
Abstract
Introduction
M
We describe a new and sensitive proteomic technique for specific identification of S-glutathionylated proteins in monocytes and macrophages. We show that this reversible post-translational modification occurs on a large number of proteins and may alter expression, activity, and/or degradation of these proteins, thereby affecting a vast array of cellular functions. This newly identified redox-sensitive regulatory network may provide a mechanistic link between metabolic stress and the well-documented changes in the functional phenotype of monocytes and macrophages observed in patients with metabolic disorders. Finally, we provide the first evidence for sexual dimorphism in metabolic stress-induced thiol oxidation in macrophages.
ROS-mediated oxidative modification of protein thiols has been studied extensively for its role in signal transduction and regulation of various cellular processes, including proliferation, transcription and translation, and apoptosis (38). Protein thiol oxidation products include (i) sulfenic, sulfinic, or sulfonic acids, (ii) S-nitrosothiols, (iii) sulfenylamide, (iv) intra- and intermolecular disulfides, and (v) mixed disulfides with low-molecular-weight thiols, including glutathione and cysteine (30, 67). However, protein S-glutathionylation (and deglutathionylation) has emerged as a bona fide signaling mechanism (54, 55) due to the (enzymatically) reversible nature of the protein S-glutathionylation reaction, the specificity and selectivity of cysteine residues modified, and the ability of S-glutathionylation to modulate protein function. To date, only a few S-glutathionylated proteins have been validated as bona fide redox signaling transducers based on five criteria originally outlined by Mieyal et al. (42). We recently identified three proteins critical for monocyte migration that fulfill these criteria: actin (36, 62), mitogen-activated protein kinase phosphatase 1 (MKP-1) (32), and 14-3-3ζ (31). Others reported that phosphatase and tensin homolog deleted from chromosome 10 (PTEN) (11), protein tyrosine phosphatase 1B (PTP1B) (50), Na-H exchanger isoform 1 (NHE1) (33), caspase-1 (41), and paraoxonase 1 (PON1) (52) were found to be S-glutathionylated in monocytes and macrophages of varying lineages (11, 33, 41, 50, 52). These data raise the question whether protein S-glutathionylation is limited to only select proteins and specific pathways or whether this unique post-translational modification may play a broader functional role in monocytes and macrophages. In support of the latter hypothesis, we found that metabolic stress increases global S-glutathionylation in peritoneal macrophages from mice fed a high-fat diet (HFD) (47). Increased S-glutathionylation correlated with functional changes in these macrophages, including the hypersensitization to chemokine-induced adhesion and accelerated monocyte chemotaxis, a phenomenon we termed metabolic priming of monocytes (47, 62).
To identify S-glutathionylated proteins in macrophages exposed to metabolic stress, we developed a novel, specific, and highly sensitive proteomic approach. Previous studies have used global labeling techniques to identify individual S-glutathionylated proteins (10, 15, 27, 37, 38); however, major limitations of these approaches include a lack of sensitivity and the use of either supraphysiological concentrations of oxidants such as hydrogen peroxide (H2O2) or nonphysiological oxidants such as diamide to induce detectable changes in protein S-glutathionylation. The redox proteomic technique we developed is sensitive enough to identify S-glutathionylated proteins in both a human monocytic cell line and in murine peritoneal macrophages and to monitor changes in the level of modification in response to metabolic stress. Our study demonstrates that in monocytic cells, metabolic stress promotes the S-glutathionylation of over 600 proteins, and we provide the first report of global cellular changes in S-glutathionylation in response to (patho)physiological stimuli both in vitro and in vivo. Furthermore, our data suggest that S-glutathionylation of reactive protein thiols may serve as a mechanism for cells to sense and rapidly respond to changes in the extracellular environment by altering activities and/or expression levels of a large number of proteins. This novel mechanistic link between changes in the metabolic environment and global alterations in macrophage signaling pathways and functionalities may help explain the plasticity of macrophages and their adaptability to vastly different and dynamic microenvironments.
Results
Strategy for specific labeling of S-glutathionylated proteins
Current proteomic techniques designed to detect S-glutathionylated proteins lack either the sensitivity and/or the specificity for protein-GSH mixed disulfides. To address both limitations, we designed a new strategy (Fig. 1) that involves a novel enrichment technique based on iodoacetamide conjugated to desthiobiotin (IAM-Desthiobiotin) labeling, followed by high-performance liquid chromatography–electrospray ionization tandem mass spectrometry (HPLC-ESI-MS/MS), to identify and quantify S-glutathionylated proteins.
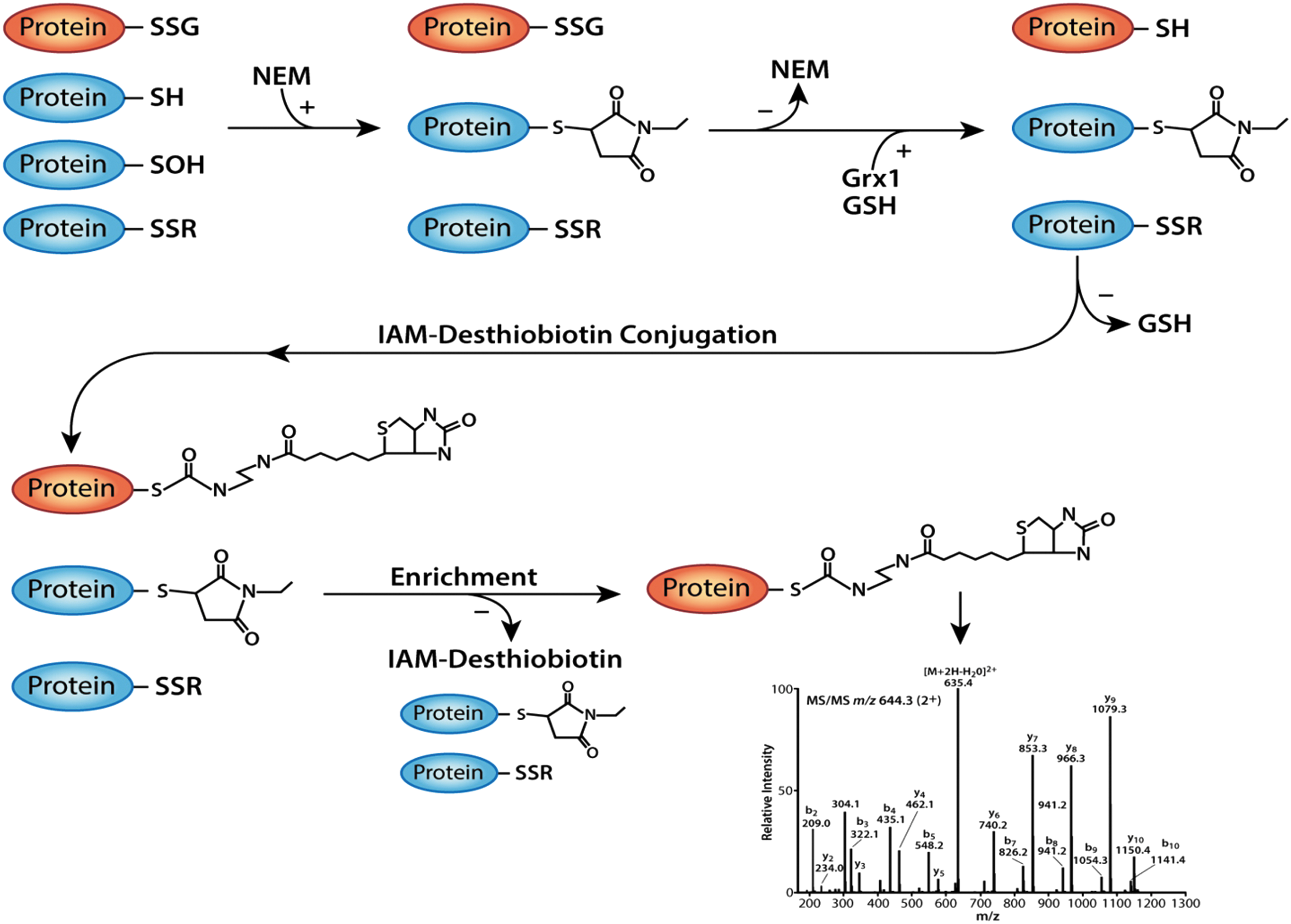
In the first step, all intracellular free thiols and sulfonic acid groups were alkylated by treating intact cells before and during lysis with the cell-permeable alkylating agent, N-ethylmaleimide (NEM). NEM renders thiols unreactive and reduces false-positive signals due to transthiolation and disulfide formation (18) that can occur with free thiols (-
To avoid inactivation of Grx1, unreacted NEM was removed from the samples before the addition of the Grx1 enzyme (Fig. 1). In addition, excess GSH was added to the deglutathionylation reaction to recycle the active site of Grx1 and maintain its catalytic activity (19). GSH was removed after completion of the reaction to prevent interference with the thiol-reactive label, IAM-Desthiobiotin (Fig. 1). IAM is more selective for the thiolate anion than NEM and results in lower nonspecific labeling, making IAM-Desthiobiotin a highly specific and selective labeling reagent for the reactive thiols generated after Grx1 reduction. IAM-Desthiobiotin-labeled proteins were enriched using streptavidin-coated agarose beads (Fig. 1). DES weakly interacts with the biotin moiety, allowing for the elution of DES-labeled proteins from streptavidin under mild conditions (28), minimizing protein loss due to denaturation and aggregation. After elution, samples were subjected to mass spectrometry analysis (see the Materials and Methods section for more details).
Protein S-glutathionylation in THP-1 monocytes in response to metabolic stress and H2O2
We showed previously that metabolic stress (and H2O2 treatment) sensitizes monocytes to chemokine activation, converting these cells into a hypermigratory proatherogenic phenotype (32, 62), a process we termed metabolic priming. We went on to demonstrate that metabolic priming is mediated by S-glutathionylation of proteins involved in cell signaling, cytoskeletal remodeling, and cell migration (31, 32, 62), suggesting that protein S-glutathionylation might be a global molecular mechanism by which monocytes and macrophages alter the proteome to respond to cellular stimuli. To test this hypothesis, THP-1 monocytes were cultured in the absence or presence of either metabolic stress (low-density lipoprotein + high D-glucose [LDL+HG]) or thiol oxidative stress (H2O2), and lysates from these cells were analyzed to identify and semiquantify S-glutathionylated proteins.
In three independent labeling experiments, HPLC-ESI-MS/MS analysis identified a total of 636 proteins that were S-glutathionylated in lysates from THP-1 monocytes. With the exception of a single study using diamide as an oxidant (57), this is one of the few studies to identify over 100 S-glutathionylated proteins using a proteomic screen. Due to the surprisingly large number of identified proteins, we focused our subsequent analysis on those 133 S-glutathionylated proteins that were reproducibly identified in each of the three independent experiments (Supplementary Table S1).
Of these 133 proteins, 115 were detected in either unstressed or metabolically stressed THP-1 monocytes or both (Fig. 2A). Of these 115, 16 proteins (14%) were S-glutathionylated in unstressed THP-1 monocytes, but not in metabolically stressed cells, while only seven proteins (6%) were identified as being detected only in lysates of metabolically stressed THP-1 cells, but not in unstressed cells, that is, de novo S-glutathionylated (Fig. 2A). However, the majority of S-glutathionylated proteins (92 or 81%) were identified by LC-MS/MS under both conditions. These data indicate that most proteins that are S-glutathionylated in monocytes under metabolic stress are also S-glutathionylated to some degree under resting conditions. However, metabolic stress altered the level of S-glutathionylation in many of these proteins. Levels of S-glutathionylation in response to metabolic stress remained unchanged (or inconsistent between experiments) in 63% of the 115 proteins (Supplementary Table S1, “ = ”) and increased in 29% of proteins (Supplementary Table S1, “↑”). Surprisingly, 9% of proteins showed decreased S-glutathionylation levels under metabolic stress (Supplementary Table S1, “↓”). Taken together, the observed changes in protein S-glutathionylation suggest that metabolic stress targets specific proteins for increased S-glutathionylation, yet others for decreased S-glutathionylation. These data support the concept of a global, but highly controlled and specific, response to metabolic stress.
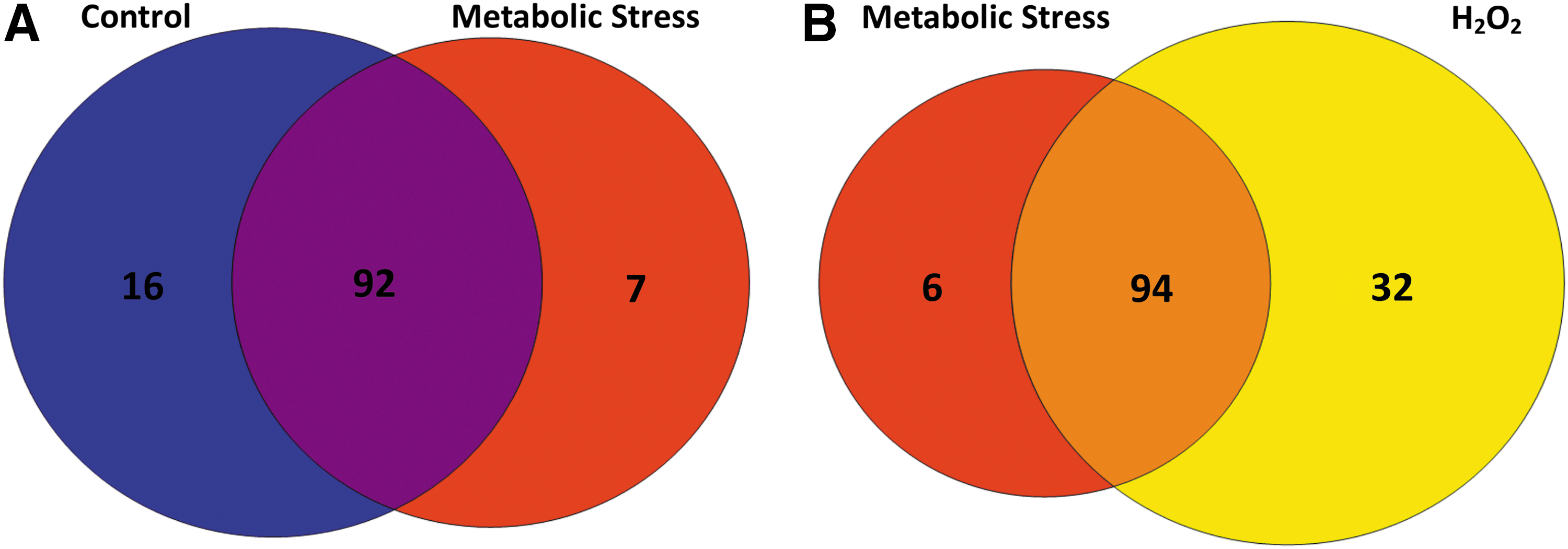
When we compared protein S-glutathionylation between metabolically stressed THP-1 monocytes and H2O2-treated cells, we found that the vast majority of the proteins (71%) were S-glutathionylated under both conditions (Fig. 2B), indicating that H2O2 may be the oxidant responsible for mediating S-glutathionylation induced by metabolic stress. Twenty-four percent (32 proteins) of identified proteins were detected in lysates only H2O2 treated and 5% (six proteins) were exclusive to cells exposed to metabolic stress (Fig. 2B). These data suggest that while H2O2 can mimic the effects of metabolic stress (62), high levels of (exogenous) H2O2 target additional proteins that do not appear to be S-glutathionylated in cells exposed to metabolic stress.
Using Ingenuity® Pathway Analysis (IPA®) software and UniProt, we were able to group the 133 proteins (Supplementary Table S1) by specific location (Supplementary Table S2) and function (Supplementary Table S3). The vast majority of the 133 proteins (74%) were predicted to be cytoplasmic, while 14% were predicted to be nuclear (Supplementary Table S2); this distribution did not appear to depend on whether S-glutathionylation of the protein was altered by metabolic stress or not. Of the 24 functions associated with the 133 proteins, the top six functional classes were of oxidoreductases, chaperones, ion-binding proteins, nucleotide-binding proteins, ribonucleoproteins, and translation regulators (Supplementary Table S3).
To confirm that IAM-Desthiobiotin-labeled proteins identified with our enrichment technique were S-glutathionylated (Fig. 1), we labeled metabolically primed THP-1 monocytes with biotin-conjugated glutathione ethyl ester (BioGEE, see the Materials and Methods section for details). Thirteen proteins were selected for identification by BioGEE either because others had previously identified them as S-glutathionylated proteins (15, 21, 23, 31, 34, 53, 64) or, in our initial screen, they exhibited consistent and robust S-glutathionylation levels (based on summed intensities of the fragments in the product ion spectra). All 13 proteins selected were detected after BioGEE labeling and streptavidin affinity purification (Fig. 3). Furthermore, these proteins were no longer detectable when lysates were treated with dithiothreitol (DTT), indicating that these proteins were indeed S-glutathionylated in THP-1 monocytes.

To assess the contribution of H2O2 to protein S-glutathionylation in monocytes, we examined if overexpression of H2O2-scavenging catalase would block BioGEE conjugation to PKM, β-actin, and histone H3 in THP-1 monocytes. Overexpression of catalase by either adenovirus infection or treatment with polyethylene glycol (PEG)-conjugated catalase resulted in decreased BioGEE labeling of both PKM and β-actin in metabolically stressed cells (Fig. 4). Importantly, BioGEE labeling of both PKM and β-actin was also reduced by catalase overexpression in cells under normal culture conditions in the absence of metabolic stress, indicating that protein S-glutathionylation in resting cells may also require H2O2 (Fig. 4A). These data show that catalase can selectively block protein S-glutathionylation in monocytes and suggest that H2O2 is required for S-glutathionylation of proteins such as PKM and β-actin in THP-1 cells. Interestingly, BioGEE labeling of histone H3 was not affected by catalase overexpression (Fig. 4).

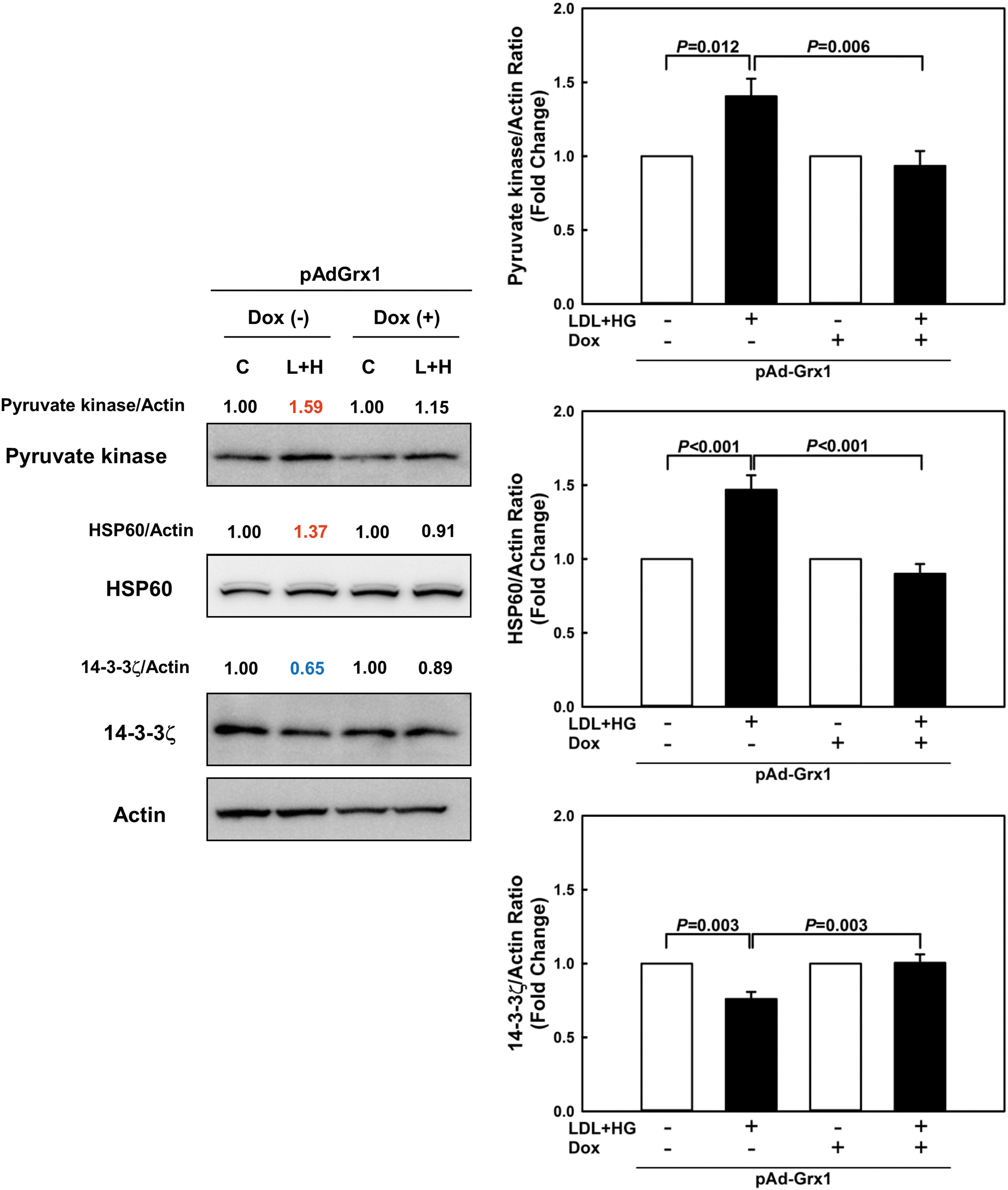
Protein S-glutathionylation can affect expression levels, activity, and/or intracellular localization of proteins (9, 32). We therefore explored whether metabolic stress in THP-1 monocytes altered expression levels of these 13 validated proteins and examined whether these changes could be prevented by overexpressing Grx1. Compared with vehicle-treated cells, metabolically stressed THP-1 monocytes showed a significant increase in the expression of pyruvate kinase and heat shock protein 60 kDa (HSP60) and a significant decrease in 14-3-3ζ protein levels (Fig. 5). None of the other proteins showed any significant change in expression levels. Overexpression of Grx1 completely prevented the changes in protein levels induced by metabolic stress (Fig. 5). These results suggest that metabolic stress-induced S-glutathionylation alters the expression levels of key proteins involved in cell signaling and monocyte metabolism, which may directly impact monocyte functions.
Protein S-glutathionylation in primary peritoneal macrophages
After validating our labeling strategy in protein S-glutathionylation in THP-1 monocytes, we examined whether protein S-glutathionylation also occurs in peritoneal macrophages of mice exposed to long-term metabolic stress. To our knowledge, this is the first time a proteomic analysis of S-glutathionylated proteins has been applied to primary cells isolated from an animal model mimicking human disease. Both male and female atherosclerotic-prone LDL receptor-null (LDLR−/−) mice were fed an HFD for 10 weeks to induce hyperlipidemia and mild hyperglycemia (48). As a control, LDLR−/− mice received a defined low-fat maintenance diet (MD). Our previous studies in this murine atherosclerosis model showed that peritoneal macrophages isolated from LDLR−/− mice exhibit increased thiol oxidative stress, increased protein S-glutathionylation, and accelerated macrophage chemotactic activity, which all directly correlated with the extent of atherosclerotic lesion formation (48, 62). In the current study, after 10 weeks, male and female mice on the HFD developed hyperlipidemia and mild hyperglycemia, although blood glucose elevations did not reach statistical significance (Supplementary Table S4). In contrast to male mice, HFD-fed females maintained the same body weight and plasma triglyceride levels as MD-fed females, despite developing severe hypercholesterolemia, recapitulating findings from our previous study (48).
Using IAM-Desthiobiotin-labeled lysates from primary peritoneal macrophages, which had to be pooled by sex and diet due to the low amount of protein, we identified a total of 127 S-glutathionylated proteins in males and females (Supplementary Table S5). Of the 127 IAM-Desthiobiotin-labeled proteins, 67 S-glutathionylated proteins (53%) were found in lysates from both male and female mice (Fig. 6A). Surprisingly, we detected 53 IAM-Desthiobiotin-labeled proteins (42%) in macrophage lysates that were unique to females and not identified in macrophage lysates from male mice. In fact, we detected nearly twice as many IAM-Desthiobiotin-labeled proteins (120) in macrophages from female mice when compared with male mice (74) (Fig. 6A). These data suggest a potential sexual dimorphism in the effect of metabolic stress on protein S-glutathionylation in primary macrophages. As with THP-1 cells, the majority of the proteins (46 or 62%) found in male mice were identified in macrophages from both MD-fed and HFD-fed mice (Fig. 6B). However, 16 of these 46 proteins (35%) showed increased IAM-Desthiobiotin labeling in response to metabolic stress, that is, HFD feeding, 7 (15%) showed decreased IAM-Desthiobiotin labeling, and IAM-Desthiobiotin labeling remained unchanged in 50% of the 46 proteins (Supplementary Table S5). These findings indicate that metabolic stress has divergent effects on the level of S-glutathionylation of these proteins. In macrophages from male mice, 17 of 74 IAM-DES-labeled proteins (23%) were only found in response to HFD, that is, were S-glutathionylated de novo in response to metabolic stress, and only 11 proteins (15%) were found exclusively in macrophages from MD-fed mice (Fig. 6B).
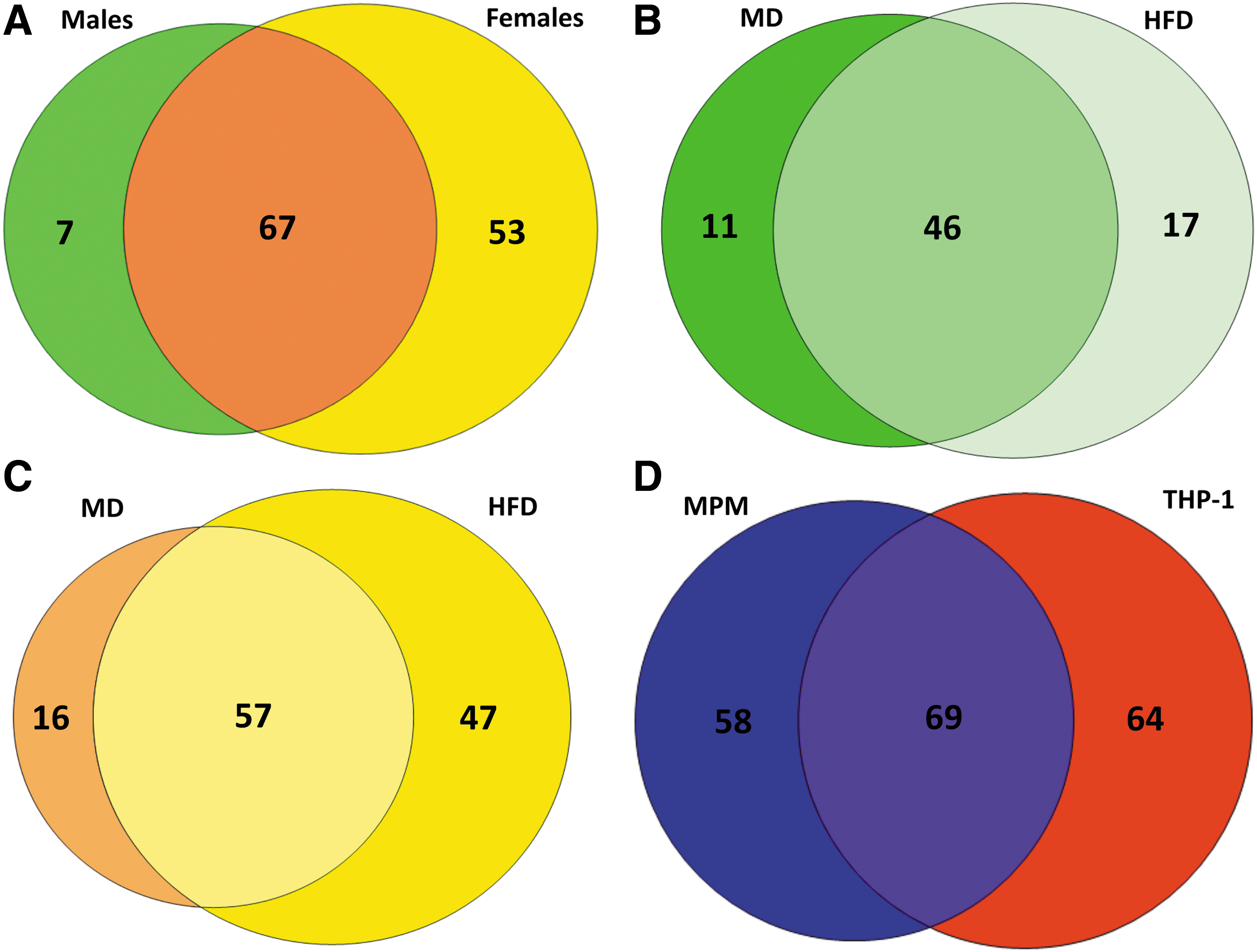
In contrast, macrophages from female mice showed a much higher level of de novo IAM-Desthiobiotin-labeled proteins, that is, S-glutathionylated proteins only detected in response to HFD (47 proteins or 39% vs. only 23% in males, Fig. 6C). These data strongly suggest that monocytes and macrophages in female mice show a unique spectrum of S-glutathionylated proteins in response to metabolic stress, which may alter macrophage function differently in females compared with male mice. Only selected proteins were found to be S-glutathionylated exclusively in macrophages from MD-fed mice (Fig. 6B, C: males: 15%, females: 13%). Together, these findings also suggest that most proteins that are susceptible to S-glutathionylation in primary (murine) macrophages are already modified under homeostatic conditions and only change their level of modification in response to metabolically stressed environments. This finding is consistent with our results in THP-1 monocytes.
When comparing IAM-Desthiobiotin-labeled proteins identified in mouse peritoneal macrophages with those found in human THP-1 monocytes, 36% of the proteins (69 of 191 proteins) were identified in both cell types, while 30% (58 proteins) were unique to mouse peritoneal macrophages and 34% (64 proteins) were unique to THP-1 cells (Fig. 6D). More importantly, 52% of the IAM-Desthiobiotin-labeled proteins identified in THP-1 monocytes (69 of 133 proteins) were also identified in mouse peritoneal macrophages, while 54% (69 of 127 proteins) of IAM-Desthiobiotin-labeled proteins identified in mouse peritoneal macrophages were also identified in THP-1 monocytes (Fig. 6D). These data suggest that the pattern of thiol oxidation in response to metabolic stress appears to be largely conserved between murine and human monocytic cells.
Identified functional roles of S-glutathionylated proteins
We used IPA and STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis to gain insights into the potential functional consequences of dyslipidemia-induced alterations to the S-glutathionylation status of the macrophage proteome. IPA analysis grouped the 127 IAM-DES-labeled proteins identified in murine peritoneal macrophages by specific function (Supplementary Table S6) and location (Supplementary Tables S7 and S8). Of the 36 functions associated with these 127 proteins, the most abundant functionalities were oxidoreductases, structural proteins, chaperones, ion-binding proteins, nucleotide-binding proteins, and peptidases (Supplementary Table S6), very similar to the main functions associated with S-glutathionylated proteins found in THP-1 monocytes (Supplementary Table S1). As with THP-1 monocytes, the vast majority of the proteins identified in macrophages from both female (76%) and male mice (80%) were located in the cytoplasm, while only 4% IAM-Desthiobiotin-labeled proteins were localized in the nucleus of macrophages from either sex (Supplementary Tables S7 and S8).
To determine which specific processes in male versus female macrophages would be affected in response to HFD feeding, we performed STRING analysis. The protein network for all S-glutathionylated proteins in female macrophages differs dramatically from that for male macrophages (Supplementary Figs. S3 and S4). The two most significant processes showing the highest enrichment scores for increased protein S-glutathionylation in macrophages of female mice were Cell Differentiation (p = 8.9 × 10−5) and Inflammatory Response (p = 8.8 × 10−4) (Fig. 7), while the two most significant processes for male macrophages were H2O2 Metabolism (p = 3.7 × 10−6) and Response to Stress (p = 6.4 × 10−5) (Fig. 8).

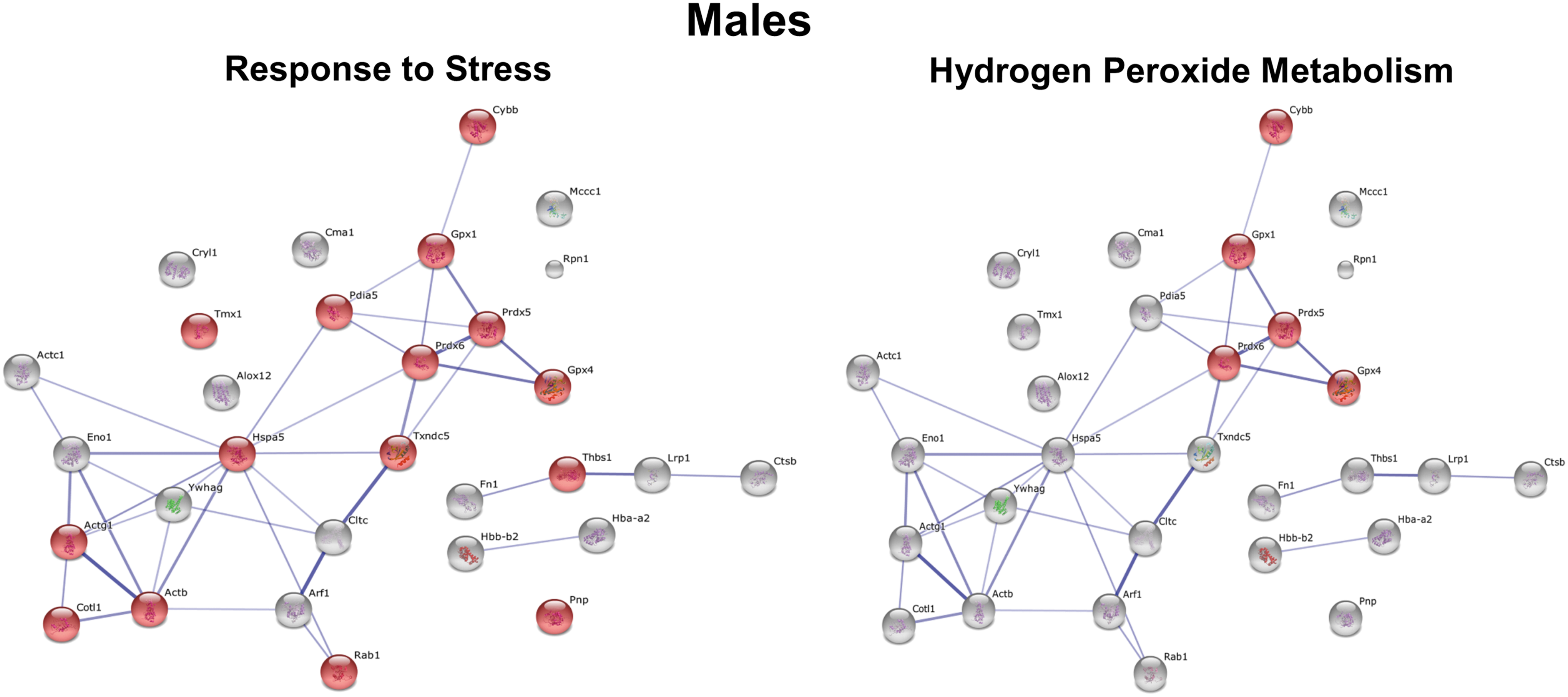
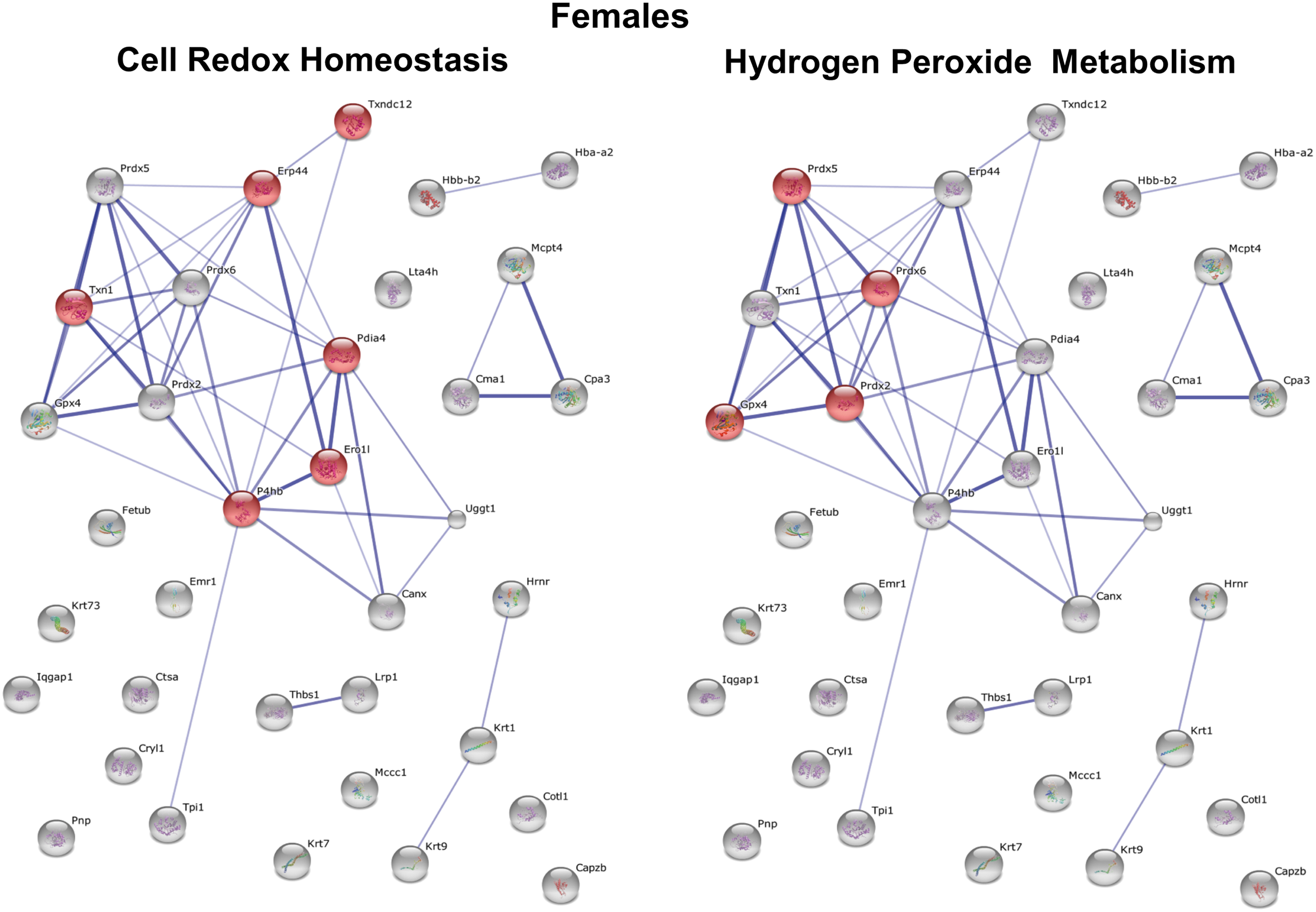
Interestingly, we also identified a number of functional clusters for proteins with decreased S-glutathionylation in macrophages from female mice, the top two clusters being Cell Redox Homeostasis (p = 6.1 × 10−6) and H2O2 Metabolism (p = 4.3 × 10−6) (Fig. 9). In contrast, no biological processes showed significant enrichment scores in macrophages from male mice for proteins with decreased S-glutathionylation. Together, these findings suggest that metabolic stress-induced thiol oxidation and protein S-glutathionylation affect not only different proteins but also distinct processes in macrophages from male and female mice. In fact, metabolic stress in male and female macrophages had exactly opposite effects on proteins involved in H2O2 metabolism (increased protein S-glutathionylation in males, but decreased S-glutathionylation in females), which includes proteins involved in antioxidant defense, the detoxification of H2O2, and redox signaling.
Discussion
We have developed a novel, sensitive, and selective technique to identify cellular S-glutathionylated proteins and utilized this labeling technique to examine metabolic stress-induced alterations in the S-glutathionylation state of proteins in monocytes and macrophages. This method allowed us to identify over 600 proteins in THP-1 monocytes and monitor changes in their S-glutathionylation state in response to (patho)physiologically relevant conditions. Furthermore, this technique allowed us to identify S-glutathionylated proteins in lysates of primary macrophages isolated from dyslipidemic and mildly hyperglycemic atherosclerosis-prone mice. To our knowledge, this is the first study identifying over 100 S-glutathionylated proteins in primary cells from an in vivo model of metabolic stress and human chronic inflammatory disease. Previous proteomic studies have typically found fewer than 30 S-glutathionylated proteins, and S-glutathionylation was induced by either supraphysiological concentrations of H2O2 or nonphysiological agents such as diamide (15, 16, 37). Importantly, our report is also the first to identify a potential sexual dimorphism associated with (thiol) oxidative stress in monocytes and macrophages.
There are three main reasons for the unprecedented sensitivity and selectivity of our technique. First, we used cell-permeable NEM to alkylate all free intracellular thiols to minimize both the loss of existing S-glutathionyl residues through transthiolation reactions (18) during cell lysis and any nonspecific IAM-Desthiobiotin labeling. Second, the use of an enzymatic reductant, Grx1, which is highly selective for mixed disulfides between protein thiols and glutathione, ensured that only thiols derived from S-glutathionylated proteins would be conjugation with IAM-Desthiobiotin. Last, we used a highly sensitive thiol-reactive probe coupled to DES rather than a biotin tag to increase the labeling efficiency and to allow for the elution of labeled proteins from the streptavidin-coated beads under mild conditions, thereby increasing the overall yield in IAM-Desthiobiotin-labeled proteins (28). Coupling this highly efficient labeling method to mass spectrometry analysis dramatically increased both selectivity and sensitivity of this proteomic approach. Thirteen proteins were selected for validation based on previous reports of S-glutathionylation and their robust mass spectrometry signal. The validation of these 13 selected IAM-Desthiobiotin-labeled proteins as bona fide S-glutathionylated proteins confirmed that our proteomic technique is not only sensitive and highly selective but also accurate and reliable.
Protein S-glutathionylation occurs under conditions of increased ROS production that leads to oxidation of the thiolate, producing proteins with sulfenic, sulfinic, and sulfonic acid residues (26, 42). These oxidized thiol groups, particularly sulfenic acids, can react with glutathione (GSH) to form mixed disulfides or S-glutathionylated proteins (26). Our data showing that catalase overexpression significantly represses both PKM and β-actin S-glutathionylation suggest that H2O2 mediates the S-glutathionylation of at least some proteins in monocytes. However, histone H3 S-glutathionylation was unaffected by catalase overexpression (Fig. 4). One possibility is that transgenic catalase may only prevent S-glutathionylation of cytosolic protein and was unable to translocate into the cell nucleus to block H3 S-glutathionylation. Alternatively, H3 S-glutathionylation may be regulated by an H2O2-independent mechanism, for example, by glutathione-S-transferase enzymes (26). Nevertheless, our data strongly suggest that at least some proteins in monocytes and macrophages are S-glutathionylated in an H2O2-dependent manner.
One limitation to our approach is inherent to nontargeted mass spectrometry, in that IAM-Desthiobiotin-conjugated peptides were only detected for high-abundance proteins, including actin, actin-related protein 3, GAPDH, HSP60, lactate dehydrogenase, ER60 protein, elongation factor 2, phospholipid glutathione peroxidase, and Grx. Identification of specific cysteine residue(s) labeled was not possible for the vast majority of IAM-Desthiobiotin-tagged proteins due to lack of sufficient sequence coverage. Therefore, future studies will focus on immunoprecipitation of candidate proteins and subsequent targeted tandem MS analysis, which should permit the detection of S-glutathionylated cysteine residue(s) (6, 8). A second more general limitation to this study is the THP-1 monocytic cell line used to validate the labeling technique. These monocytic cells may have limited clinical relevance with regard to metabolic and thiol oxidative stress in human monocytes and macrophages. Although we confirmed our findings in vivo in macrophages from a murine model of diet-induced dyslipidemia and atherosclerosis, validation in human monocytes and patients is still needed.
Originally, S-glutathionylation was considered a protective mechanism by cells to avoid irreversible oxidation of protein thiols and conserve intracellular glutathione during oxidative stress (54, 61). However, our group previously found that S-glutathionylation of MKP-1 not only inactivates this key phosphatase but also results in its proteasomal degradation, demonstrating that monocytes and macrophages respond to changes in their metabolic environment by altering both MKP-1 activity and expression levels (32). Of the 13 S-glutathionylated proteins we selected for validation, metabolic stress induced changes in protein expression levels in three of these proteins: pyruvate kinase, HSP60, and 14-3-3ξ. Furthermore, these metabolic stress-induced changes in protein levels were mitigated by overexpression of Grx1, the thiol transferase that catalyzes the reduction of mixed disulfides between protein thiols and GSH. These data support previous findings from our group and others that protein S-glutathionylation can regulate enzymatic activity as well as expression levels and degradation rates of proteins such as crystalline (72), MKP-1 (32), and 14-3-3ξ (31). Our data suggest that regulation of protein expression, activity, and stability by S-glutathionylation may be common to a much larger number of (redox-sensitive) proteins than previously known.
In silico analyses (IPA) of S-glutathionylated proteins identified in our study suggest that protein S-glutathionylation affects proteins involved in a wide array of cellular functions, including cell adhesion and migration, cell signaling, transcription, translation, and energy metabolism to name only a few. The vast number of proteins and protein functions affected by metabolic stress-induced S-glutathionylation, along with the dramatic changes in global protein S-glutathionylation in response to metabolic stress, suggests a broader role for the redox-dependent post-translational modification in altering the cell's overall functional phenotype.
One rather unexpected discovery from our study is the sexual dimorphism we found with regard to the effect of metabolic disorders on protein S-glutathionylation in murine peritoneal macrophages isolated from dyslipidemic and mildly hyperglycemic mice. While sexual dimorphism is well recognized in atherosclerosis and cardiovascular disease (43, 44), as well as in the immune system (4, 45), to our knowledge, this is the first evidence that metabolic stress-induced thiol oxidation may regulate and modify proteins differently in cells from males and females. While the underlying mechanism is not clear at this time, our findings may help explain the sexual dimorphism reported in numerous mouse models of metabolic diseases, particularly murine models of atherosclerosis (12, 22). Interestingly, male LDLR−/− mice are more prone to diet-induced atherosclerosis than female mice (59), yet the number of S-glutathionylated macrophage proteins we identified was nearly twice as large in females as males. This raises the intriguing possibility that the sexual dimorphic response of macrophages to metabolic stress-induced S-glutathionylation may contribute to relative atheroprotection observed in female LDLR−/− mice. This hypothesis is supported by the observation that the proteins involved in the H2O2 Metabolism cluster (glutathione peroxidases 1 and 4, peroxiredoxins 5 and 6, cytochrome b-245) showed increase S-glutathionylation in male macrophages, whereas in females, the proteins in the same clusters (glutathione peroxidase 4, peroxiredoxins 2, 5, and 6) showed decreased levels of S-glutathionylation in response to metabolic stress. Considering that S-glutathionylation is likely to affect (and inhibit) the activity of these key redox enzymes, altering the overall activity of this functional cluster of antioxidant/redox signaling proteins may have a profound impact on macrophage functions and thus may help explain the relative resistance of female LDLR−/− mice to atherosclerosis.
In summary, we describe a new and sensitive proteomic technique for the specific identification of S-glutathionylated proteins in monocytes and macrophages. Our findings show that this reversible post-translational modification occurs on a large number of proteins and may alter expression, activity, and/or degradation of these proteins, thereby affecting a vast array of cellular functions. This newly identified redox-sensitive regulatory network may explain how cells respond to changes in their metabolic environment and thus provides a mechanistic link between metabolic stress and the well-documented changes in the functional phenotype of monocytes and macrophages observed in patients with metabolic disorders (7, 17, 65). Applications of this technique to other cell types will provide new insights into their redox-regulated signaling pathways and the responses of these cells to pathophysiological conditions and metabolic diseases.
Materials and Methods
Animals
LDLR−/− mice on a C57BL/6J background (The Jackson Laboratories) were housed and maintained in colony cages on a 12-h light/12-h dark cycle. All studies were performed with the approval of the UTHSCSA Institutional Animal Care and Use Committee. At 18–24 weeks of age, male and female mice were randomized into two groups: MD (AIN-93G, Bio-Serv) or HFD (21% fat wt/wt and 0.15% cholesterol wt/wt; AIN-76A; Bio-Serv). Mice were maintained on their respective diets for 10 weeks. Fasted body weights and blood glucose were assessed at baseline, every 4 weeks during the dietary regimen, and immediately before sacrifice.
Peritoneal macrophage isolation and purification
Peritoneal macrophages were isolated by peritoneal lavage and subsequently purified using negative selection with mouse pan B (B220) and mouse pan T (Thy 1.2) magnetic beads (Dynabeads®). After purification, ∼95% of the cell population was CD68 positive, a monocyte/macrophage lineage marker, as measured by FACS analysis. Purified peritoneal macrophages were washed once with 1 mM NEM in phosphate-buffered saline (PBS) and stored at −80°C until labeling. Purified peritoneal macrophages from five mice of the same sex and dietary regimen were pooled for protein S-glutathionylation labeling and detection.
Cell culture
THP-1 monocytes were cultured at 37°C for 24 h in complete growth media, RPMI 1640 medium (Hyclone and Cellgro®), containing 10% fetal bovine serum, 2% glutamax, 1% sodium pyruvate (Cellgro), 1% penicillin/streptomycin (Cellgro), 1% HEPES, and 0.1% 2-mercaptoethanol. For metabolic priming of THP-1 cells, complete growth media were supplemented with either vehicle or freshly isolated native human LDL (100 μg/ml) plus HG (20 mM) for 20 h. For H2O2 treatment of THP-1 monocytes, cells were treated with 1 mM H2O2 for 15 min.
LDL isolation
LDL was freshly isolated by ultracentrifugation from pooled plasma from healthy blood donors. Freshly isolated LDL was purified by gel filtration chromatography, filter sterilized, and characterized as described previously (2, 68).
Overexpression of Grx1 and catalase
To overexpress human Grx1 in THP-1 monocytes, we used a doxycycline-inducible Tet-On adenoviral system expressing a Grx1-EGFP fusion protein (47, 62). THP-1 monocytes were incubated for 24 h with adenoviruses (multiplicity of infection = 25) in complete growth media. Transgene expression was induced by adding Dox (1 μg/ml; Sigma) for 24 h. For adenoviral delivery of human Catalase, THP-1 cells were infected with adenovirus particles (5 × 105 viral particles/cell) for 20 h. Adenovirus encoding human Catalase was a kind gift from Dr. Markus Bachschmid (University of Boston). For PEG-Catalase overexpression, THP-1 cells were treated with 250-U/ml Catalase-polyethylene glycol (Sigma) or an equal concentration of PEG-Mw 4600 (Sigma) for 20 h.
Labeling of S-glutathionylated proteins
Human monocytic THP-1 cells
After treatment, THP-1 cells were initially washed and incubated with 1× PBS containing 10 mM NEM before lysis. NEM was used for all pre-and postlysis steps to block all reactive thiols that would potentially be labeled by IAM-Desthiobiotin and thus produce false-positive signals. NEM was added prelysis to avoid thiol oxidation during cell lysis and other processing steps and to avoid loss of protein thiol-GSH mixed disulfides due to transthiolation and disulfide formation during sample preparation (18). NEM is a commonly used thiol-alkylating agent because of its high reactivity and specificity for thiols at physiological pH (6.5–7.5); at higher pH (>7.5), NEM reacts with lysines, arginines, histidines, and tyrosines (37, 46) and has been reported to also react with sulfenic acids (49) and possibly with sulfinic acids (29, 49). NEM has proven to be a better overall alkylating agent than IAM based on its ability to react with free thiols and sulfenic acids faster, more efficiently, and within a broader effective pH range (49, 51). Sulfonic acids (-
To label free thiols, IAM-Desthiobiotin (5 mM, Invitrogen) was added and incubated for 90 min at room temperature (RT) in the dark, pH 7.5. IAM is also a thiol-alkylating agent, but it reacts much slower than NEM. However, IAM is more selective for the thiolate anion and results in lower nonspecific labeling compared with NEM, making IAM-DES a highly specific and selective labeling reagent for the reactive thiols generated after Grx1 reduction. IAM requires a more alkaline pH range (7.5–8.5) for optimum reactivity and reacts more readily with cysteines at their pKa around pH 8.0 (46, 71). At higher pH values and temperatures, IAM can alkylate methionines, lysines, arginines, tyrosines, histidines, and the N-terminal and C-terminal carboxyl (20, 35, 70).
Samples were then passed through a desalting column to remove excess IAM-Desthiobiotin, and proteins binding nonspecifically to the agarose beads were removed using biotin-blocked streptavidin-coated agarose beads (Invitrogen). Lysates were then centrifuged at 13,000 g and supernatant was transferred to a new tube. IAM-Desthiobiotin-labeled proteins were enriched using streptavidin-coated agarose beads (Fig. 1), and labeled proteins were purified from lysates in sequential pulldown steps using 50 μl of fresh streptavidin-coated agarose beads for each step (150 μl total): 2 h of incubation, followed by an overnight (ON) incubation, and a final 6-h incubation. DES binds to streptavidin with a weaker affinity than biotin and, therefore, allows for the elution of DES-labeled proteins from streptavidin under mild conditions (28), minimizing protein loss due to denaturation and aggregation. Beads were pelleted for 1 min at 3000 rpm (∼1000 g) and subsequently washed thrice in lysis buffer before elution of the proteins. Proteins were eluted using 5× sodium dodecyl sulfate (SDS) loading buffer (30 μl) and 20 mM biotin (7.5 μl), combined, and stored at −20°C until further analysis.
Primary murine peritoneal macrophages
Purified murine peritoneal cells were labeled similarly to THP-1 cells (described above), except that due to the limited protein concentration in the primary macrophage samples, the 1D electrophoresis purification step (see below) was omitted and the IAM-Desthiobiotin-labeled proteins were subjected to on-bead digestion immediately before analysis by mass spectrometry as described below.
1D electrophoresis
Before gel loading, streptavidin-coated agarose beads with IAM-Desthiobiotin-labeled proteins were thawed and incubated at RT for 30 min and then boiled for 5 min. To determine the labeling and pulldown efficiency, 20 μl of sample was loaded onto a 12% SDS-polyacrylamide gel electrophoresis (PAGE) gel, separated by gel electrophoresis, and transferred to a polyvinylidene difluoride membrane. Membranes were blocked in 1× TBS containing 5% bovine serum albumin and incubated with either an antibiotin–horseradish peroxidase (HRP) antibody or streptavidin-HRP (ON, 4°C, 1:100; Cell Signaling). Blots were then developed using an enhanced chemiluminescence kit (Pierce).
For mass spectrometry analysis, 40 μl of IAM-Desthiobiotin-labeled proteins (one-third of the final volume eluted from the streptavidin-coated agarose beads) was loaded onto a 12% gel and allowed to run 1 cm into the separating region. The gel was fixed for 30 min in 10% methanol and 7% acetic acid and stained for 1h with Coomassie Blue (8.5% phosphoric acid, 5% methanol, 0.12% Coomassie Blue G-250, 0.132% ammonium sulfate). The gel was destained using the fixative solution until background was sufficiently removed and then imaged on the GS-800 calibrated densitometer (Bio-Rad). The gel was stored at 4°C in water until further processing.
Mass spectrometry
THP-1 cells
Briefly, after destaining, each lane of the 12% SDS-PAGE gels was divided into seven slices. Proteins in each slice were digested with trypsin (Promega), and the digests were analyzed by capillary HPLC-ESI-MS/MS on a Thermo Fisher LTQ Orbitrap Velos mass spectrometer. Online HPLC separation of the digests was accomplished with an Eksigent/AB Sciex NanoLC-Ultra 2-D HPLC system. Precursor ions were acquired in the Orbitrap in centroid mode at 60,000 resolution (m/z 400); data-dependent, collision-induced dissociation spectra of the six most intense ions in the precursor scan were acquired at the same time in the linear trap. Mascot (version 2.4.1; Matrix Science) was used to search the spectra searched against the human subset of the NCBInr_20130102 database combined with a database containing sequences for common contaminants. Postprocessing of the Mascot results (including determination of probabilities of peptide assignments and protein identifications) was accomplished with Scaffold (Proteome Software). Further protein functionality analysis was performed using Ingenuity Software program (IPA, Qiagen;
Peritoneal macrophages
Samples were subjected to on-streptavidin bead trypsin digestion. HPLC-ESI-MS/MS analysis of the digests was conducted as described above.
The Scaffold results were exported to Excel using the following parameters: (i) quantitative value based on the summed intensities of the fragments in the product ion spectra (called total ion current in scaffold) without normalization; (ii) 99% protein threshold; (iii) 95% peptide threshold; and (iv) one peptide minimum. The summed product ion current value provides an indication of the relative abundance of the peptide and is used for semiquantitative analysis. The ratios of summed product ion currents for proteins found in THP-1 cells treated with LDL+HG to the untreated cells were used to detect the differences between the two conditions. The result categories are increased if the ratio was equal to or greater than 1.5 in at least two experiments; decreased if the ratio was equal to or less than 0.5; no clear pattern/no difference if the ratio was between 0.5–1.5 or in fewer than two experiments; and no data if values were only found in H2O2-treated samples. When a protein was not detected in one of the paired samples, a value 0.1 was used to permit ratio calculations. For analysis of THP-1 cells, keratin, trypsin, and nonspecifically bound proteins were removed from consideration, while keratin was kept for the primary cell analysis.
Validation of protein S-glutathionylation by Western blot analysis
THP-1 monocytes were preincubated for 1 h in complete growth media containing 250 μM BioGEE (Life Technologies) (41), and then treated with LDL+HG for 20 h. Cells were washed with ice-cold PBS and lysed in RIPA lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% sodium deoxycholate) supplemented with protease inhibitors and 10 mM NEM to block further thiol oxidation. To confirm the specificity of BioGEE labeling of S-glutathionylated proteins, one aliquot of the lysates (500 μg protein) was reduced using 10 mM DTT for 20 min at 65°C before incubation with streptavidin-conjugated agarose (Invitrogen). Lysates were incubated for 1 h at 4°C with streptavidin-conjugated agarose beads. Beads were then rinsed thrice with lysis buffer. S-glutathionylated proteins were released from the beads with SDS sample buffer containing 10 mM DTT. Samples were then separated by 10% or 12% SDS-PAGE, and levels of BioGEE-labeled protein were evaluated by Western blot analysis using the following antibodies: 14–3-3δ/ζ, 14-3-3γ, β-tubulin, GAPDH, HSP90β, protein S100α, and pyruvate kinase (all from Pierce/Thermo Scientific); ATP synthase subunit alpha (ATP5A1), glutathione peroxidase 4, and histone H3 (all from Abcam); and β-actin, cofilin, and HSP60 (all from Santa Cruz). Proteins were detected by chemiluminescence on a KODAK Image Station 4000MM.
Statistics
Data were analyzed using Student's t-test or ANOVA for multigroup comparisons. ANOVA and post hoc analyses were performed using the least significant difference method (SigmaStat; Systat Software). All data are presented as mean ± SE of at least three independent experiments unless stated otherwise. Results were considered statistically significant at the p < 0.05 level.
Footnotes
Acknowledgments
This work was supported by grants to R.A. from the NIH (HL-70963 and HL115858) and the AHA (0855011F). S.U. was supported by a fellowship from the Translational Science Training (TST) Across Disciplines program at the University of Texas Health Science Center at San Antonio, with funding provided by the University of Texas System's Graduate Programs Initiative. H.S.K was supported by a grant from the National Research Foundation of Korea (NRF-2014R1A5A2009392). This work received computational support from Computational System Biology Core, funded by the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health. The authors would like to thank Kevin Hakala, the former Proteomics Technical Directors of the Institutional Mass Spectrometry Core Laboratory at UTHSCSA, and Ana Carrera for their technical assistance. Mass spectrometry analyses were conducted in the proteomic component of the UTHSCSA Mass Spectrometry Laboratory, supported by UTHSCSA and NIH grant 1S10RR031586-01 (STW). The authors would also like to thank Dr. Markus Bachschmid, Boston University, for providing adenoviruses carrying catalase or EGFP.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.