
Abstract
Introduction
V
Salusin-β is identified as a cardiovascular bioactive peptide of the 20-amino acid, has mitogenic effects, and is translated from an alternatively spliced mRNA of TOR2A, a gene encoding a protein of the torsion dystonia family (36). The initial 18 amino acids of human salusin-β have high homology with the N-terminal sequence of rat salusin (40). Previous studies in our lab showed that central salusin-β contributed to sympathetic activation, arginine vasopressin release, and hypertension, and plasma salusin-β level was increased in renovascular hypertensive rats (8, 28, 39, 46). Recently, we found that salusin-β promoted VSMC proliferation and vascular fibrosis, and increased circulating salusin-β contributed to hypertension and vascular remodeling (38). Patients with diabetes mellitus, coronary artery disease, and cerebrovascular disease displayed a distinct increase in plasma salusin-β levels, and salusin-β may be employed as a promising candidate biomarker for predicting systemic vascular diseases (14). Salusin-β is widely distributed in a host of tissues, including heart, aorta, and left internal mammary artery (3), and has been implicated in atherosclerosis (43) and angiogenesis after myocardial ischemia reperfusion injury (27). Salusin-β has been found to have pro-atherosclerotic effects that are associated with the formation of human macrophage foam cells (43). However, the pathogenic role and potential therapeutic implication of salusin-β in the VSMC migration and neointima formation responses to vascular injury are unknown. We hypothesized that salusin-β may contribute to the VSMC migration and intimal hyperplasia after vascular injury. The present study was designed to determine the effects of salusin-β on VSMC migration and intimal hyperplasia after vascular injury and the possible link with the reactive oxygen species (ROS)-mediated downstream signaling pathway.
Our data provide a novel view that salusin-β promotes migration of vascular smooth muscle cells (VSMCs) and contributes to the exacerbated intimal hyperplasia postinjury. NADPH oxidase 2 (NOX2)-derived reactive oxygen species (ROS) were important initiating factors for salusin-β-induced p65-NFκB nuclear translocation, which positively regulates the activity and expression of MMP-9 and thus induces VSMC migration. Knockdown of salusin-β attenuates the vascular injury-induced NOX2 expression, ROS production, intimal thickening, and intimal hyperplasia. Our findings provide new insights that the intervention of salusin-β may be a promising therapeutic strategy for some vascular diseases such as atherosclerosis and stent restenosis.
Results
Expression of salusin-β in injured carotid arteries in rats
Moderate intimal hyperplasia developed in ligated carotid arteries 4 weeks after injury and became much more severe 8 weeks later. No obvious intimal hyperplasia was found in sham-operated carotid arteries (Fig. 1A). Injury caused an increase in neointima thickness and a reduction in lumen diameter in injured carotid arteries (Supplementary Fig. S1; Supplementary Data are available online at

Effect of salusin-β on the migration of VSMCs
VSMC migration is an essential component for neointima formation in response to vascular injury (34). The effects of salusin-β on the aortic VSMC migration in vitro were evaluated with both scratch-wound assay and Boyden chamber chemotaxis assay. Salusin-β potently stimulated VSMC migration, including increased migrated distance (Fig. 2A, C) and number of migrated cells (Fig. 2B, D), almost reaching its maximal effects at the concentration of 10 nM. Therefore, this concentration of salusin-β was used in in vitro studies.

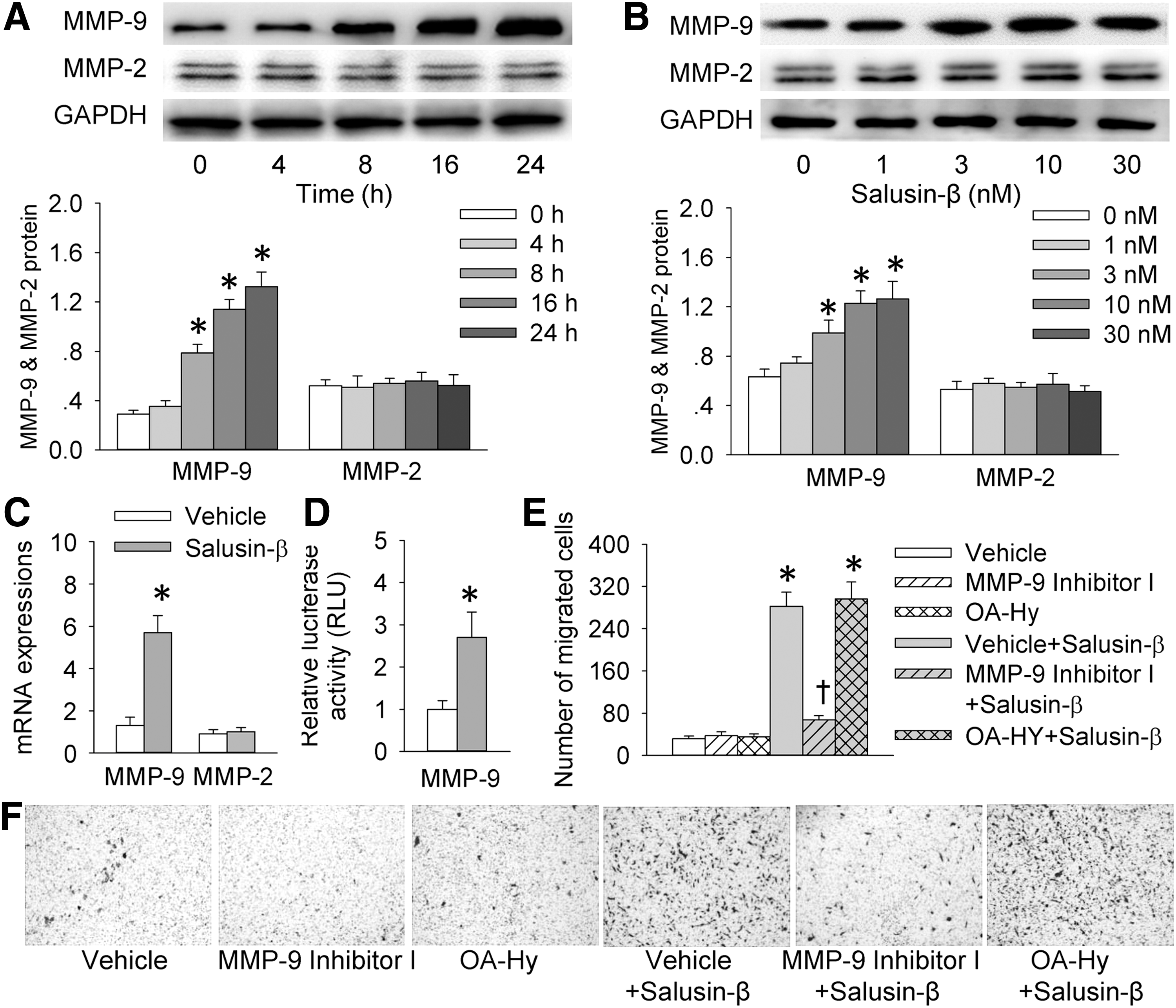
MMP-9 mediates the effect of salusin-β on VSMC migration
Activation of MMPs plays a key role in ECM degradation and remodeling, thus facilitating VSMC migration (32). Salusin-β increased MMP-9 expression in VSMCs in a time-related and dose-related manner, but it had no significant effect on MMP-2 expression (Fig. 3A, B). Similarly, salusin-β increased MMP-9 mRNA levels but not MMP-2 levels in VSMCs (Fig. 3C). Luciferase reporter gene assay showed that salusin-β enhanced the reporter activity of the MMP-9 promoter construct (Fig. 3D). Importantly, salusin-β-induced VSMC migration was prevented by MMP-9 inhibition rather than by MMP-2 inhibition (Fig. 3E, F).
Effect of salusin-β on p65-NFκB nuclear translocation in VSMCs
Translocation of p65 of nuclear factor kappa beta (NFκB) from cytoplasm to nucleus is recognized as a prerequisite for transcription. The phosphorylation and the subsequent degradation of the inhibitor of NFκBα (IκBα, an endogenous inhibitor of NFκB) are required for p65 nuclear translocation (45). Incubation of VSMCs with salusin-β promoted the phosphorylation and degradation of IκBα (Fig. 4A). Salusin-β induced the translocation of p65 of NFκB from cytoplasm to nucleus in a time-dependent manner (Fig. 4B).

Roles of p65-NFκB in MMP-9 expression and VSMC migration responses to salusin-β
The p65-NFκB signaling is a key mediator in the MMP-9 transcription (9). Salusin-β significantly increased binding activities of the NFκB motifs in the MMP-9 promoter (Fig. 5A). Knockdown of NFκB with small interfering RNA (siRNA) suppressed the salusin-β-induced MMP-9 expression (Fig. 5B). Similarly, treatment with Bay 11-7082, an inhibitor of NFκB, reduced the salusin-β-induced MMP-9 expression (Fig. 5C). Importantly, VSMC migration response to salusin-β was prevented by knockdown of NFκB with siRNA (Fig. 5D) or by inhibition of NFκB with Bay 11-7082 (Fig. 5E). However, salusin-β-induced MMP-9 expression was not affected by LY294002 (a PI3K inhibitor), MK-2206 (an Akt inhibitor), or U0126 (an extracellular signal-regulated kinase [ERK] inhibitor) (Supplementary Fig. S2), suggesting that PI3K/Akt and ERK signaling pathways are not responsible for the MMP-9 expression response to salusin-β in VSMCs.

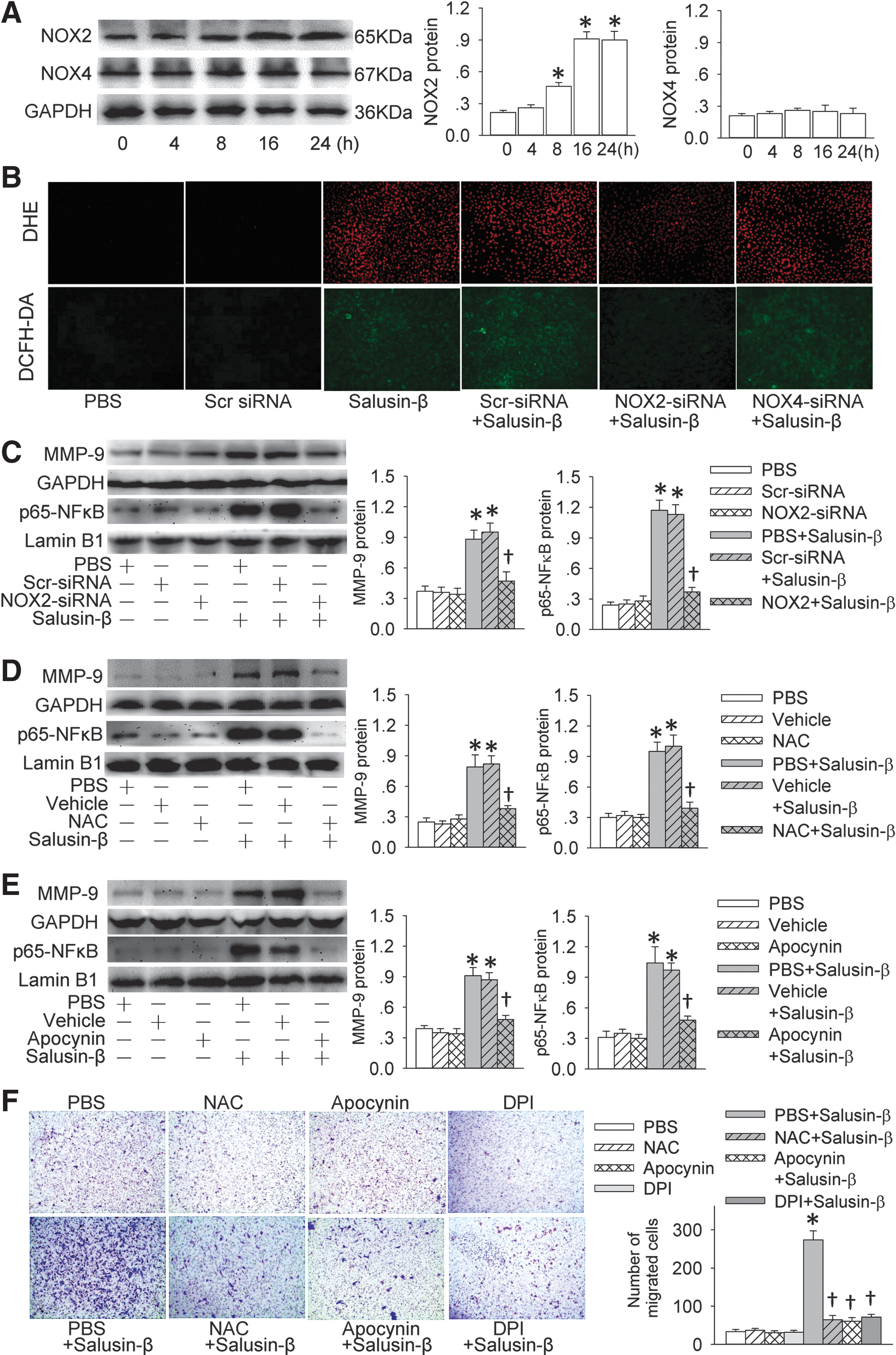
Roles of NOX2-derived ROS in the effects of salusin-β in VSMCs
Salusin-β had no significant effect on NOX4 expression, but it increased NADPH oxidase 2 (NOX2) expression in a time-dependent manner (Fig. 6A). It increased the production of superoxide anions, which was prevented by NOX2-siRNA transfection but not by NOX4-siRNA transfection (Fig. 6B and Supplementary Fig. S3). Moreover, the knockdown of NOX2 with NOX2-siRNA inhibited salusin-β-induced MMP-9 expression and p65-NFκB nuclear translocation (Fig. 6C). The specificity and the effectiveness of the siRNAs were confirmed by the downregulation of relevant target protein expression (Supplementary Fig. S4). Salusin-β-induced MMP-9 expression and p65-NFκB nuclear translocation were prevented by the treatment with N-acetyl-
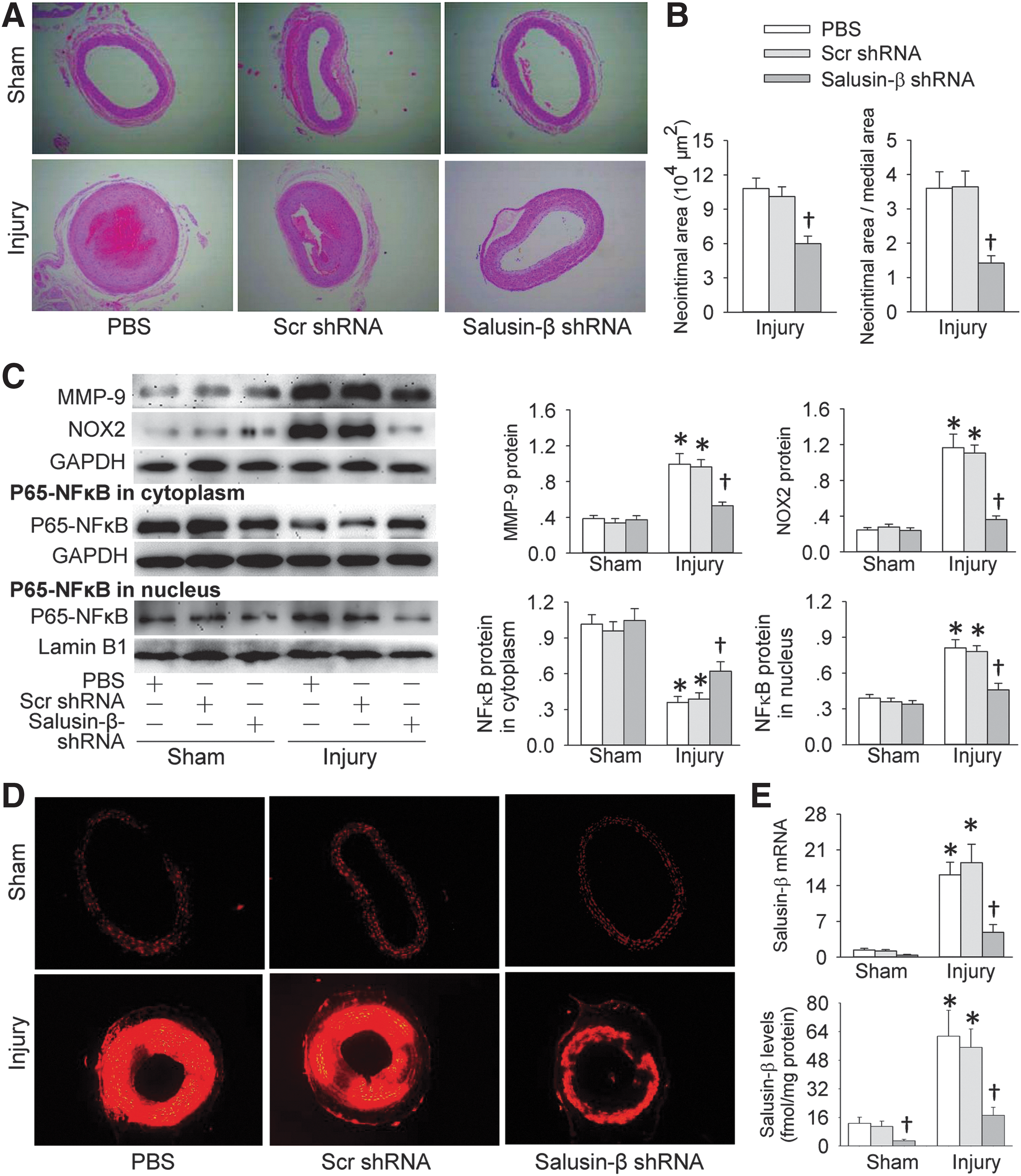
Salusin-β silencing reduces neointima formation and ROS production in injured carotid arteries in rats
Adenoviruses harboring shRNA-targeted salusin-β (Ad-salusin-β shRNA) were used to determine whether knockdown of salusin-β in rats reduces intimal hyperplasia in injured carotid arteries. An intravenous injection of Ad-salusin-β shRNA was administered 4 weeks postinjury when moderate neointima had formed. All the measurements were conducted 4 weeks after the injection. Ad-salusin-β shRNA reduced the neointima area and the ratio of neointima area to medial area in injured carotid arteries (Fig. 7A, B). It decreased the neointima thickness and increased the lumen diameter in injured carotid arteries of rats (Supplementary Fig. S9). It prevented the injury-induced MMP-9 and NOX2 expression as well as p65-NFκB nuclear translocation (Fig. 7C). Dihydroethidium (DHE) fluorescent dye showed that the production of superoxide anions in the injured carotid arteries was increased in the rats, which were prevented by Ad-salusin-β shRNA (Fig. 7D and Supplementary Fig. S10). On the other hand, salusin-β mRNA and protein expressions were increased in injured carotid arteries of rats, which were attenuated by Ad-salusin-β shRNA (Fig. 7E).
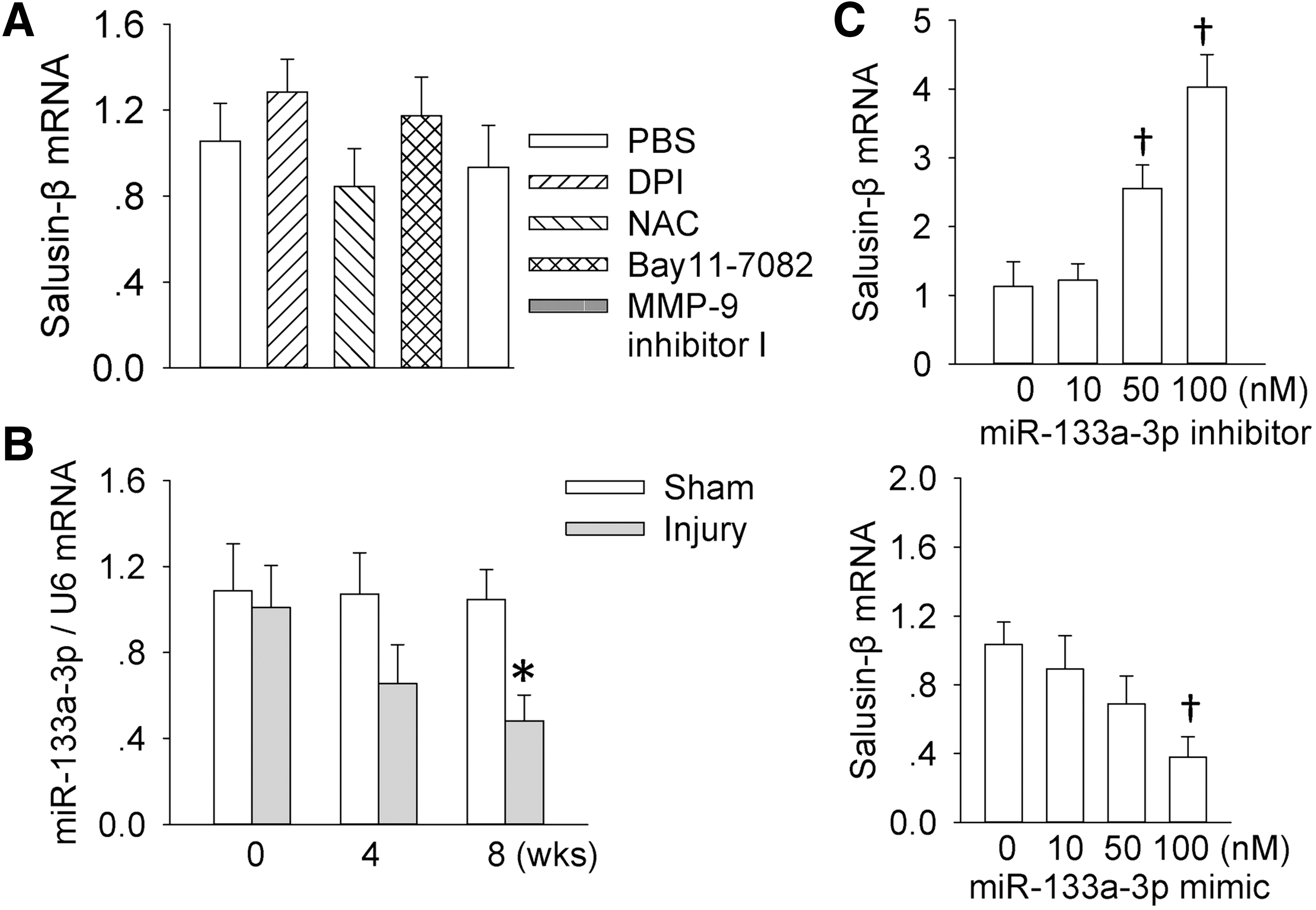
Reduced miR-133a-3p expression contributes to the salusin-β upregulation in injured carotid arteries of rats
An interesting question is why salusin-β is upregulated in injured carotid arteries of rats. We found that salusin-β mRNA expression in the VSMCs was not affected by the treatment with a flavin-containing enzyme, including NADPH oxidase DPI, a free-radical scavenger NAC, an NFκB inhibitor Bay-11-7082, or an MMP inhibitor I (Fig. 8A), suggesting that the salusin-β expression is not regulated by ROS and/or NFκB. On the other hand, microRNAs (miRNAs) are recognized as a class of single-stranded nucleotide noncoding RNAs and they exert negative regulation of targeted gene expressions by targeting the 3′-UTR of specific mRNAs. Computational miRNA target analysis showed that miR-133a-3p is able to bind to the salusin-β mRNA 3′-UTR, implying that salusin-β may be a possible molecular target for miR-133a-3p. Recently, it has been found that miR-133a inhibits the migration of esophageal cancer and gastric cancer cells (26, 33). Thus, we further investigated whether miR-133a-3p is involved in the expression of salusin-β. We found that miR-133a-3p was downregulated in injured carotid arteries during the 4–8 weeks postinjury (Fig. 8B). Transfection of miR-133a-3p mimic diminished but miR-133a-3p inhibitor increased the mRNA expression of salusin-β in VSMCs (Fig. 8C). These results indicated that miR-133a-3p is a negative modulator of salusin-β expression, and reduced miR-133a-3p expression contributes to the overexpression of salusin-β in injured carotid arteries.
Effects of salusin-β in carotid VSMCs of rats
We further examined the effects of salusin-β in carotid VSMCs in vitro, because all the in vitro studies mentioned earlier were carried out in aortic VSMCs. Salusin-β promoted VSMC migration and ROS production in carotid VSMCs (Supplementary Fig. S11). It increased NOX2 and MMP-9 expressions and caused p65-NFκB nuclear translocation in carotid VSMCs (Supplementary Fig. S12). These results were in agreement with the findings in aortic VSMCs.
Discussion
VSMCs are mainly housed in the tunica media in the physiological state and play essential roles in vascular homeostasis (4). Resting VSMCs have initiated the proliferation and migration in response to cytokines and growth factors, which contribute to the progression of vascular occlusion diseases such as atherosclerosis, hypertension, diabetes, and restenosis after angioplasty (31). The media-to-intima migration of VSMCs is associated with exaggerated restenosis rates after angioplasty and atherosclerosis (5). It is known that the salusin-β immunoreactivity was dominantly expressed in the VSMCs and fibroblasts in coronary atherosclerotic lesions (43). We found that salusin-β was upregulated in injured carotid arteries in the rat carotid artery ligation model. Salusin-β promoted the migration of aortic VSMCs and carotid VSMCs in vitro. Knockdown of salusin-β in rats attenuated the injury-induced neointima formation, ROS production, NOX2 and MMP-9 expressions, and nucleus translocation of p65-NFκB. These findings indicate that salusin-β is crucial in the VSMC migration, neointima formation, and vascular remodeling after vascular injury. Intervention of salusin-β may be a strategy for preventing the excessive migration of VSMCs and neointima formation in atherosclerosis and stent restenosis.
MMPs, a family of zinc proteinases, are key components in the control of degradation of ECM proteins during the development of atherosclerosis or vascular injury (41). The degradation of ECM, which is mediated by MMPs, is required for the migration of VSMCs and plays a vital role in the pathogenesis of cardiovascular diseases (15, 18). On the other hand, MMP synthesis is regulated primarily at the transcriptional level, and NFκB is a key transcription factor in the production of MMP-9 via its subunit p65 binding to MMP-9 promoter. NFκB proteins are localized in the cytoplasm that is bound to the inhibitory protein IκBα in most unstimulated cells. The IκBα is phosphorylated and degraded on stimulation, and then results in the release of NFκB dimers from the inhibitory complex and the translocation of NFκB to the nucleus, where it stimulates the transcription of related genes (11, 16). The present study reveals that salusin-β exerts a positive regulation of VSMC migration via increased MMP-9 expression. Salusin-β stimulates a cascade of processes, including the IκBα phosphorylation and degradation, the nucleus translocation of p65-NFκB, the recruitment of p65 to MMP-9 promoter, the increases in MMP-9 expression and activation, the VSMC migration, and the neointimal formation after vascular injury.
NFκB is considered a redox-sensitive transcription factor, and intracellular redox status has a major role in the control of NFκB activity (25). Arterial injury induces immediate profound vascular oxidative stress and NFκB activation (37). It has been found that oxidized high-density lipoprotein induces the proliferation and migration of VSMCs by promoting the production of ROS (42). A major source for ROS in the cardiovascular–renal system is a family of NOXs (7, 30). NOX1/NOX2 promotes the development of endothelial dysfunction, hypertension, and inflammation, whereas NOX4 may have a role in protecting the vasculature during stress (23). Lipopolysaccharide (LPS) stimulates NOX2-derived ROS production via ERK and induces various types of MMP expression and migration of macrophages (22). In the present study, we found that salusin-β stimulated the NOX2 expression in VSMCs, and increased the superoxide anion production, which was attenuated by silencing of NOX2 but not by silencing of NOX4. The salusin-β-induced VSMC migration was prevented by the treatment with an ROS scavenger NAC or an NADPH oxidase inhibitor apocynin or by silencing of NOX2. On the other hand, salusin-β-induced MMP-9 expression and p65-NFκB nuclear translocation were prevented by silencing of NOX2, or a scavenger of ROS, or an inhibitor of NADPH oxidase. These results indicate that NOX2 is a major downstream mediator in the oxidative stress and VSMC migration responses to salusin-β, and salusin-β elicits NFκB activation and MMP-9 expressions via NOX2-dependent ROS production. The findings were further supported by the findings that the increased NOX2 expression and ROS production in injured carotid arteries were prevented by knockdown of salusin-β with Ad-shRNA in rats.
It is interesting to know whether the ROS production is involved in the injury-induced salusin-β upregulation. We found that scavenging ROS or inhibiting NOX, NFκB, or MMP had no significant effect on the salusin-β mRNA expression, suggesting that the expression of salusin-β is not regulated by ROS and/or NFκB. It is known that miRNAs are a class of single-stranded nucleotide noncoding RNAs, and they exert negative regulation of targeted gene expressions by targeting the 3′-UTR of specific mRNAs. Computational miRNA target analysis showed that miR-133a-3p is able to bind to the salusin-β mRNA 3′-UTR, implying that salusin-β may be a possible molecular target for miR-133a-3p. It has been found that miR-133a inhibits the migration of esophageal cancer and gastric cancer cells (26, 33). Thus, we further investigated whether miR-133a-3p is involved in the expression of salusin-β. We found that miR-133a-3p was downregulated in injured carotid arteries during the 4–8 weeks postinjury. Transfection of miR-133a-3p mimic diminished but miR-133a-3p inhibitor increased the mRNA expression of salusin-β in VSMCs. These results suggested that miR-133a-3p is a negative modulator of salusin-β expression, and reduced miR-133a-3p expression may be responsible for the overexpression of salusin-β in injured carotid arteries.
In conclusion, salusin-β promotes VSMC migration via NOX2-derived ROS production, IκBα phosphorylation and degradation, nucleus translocation of p65-NFκB, recruitment of p65 to the MMP-9 promoter, and the MMP-9 expression and activation. Salusin-β plays an important role in the neointima formation and intimal thickening after vascular injury. Intervention of salusin-β may be a potential therapeutic strategy for some vascular diseases such as atherosclerosis and stent restenosis.
Materials and Methods
Animals
Experiments were performed in male Sprague–Dawley rats. Procedures were approved by the Experimental Animal Care and Use Committee of Nanjing Medical University and conformed to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH publication, 8th edition, 2011). The rats were housed in a temperature-controlled room with a 12-h light/dark cycle and with standard chow and tap water ad libitum.
Animal model of vascular injury
Unilateral carotid artery ligation was performed to induce vascular injury in 8-week-old rats, as previously described (29). Briefly, rats were anaesthetized with sodium pentobarbital (50 mg/kg, i.p.). The adequacy of anesthesia was determined by the absence of corneal reflexes and paw withdrawal response to a noxious pinch and surgical manipulation. A midline incision was made in the ventral surface of the neck. Bilateral carotid arteries were isolated. The left common carotid artery was ligated with a 6.0 silk suture that was proximal to the carotid bifurcation, and the right one was not ligated and served as a control. Acute experiments were carried out after 4 or 8 weeks.
Hematoxylin–eosin staining, immunohistochemistry, and dual immunofluorescence
The rats were euthanized with an overdose of pentobarbital sodium (150 mg/kg, iv), and carotid arteries were harvested for histological, immunohistochemical, and molecular biological analyses. To exclude unspecific changes caused by the ligature itself, the most proximal part of the carotid artery to the ligation was discarded. The arteries were fixed in 4% paraformaldehyde solution, embedded in paraffin, and finally transversely cut into 5-μm sections. The serial sections were subjected to hematoxylin–eosin (HE) or immunohistochemical staining for morphometric assay after deparaffinization and rehydration. After routine HE staining, the morphological features of the vessel wall in each section were photographed and analyzed with computer-based Image Pro Plus software (version 6.0; Media Cybernetics) to calculate the ratio of neointima to media area, an indicator of neointima formation. The neointimal layer was considered as the distance from the vessel lumen to the internal elastic fibers, and the media layer was defined as the area ranging from the internal to the external elastic fibers.
For immunohistochemical staining, paraffin-embedded vascular sections were rehydrated and immersed in 3% hydrogen peroxide for 15 min to quench the endogenous peroxidase. The sections were blocked in 10% goat serum for 1 h at room temperature and then incubated with the primary antibody against the salusin-β overnight at 4°C. The immune positive signals were detected with DAB substrate-chromogen solution for 5 min by following the manufacturer's suggestions. Finally, the sections were counterstained with hematoxylin.
For dual immunofluorescence, the paraffin-embedded sections were permeabilized by using 0.1% Triton X-100 for 10 min after deparaffinization and rehydration. The sections were washed in phosphate-buffered saline (PBS) containing 10% goat serum and co-incubated with primary antibodies rabbit anti-salusin-β and mouse monoclonal anti-SM α-actin for 24 h at 4°C. The secondary tetramethylrhodamine (TRITC)-conjugated goat anti-rabbit IgG (1:400) or fluorescein isothiocyanate (FITC)-conjugated monkey anti-mouse IgG (1:200) was employed, respectively. Nuclei were stained with 4′,6-diamidino-2-phenylindole after immunofluorescence staining. The fluorescence signal was obtained by using a fluorescence microscope (DX51; Olympus, Tokyo, Japan).
Culture of primary VSMCs
The primary culture of VSMCs from the rat aorta and carotid artery was, respectively, performed with enzymatic digestion. VSMCs were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in a 5% CO2 humidified incubator. Cells in the second to sixth passages were used, and cells at 80–90% confluence were growth arrested by incubating them in serum-deprived DMEM for 24 h before stimulation (1).
Western blot
Protein extractions in tissues or cells and western blotting were carried out as described in our previous reports (38). In brief, the protein lysates were obtained by RIPA lysis buffer and the protein concentration was quantified with the BCA method. The equal amounts of proteins were separated in SDS-PAGE and electro-transferred onto a PVDF membrane; the membrane was then blocked with 5% nonfat milk for 60 min at room temperature, followed by overnight incubation with the indicated primary antibodies at 4°C. The PVDF membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 60 min at room temperature, and the positive bands were visualized by using the enhanced chemiluminescent reagent (Merck Millipore Corporation, Billerica, MA). GAPDH served as an internal control.
Real-time PCR
Total RNA was separated by using a TRIzol reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer's protocols. RNA concentrations and purity were assessed by the measurement of optical density at 260 and 280 nm. Reverse transcriptase reactions were then performed by using the PrimeScript® RT reagent Kit according to the manufacturer's instructions. Real-time PCR was performed by using Quantitative PCR with SYBR Premix Ex Taq TM (Takara, Otsu, Japan) and ABI PRISM 7500 sequence detection PCR system (Applied Biosystems, Foster City, CA). The samples were relatively quantified by normalizing the targeted gene level to that of the internal control by the ΔΔCt method. The sequences of primers were listed in the Supplementary Tables (Table S1).
VSMC migration and invasion analysis
VSMC migration was evaluated by scratch-induced wound-healing assay (10). VSMCs were seeded into six-well plates and the near-confluent VSMCs in six-well plates were wounded by scraping with a standard 1 ml pipette tip to make a gap across the diameter of the wells. The images of cell migration across the wound were photographed, calculated, and expressed as a percentage of the distance migrated in control wells within each experiment. VSMC invasion was determined by a Boyden chamber assay (47). VSMCs were seeded onto the upper surface of an 8-μm pore size chamber, and a culture medium with or without salusin-β. After 24 h of incubation, the cells that migrated to the lower surface of the filter were fixed by methanol and stained with 1% crystal violet. The number of stained cells from four randomly chosen fields was counted in triplicate independent experiments.
siRNA transfections
VSMCs were cultured at an initial density of 1 × 105 cells/ml to form a monolayer and seeded onto six wells on the day before the transfection. At 24 h after the VSMCs were sub-cultured (30–40% confluent), the siRNA sequences against the rat p65 subunit of NFκB or a scrambled siRNA (Scr-siRNA) (100 nmol/L; Santa Cruz Biotechnology, Santa Cruz, CA) were transiently transfected into the VSMCs by using Lipofectamine (Invitrogen, Carlsbad, CA) while following the manufacturer's protocols (24).
Reporter gene transfection and luciferase activity assay
A 1.3-kb fragment of 5′-flanking region of the rat MMP-9 gene (Gen-Bank/EMBL databases, accession no. U36476) was cloned to SacI/HindIII site of the pGL2-basic vector containing a polyadenylation signal upstream of the luciferase gene (44). The firefly luciferase reporter of NFkB containing a TA promoter was purchased from Beyotime (pNF_BTA-luc; Beyotime Biotechnology, Shanghai, China). VSMCs were plated in 24-well plates before transfection and grew to about 70% confluence. The luciferase reporter gene was transfected to cells by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After transfection for 6 h, cells were pretreated with or without salusin-β for an additional 18 h. VSMCs were harvested, and the luciferase activity was measured by using a dual luciferase reportor gene assay kit (Beyotime Biotechnology). The luciferase activity was normalized to the internal control.
Chromatin immunoprecipitation
Chromatin immunoprecipitation assay was conducted with a standard protocol and kit (Millipore Corporation) as previously described (49) to evaluate whether NFκB-p65 binds to the MMP-9 promoter in salusin-β-stimulated VSMCs. In short, the VSMCs challenged by salusin-β were subjected to incubation of 1% formaldehyde for 10 min for proteins with DNA cross-linking. The crosslinking was stopped with 125 mM glycine, and then the samples were washed, scraped, and collected. The cross-linked chromatin was sonicated to an average size of 400–600 bp. The DNA fragments were immunoprecipitated with 5 μg of anti-p65 antibodies (Santa Cruz Biotechnology) or a rabbit monoclonal control IgG (Santa Cruz Biotechnology). The precipitated DNA fragments were amplified by PCR with the primers (positive primers) encompassing the p65 site in the MMP-9 promoter: 5′-TGCCCCTGAGGCTTCCCCAA-3′ (forward); 5′-TGGAGCCCCTCCCCACACTG-3 (reverse).
Measurement of ROS generation in vivo and in vitro
Intracellular ROS in VSMCs were determined with two fluorescent probes, DHE and DCFH-DA, respectively (13, 21). The confluent VSMCs were transfected with Scr-siRNA, NOX2 siRNA, or NOX4 siRNA for 6 h before incubation of salusin-β for another 24 h. The VSMCs were then fixed and loaded with DHE (10 μmol/L) or DCFH-DA (10 μmol/L) for 30 min in a light-protected humidified chamber. The fluorescence was obtained with a fluorescence microscope and quantified with the IMAGE-PRO PLUS 6.0 by using the same parameters.
Pretreatment with adenovirus construction salusin-β shRNA plasmids in vivo
Three adenoviral constructs carrying shRNA against salusin-β and a scrambled shRNA (a negative control) were designed and constructed by Genomeditech Co. (Shanghai, China). The adenovirus harboring shRNA sequence-targeted salusin-β that downregulated the salusin-β expression by 75% was selected for the present study (Table S2). Gene transfer of salusin-β in vivo was carried out 4 weeks after carotid arteries ligation to induce vascular injury or sham operation in rats. The rats were, respectively, subjected to the intravenous injection of PBS, adenovirus expressing scrambled shRNA, or adenovirus expressing salusin-β-shRNA (2 × 1011 plaque-forming units/ml, 100 μl) via tail vein. Two weeks later, the injection was repeated again to ensure the adequate downregulation of salusin-β in the carotid arteries (12, 48). All measurements were conducted 4 weeks after the first intravenous injection (8 weeks after carotid arteries ligation or sham operation).
Chemicals and antibodies
Human salusin-β was obtained from Phoenix Pharmaceuticals (Belmont, CA). Cell culture supplies were purchased from Costar (Corning, Inc., Cypress, CA). MMP-9 Inhibitor I was purchased from Santa Cruz Biotechnology. OA-Hy and U0126 were purchased from Calbiochem (San Diego, CA). NF-κB inhibitor BAY 11-7082, N-acetyl-
Antibodies against MMP-9, MMP-2, GAPDH, and Lamin B1 and indicated HRP-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology. Antibodies against p65, IκBα, and phosphor-IκBα were obtained from Cell Signaling Technology (Beverly, MA). Antibodies against NOX2 (ab180642) and NOX4 (ab60940) were purchased from Abcam (Cambridge, MA). The secondary TRITC-conjugated goat anti-rabbit IgG or the FITC-conjugated monkey anti-goat IgG was acquired from Life Technologies. The negative control for rabbit IgG was obtained from Sigma Chemical Co.
Statistical analysis
Comparisons between two groups were made by Student's t-test. One-way or two-way ANOVA followed by post hoc Bonferroni test was used when multiple comparisons were made. All data were expressed as mean ± SE. A value of p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors are grateful for the generous support of the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine. This study was supported by the National Natural Science Foundation of China (31171095, 31271213, 91439120, and 31571167).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.