
Abstract
Introduction
T
It is known that stunned myocardium involves an impairment of the calcium (Ca2+) homeostasis, accompanied by an increase of oxidative stress and damage (20, 23, 49). Several authors have pointed out that during reperfusion there is a notable increase in the mitochondrial superoxide radical anion (O2 −) and hydrogen peroxide (H2O2) production, which leads to cell damage (7, 23, 48). This mitochondrial production of O2 − and H2O2 occurs both in physiological (12, 9) and pathophysiological conditions, such as ischemia and reperfusion (I/R) injury (30, 41, 48).
Although the thioredoxin system and particularly thioredoxin1 (Trx1) exerts a protective effect against injury by ischemia and reperfusion, reducing the infarct size, there is not enough experimental evidence that this cardioprotection is extended to myocardial stunning. Our novel results strongly suggest that Trx1 ameliorates systolic and diastolic dysfunction of myocardial stunning, including isovolumic relaxation and myocardial stiffness, by reducing the free radical-mediated damage in ventricular and mitochondrial function. The description of these new regulatory mechanisms in myocardial stunning opens the possibility to new therapeutic strategies in the ischemia/reperfusion injury.
We have previously demonstrated, in isolated rabbit hearts, that stunned myocardium is associated to a mitochondrial dysfunction called “complex I syndrome” with a decrease in tissue and mitochondrial O2 consumption and an increase of H2O2 and peroxynitrite (ONOO−) productions (48). Accordingly, Demaison et al. noted that mitochondrial dysfunction is part of the deleterious mechanism of the stunned myocardium (15, 16). Recently, Lou et al. (31) also demonstrated that some protective interventions, such as ischemic postconditioning, are capable of reverting postischemic ventricular dysfunction due to an improvement of mitochondrial function through the activation of the reperfusion injury salvage kinase pathway. These studies show a relationship between the ventricular and mitochondrial function, and the proteins involved in the protective mechanisms.
Due to the aforementioned reference to oxidative stress in the stunned myocardium, it is important to study the role of antioxidant systems in this pathophysiological entity. In this sense, thioredoxin (Trx) takes part in one of the most important cellular antioxidant systems known to date (33). Particularly, Trx1 exerts a protective effect against I/R injury, reducing the infarct size (1, 56). However, there is not enough experimental evidence, at least to our knowledge, that this cardioprotection is extended to postischemic ventricular dysfunction.
Yoshioka et al. reported that a deficiency in the thioredoxin-interacting protein (TXNIP) improves the recovery of mitochondrial and ventricular function of the stunned myocardium (54), but they did not show a specific effect of Trx1 on ventricular function. Furthermore, they used a transgenic model, with a TXNIP deficiency that has normal myocardial Trx1 activity and abnormal mitochondria morphology (55, 56). Due to these reasons, the aim of this work was to study for the first time the role of Trx1 in postischemic ventricular dysfunction. We evaluated the behavior of the systolic and diastolic ventricular function during the stunned myocardium in transgenic mice with cardiac-specific overexpression of Trx1 and a dominant negative mutant (C32S/C35S) of Trx1 (DN-Trx1) mice, in which the activity of endogenous Trx1 is diminished.
Both components of diastolic function, isovolumic relaxation and myocardial stiffness, were evaluated. An additional goal of this study was evaluate whether the stunned myocardium also induces changes in the mitochondrial function, in the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a), and total and phosphorylated phospholamban (PLB) expression.
Results
Figure 1 shows the behavior of ventricular function in Wt and both transgenic mice. The left ventricular developed pressure (LVDP, Panel A) and the first derivative of LV pressure (LV + dP/dtmax, Panel B) represent the contractile state. No differences were observed in the LVDP at baseline conditions among the groups. However, a significant decrease of the LVDP was observed at 30 min of reperfusion in Wt and DN-Trx1 mice compared with Trx1 mice (Wt: 27.1 ± 6.3; DN-Trx1: 29.2 ± 7.1 mmHg vs. Trx1: 57.4 ± 4.9; p < 0.05). Thus, Trx1 mice showed a significant improvement in the recovery of the contractile state at 30 min of reperfusion, and this beneficial effect was abolished in the DN-Trx1 mice. The LV+dP/dtmax exhibited similar behavior to the LVDP (Panel B).

The left ventricular end diastolic pressure (LVEDP, Panel C) reflected a significant increase of myocardial stiffness at 30 min of reperfusion in the Wt group (24.5 ± 4.8 mmHg). This deleterious effect was exacerbated in DN-Trx1 mice (37.7 ± 5.5 mmHg, p ≤ 0.05 vs. Wt and Trx1), and it was clearly attenuated in the Trx1 mice (11.8 ± 2.9 mmHg; p ≤ 0.05 vs. Wt and DN-Trx1). In regards to t63 (Panel D), a decrease in relaxation rate at the onset of reperfusion (1.5 min) was observed in the Wt and DN-Trx1 groups (63.3 ± 3.2 and 65.4 ± 5.2 ms). These antirelaxant effects were not observed in Trx1 mice (Trx1: 51.4 ± 1.9 ms, p < 0.05 vs. Wt and DN-Trx1). At the end of reperfusion (30 min), both the Wt and the Trx1 groups returned to t63, similar to the preischemic values (Wt: 52.1 ± 2.1 and Trx1: 47.5 ± 2.5 ms).
However, in the DN-Trx1 group, the decrease in relaxation rate was exacerbated compared with the Wt and the Trx1 mice (78.2 ± 9.8 ms, p < 0.05 vs. Wt and Trx1). Figure 2 shows the expression of PLB and SERCA2a proteins, both of which are associated with myocardial relaxation. Total PLB and SERCA2a did not show significant changes, either among their baseline values or after early reperfusion (1.5 min) in the different groups studied. Phosphorylation of PLB at Thr17 residue increased in Wt and DN-Trx1 groups at reperfusion compared with preischemic values (Wt: 1.76 ± 0.22; DN-Trx1: 1.35 ± 0.12, p < 0.05 vs. respective baseline values, Panels A and E). In the Trx1 mice, an increase in the Thr17 phosphorylation (1.38 ± 0.17, p < 0.05 vs. Wt and DN-Trx1) was already observed in baseline conditions. However, no changes were observed during reperfusion (Panel C).

Heart mitochondrial function after I/R was evaluated by the determination of state 3 (active) and state 4 (resting) rates of O2 consumption (Table 1 and Fig. 3). State 3 O2 uptake supported by malate–glutamate was significantly impaired after I/R in Wt mice (13% decrease) and in DN-Trx1 mice (33% decrease, p < 0.001). On the contrary, mitochondria from Trx1 mice did not show a significant change in active respiration after I/R. After I/R, Trx1 mice showed state 3 respiration values that were significantly higher than in Wt (24%, p < 0.05) and DN-Trx1 mice (47%, p < 0.001). With regard to state 4 respiration, the O2 consumption rate without ADP remained almost unchanged after I/R in Wt, Trx1, and DN-Trx1 mice. Interestingly, Trx1 mice showed increased state 4 values after I/R.
Basal Wt (0/0), n = 7; Basal Trx-1 and DN-Trx-1 (0/0), n = 5; I/R (15/30), n = 6. aTrx-1 (15/30) versus Wt (15/30) (p < 0.05).
DN-Trx-1 (15/30) versus Trx-1 (15/30) (p < 0.001).
DN-Trx-1 (15/30) versus DN-Trx-1 (0/0) (p < 0.001).
I/R, ischemia/reperfusion; RC, respiratory control.
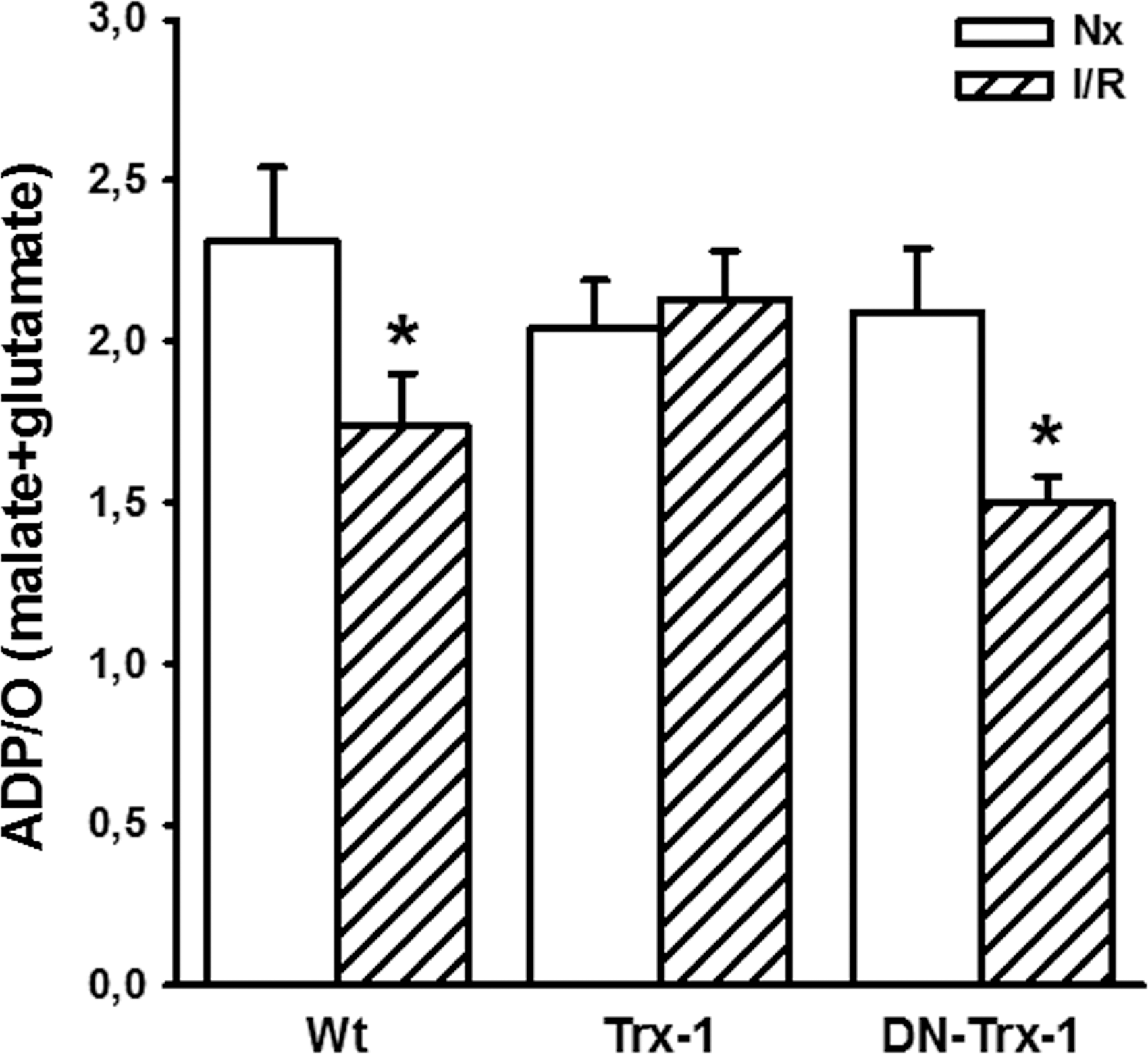
Considering the respiratory control (RC) values, using the ratio between state 3/state 4 respiration, after I/R, a slight decrease (16%) in the Wt and Trx1 groups and a moderate decrease (31%) in the DN-Trx1 mice were detected. The ADP/O (Fig. 4) ratio is an indicator of the efficiency of oxidative phosphorylation, that is, the ATP production coupled to O2 consumption. The I/R process produced a significant decrease in ADP/O ratios for Wt and DN-Trx1, 25% and 28%, respectively; whereas the Trx1 mice showed unchanged ADP/O values. The effects observed in state 3 and state 4 respirations, RC, and ADP/O ratios in the comparison between Wt and Trx1 mice are interpreted as evidence of an effective protection of Trx1 in the oxidative damage after mouse heart I/R. Table 2 shows mitochondrial O2 consumption in the presence of oligomycin, an inhibitor of ATP synthesis.
Nx Wt (0/0), n = 7; Nx Trx-1 and DN-Trx-1 (0/0), n = 5; I/R (15/30), n = 6 for each group.
p < 0.05 versus DN-Trx-1 (0/0); ** p < 0.05 versus Wt (15/30); and DN-Trx-1 (15/30).
No significant changes between baseline conditions and after I/R in Wt and Trx1 groups were observed, but only the DN-Trx1 group I/R produced a significant decrease. However, Trx1 mice showed an increase in state 4o values after I/R compared with Wt and DN-Trx1 groups (Table 2). In presence of the uncoupler carbonylcyanide-3-chlorophenylhydrazone (m-CCCP), we observed similar behavior regarding mitochondrial O2 consumption in malate–glutamate state. After I/R, a nonsignificant decrease in state 3u values in the Wt in relation to their baseline value (14%) and a significant decrease in the DN-Trx1 group (27%, p < 0.05) were observed. In the Trx1, there were no differences between baseline conditions and after I/R. After I/R, Trx1 mice showed state 3u respiration values that were significantly higher than in Wt (23%, p < 0.05) and DN-Trx1 mice (29%, p < 0.01).
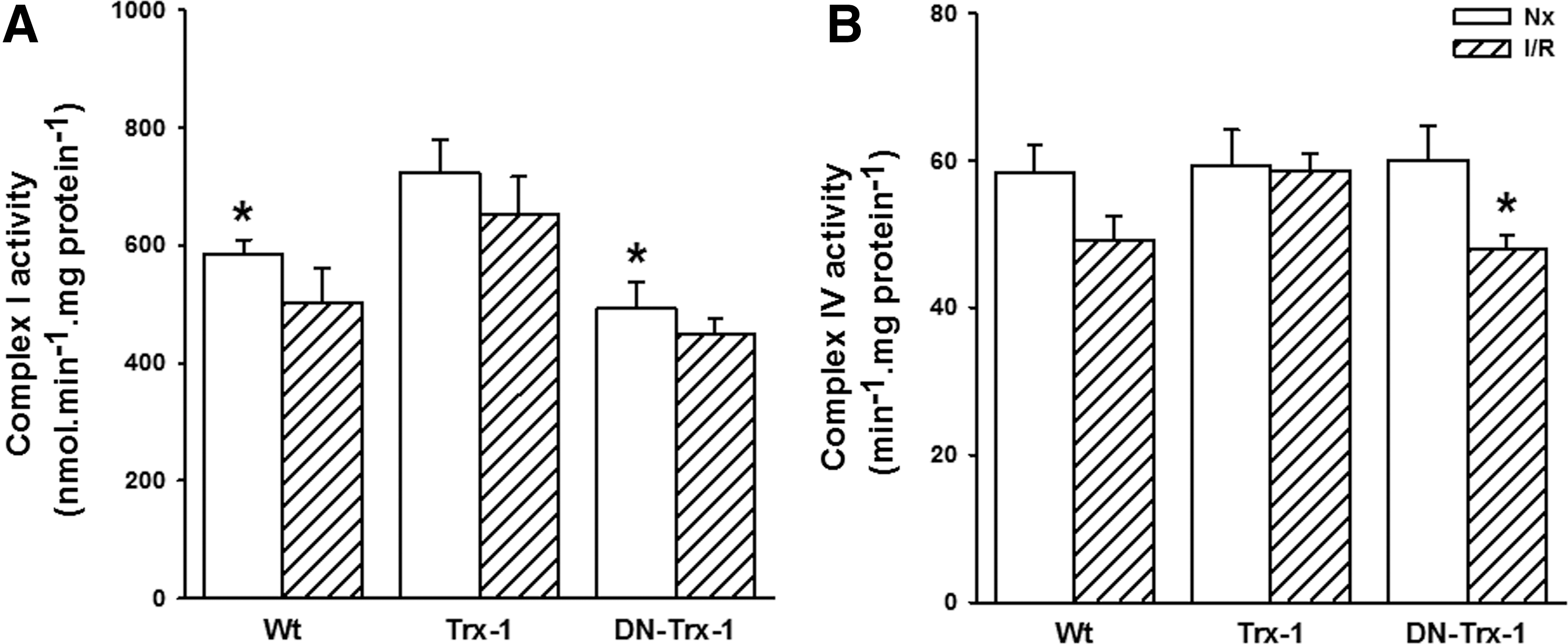
The respiratory impairment of mouse left ventricle mitochondria was further investigated by assaying the activity of mitochondrial respiratory complexes (Fig. 5). Complex I, which has been reported as selectively damaged after I/R in rabbits (48), showed a nonsignificant decrease of 14% in the Wt group, 9% in DN-Trx1 mice, and 10% in the Trx1 mice (Panel A). Interestingly, baseline values of complex I activity were increased in Trx1 mice. Their results were significantly higher than in Wt (27% <0.05) and DN-Trx1 (47%, p < 0.01) mice. Complex IV activities slightly decreased by I/R in Wt and Trx1 mice. However, the DN-Trx1 group showed an inhibition of 20% after the I/R process (Panel B).
The emission of H2O2 was assessed in energized isolated mitochondria using complex I substrates to establish state 4 (Fig. 6). The rates obtained for the Wt and Trx1 mice were similar, whereas baseline values for DN-Trx1 were higher than for the other two groups. After I/R, an increase was observed for Wt (29%) and DN-Trx1 mice (47%); meanwhile, in Trx1 mice, a slight but nonsignificant H2O2 formation increase (14%) was detected.

Finally, we also measured mitochondrial aconitase activity to assess the production of oxidant and nitrating species in vivo (Table 3). No significant changes in activity of this enzyme were observed after stunning in Wt, Trx1, and DN-Trx1 groups.
(0/0): normoxic conditions; 15/30: 15 min of ischemia and 30 min of reperfusion. n = 5 per group.
Discussion
In the present study, we have demonstrated that the overexpression of Trx1 in transgenic mice attenuates systolic and diastolic postischemic ventricular dysfunction (stunned myocardium), considering both isovolumic relaxation and myocardial stiffness. Consistent with these results, we observed that the beneficial effect was abolished in DN-Trx1 transgenic mice, in which the activity of endogenous Trx1 is reduced (50). Even more, in these mice, an exacerbation in myocardial stiffness and isovolumic relaxation alterations were observed, compared with the Wt mice at the end of reperfusion. At least to our knowledge, only Yoshioka et al. (54) studied the effects of the Trx1 system in a model of pure myocardial stunning, in the absence of necrosis. However, this study has profound differences with ours.
First, the aforementioned authors used a transgenic model with a TXNIP deficiency, but in a previous work of the same authors (55) they were unable to demonstrate that TXNIP KO increases Trx1 activity. For these reasons, TXNIP KO mice are not comparable to our experimental model, where there is a clear increase of Trx1 expression and activity; second, TXNIP KO mice have abnormal mitochondria in the heart, which cannot be explained by Trx1 since transgenic mice have normal mitochondria (55). Finally, as previously mentioned, these authors only evaluated the systolic function. Taken together, we can conclude that our study is the first to show a direct effect of cytosolic Trx1 on systolic and diastolic ventricular function, the signaling pathway involved in relaxation impairment, and their relationship to mitochondrial function.
The beneficial effects on the ventricular diastolic function were accompanied by normalization in the PLB phosphorylation at Thr17, which was increased in the Wt and the DN-Trx1 mice at the onset of reperfusion. Moreover, mitochondrial function was altered in Wt mice during late reperfusion, shown by a decrease in the mitochondrial O2 consumption in state 3, and accompanied by a slight drop of complex I activity, after I/R. In DN-Trx1 mice, this alteration after I/R was exacerbated in both O2 consumption and complex I activity, in accordance with the exacerbation of diastolic dysfunction at 30 min of reperfusion. Conversely, overexpression of Trx1 was associated to a slighter drop in the complex I activity without changes in O2 consumption in mice hearts subjected to a stunning protocol.
Thus, we showed that Trx1 overexpression exerts cardioprotective effects on the stunned myocardium, including a modification in the phosphorylation of PLB and an improvement in the mitochondrial function.
In this study, the behavior of the ventricular function at the onset of reperfusion was accompanied by changes in the phosphorylation of the PLB in the Thr17 residue, without changes in the SERCA2a and total PLB expression. First in relation to transgenic (TG) overexpressing Trx1 and then in Wt and DN-Trx1 mice, our data show that in Trx1 mice, the hearts have a higher phospholamban phosphorylation (p-PLB) Thr17 phosphorylation in comparison to Wt and DN-Trx1 at baseline condition. This PLB behavior before a certain intervention, in our case ischemia, was also shown by Catalucci et al. (13). These authors, and in concordance with our findings, demonstrated that at baseline conditions, a higher p-PLB Thr17 phosphorylation is accompanied by a greater Ca2+ reuptake by the sarcoplasmic reticulum (13), but in their case they used a transgenic model with Akt overexpression. Therefore, our mice probably, which at baseline conditions have a higher PLB phosphorylation, avoid, at least partially, a Ca2+ overload during reperfusion when subjected to a protocol of I/R. This may justify the lack of isovolumic relaxation impairment at the onset of reperfusion. Second, the behavior of p-PLB Thr17 in Wt and DN-Trx1 after ischemia is different. It was observed that unlike TG Trx1, there was an increase in p-PLB Thr17 in Wt and DN-Trx1 at the onset of reperfusion (1.5 min). These changes were accompanied by a slowing of isovolumic relaxation rate. This increase in early reperfusion phosphorylation with a deleterious functional repercussion are consistent with those who showed that increases in PLB phosphorylation (Thr17) at the onset of reperfusion try to correct the Ca2+ homeostasis alteration that occurs, thus normalizing relaxation in the last stages of reperfusion (36, 37, 44).
It has been widely shown that an increase in both, the expression and activity of SERCA2a, could prevent the Ca2+ overload that occurs during reperfusion after the I/R episode (27, 40, 45, 46). Guo et al. showed that I/R induced endoplasmic reticulum stress, SERCA2a dysfunction, and subsequent impairment in ventricular function (22). Moreover, Kuster et al. (29) demonstrated in cardiomyocytes that exposure to H2O2 100 mM produces a systolic dysfunction that is characterized by reduced contractility and inhibition of SERCA2a. Taken together, and given that in our Trx1 mice we have a better redox balance, we could assume an improvement in SERCA2a function and, as a consequence, decreased Ca2+ overload after I/R.
Due to the aforementioned, our data suggest that an increase of the cell antioxidant defenses in baseline conditions would prevent the relaxation impairment after ischemia, in early reperfusion in hearts subjected to a protocol of myocardial stunning. This increase in antioxidant defenses would be given in our experimental model by Trx1 overexpression.
The mitochondrial dysfunction observed in this study, also termed “complex I syndrome” is characterized by a reduction of O2 uptake, malate–glutamate mitochondrial respiration, and complex I activity. An augmentation of protein nitration and oxidations products, and increased O2 − and H2O2 production rates were observed (48). Previous reports demonstrated that complex I is the major target of mitochondrial damage after I/R injury (21, 48). In this study, we observed a slight drop of 18% in complex I activity at the end of reperfusion in Wt mice. In this condition, a failure in myocardial function is evidenced, not only in the contractile phase, as expected, but also in both components of the diastolic phase: isovolumic relaxation and myocardial stiffness. In parallel, there is a mitochondrial dysfunction, with decreased mitochondrial O2 consumption, ADP/O ratio, and mitochondrial complexes activity.
The results reported here describing myocardial and mitochondrial dysfunction in mouse stunned myocardium are similar to the ones previously reported in rabbit stunned hearts (48). This protective role of Trx1 also supports the concept that the mechanism of I/R involves an increased rate of free radical-mediated reactions that lead to a condition of oxidative stress. In the reperfusion, there is an abrupt change in cellular O2 levels and a fully reduced mitochondrial respiratory; these facts result in a primary burst of O2 − generation (4, 48, 51). This primary product (O2 −) is rapidly dismutated to H2O2, which, in turn, generates hydroxyl radical (HO•). This last species is capable of initiating free radical-mediated reactions, called lipoperoxidation, in which organic hydroperoxides (ROOH) are produced (32).
The first part of the oxidative stress description occurring after I/R protocol is explained by the overproduction of H2O2 and ROOH, which are relatively stable products (32). The second part implies the oxidation of thiol groups with regulatory and signaling functions (39). The properties of the thioredoxin system that reduces the production of H2O2, ROOH, and disulfide groups at fast rates clearly explain the biochemical mechanism of the antioxidant effect (39). Regarding complex I, we also measured mitochondria-specific markers of oxidative stress such as aconitase activity, trying to provide strong support for in vivo mitochondrial ROS production. Unfortunately, in our model of short ischemia (myocardial stunning), no changes were observed regarding complex I activity and aconitase.
This behavior could be related to the fact that values are in the nanomolar range, and it is unlikely that aconitase would be damaged and inactivated significantly in this short ischemia–reperfusion model of myocardial stunning. This behavior in aconitase activity was also shown by other authors (3, 26). In this sense, Bulteau et al. (3) using a rat model subjected to 30 min of ischemia and 60 min of reperfusion showed an reversible inactivation of aconitase activity. They demonstrated that this activity decreased 65% compared with a control group at 5 min of reperfusion, but at 15 min of reperfusion these values reached similar to preischemic values. In a similar manner, Koga et al. (26), although using 15 min of ischemia, the same as our protocol, only observed changes in aconitase activity at 5 min of reperfusion.
Taking this information as a whole, it may be that changes in this enzyme activity occur in early reperfusion. This may explain why when we measure it at 30 min of reperfusion, we cannot evidence a decrease in its activity.
An interesting finding of this study was that mice overexpressing Trx1 show an increased (24%) activity of mitochondrial complex I in baseline conditions; meanwhile, in DN-Trx1, animals show a decrease (16%) in this activity. The mechanism underlying these observations may comprise protein S-glutathionylation, the reversible conjugation of GSH to cysteines within a protein. S-glutathionylation has been shown to play an important role in modulating mitochondrial function and morphology (17, 24, 34). It has been shown that several subunits of the Complex I are crucial for regulation by S-glutathionylation (14, 25, 35). This covalent modification of Complex I leads to the inactivation of this protein. Moreover, Complex I from heart and other tissues has been shown to be susceptible to regulation by glutathionylation reactions (25, 42).
It has also been shown that subunit Ndufa11 suffers S-glutathionylation in the isolated perfused heart mice model subjected to I/R (28). The increase in S-glutathionylation in GSH-depleted conditions, that is, DN-Trx1 mice, might be surprising at first. However, given that GSH is a cofactor for GSH reductase-catalyzed deglutathionylation, the reduction of the levels of GSH could limit GSH reductase activity, and therefore enhance S-glutathionylation. During oxidative stress, cysteines are among the most vulnerable with regard to oxidative modifications. GSH, an antioxidant component, is present at millimolar concentrations in the cells (1–10 mM), and the conjugation of GSH to oxidized cysteines acts as a cytoprotective mechanism to prevent oxidation. Of note, S-glutathionylation occurs not only in response to overt oxidative injury but also in pathophysiological states, and in settings where ratios of GSH to oxidized GSH (GSSG) are low (i.e., 100:1 vs. 3:1).
It has been shown that mitochondrial thiols maintain the steady-state levels of mitochondrial H2O2, cellular redox homeostasis, and cytosolic redox-sensitive signaling modulation; changes in these thiols could affect transcription, growth, and finally modulate the behavior in cell survival pathways. Mitochondria are able to generate second messengers (redox: H2O2 and NO; energy: ATP) that are involved in the regulation of redox-/energy-sensitive cell signaling pathways; in this way, it is possible to generate physiological actions between mitochondria and other proteins (52, 53). Contrasting with this, several molecules can translocate into the mitochondria and perform redox changes in other organelles. The redox environment could be regulated by communication between mitochondria and other cell components (52).
Thus, changes in the redox balance in the cytosol, for example, Trx1 overexpression, could produce regulatory changes in mitochondria that were evidenced, in this work, through state 3 oxygen consumption (active respiration), ADP/O ratio, H2O2 production, and complex I activity.
In summary, we have demonstrated that Trx1 overexpression has a clear protective effect on the stunned myocardium, not only on the contractile state but also on the two diastolic components: isovolumic relaxation and myocardial stiffness. Furthermore, the improvement in isovolumic relaxation rate reflected a decrease in the p-PLB; and the attenuation of myocardial stiffness involved a clear improvement of mitochondrial dysfunction, evidenced by almost unchanged rates of O2 consumption, ATP production, and complexes activity.
Materials and Methods
Animal care
The experimental protocol was approved by the Animal Care and Research Committee of the University of Buenos Aires (UBA #0037016/2010). Mice were housed in ventilated cages with a 12 h light/dark cycle and controlled temperature (20–22°C), and they were fed with normal chow and water ad libitum.
Transgenic mice
We have used the transgenic mice from the same colonies of Prof. Junichi Sadoshima, who generously donated these mice to us. Two transgenic mice models were used: (1) transgenic mice with cardiac-specific overexpression of Trx1 (Trx1) generated on an FVB background using the α-myosin heavy chain promoter to achieve cardiac-specific expression (2, 50), and (2) DN-Trx1 was generated by mutation of 32Cys and 35Cys of hTrx1 to Ser using QuikChange (Stratagene). This redox inactive mutant of Trx1 works as a dominant negative for endogenous Trx1 in mice hearts (50).Wild-type (Wt) mice were also used as the control group.
Isolated mice hearts
The hearts of 3-month-old male mice weighting 24.2 ± 1.5 g were used. Mice were anesthetized by an intraperitoneal injection of sodium pentobarbital (150 mg/kg) and sodium heparin (500 IU/kg, bolus i.p). After anesthesia, hearts were excised and the aorta was immediately cannulated with a 21-gauge cannula. Hearts were rapidly excised and perfused according to the Langendorff technique. We performed 20 min of stabilization, 15 min of global ischemia, and 30 min of reperfusion. Hearts were perfused with Krebs bicarbonate buffer containing (in mM): NaCl 118.5, KCl 4.7, NaHCO3 24.8, KH2PO4 1.2, Mg SO4 1.2, CaCl2 1.5, and glucose 10, bubbled with 95% O2 and 5% CO2 (pH 7.40) at 37°C as previously described (43). Two electrodes were sutured and connected to a pacemaker to produce a constant heart rate of 472 ± 3 beats/min.
The coronary perfusion pressure (CPP) was monitored through a pressure transducer that was connected to the perfusion line. Hearts were perfused at a constant flow of 4.01 ± 0.20 ml/min, which was adjusted to obtain a CPP of 73 ± 3 mmHg in the initial stabilization period and maintained constant throughout the experiment. LVDP and the maximal rate of rise of left ventricular pressure (LV + dP/dtmax) were determined as contractile state indexes. Isovolumic relaxation rate was analyzed using t63, which is defined as the time required for the left ventricular pressure to fall to 63%. LVEDP, a myocardial stiffness index, was also measured.
Mitochondrial isolation and mitochondrial membrane preparation
Heart mitochondria were obtained from mouse heart homogenates by differential centrifugation in a refrigerated centrifuge (model RC5S; Sorvall-Instruments-Du Pont). Left ventricles were excised, washed, and minced in ice-cold STE buffer containing 250 mM sucrose, 10 mM Tris-HCl, and 2 mM EGTA, at pH 7.4. A brief digestion was performed in STE medium supplemented with 0.5% (w/v) fatty acid-free BSA, 5 mM MgCl2, 1 mM ATP, and 2.5 UI/ml type XXIV bacterial proteinase. After 4 min at 4°C, hearts were homogenized with a small Potter-Elvejhem glass-Teflon homogenizer after the addition of five volumes of STE buffer, and they were centrifuged at 8000 g for 10 min. The obtained pellet was resuspended in ice-cold STE buffer and centrifuged at 700 g for 10 min. The pellet was discarded, and mitochondria were precipitated by two 10 min centrifugations at 8000 g.
Finally, mitochondria were resuspended at about 20 mg protein/ml in STE buffer. The whole procedure was carried out at 0–4°C (38). It is to be remarked that this procedure allowed the isolation of 1.5–2.1 mg of heart mitochondrial protein from a single mouse. Mitochondrial membranes were obtained by two cycles of freezing and thawing of the mitochondrial preparation, followed by homogenization by passage through a 29 G hypodermic needle (11). Protein concentration was measured by the Folin reagent using BSA as a standard.
Mitochondrial O2 consumption
Mitochondrial O2 uptake was determined polarographically with a Clark-type electrode (Oxytherm, Hansatech Instruments Ltd.) in a 1.0-ml chamber at 30°C in an air-saturated reaction medium ([O2] = 220 μM). Heart mitochondria were suspended, at 0.2–0.3 mg protein/ml, in a respiration buffer consisting of 120 mM KCl, 5 mM KH2PO4, 1 mM EGTA, 20 mM HEPES and 1 mg/ml fatty acid-free BSA, pH 7.40, 2 mM malate, and 5 mM glutamate as substrates without (state 4) or with the addition of 0.5 mM ADP (state 3) (10). Respiration is expressed in ng-at O/min × mg protein. RC was calculated as the ratio of state 3/state 4 respiration rates. Oligomicin (0.2 μM) and carbonyl cyanide m-chlorophenylhydrazone (m-CPPP, 1 μM) were used to set state 4o and state 3u. These measurements were performed at baseline conditions (0/0) and after 30 min of reperfusion (15/30).
Activities of mitochondrial respiratory complexes
The enzymatic activities of mitochondrial complexes I and IV were determined spectrophotometrically (Beckman DU 7400 Spectrophotometer) at the a-band of cytochrome c (550 nm, E = 19 mM −1 cm−1) at 30°C. With mitochondrial membranes suspended in 100 mM KH2PO4/K2HPO4, pH 7.40, complex I activity was determined as NADH-cytochrome c reductase. Mitochondrial membranes were added with 0.20 mM NADH as a substrate, 25 μM cytochrome c3+ and 0.5 mM KCN. Enzymatic activities are expressed as nmol cytochrome c2+/min × mg protein.
Complex IV (cytochrome oxidase) was determined in the same buffer that was supplemented with 50 μM cytochrome c2+. Reduced cytochrome c was prepared by reduction of cytochrome c3+ with Na2S2O4, followed by Sephadex G-25 chromatography. The rate of cytochrome c2+ oxidation was calculated as the pseudo-first-order reaction constant k′/mg protein. These measurements were performed at baseline conditions (0/0, n = 4 per group) and at 30 min of reperfusion (15/30, n = 5 per group).
Western blots
Hearts samples (n = 6 per group) were homogenized at 0°C for 2 min in 20 mM Tris, 30 mM NaCl, 0.1% SDS, 1% Triton, 0.2 mM DTT, pH 7.40, protease, and phosphatase inhibitors at a ratio of 330 μl every 100 mg of heart using a PRO 200 Scientific homogenizer. Homogenates were centrifuged at 800 g for 10 min at 0–4°C. The supernatant protein was quantified with the Bradford reagent. Homogenate proteins, 50 μg of each sample, were separated by electrophoresis in 16% Tricine-SDS-PAGE gels, transferred to a PVDF membrane (Thermo Fisher Scientific Inc.), and blocked with 5% BSA for 2 h at room temperature. Subsequently, the samples were incubated with anti-PLB (1:5000) (Badrilla Ltd., West Yorkshire, United Kingdom), phosphorylated PLB Threonine 17 residue (Thr 17; 1: 3000) (Badrilla Ltd.), and, finally, anti-SERCA2a (Thermo Scientific) (1:1000) antibodies overnight at 4°C with agitation. Later, the samples were incubated with antirabbit secondary antibody conjugated with horseradish peroxidase (HRP; 1:20000) (EMD Millipore Corporation), for 1 h at room temperature. The membrane was developed with photographic plates (Eastman Kodak Company) and Super Signal West picochemiluminescent substrate (Thermo Scientific). These protein expressions were quantified by densitometry with Image Gauge 4.0 software (FUJIFILM Holdings Corporation), and data were expressed as relative to GADPH expression (1:3000) (Cell Signaling Technology, Inc.). These measurements were performed at baseline conditions (0/0) and at 1.5 min of reperfusion (15/1.5).
Hydrogen peroxide production
Hydrogen peroxide (H2O2) production was determined fluorometrically at 365–450 nm (Hitachi F-3010 Spectrofluorometer) using the scopoletin–HRP assay (8), at 30°C. The reaction medium consisted of mannitol 230 mM, sucrose 70 mM, 30 mM Tris-HCl, pH 7.40, 7 mM succinate, 0.6 μM Cu, Zn-SOD, 1 μM HRP, 1 μM scopoletin, and heart coupled mitochondria (0.02–0.05 mg protein/ml), with or without the addition of 10 μM catalase. A calibration curve was performed using H2O2 (0.05–0.35 μM) to express the fluorescence changes as nmol H2O2/min. mg protein. Only the fluorescence change inhibited by catalase addition was considered to calculate H2O2 production.
Aconitase activity
Aconitase activity was measured spectrophotometrically in mitochondrial samples (19). Freshly isolated mitochondria samples were sonicated (four bursts of 30 s ON and 60 s OFF) followed by centrifugation at 8250 g for 10 min at 4°C. Specific activity of the mitochondrial aconitase present in the supernatant was measured by monitoring the conversion of sodium citrate to cis-aconitase at 37°C (240 nm, ɛ = 3.6 mM −1cm−1) (18, 47). The reaction medium contains 100 mM Tris-HCl buffer (pH 7.4), 1 mM sodium citrate, and 0.05–0.08 mg mitochondrial protein/ml. The activity is expressed as nmol/min × mg protein. These measurements were performed at basal conditions (0/0) and at 30 min of reperfusion (15/30).
Statistics
Results are expressed as mean ± standard error of the mean. Ventricular function and Western blot: Inter-group comparisons were performed using analysis of variance and then the Bonferroni test for multiple comparisons. p < 0.05 was considered statistically significant. Mitochondrial function: Student–Newman–Keuls test was used to analyze the significance of differences. Figures and tables include the significance in the differences both within groups (i.e., 15/30 vs. 0/0) and among groups in the same condition (i.e., 0/0 or 15/30).
Footnotes
Author Disclosure Statement
No competing financial interests exist.