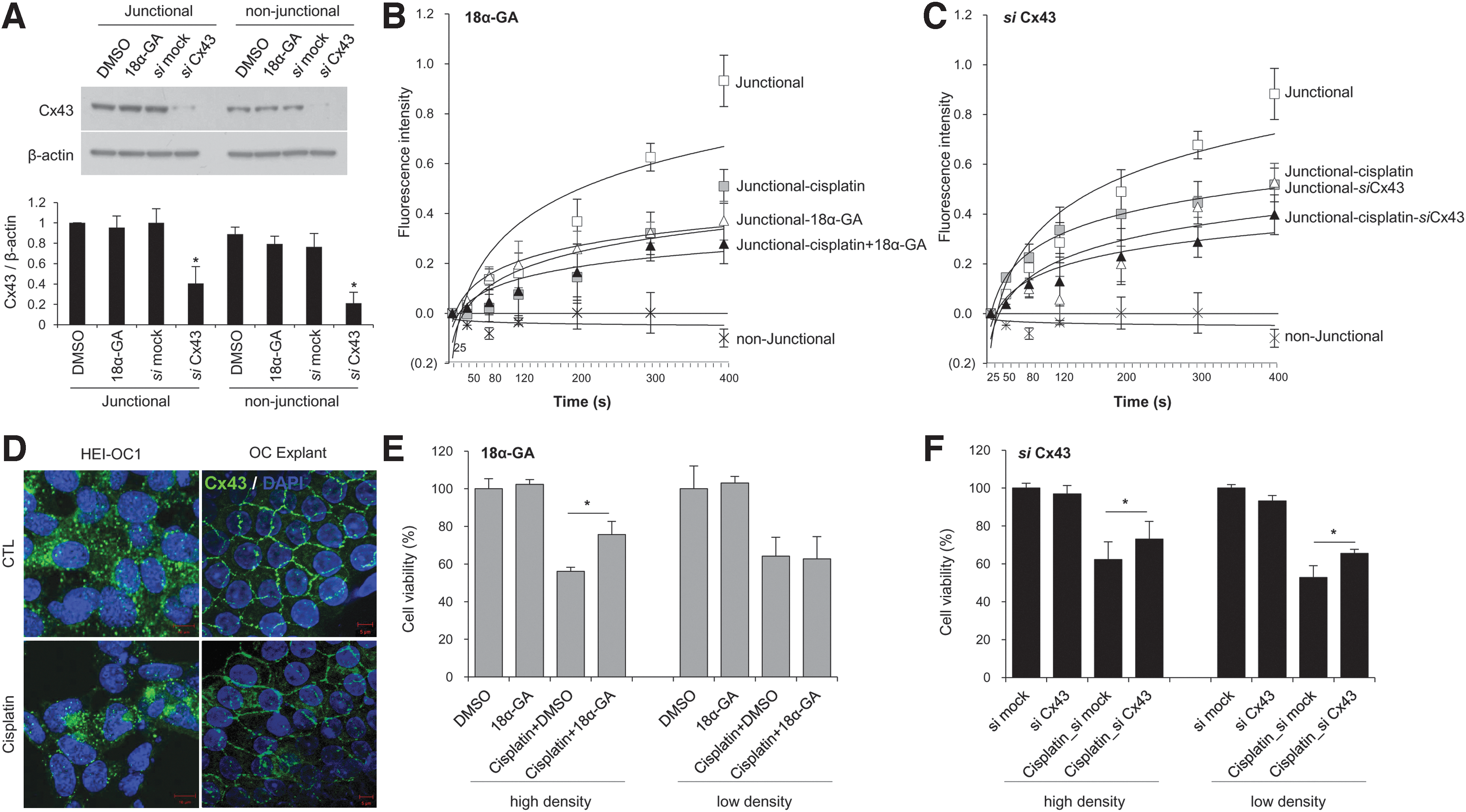
To study the impact of cisplatin on GJIC in auditory cells, we performed FRAP assays. Following treatment with 10 μM cisplatin, the relative intensity of fluorescence significantly decreased by 53.5% ± 9.54% compared with that observed in nontreated cells and decreased further following the addition of 18α-GA (from 60.1% ± 1.1% to 69.9 ± 9.4%) and knockdown of Cx43 (from 40.3 ± 8.8% to 54.9 ± 8.9%) (Fig. 2B, C). Immunofluorescent staining of Cx43 revealed that there were fewer intercellular gap junction plaques between adjacent cells at the plasma membranes in both cisplatin-treated cells and rat cochlear explant cultures (Fig. 2D).
Next, we assessed whether Cx43 siRNA and 25 μM 18α-GA affected the degree of cytotoxicity. At high cell density culture, cisplatin-induced cell death was decreased in both the Cx43 knockdown and 18α-GA treatment conditions (Fig. 2E, F). This suggests that gap junctions have a proapoptotic role in cisplatin-induced injury in this auditory cell line. Our previous studies demonstrated that gap junctions have bystander effect, and 18α-GA, a gap junction blocker, has protective effects against cisplatin. This protective effect of 18α-GA is mediated by activation of the mitogen-activated protein kinase (MAPK) pathway (28). Recently, it was reported that gap junction blockers, particularly carbenoxolone (CBX), significantly reduced cell death by attenuating oxidative stress (13, 25, 57). Considering that ototoxicity is primarily linked to oxidative stress, we evaluated the antioxidant potential of gap junction blockers with oxidative damage (4-HNE, NOX3, CM-H2DCFDA), antioxidant enzyme (SOD2) markers in HEI-OC1 cells. Previous studies have reported that NOX3 isoform and 4-HNE are highly expressed in the cochlea (29, 34), where they can mediate oxidative stress from ROS generation under stressful conditions (24). Furthermore, SOD2, which is expressed in the mitochondrial matrix, is thought to play an important role in controlling ROS and also related with auditory cell death from oxidative stresses (21). When HEI-OC1 cells were cotreated with 10 μM cisplatin and the gap junction blocker, 18α-GA, for 24 h, both the expression of 4-hydroxynonenal-modified protein (a membrane lipid oxidation product) and the activation of a fluorescent probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA), to detect ROS formation significantly decreased compared with cells without 18α-GA treatment (Supplementary Fig. S2). However, there were no changes in NOX3 and SOD2 levels. Further studies are needed to determine the major signaling events associated with the cisplatin-induced bystander effect and to understand the relationship between gap junction blockers and oxidative stress.
In low cell density cultures that simulate the nonjunctional conditions, treatment with 18 α-GA had no effect on cisplatin-induced toxicity (Fig. 2E). However, interestingly, the cells in which Cx43 was knocked down displayed higher cell viability compared with mock-transfected cells under cisplatin regardless of junctional or nonjunctional conditions (Fig. 2F). Therefore, we hypothesized that the protective effect of Cx43 knockdown may not only depend on GJIC but also may be attributable to Cx43 itself or a hexameric complex, hemichannel. To test this hypothesis, we studied whether Cx43 could affect cisplatin-induced cell death in a gap junction-independent manner.
Cx43 hemichannels are not changed by cisplatin treatment
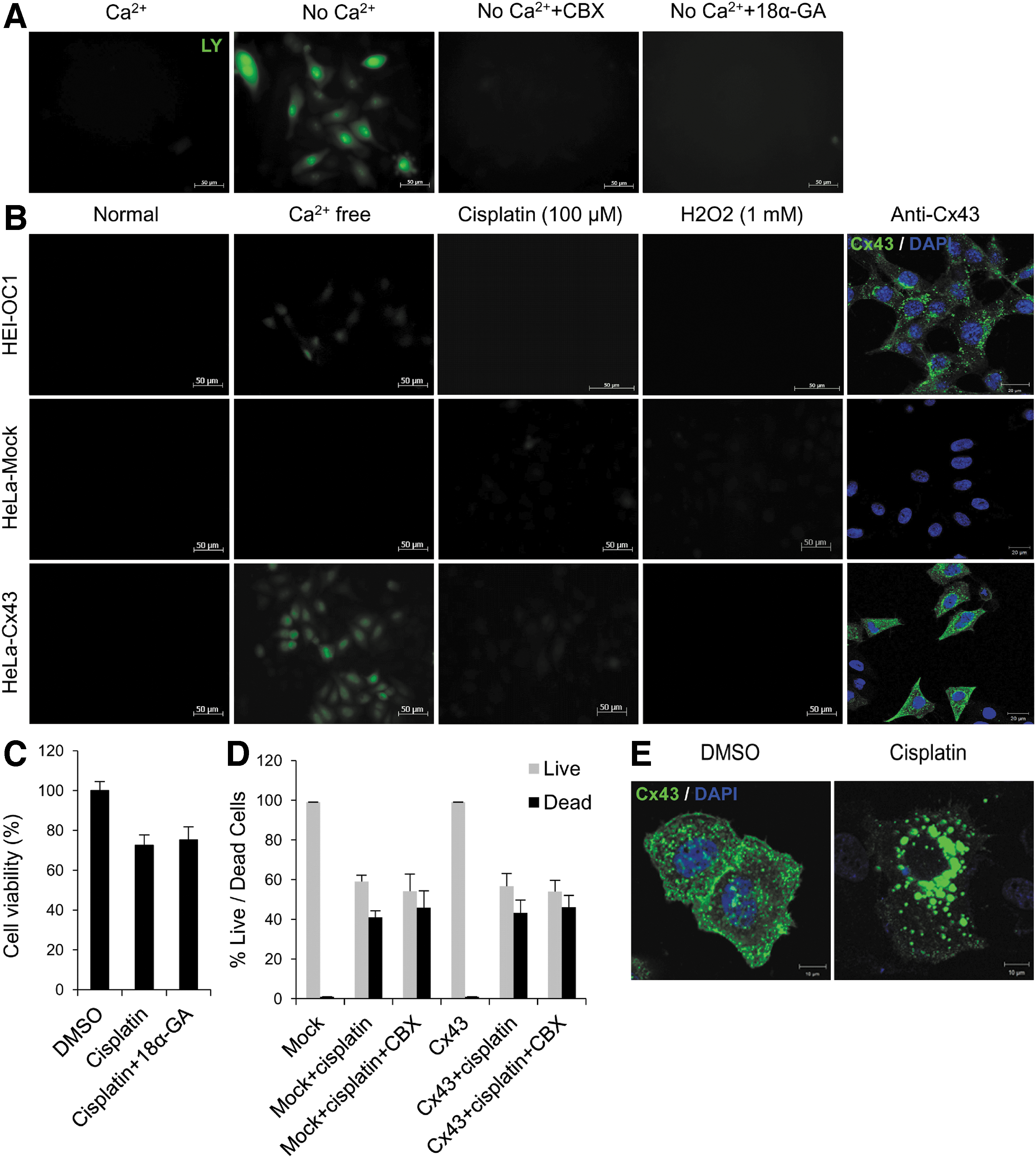
To investigate the role of hemichannels, the LY dye transfer method was used in HEI-OC1 cells and Cx43-transfected HeLa cells. It has been reported that hemichannel activity can be regulated by the extracellular Ca2+ concentration and that the opening is induced under low Ca2+ conditions (50). The functional hemichannels were confirmed by passage of LY dye into cells in the absence of Ca2+ at low cell density, minimizing cell contacts. The LY uptakes were inhibited in cells pretreated with hemichannel blockers, 18α-GA and CBX (Fig. 3A). These results indicate that Cx43 acts as a hemichannel in a gap junction-independent manner. We then examined whether cisplatin or oxidative stress (via addition of hydrogen peroxide, H2O2) could induce hemichannel opening. As shown in Figure 3B, no LY uptake was observed in the presence of either 100 μM cisplatin or 1 mM H2O2, and cotreatment with 18α-GA or CBX did not affect cell viability in cisplatin-treated cells at low cell density culture (Fig. 3C, D). In high-density cell culture condition, cell viability of Cx43-transfected HeLa cells was higher in the CBX and cisplatin cotreatment group than the group treated with cisplatin alone (Supplementary Fig. S3). These outcomes imply that the opening of Cx43 hemichannels is not related to cisplatin-induced cell death. Furthermore, we found that Cx43s were distributed in a scattered pattern in the cytoplasm and plasma membranes, whereas treatment with cisplatin caused significant condensation of Cx43s close to the nuclei (Fig. 3E). Thus, cisplatin also inhibits the trafficking of Cx43 to the plasma membrane, influencing the properties of hemichannel.
Cx43 mediates cisplatin-induced toxicity through a gap junction-independent mechanism
Although HEI-OC1 cells with low confluence underwent less cisplatin-induced cell death by Cx43 siRNA, cell density control was insufficient to prevent all cell-to-cell contacts. Accordingly, we used two methods to rule out the presence of gap junctions between coupled cells at low cell density culture. The constructs encoding full-length (FL) sequence, NT (aa 1–256), and the cytoplasmic CT domain (aa 243–382) of Cx43 were used in Cx-deficient HeLa cells (35) (Fig. 4A). After transfecting HeLa cells for 24 h with the three constructs, reverse transcription–polymerase chain reaction (RT-PCR) was performed using primer pairs for both upstream and downstream directions (T7 and Bovine growth hormone [BGH] primers). Amplified PCR products of sizes, 1307, 922, and 574 bp, were observed, while empty vector-transfected HeLa cells displayed a size of 157 bp. The nontransfected HeLa cells displayed no expression of any PCR products (Supplementary Fig. S4A). Immunofluorescence study showed that Cx43-CT was placed in the cytosol and nucleus, whereas Cx43-NT was mainly localized at the plasma membrane (Fig. 4A). Cisplatin-induced cell death was significantly enhanced in cells transfected with Cx43-FL (78.7% ± 8.5%), NT (85.9% ± 3.9%), and CT (83.3% ± 3.4%) transfection compared with mock-transfected cells (91.0% ± 3.3%), and there was no significant difference in the decrease of cell viability between partially sequenced Cx43-transfected cells and Cx43-FL-transfected cells (Fig. 4B). These findings indicate that nonjunctional Cxs play a similar role to junctional Cxs in the apoptotic point of view.
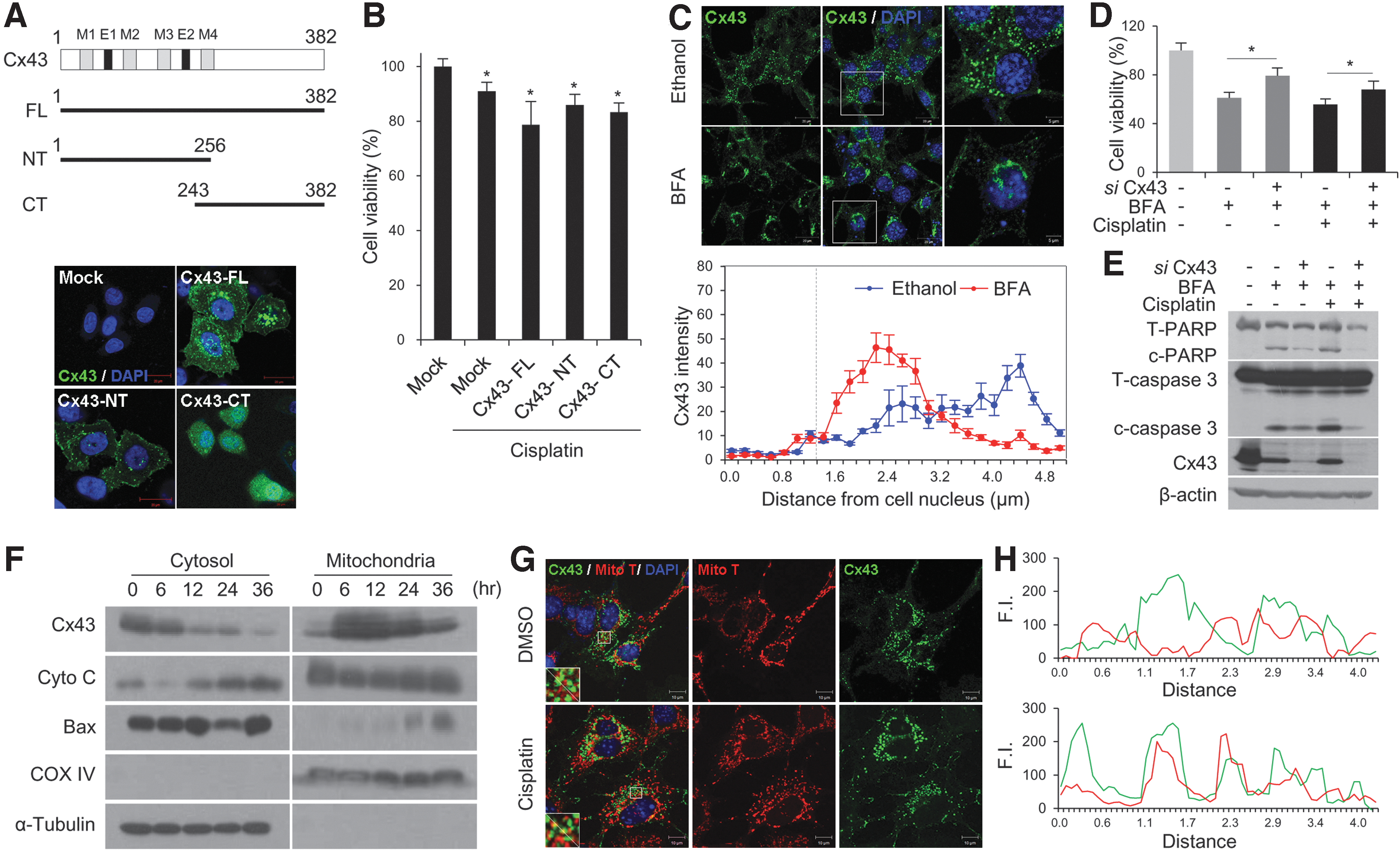
Newly synthesized Cxs oligomerize into a hemichannel at the trans-Golgi network after exiting the endoplasmic reticulum and are then transported along microtubules to the plasma membrane (12). Brefeldin A (BFA) is a natural fungal metabolite that blocks the transfer of Cxs from the Golgi complex to the plasma membrane, in turn preventing the formation of gap junctions (44). Decreased gap junction formation and GJIC have been observed following the treatment with vesicular trafficking inhibitors, such as monensin and BFA (8, 47). As shown in Figure 4C, after the treatment of BFA, HEI-OC1 cells no longer had Cx43s at junctional regions and appeared to internalize from the plasma membrane. Mean fluorescence intensity of Cx43 tended to concentrate near the nucleus in the presence of BFA (Fig. 4C). Under the nonjunctional condition, Cx43 knockdown reduces cisplatin-induced cell death regardless of BFA accompanied by less cleaved caspase-3 and poly (ADP-ribose) polymerase (PARP) (Fig. 4D, E). These data show that nonjunctional Cx43 accelerates cell death in response to cisplatin.
Additionally, we attempted to identify mitochondrial Cx43 in HEI-OC1 cells using differential centrifugation. It is well known that cytochrome C release into the cytosol due to mitochondrial dysfunction plays a significant role in ototoxicity (3). In addition, recent reports have shown that Cx43 is also present at the mitochondria where it may play a critical role in the regulation of cell death independently of gap junctions (35). We discovered abundant expression of Cx43 in the mitochondrial fraction and noticed colocalization of Cx43 and mitochondria (Fig. 4F, G). Following incubation with cisplatin, Cx43 translocation from the cytoplasm to the mitochondria was increased in a time-dependent manner, which was concomitant with the translocation of cytochrome C (Fig. 4F). This suggests that Cx43 translocation into mitochondria may play a significant role in promoting auditory cell death through a nonjunctional pathway. Taken together, these data suggest that both junctional and nonjunctional Cx43 may be involved in the cisplatin-induced proapoptotic pathway.
A gap junction blocker attenuates cisplatin-induced hearing loss in rats
Based on the above in vitro findings, we hypothesized that temporal inhibition of gap junctions during cisplatin exposure would reduce the extent of ototoxicity in animal models. In ultrastructural analyses using transmission electron microscopy (TEM), a number of intercellular gap junction plaques were observed in the plasma membranes of adjacent supporting cells in the normal cochlea of Sprague-Dawley rats (Fig. 5A). We used CBX to investigate the potential otoprotective effects of gap junction blockers in animal models. CBX is a water-soluble hemisuccinate derivative of 18β-glycyrrhetinic acid and it has been widely used to inhibit GJIC to investigate the role of gap junctions. The rats were intraperitoneally injected with 50 mg/kg CBX, daily for 2 days, and the expression of Cx43 in the cochlea was assessed by immunohistochemistry and Western blot. Multiple isoforms of Cx43 were found to exist by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis, including a nonphosphorylated (P0) and two phosphorylated isoforms (P1 and P2) (7). The inactive Cx43 P0 progresses to active P1 or P2 isoforms, which is related to the structure of gap junctions (33). It has been reported that inhibitors of Cx43 trafficking or gap junction blockers result in loss of the higher Cx43-P1 or P2 form (37). As mentioned above, expression of Cx43 was not affected by CBX, but levels of Cx43-P1 and P2 were observed to decrease (Fig. 5B). These results indicate that CBX inhibits the function of gap junctions without altering the expression of Cx43 protein (Supplementary Fig. S4B).
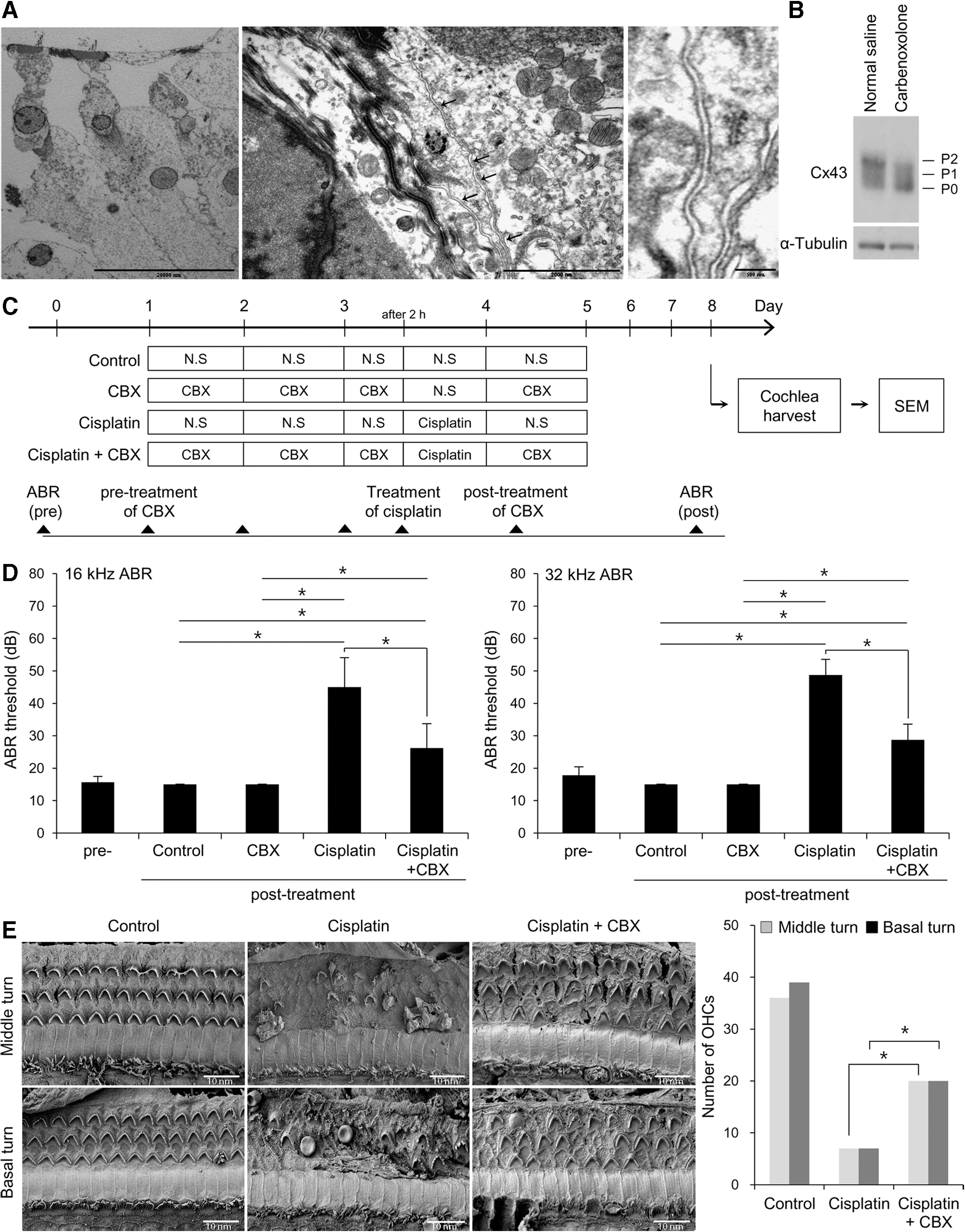
The pretreatment hearing thresholds at 16 and 32 kHz in the normal saline, cisplatin, CBX, and cisplatin+CBX-treated groups were 15.7 ± 1.8 dB and 17.8 ± 2.6 dB, respectively. The hearing thresholds at 16/32 kHz after 5 days of treatments were 15.0 ± 0.08 dB/15.0 ± 0.08 dB in the normal saline group, 45.0 ± 9.1 dB/48.8 ± 4.8 dB in the cisplatin group, and 26.3 ± 7.5 dB/28.8 ± 4.8 dB in the cisplatin+CBX group, respectively (Fig. 5D). Scanning electron microscopy (SEM) revealed that the loss of stereocilia was significantly prevented in both middle and basal turns in the cisplatin+CBX group compared with that observed in the group treated with cisplatin alone (Fig. 5E). Together with the functional findings, this indicates a preventive effect of gap junction blocker against cisplatin-induced cochlear damage.
Discussion
One of the major side effects of cisplatin use as a chemotherapeutic agent is ototoxicity, which includes hearing loss and balance disturbance. Therefore, a therapeutic strategy to reduce ototoxicity following cisplatin treatment is urgently required. Despite several studies on the mechanism of cisplatin-induced cytotoxicity, the ototoxic pathophysiology remains unclear, and most studies have focused on the intracellular mechanisms. Cx-mediated GJIC plays a pivotal role in the regulation of endocochlear potential by the K+ transport system for hearing (56). On the other hand, our study indicated that Cx43 and gap junctions have a proapoptotic effect under ototoxic conditions. Blocking gap junctions reduces cisplatin-induced ototoxicity in in vitro and in vivo studies.
Effect of gap junctional Cx43
Several lines of evidences have supported the pathologic role of gap junctions. Roh et al. showed that inhibiting astrocyte activation via the gap junction uncoupler, CBX, during the induction phase of spinal cord injury-induced neuropathic pain could diminish behavioral signs, including hyperalgesia and allodynia (40). However, because the neuroprotective effect of CBX did not occur during the maintenance phase, astrocyte gap junctions may play a role in the early phase of astrocyte activation (40). Patel et al. also showed that Cx32 gap junction inhibition could prevent the spread of injury in drug-induced liver toxicity (35). Although the precise relationship between gap junctions and cell death is not yet clear, Ca2+, inositol 1,4,5-triphosphate, cyclic adenosine monophosphate (cAMP), guanosine monophosphate, and adenosine triphosphate are possible molecules that pass through gap junctions (9). CBX is a water-soluble hemisuccinate derivative of 18β-glycyrrhetinic acid, and it has been widely used to inhibit GJIC to investigate the role of the gap junction. However, another compound, 18α-GA, has low solubility and has to be dissolved in dimethyl sulfoxide (DMSO), chloroform, or ethanol. Therefore, we used CBX as a gap junction blocker in animal experiments, and no significant change in cochlear function was observed in normal rats following administration of CBX (Fig. 5D). The other major mechanism of both 18α-GA and CBX includes inhibition of 11beta-hydroxysteroid dehydrogenase type 1 (11β–HSD1) (6). Inhibition of 11β–HSD1 potentially affects the main components of metabolic syndromes, such as insulin resistance, testosterone production, and inflammation (4), by catalyzing the conversion of cortisone to cortisol, most notably in brain liver, adipose tissue, brain, and lung (18). These are crucial factors in the activity of mineralocorticoids and glucocorticoids. Mineralocorticoids and glucocorticoids are potentially related to hearing disorders and middle-ear inflammation (27). Therefore, we tested the effect of the 11β–HSD1-specific inhibitor, BVT.2733, in HEI-OC1 cells in vitro. Although the effect of BVT.2733 was less than that observed in the following treatment with 18α-GA or other gap junction blockers on cisplatin-induced toxicity, cell viability was increased by pre- and cotreatment with BVT.2733 compared with DMSO control in a dose-dependent manner (5, 10, 15, and 25 μM) (Supplementary Fig. S5D). Thus, inhibition of 11β–HSD1 may also participate in the protective effect of 18α-GA and CBX. To clarify the main roles of gap junction blocker and to exclude other minor functions, our study used the dye coupling test such as FRAP and SLDT. Other tests that are able to evaluate the functional level of gap junction, the conductivity/permeability properties of gap junction, or the permeable factors related to cisplatin-induced auditory cell death may also be helpful.
In the cochlea, five Cx isoforms are differentially expressed, including Cx43, Cx31, Cx30, Cx29, and Cx26, depending on the area of cochlea. Specifically, Cx43 is the most widespread Cx member in the human body and is expressed primarily in the organ of Corti, spiral limbus, and spiral ligament cells of the cochlea (53). Although pharmacological gap junction blockers exhibited protective effects on cisplatin-induced auditory cell death, their status as a nonspecific gap junction blocker implies that the next step requires exploration of other Cx proteins. Several studies have shown that other types of Cx proteins are also associated with accelerated cytotoxicity and that the specific effects may vary depending on the type of insult (1, 9, 11). Reasons for the observed differences might include their different gating properties as well as the permeability and conductance of the Cx-mediated gap junctions. Kanaporis et al. found that Cx43-derived gap junction channels were significantly more permeable to cAMP compared with Cx40 or Cx26, indicating that cAMP transport might be determined based on Cx protein type (22). Although we have focused on Cx43 in our study, further investigation of other Cx proteins is required.
Effect of nonjunctional Cx43
The present data, along with those from our previous study using HEI-OC1 cells, indicate that Cx43-mediated gap junctional communication has a proapoptotic effect called the bystander effect under ototoxic condition; however, this is a contradictory result (28). We found that gap junctional communication was significantly decreased during cisplatin treatment; nevertheless, knockdown of Cx43 efficiently protected auditory cells against cisplatin-induced apoptosis (Fig. 2C, F). Therefore, we hypothesized that the protective effect of Cx43 knockdown may not only depend on gap junctional communication but also may be attributable to Cx43 on its own or in a hexameric complex referred to as a hemichannel. To test this hypothesis, we studied whether Cx43 could affect cisplatin-induced cell death in a gap junction-independent manner. In this study, we provide evidence that Cx43 significantly promotes cisplatin-induced cell death in a gap junction-dependent and independent manner. We propose that Cx modulation may have an impact on strategies to reduce its ototoxic effect. Several lines of evidence support the idea that Cx43 affects cytotoxicity via gap junction-dependent and independent mechanisms. First, Cx-expressing auditory cells were affected by Cx43 knockdown and pharmacological gap junction inhibition in both high (junctional) and low cell density (nonjunctional) cultures. Second, the nonchannel-forming domain of Cx43 (Cx43-CT) enhanced cell death in response to cisplatin. Third, under conditions that disturbed the trafficking of Cx43 to the plasma membrane, there was a significant reduction in cell death following Cx43 knockdown. Fourth, the cisplatin-induced apoptotic pathway was involved in the increased mitochondrial Cx43 level.
Several lines of evidence have suggested that Cx43 itself may regulate cell physiology independent of gap junctions (9). The mechanism by which Cx43 itself promotes cell death is unclear, but it has been reported that Cx can directly regulate the genes involved in apoptosis (10), DNA synthesis (10), cell growth (14, 20, 30, 32, 58), and interaction with apoptotic factors such as B-cell lymphoma 2 (Bcl-2) and apoptosis signal-regulating kinase 1 (15, 23). Furthermore, recent studies in cardiac myocytes and retinal endothelial cells have shown that Cx43 also exists in the mitochondria. Sun et al. also found a region where Cx43 directly interacts with Bax to permeabilize the mitochondrial membrane and perhaps to affect apoptosis (49). In this study, we have identified abundant mitochondrial Cx43 in HEI-OC1 cells and shown that it translocates from the cytosol to the mitochondria following cisplatin treatment, which in turn triggers the release of cytochrome C into the cytosol. This suggests that Cx43 translocation may play a significant role in promoting auditory cell death through a nonjunctional pathway. Mitochondrial pathways are important for the cell fate decision in response to damage, which is mainly regulated by the Bcl-2 family proteins, such as Bcl-xL, Bak, Bax, and Bcl-2 (17). There is a possibility that mitochondrial Cx43 may interact with proapoptotic Bcl-2 proteins in the presence of cisplatin. A more in-depth study on possible links between Cx43 and cisplatin-induced cytotoxicity should be performed to accurately understand the underlying molecular mechanisms.
Based on our data with the literatures discussed above, we proposed that the downregulation of gap junction function and Cx43 level may contribute to protect auditory cell death (see Fig. 6).

Cx43-derived gap junction and nonjunctional conditions
In the present study, we controlled cell densities to model junctional and nonjunctional conditions because gap junctions are formed by cell-to-cell contact. Currently, there are several ways to determine the presence of gap junction function, such as FRAP, SLDT, fluorescence resonance energy transfer (FRET), dye transfer assay (MDTA), preloading assay, and microinjection. Both FRAP and SLDT techniques were used to demonstrate a significant difference in the low and high cell density cultures in the present study. FRAP technique is noninvasive, faster than others, and provides quantitative kinetic data. Hong et al. also manipulated the cell densities to determine the effects of GJIC (19). Accordingly, HEI-OC1 cells were cultured at low densities to prevent the formation of gap junctions, but cell density control was not enough to fully prevent cell-to-cell contact. We used two methods to rule out the presence of gap junctions between coupled cells. Gene transfection was used to express the CT domain of Cx43 that has no channel-forming capacity and found that in those cells, Cx43 was only expressed in the cytosol. We showed that the CT domain of Cx43 induces more auditory cell death compared with wild-type cells in the presence of cisplatin. Behrens et al. also demonstrated the role of Cx43 in a gap junction-independent manner using Cx43 CT (amino acids (aa) 257–382)-expressing cells (2). Additionally, we used BFA to disrupt the association of Cx43 with the plasma membrane. We show here that Cx43 knockdown reduces cisplatin-induced cell death regardless of BFA treatment. Taken together, these data show that we were able to demonstrate that nonjunctional Cx43 accelerates cell death in response to cisplatin. However, Cx43 hexamer/nonjunctional hemichannel does not have a proapoptotic role following exposure to cisplatin, which might be caused by internalization of Cx43 proteins.
In conclusion, Cx43 plays a proapoptotic role in cisplatin-induced auditory cell death through gap junctional-dependent as well as independent pathways. Cx-mediated signaling may be a valuable target for preventing strategy against drug-induced ototoxicity.
Materials and Methods
Cell culture and treatments
HEI-OC1 cells, a conditionally immortalized cochlear epithelial cell line, and HeLa cells, human epithelial carcinoma cell line, were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Grand Island, NY) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO). HEI-OC1 cells were incubated at 33°C in a humidified atmosphere containing 10% CO2, and HeLa cells were incubated at 37°C in a 5% CO2. Cisplatin (Sigma-Aldrich) was used to induce auditory cell death. The gap junction inhibitors, 18α-GA, CBX, and 2-APB, were used to block GJIC between the cells. BFA was used to inhibit the assembly of gap junctions (Sigma-Aldrich), whereas BVT.2733 was used to inhibit 11β–HSD1.
Ex vivo culture and treatment
OC explants from Sprague-Dawley rats at postnatal day 7 were isolated as previously described (45). Explants were maintained in DMEM, including 10% FBS and 0.06 mg/ml penicillin, at 37°C with 5% CO2. After 24 h, explants were exposed to the same medium containing 5 μM cisplatin for 2 days.
Plasmid constructs and transfection
RT-PCR was performed using standard protocols. Primer sequences for the FL of Cx43 were 5′-TTAGGCTAGCATGGGTGACTGGAGCGCCTTAGG-3′ (forward primer) and 5′-TTAGGCTAGCATGGGTGACTGGAGCGCCTTAGG-3′ (reverse primer). After amplification of human complementary DNA (cDNA), the Cx43 product was cloned into pcDNA 3.1 (-) (5427 bp) (Invitrogen, Eugene, OR) using NheI and EcoRV restriction enzyme sites according to the manufacturer's instructions. The cDNA fragment encoding the NT (amino acids 1–256) and CT domains (amino acids 243–382) were synthesized by PCR using human Cx43 cDNA and were then cloned into the pcDNA 3.1 (-) plasmid using XbaI and EcoRV restriction enzymes. Primer sequences for each domain were as follows: CX43-NT: 5′-TTAGTCTAGAATGGGTGACTGGAGCGCCTTAG-3′ (forward primer) and 5′-ACATGATATCCTAGGGCTCAGCGCACCACTGGTC-3′ (reverse primer) and CX43-CT: 5′-TTAGTCTAGAATGGAGCGACCCTTACCATGCGA-3′ (forward primer) and 5′-ACATGATATCCTAGATCTCCAGGTCATCAGGC-3′ (reverse primer). Plasmid DNA was prepared with a plasmid mini preparation kit (Solgent, Daejeon, Korea) and plasmid constructs were verified by PCR with the universal primers (T7, BGH) and Sanger sequencing (Macrogen, Seoul, Korea). The empty pcDNA3.1 (-) construct was used as an empty vector control. To silence the expression of Cx43, siRNA was used (Santa Cruz Biotechnology, Santa Cruz, CA). A scrambled siRNA oligo was used as the control. Lipofectamine 2000 or RNAi MAX was used for all transfections (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Fluorescence recovery after photobleaching
Monolayered HEI-OC1 cells were plated in 35-mm glass bottom dishes and washed with phosphate-buffered saline (PBS) and calcein acetomethoxy derivative (calcein AM; 1 μM; Molecular Probes, Eugene, OR) in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; GIBCO, Carlsbad, CA) was loaded for 15 min in cell culture dishes. The ROI was photobleached with a short burst intense light from an argon laser, and fluorescence was monitored every 10 s for 400 s. The same photobleach intensity and monitoring time were used for all samples obtained from FRAP testing. All data values were normalized to 1 by prebleaching fluorescence intensity and again normalized to 0 by postbleaching fluorescence intensity values that displayed similar postbleached intensity ranges.
SLDT technique
Cells were plated in 35-mm dishes and cultured for 24 h. After washing with PBS containing Ca2+ and Mg2+, 0.05% LY (Sigma-Aldrich) was added and then randomly scraped with a surgical blade. After incubating for 5 min at 33°C, cells were washed with PBS and observed using a fluorescence microscope. The number of LY dye-coupled cells was counted.
LY dye uptake
HEI-OC1 cells, HeLa cells transfected with Cx43 or empty vector, were incubated with 2 mM LY in full nutrient media, Ca2+-free media, 100 μM cisplatin, and 1 mM H2O2-containing media. To evaluate the effect of hemichannel blockers, cells were pretreated with 18α-GA or CBX for 2 h, and LY uptake was measured by an Axiovision inverted microscope (Carl Zeiss AG, Gottingen, Germany).
Animal experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee of Ajou University School of Medicine. Twenty four Sprague-Dawley rats (male, 7 weeks old) were purchased from Orient Bio, Inc. (Seongnam, Korea). Animals were housed in temperature (22°C ± 2°C) and humidity-controlled rooms under a 12-h light/12-h dark cycle. Animals were randomly divided into four groups (n = 6 per group), which were (a) normal saline, (b) cisplatin, (c) CBX, and (d) cisplatin+CBX-injected groups. Cisplatin (16 mg/kg) was administered by intraperitoneal injection. CBX (50 mg/kg) was administered every 24 h for 4 times by intraperitoneal injection. As for cisplatin, the injection was done 2 h after the third CBX administration. An equivalent volume of 0.9% normal saline was injected into the sham group. Hearing function was evaluated by auditory brainstem response (ABR; Tucker-Davis Technologies, Gainesville, FL) before the first CBX injection and again 8 days after cisplatin administration. After the rats were sacrificed, cochleae were processed for use in the morphological study (Fig. 5C).
ABR test
The ABR was tested using the BioSig 32 System (Tucker-Davis Technologies) to estimate hearing thresholds, as previously described (5). The thresholds were determined using the lowest level of intensity at which a clear waveform was visible in the evoked trace. The frequency-specific ABR in response to tone burst stimuli was recorded at 16 and 32 kHz. Rats were excluded if their hearing thresholds exceeded 25 dB at frequencies of 16 or 32 kHz before beginning the experiments. All ABR results were measured in the same time and interpreted independently in a blinded manner by three people.
Transmission electron microscopy
Tissues were fixed with 2.5% glutaraldehyde in 0.1 M PBS, pH 7.4, and were postfixed in 1% osmium tetroxide. After being dehydrated through a graded alcohol series, tissues were embedded in Spurr's epoxy resin. The ultrathin sections were collected on grids and stained with uranyl acetate and lead citrate, and specimens were observed under a TEM (EM 902A; Carl Zeiss MicroImaging GmbH).
Scanning electron microscopy
Animals were anesthetized with an intraperitoneal injection of Zoletil 50 (0.1 cc/100 g; Virbac Laboratoire) and Rompun 2% (0.02 cc/100 g; Bayer Korea). The temporal bones were removed and fixed with 4% glutaraldehyde solution at 4°C for 12–24 h. After washing with PBS, the end-organ surfaces were prepared for SEM and dehydrated serially in acetone. The specimens were immersed in hexamethyldisilazane, air-dried, and finally sputter coated. Photographs were taken with a JSM-5410 LV SEM camera (Jeol USA, Peabody, MA).
Live/dead cell staining
Live/dead cell staining was performed according to the manufacturer's protocol (Invitrogen, Molecular Probes, Carlsbad, CA). Briefly, cells were washed with PBS, and a mixed solution of calcein AM and ethidium homodimer-1 (EthD-1) was added to cells. After incubation for 30 min at room temperature, cells were imaged under an Axiovision fluorescence microscope (Carl Zeiss AG).
Endogenous ROS measurement
Intracellular production of ROS was measured using CM-H2DCFDA (Molecular Probes, Invitrogen). For CM-H2DCFDA staining, cells were incubated with 5 μM dye in prewarmed HBSS for 30 min at 33°C in the cell culture incubator. The cells were washed thrice with HBSS and then examined by an Axiovision inverted fluorescence microscope (Carl Zeiss AG).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay
Cells were seeded in a 96-well microtiter plate and allowed to adhere for 24 h before treatment with cisplatin±agents, following which 20 μM of MTS was added to each well. The plate was incubated for 4 h at 33°C, and then absorbance (490 nm) was measured using a microplate reader (Bio-Rad Model 680, Hercules, CA). All assays were repeated at least three separate times.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 20 min and permeabilized in 0.2% Triton-X 100/PBS for 10 min at room temperature. After blocking with 1% bovine serum albumin (GenDEPOT, Barker, TX) in PBS, cells were processed for indirect immunofluorescence using primary and secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or cyanine 3 (Cy3) (Jackson ImmunoResearch Laboratories, West Grove, PA). Mitochondria were observed using MitoTracker® Red CMXRos (Molecular Probes, Invitrogen), according to the manufacturer's instructions, and 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Molecular Probes) was used to visualize nuclei. Cells were analyzed by confocal microscopy using a Zeiss LSM 710 microscope (Carl Zeiss Microscopy, Jena, Germany). For quantification of fluorescence intensity between two different channels, the ZEN software (Carl Zeiss Microscopy) was used. A line of interest was drawn in the image and fluorescence intensity was measured across an end-to-end line. The intensity profiles were exported to a Microsoft Excel spreadsheet and displayed as a histogram.
Western blot
Total protein was extracted from cells with RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris buffer) and Xpert Protease Inhibitor Cocktail Solution (GenDEPOT). Mitochondrial and cytosolic fractions were extracted using a mitochondrial isolation kit (Pierce, Rockford, IL) in accordance with the manufacturer's instructions. Lysates were incubated at 4°C for 30 min and the protein concentration was determined using a DC protein assay kit (Bio-Rad Laboratories). Proteins were separated by SDS-PAGE and transferred onto a polyvinylidene fluoride (Millipore, Billerica, MA) membrane. Membranes were blocked with 5% skimmed milk in 0.05% Tween 20/PBS (PBST) for 1 h and then incubated with the relevant primary antibodies at 4°C overnight. After washing in PBST, the membranes were probed with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature, and protein bands were visualized using chemiluminescence solution (GenDEPOT). The following antibodies were used: rabbit anti-Connexin 43 (Cat. No. 3512; Cell Signaling), rabbit anti-PARP (Cat. No. 9542; Cell Signaling), rabbit anti-Caspase-3 (Cat. No. 9662; Cell Signaling), rabbit anti-β-actin (Cat. No. 4970; Cell Signaling), rabbit anti-COX IV (Cat. No. 4850; Cell Signaling), mouse anti-α-tubulin (Cat. No. sc-32293; Santa Cruz Biotechnology), mouse anti-cytochrome C (Cat. No. CTC05; Enzo Life Sciences, Lorrach, Germany), mouse anti-4-HNE (Cat. No. ab48506; Cambridge, MA), rabbit anti-NOX3 (Cat. No. SAB4502060; Sigma-Aldrich), and mouse anti-SOD2 (Cat. No. sc-133134; Santa Cruz Biotechnology).
Quantitative RT-PCR
Total RNA was isolated from cells using an Easy-BLUE RNA extraction kit (iNtRON Biotechnology, Gyeonggi-do, South Korea). Standard reverse transcription was performed with an amfiRivert cDNA Synthesis kit (GenDEPOT) according to the manufacturer's instructions. Quantitative RT-PCR (qRT-PCR) measurements were performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems; Life Technologies, Carlsbad, CA) and SYBR Green I qPCR kit (Takara, Tokyo, Japan) according to the manufacturers' instructions. The cDNA was amplified with the following primers: 5′-CCTTGGCCTCATAGACACAGAAAC-3′ and 5′-GGAGTCAAAGGCGATTTGTCA-3′ for mouse 11beta-hydroxysteroid dehydrogenase type 1 (11β–HSD1) and 5′-AACGGGAAGCCCATCACC-3′ and 5′-CAGCCTTGGCAGCACCAG-3′ for mouse GAPDH. With normalization to GAPDH, the relative gene expression was analyzed using the comparative threshold cycle method (Applied Biosystems; Life Technologies). The expression of the genes of interest was expressed as fold change over control.
Statistical analysis
All values are expressed as mean (±standard deviation). Statistical significance between the groups was calculated by the Mann–Whitney U test using SPSS software version 12.0 (SPSS, Inc., Chicago, IL). Probability values (p-value) <0.05 were considered statistically significant.
Footnotes
Acknowledgments
We appreciate Dr. Chunjie Tian's contribution for the animal studies. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education Science and Technology (NRF-2013R1A2A2A01008325 and NRF-2015R1A2A15055956).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.