
Abstract
Introduction
B
Breast Cancer Classification: Subtype Approach
While a determined approach to aggressive screening implementation for early detection of breast cancer serves highly advantageous in providing profuse treatment strategies and improved prognosis among patients, metastatic breast cancer treatment is replete with challenges and shortcomings owing to its diverse heterogeneity and paucity of effective adjuvant chemotherapeutics. Therapeutic opportunities at this stage are further confounded by factors, such as age, genetic background and medical history of the patient, molecular subtype of breast cancer, and node status, and hence it is necessary that risk prediction models incorporate these factors in addition to the molecular genetics underlying breast cancer for more thorough comprehension and ability to predict suitable therapeutic intervention (44, 107, 260). From an era of localized treatment strategies restricted to mastectomy serving the universal panacea, the advent of genetic tools has aided in harnessing target-specific therapies to address this arduous and immensely heterogeneous disease. Relentless research and perseverance in this direction have fueled headway in both systemic and locoregional therapeutic strategies, finally obliterating the drastic Halstedian model measures for treatment of cancers. Neoadjuvant and systemic chemotherapeutic approaches are now being employed as standard care practices to complement local therapy owing to their prodigious benefits in curtailing tumor progression, thereby possessing immense potential in limiting tumor spread (233).
It is now well established that invasive breast cancer is a multifaceted disease characterized by innate intra- and intertumoral heterogeneity. Several articles by imminent scientific groups have identified distinct molecular signatures for predicting metastatic potential of these breast carcinomas. In a strictly scientific scenario, breast cancers are categorized on the basis of receptors displayed on the tumor cells. These refer to the estrogen (ER), progesterone (PR), and human epidermal growth factor receptor 2 (HER) or ERB-B2 receptors. Microarray gene expression profiling and sequencing platforms have led to identification of succinct trademark biomarkers to classify invasive ductal carcinoma into distinct subtypes through strict hierarchical clustering and dendrogram analysis. There are four primary subtypes of breast cancers that are clinically adopted—luminal A, luminal B, HER2-related or HER2-enriched (HER2-E), and basal-like ductal cancer. Additional subdivisions recognized include the poorly defined normal and claudin-low subtypes as well as others as proposed by Curtis et al. using molecular transcriptomic and microarray-based approaches (50, 229, 236). This immense heterogeneity among the diverse classes is concurrent with varied prognosis, survival outcomes, and sensitivity to therapeutics.
Tumor cells displaying the ER and PR steroid receptors are amenable to antiestrogen therapies with tamoxifen (estrogen receptor modulator) and aromatase inhibitors (causes estrogen diminution), pioneering the era of targeted therapy in the late 20th century and continuing to persist as the sentinel adjuvant treatment strategy. A vast majority of metastatic breast cancers comprise the TNBCs, characterized by loss of ER, PR, and atypical HER expression. They are chiefly basal in nature and especially correlate with poor prognosis and survival outcomes.
TNBC: advances and pitfalls
As mentioned above, TNBCs comprise an elfin, yet significant, populace among the breast cancer subtypes. They account for about 20% of all breast cancers and are characterized chiefly by the absence of ligand receptors on their cell membranes (8). This results in constitutive activation of the signaling components downstream, independent of ligand binding, thereby rendering them refractory to the current plethora of tyrosine kinase inhibitors. Obviously, the pathology is associated with rather poor clinical outcomes, in spite of exploiting the current battery of chemotherapeutic, adjuvant, and locoregional therapies. Although TNBCs initially do respond to chemotherapy and surgical removal of the neoplasms has rendered favorable clinical effect in the preliminary stages, their aggressiveness and grim outcomes are particularly imposed through the inability of these treatment strategies to account for complete elimination of all the tumor cluster populations, thereby causing patients to succumb to TNBC-associated relapse (8, 35, 208).
TNBCs were once morphologically characterized as a unique classification among the molecular subtypes of breast cancer that manifested itself with a typically basal phenotype. However, recent observations indicate that although a vast majority of these cells conform to a basal morphology, they are assorted in a milieu of heterogeneous cancer cells identified with a vast array of molecular phenotypes. Independent research groups have thus painted additional classifications to the TNBC group by applying bioinformatics and gene ontology platforms, as well as immunohistochemical approaches to assess molecular portraits of metastatic breast cancer cell lines and patient tumor samples, as well as microarrays. One research group divides TNBC majorly into basal-like cells, further comprising a small but significant population of the luminal and mesenchymal subtypes too, which also included the Her2-enriched, claudin-low, and few subpopulations that were predicted to be normal-like mammary tumors (198). It is paradoxical that one would expect Her2-enriched populations to exist in tumors classified as negative for the ERBB2 receptor through immunohistological analyses. However, these represent a very small population identified through whole genome sequencing arrays that are discerned to significantly contribute to the aggressive phenotype mirrored by TNBC subtype. In parallel, another research group reported an even more complex portrait of TNBCs, classifying them into seven distinct subtypes that included two basal-like subfamilies, basal-like 1 and basal-like 2, immunomodulatory, mesenchymal, mesenchymal stem like, luminal androgen receptor, and unstable (134). This results in a more dismal and confounding picture in terms of developing effective treatment strategies, adding to the distressing challenge imposed by its invasive and aggressive nature due to the distinct gene ontologies and pathway classifications that these cells are classified into. These independent observations broadly converge on assigning an enhanced molecular categorization of TNBC. The outcome of these studies provides clinicians with a more rigid signature to determine the disposition of the tumor and furnish more effective treatment alternatives (113, 131).
There are reports indicating several mutations in genetic loci on the human genome, which can now be identified as significant risk predictors of breast cancer (161). However, the association with TNBC is far lower with only 25 significant predictors of TNBC currently identified (205). The 19p13.1 locus herein seemed to have a particularly strong correlation with conferring risk of developing breast cancer of the TNBC phenotype. This interesting association was observed in the moiré of increased risk of TNBC and mutational events targeting the 19p13.1 locus. A hotspot for mutations, this locus seems to specifically restrict itself in altering pathways and regulating genes that are unique to amalgamate in promoting the development of tumors defined the TNBC phenotype, but not the hormone-positive subtypes (241). Although the authors maintain that the mechanism underlying the favored association for this loci with respect to TNBC development is unknown, it does present an intriguing notion that the different tumor subtypes may indeed materialize through distinct pathways radiated through modification of specific loci within the chromosomal makeup; thus, pressing for supplementary studies in this avenue.
In most studies, one associates the aggressive TNBC subtype by expression of basal and claudin-low populations (196). Moreover, these tumors characterized by their basal phenotype are strongly associated with poor prognosis and further exhibit an enhanced tendency to present with BRCA1 mutations, exhibiting enhanced tendency of cancer progression as a result of genome instability and involvement of the DNA damage pathways (77, 95, 130, 134). Studies have also indicated a strong molecular similarity between the TNBC intrinsic phenotype and properties attributed to tumor-initiating cancer stem cells (CSCs) (100, 202). Thus, they present with poor prognosis and overall survival rates, which might probably be meted out through chemotherapeutic advents that explicitly target these cell subtypes.
Recent studies therefore propose novel therapeutic opportunities for targeting in this avenue, owing to a clearer understanding of the etiology of TNBC. Combination therapies to counter for activation of parallel pathways as well as use of synthetic lethality opportunities in TNBC have proven fruitful. Some of the current strategies employed to combat TNBC have been embedded herewith, keeping in mind the molecular characterization of the targeted population (Table 1). We can therefore hope for a more promising future through substantial developments in chemoadjuvants and novel therapeutic windows such as immunological tools to address this burgeoning matter.
Drugs identified through screens on the basis of their molecular functions/cellular targets and under clinical evaluation or approved for treatment of TNBC
EGF, epidermal growth factor receptor; HDAC, histone deacetylase; mTOR, mechanistic target of rapamycin; PARP, poly (ADP-ribose) polymerase; TNBC, triple negative breast cancer; VEGF, vascular endothelial growth factor.
Systematization of Breast Cancers: The Pathway Approach
While morphological features and immunohistochemical analysis-based classifications initially served to determine tumor grade and mode of treatment, it is now well recognized that breast cancer is characterized by distinct gene signature patterns and trademark molecular pathways. Moreover, intrinsic genetic mutations and ectopic expression of genes causing activation of diverse signaling cascades are typically manifested, making it difficult to comprehend whether these distinct molecular pathways predict tumor subtypes or there exists no preference for the different tumors to adopt particular pathways for sustaining tumor progression.
Several pathway-specific alterations have been determined as critical factors predisposing to breast cancer. In this study, we discuss vital signaling cascades that are strongly correlated with metastatic breast cancer. We also begin with providing insight into a critical role for epithelial-to-mesenchymal transition (EMT) in association with metastatic breast cancer so as to impart more clarity in the vital reprogramming of cancer cells through activation of deregulated pathways and the consequences therein. The pathways are restricted to two broad categories, compartmentalizing the typical receptor-mediated signaling pathways independent from additional factors within signal transduction channels—some of which have been only recently discovered to serve as dynamic effectors of metastasis and stemness in cancers—and the other that comprises a diverse subgroup of signaling nodes that contribute to metastatic potential of tumors through receptor-independent intrinsic pathways that are equally necessary for the establishment and maintenance of systemic tumor populations.
EMT plasticity in breast cancers
EMT refers to a distinct physical switch in the tumor cell morphology that causes it to acquire a more invasive disposition. Epithelial cancer cells are characterized by a succinct cuboidal–columnar morphology, giving it a typical cobblestone appearance. These cells also present an inherent apico-basal polarity and adhere to the basal membrane through tight adhesion proteins. They are molecularly distinguished especially through their strong expression of E-cadherin and epithelial cytokeratin. Granted favorable stimuli, these tumor cells dedifferentiate to reactivate a primitive embryonic developmental program initiated through a change in cellular polarity, causing cells to detach from the basement membrane and invade the surrounding stromal tissue. Such invasion is particularly facilitated through the secretion of proteases as well as the launch of cellular escape mechanisms to evade programmed cell death. Tumor cells can further intravasate blood and lymph vessels, thereby attaining enhanced migratory potential, which allows them to traverse through the blood stream and ultimately deposit at a metastatic secondary niche (250). Herein, these cells can either remain in solitary clusters or undergo a change in their polarity through mesenchymal-to-epithelial transition (MET)—processes categorized as micrometastasis and macrometastasis, respectively (Fig. 1)
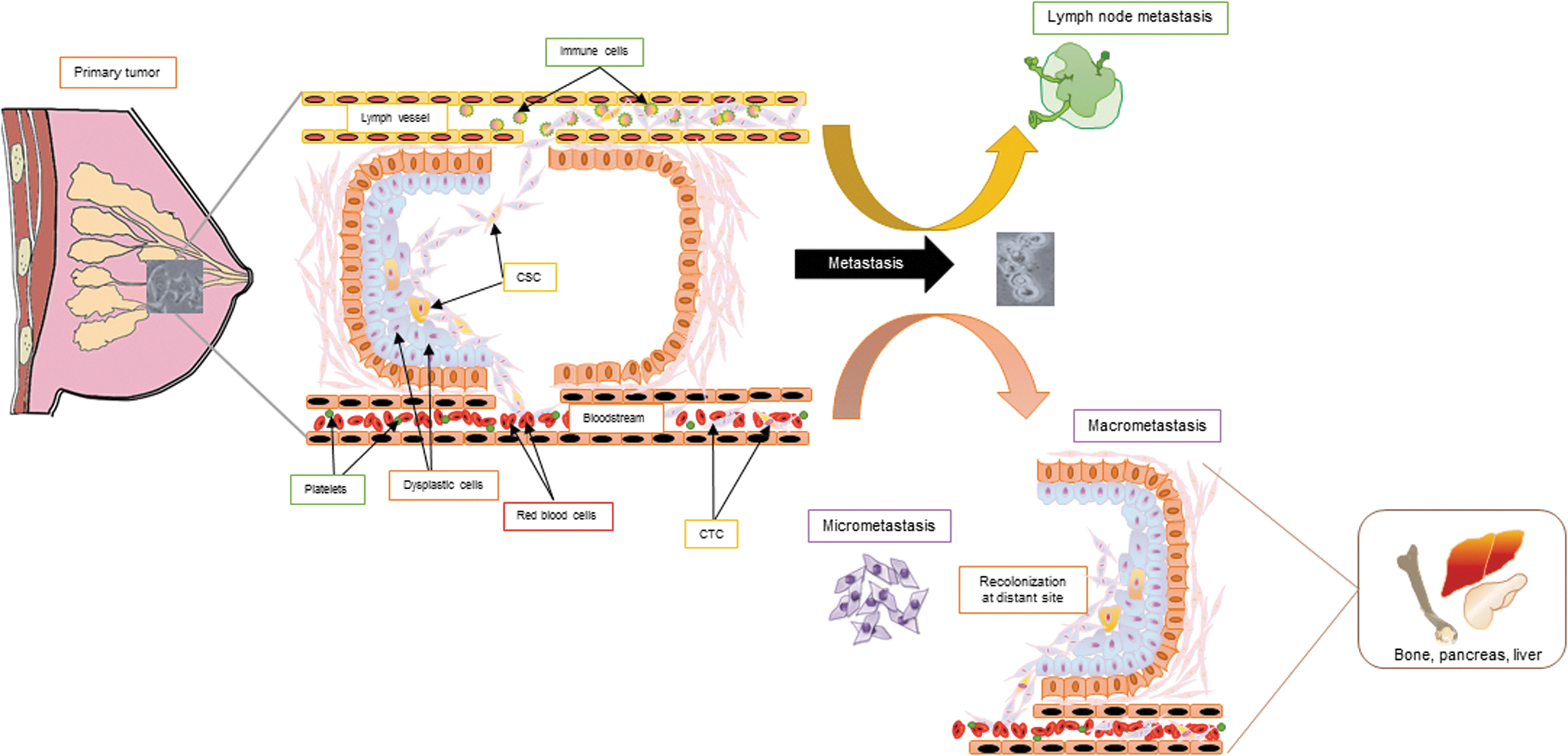
The surrounding stroma is another factor that influences the EMT plasticity of cancer cells in a profound manner. In breast cancer, stromal cells comprise the nonmalignant host cells surrounding the tumor population. They facilitate growth and development of the mammary gland. The mammary stroma comprises fibroblasts, blood vessels, adipocytes, immune inflammatory cells, and the extracellular matrix (ECM). The ECM in particular is a dynamic center that aids in further augmenting the tumorigenic potential through secretion of growth factors or cytokines. The cancer-associated mammary epithelium often displays receptors such as Frizzled, parathyroid hormone-related protein (PTHrP), and transforming growth factor-β (TGF-β), which are involved in the normal development of the mammary gland. The tumor-associated stroma, however, secretes a large number of ligands that bind these receptors and actively signal through transduction pathways, such as Wingless-related integration site (Wnt), Hedgehog, and TGF-β, leading to a considerable rise in the invasive and migratory potential of the tumor cells. The stroma is also associated with secretion of matrix metalloproteinases such as matrix metallopeptidase (MMP) and cytokines that further boost the invasive potential of cancer cells, allowing them to invade the adjacent tissue as well as supplement proangiogenic factors and immunomodulatory factors that aid in modeling the tumor microenvironment.
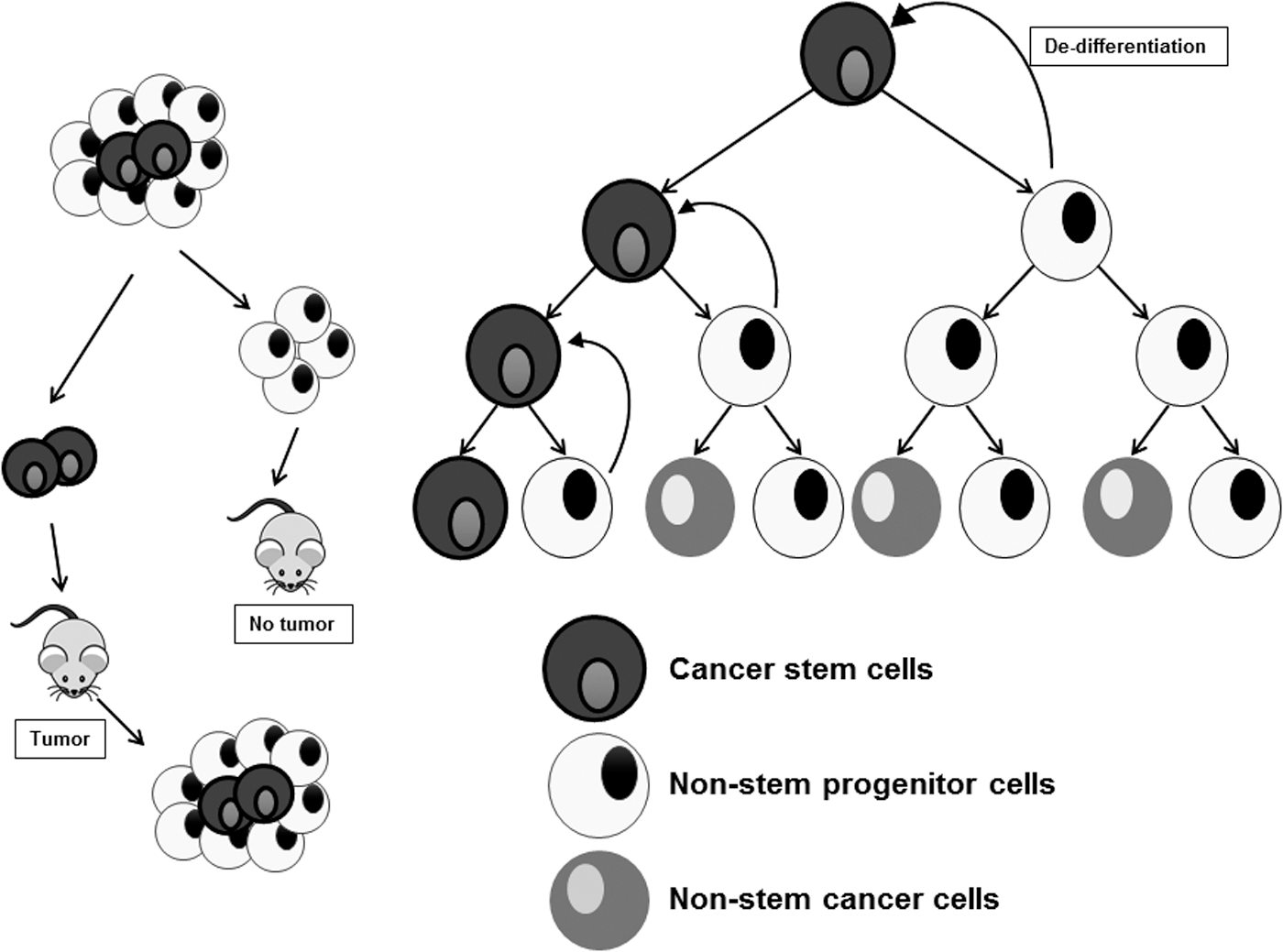
Mesenchymal cells are morphologically nonconforming to their epithelial counterparts. They are elongated with a spindle-like appearance and express typical biomarkers distinct from those expressed by epithelial cells. Moreover, these characteristics endow mesenchymal cells the distinguished ability to develop migratory potential. These metastatic mesenchymal cells were for long thought to be fibroblasts or fibroblast-like cells, thanks to their phenotypic appearance. However, mesenchymal cells did not display any of the characteristic markers inherent to the fibroblasts, and it was not until 2008 that Mani et al. proposed that these mesenchymal cells were, indeed, the CSCs (152). Through a series of ingenious experimental approaches that employed ectopic introduction of Twist/Snail transcription factors capable of inducing EMT in immortalized human mammary epithelial cells (HMLE), Mani et al. were able to illustrate that these EMT-positive mesenchymal cells expressed the CD44high/CD24low marker pattern synonymous with the stem potential. Not only did these EMT-indoctrinated cells display the numerous markers characteristic of stemness but they were also able to give rise to mammospheres, thereby providing the scientific community with the first functional link between EMT and stemness (152). The modern globally accepted CSC hypothesis was derived by Hannahan and Weinberg in their revisit of their original Hallmarks of Cancer review, Hallmarks of cancer: the next generation (97), which can be reiterated quite simply as follows (Fig. 2): • CSCs comprise a subpopulation of neoplastic cells within a tumor that have an intrinsic ability to initiate the formation of a tumor when inoculated into nude, nonobese diabetic (NOD)/severe combined immunodeficiency (SCID) mice. • These cells could arise from normal adult stem cells through acquisition of a series of aberrant mutational events that render them pro-oncogenic. • These cells possess an inherent ability to acquire mesenchymal characteristics, metastasize, and recolonize at a distant site. • They are more resistant to current chemotherapeutics targeting the non-CSCs selectively. • They can further divide to give rise to daughter cells that may either be identical to them and share the stemness characteristic as their parent or may possess traits characteristic of transit-amplifying cancer nonstem cells. However, when subjected to drug-induced stress conditions, these cancer nonstem cells can dedifferentiate and commit to a CSC fate.
It was Stephen Paget, a surgeon in London, who first evoked the concept of seed and soil, suggesting a crucial role for reestablishment of the primary tumor at a distant site through reprogramming of the cancer cell into its parental phenotypical pathology (189). This process, now widely acknowledged as MET, is recognized as a critical step for metastasis of the primary cancer to be completed. In other words, the circulating tumor cells (CTCs) must find a favorable microenvironment to seed themselves before committing to an epithelial morphology and reexpressing epithelial markers for the efficient completion of the metastatic program (209).
In human breast cancer, tumor-initiating/CSCs were first identified by virtue of their expression of the cell surface marker profile CD44high/CD24low (4). In primary breast xenografts, cells expressing these markers were enriched for their ability to initiate tumors in immune-deficient NOD/SCID mice. Interestingly, Nakshatri et al. presented evidence for a strong association of the CD44high/CD24low CSC phenotype with the basal-like molecular subtype of breast cancer tumors (171). Although, many studies on CSCs usually refer to CSCs as a subset of tumor cells with mainly mesenchymal traits (152, 207), others have shown that most basal breast cancer cell lines harbor relatively high content of both mesenchymal, also referred to as epithelial–to-mesenchymal CSC subpopulation (EMT-CSC), and epithelial CSC subpopulation with mesenchymal-to-epithelial transition traits (MET-CSC). Even in the extremely mesenchymal-like, claudin-low breast cancer subtype (a subclass of TNBCs), a significant amount of MET-like CSCs is present (172). Similarly, using a panel of isogenic single-cell clones with either EMT-like CD44high/CD24neg or MET-like CD44high/CD24low phenotypes from a TNBC cell line, Leth-Larsen et al. showed that cells that exhibited CSC-like characteristics, such as the formation of mammospheres, initiation of tumors in mice, and resistance to chemotherapy, displayed MET-like morphological traits (137). Finally, Biddle et al. also revealed that self-renewing CSCs in oral squamous cell carcinoma include two biologically distinct phenotypes. One phenotype, CD44high/ESA(EpCAM)low, was migratory and had mesenchymal traits characteristic of EMT and named (EMT)-CSC, whereas the other phenotype CD44high/ESA(EpCAM)high was proliferative, retained epithelial characteristics, and was identified as (non-EMT)-CSC (22). Taken together, these results support the existence of distinct CSC subpopulations within breast tumors with emerging evidence that the CD24low/neg EpCAMhigh MET-like CSC state could be the CSC subpopulation mainly responsible for the more malignant form of the disease.
Recent studies by two independent research laboratories have particularly impounded the role of MET in cancer progression wherein it is argued that lack of MET will render these metastatic CSCs mobile, yet unable to proliferate (184, 257). Ocana et al. speculate on the paradoxical role of the EMT-associated paired–related homeobox transcription factor 1 (Prrx1) in suppressing efficient macrometastasis and inhibiting the ability of these CTCs to acquire an inherent stemness function that is most likely to be necessary for recolonization and distant metastases in a squamous cell carcinoma model. Consistent with these findings, another study reports the peculiar role of TGF-β in promoting repression of Twist to undergo MET through activation of Id1 (Inhibitor of DNA-binding protein-1) (238). Id1 is able to occupy the promoter of Twist and thereby repress its transcription. Thus, increased expression of Id1 correlates with suppression of Twist and induction of MET. Such a concept suggests that if one were to manipulate and design a therapeutic that could target the activity of proteins critical to MET, one could indeed impair the probability of developing relapse associated with sustenance of the CSC compartment by preventing recolonization of these circulating tumor-initiating cells.
On the contrary, Weinberg and colleagues suggest that forcing mesenchymal cancer cells to undergo MET through an epigenetic reprogramming approach commits them to differentiation, paving the road to a novel approach to targeting the CSC populations to lose their tumor-initiating capacity and thereafter be vulnerable to the current battery of chemotherapeutics (194). Indeed, they demonstrate in their study that cAMP-dependent activation of protein kinase A (PKA) brought about by either forskolin (Fsk) or cholera toxin (CTx) resulted in an upregulation in the phosphorylation and subsequent activation of a histone demthylase PHF2, which reversed stemness-associated phenotype in a highly mesenchymal derivative of HMLE, NAMEC8 cells, switching them back to an epithelial cell type that was susceptible to conventional chemotherapeutics, such as doxorubicin and paclitaxel.
Weinberg and colleagues also adopted a different approach to target CSCs. Using a microarray-based approach, they were able to determine PKCα as a prominent protein capable of inducing tumor cells to acquire mesenchymal traits and stem-like phenotype. By directly subjecting the more mesenchymal CSCs to PKCα inhibitors, they observed significant cell death of only the CSC population both in vitro and in vivo without any significant adverse effect on the nonstem cell compartments (246).
Now that we have a clearer understanding of the CSCs, one can come up with at least three possible strategies to target these tumor-initiating stem cells: • Target the CSC population with chemotherapeutics that do have little to no effect on the nonstem cancer cells. • Target the CTCs to prevent the MET program, thereby rendering the cancer cells incapable of distant metastases. • Reprogram CSCs to undergo MET, thereby causing them to lose their mesenchymal and invasive properties and further target them with conventional chemotherapy.
This lends further support to the concept that cancers need to be targeted with novel chemotherapeutic strategies that selectively target the CSCs to achieve any significant success in combating the pathology.
Receptor signaling pathways in breast cancer
A plethora of receptors are activated in the events progressing to aggressive breast cancer. These comprise the bulk of TGF-β, RAS (Rat Sarcoma), Wnt, and Hippo pathways that are activated through distinct types of receptors: receptor tyrosine kinases and G-protein-coupled transmembrane receptors. While a potential role for the Sonic hedgehog deregulation has been alluded to in metastatic breast cancers, several studies need to be undertaken to complement this newly emerging gray area of research (247). Hence, we refrain from discussing the hedgehog signaling in this section. A brief note on hedgehog signaling will, however, be reviewed in the latter section (Hedgehog and Lysyl oxidase: predictors of breast cancer metastasis to the bone section). Aforementioned pathways are discussed in much greater detail in the following subsections and a simplified interactome is presented to address the complexity and cross talk inherent in these pathways (Fig. 7).

TGF-β/SMAD pathway: the strange case of Dr. Jekyll and Mr. Hyde
The TGF-β signaling pathway is considered one of the main pathways associated with EMT and breast cancer metastasis. However, its role in carcinogenesis is perplexing owing to its biphasic nature. While some reports indicate that it has a tumor-suppressive function, the vast majority of research groups have reported a critical oncogenic function for this pathway from the standpoint of breast cancer progression. The tumor-suppressive nature of this pathway has been elegantly reviewed by Derynck et al. (58). Recent studies have finally provided a chronology to this dual characteristic of TGF-β wherein it has been elucidated that although TGF-β could function as a tumor suppressor during the initial stages of carcinogenesis, it adopts a pro-oncogenic role during the later stages of tumor progression. As metastatic events are restricted to the latter chapters of cancer, our review will focus only on the contribution of oncogenic TGF-β signaling in breast cancer progression and maintenance.
Briefly, TGF-β signals through a heterotetrameric complex embodying dimeric type I and type II serine/threonine receptor kinases. The TGF-β ligand next binds to receptor complex, activating the type II receptor kinase, which subsequently phosphorylates and activates the type I receptor. Activation of the type I receptor is supervened by robust activation of the downstream effector proteins of this signaling node. Herein, SMADs (Sma and Mad-Related Family) constitute the most well-characterized mediators of TGF-β signaling network. Upon ligand-mediated activation of the type I TGF-β receptor kinase, these receptor kinases are further responsible for activation of a class of Smads known as receptor-regulated Smads (R-Smads), thereafter forming a heterotrimeric complex with the common mediator Smad (Co-Smad/Smad4). Once generated, this complex can be localized to the nucleus where it can induce transcription of TGF-β target genes. In contrast, a triennial subgroup of Smads, inhibitory Smads (I-Smad), is responsible for interacting with the type I receptor, consequently preventing the activation of R-Smads or disrupting the formation of the trimeric complex between R-Smads and Co-Smads, thereby inhibiting TGF-β-Smad signaling (Fig. 3). The importance of TGF-β-Smad signaling in mediating EMT has been exemplified using many tumor progression models. For instance, the overexpression of Smads 2 and 3was reported to induce EMT in the mammary epithelial model (200, 265), whereas the increased expression of inhibitory Smad, Smad7, was able to inhibit Smad3-induced EMT (265). In addition, the loss of Smad2 and Smad3 function has been demonstrated to contribute to lowered metastatic potential of breast cancer cells in mouse xenograft models (253).

Interestingly, several transcription factors that are critically involved in driving the EMT program, such as Snail1, Twist, and HMGA2, have been verified as bona fide partners that interact with Smad complexes (248). This was shown to induce transcriptome reprogramming through repression of genes engaged in promoting epithelial plasticity, but activation of genes specific to computing and sustaining a mesenchymal phenotype (80). For instance, the Snail1-Smad3/4 complex binds to the gene promoter of coxsackie–adenovirus receptor and E-cadherin and represses their expression in mammary epithelial cells in response to TGF-β activation (270). Although EMT-promoting Smad activation complexes are yet to be reported in orthotopic breast cancer models, activator complexes such as β-catenin-Smad2 complex have been shown to promote expression of mesenchymal genes such as α-smooth muscle actin (α-SMA) and plasminogen activator inhibitor-1 (PAI-1) in alveolar epithelial cells during TGF-β-induced EMT (123). Moreover, high expression of both, α-SMA and PAI-1, has been associated with increased tumor aggressiveness and poor clinical outcome in breast cancer patients (9, 280). Besides Smads, TGF-β has also been reported to signal through Smad-independent pathways such as mitogen-activated protein kinase (MAPK) (283) and Rho-like GTPase (21, 66) pathways. These pathways will be discussed in greater detail in the following sections.
The RAS-ERK signaling node: one axis is insufficient to drive metastasis in breast cancer
Ras proteins consist of small GTPases that regulate vital cellular processes such as cell proliferation, cell survival, and cytoskeleton remodeling. Members of the Ras superfamily are further classified into subfamilies based on their sequence composition and the cellular processes they regulate; vis-à-vis, the Rho subfamily regulates cytoskeleton dynamics, while the Ras subfamily regulates cell growth (reviewed in Wennerberg et al., 275). The Ras subfamily proteins are most commonly implicated in cancer, with up to 30% of all human tumors harboring a mutation in Ras alleles. To date, there are four known Ras isoforms, namely N-Ras, H-Ras, and splice variants K-Ras4a and K-Ras4b, encoded by three RAS genes, N-RAS, H-RAS, and K-RAS, respectively (237). Briefly, Ras isoforms contain a highly conserved G domain and a C-terminal hypervariable region. The G domain consists of five G motifs that bind to guanine nucleotides, while the C-terminal hypervariable region contains the CAAX motif, which initiates a series of post-translational modifications such as farnesylation and geranylation that facilitate its cellular localization and plasma membrane binding through the lipid tail (116). Variations in the C-terminal of Ras isoforms allow for isoform-specific modifications. Lipid modifications of Ras are determined to be absolutely essential for isoform-specific subcellular localization and function.
Ras GTPase proteins serve as critical signaling nodes, which transduce extracellular signals to intracellular signaling pathways by oscillating between their active GTP-bound state and inactive guanosine triphosphate (GDP)-bound state. This tandem switching between the two states is brought about with the aid of GTPase-activating proteins (GAPs), which append the free phosphate group onto Ras, thereby triggering Ras activation, and guanine nucleotide exchange factors (GEFs) that are responsible for triggering GTP to GDP switch resulting in Ras inactivation (Fig. 4). When GTP bound, Ras can promote the activation of RAF serine/threonine kinase, which triggers a kinase cascade involving the sequential phosphorylation of MEK1/2 and ERK1/2. Active ERK1/2, in turn, regulates numerous nuclear proteins, many of which are transcription factors, such as Myc, Fos, Ets, and ELK1. These in turn are critical orchestrators of cell proliferation and survival. When mutated, the intrinsic GTPase activity is constitutively triggered, resulting in persistently activated Ras. Despite the high frequency of aberrations within the canonical Ras/MAPK in a diverse group of solid cancers, mutations in the Ras/MAPK pathway in breast cancer are rarely observed. The pathological activation of Ras through overexpression of growth factor receptor in breast cancer, however, implies the importance of Ras in tumorigenesis of mammary cells. This also suggests that breast cancer may utilize a parallel pathway of Ras such as the PI3K/AKT/mTOR axis to promote tumorigenesis. Consequently, targeted therapy of the RAS/MAPK pathway for metastatic breast cancer has shown limited efficacy in a clinical setting (55, 166, 215).

Despite this apparent redundancy of Ras/MAPK signaling in tumorigenesis of breast cancer, a growing body of evidence suggests otherwise. For instance, increased ERK1/2 activity, a functional output of Ras activity, has been observed in metastases relative to primary breast tumors (1). In corroboration with a previous study, sustained ERK activation induces the uncoupling of two Rho GTPase effectors, RhoA and ROCK, from stress fiber formation, resulting in increased motility of the Ras-transformed cells (220). Ras could also indirectly promote metastasis through alteration of the tumor microenvironment. Leibovich-Rivkin et al. showed that in MCF-7 cells, the inflammatory cytokine, tumor necrosis factor-alpha, in cooperation with active Ras induces secretion of proangiogenic factor, CXCL-8, resulting in highly vascular tumors, which contributes to an increased metastatic potential of the tumor (135). Other mechanisms through which ERK1/2 promotes cell survival include activation of eIF4E, which augments protein translation, repression of apoptotic proteins such as Bcl-2-associated death promoter (BAD), enhances lipid metabolism, and finally boosts cellular HDAC6 expression, thereby leading to enhanced migratory potential of breast cancer cells (37, 157, 190, 221, 228, 278).
Ras is also shown to exert profound effects on cytoskeleton dynamics that results in increased invasiveness and motility of Ras-transformed cells. Using an MDA-MB-231-derived cell line, Choi and Helfman (42) reported that elevated RAS-ERK signaling in the highly aggressive MDA-MB-231-LM2 disrupted the assembly of stress fibers and focal adhesions, ultimately resulting in appreciable activation of genes associated with invasive and migratory potential, thereby contributing to lung metastases. In spite of these observations, inhibition of RAS-ERK signaling was insufficient to inhibit cell motility in LM2 cells possibly through sustained PI3K/AKT signaling. Dual inhibition of PI3K/AKT and RAS/MAPK pathways was required to inhibit cell motility, thereby suggesting that both the PI3K/AKT and the RAS/MAPK axes promote the same phenotype in tandem (42, 102, 164, 217).
The PI3K/AKT pathway in breast cancer: a torrid state of affairs
The PI3Ks are a family of intracellular lipid kinases, which are further divided into three classes—class I, class II, and class III—based on their structure and substrate specificity. Class I PI3Ks are further classified into subgroups depending on the corresponding activators for these categories. Although all classes of PI3Ks are implicated in the regulation of cell growth and survival, only class IA PI3Ks and associated downstream effectors were demonstrated to be a key orchestrator of tumorigenesis driven by oncogenic RTKs and Ras by far (284). Hence, this review would focus on the role of class IA PI3Ks in cancer.
Central to the PI3K/AKT signaling pathway, the heterodimeric class IA PI3K consists of an adaptor protein subunit (p85) and a catalytic subunit (p110). Typically, class IA PI3Ks are activated when p85 binds to phosphotyrosine residues of activated RTKs through their Src homology 2 (SH2) domains, relieving its trans-inhibitory effect on p110. Other known activators of PI3Ks include Mutant Ras, Src, and Protein kinase C (PKC). Upon activation, PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), which in turn serves to provide a docking site for numerous proteins that harbor a pleckstrin homology (PH) domain. During PI3K-AKT signaling, membrane-bound PIP3 recruits PH-containing protein, phosphoinositide-dependent kinase-1 (PDK-1), and AKT serine/threonine kinase to the plasma membrane where PDK1 phosphorylates AKT at threonine 308, thereby resulting in its activation. AKT may also be phosphorylated at the Serine 473 residue by mTORC2 and integrin-linked kinase 1 (ILK1), which are more commonly referred to as PDK2, to be fully activated (38). Acting in an opposing manner, phosphatase and tensin homolog (PTEN) tumor suppressor protein antagonizes PI3K-mediated activation of AKT by dephosphorylating PIP3 to PIP2, causing the switching off of the PI3K/AKT signaling loop. Thus, loss-of-function mutations of PTEN and/or gain-of-function mutations of PIK3CA are among the most common aberrations implicated in PI3K/AKT-driven human malignancies, especially breast cancer, thus making upstream components of the PI3K pathway attractive therapeutic targets (16, 36, 68, 74, 122, 284). Being a key regulatory kinase for a wide array of proteins, AKT activation is fundamentally responsible for diverse operations within the cell, ranging from serving as a platform for protein translation and cell survival to metabolic regulation, cellular differentiation, and migration. In the context of metastasis, the PI3K pathway is able to regulate actin rearrangements through Rac activation, leading to increased cell motility and invasive capacity of the cell (216). Moreover, active AKT is able to inhibit GSK3β, a key regulator of Snail transcription factor. Increased levels of Snail lead to repression of E-cadherin adhesion protein, which serves as a key event initiating EMT cascade (34, 153).
While activating mutations in the PI3K/AKT pathway are quite frequent in the ER, PR, and HER2-positive tumor subtypes, the TNBC subtypes seldom associate with aberrant activation of this signal transduction pathway. It is not surprising therefore that TNBCs with mutations within this pathway have been strongly correlated with enhanced chemoresistance, especially to antiestrogen therapy and aromatase inhibitors that serve as the first line of defense by blocking receptor activation. Moreover, deregulation of this pathway also renders targeted therapies ineffective and enhances resistance against pharmacological therapeutics, such as trastuzumab and lapatinib, as such drugs exert their potential upstream of PI3K activation and PTEN inhibition. To combat this tumultuous issue of resistance acquisition, combination therapy with downstream mTOR inhibitors was contemplated and shown to provide a significantly better prognosis in preclinical studies and clinical trials (84, 145, 226, 232).
Finally, recent studies have brought into light a de novo functional role in breast cancer for the most common activating mutation within this pathway—PIK3CAH1047R. Koren et al. elucidate an autochthonous role for this mutation in facilitating dedifferentiation of Lgr5 lineage-committed breast cells. This compels such Lgr5-positive lineage-restricted cells to undergo a transition and acquire stemness properties with the ability to transit into various subtypes characterized by the expression of diagnostic markers, such as luminal (keratin 8/18) and basal (keratin 5/14, smooth muscle actin), determined through FACS sorting for the respective subtypes; ergo contributing to reinforced heterogeneity and multipotency among these neoplastic cells (127). The multipotent stem-like characteristics of these transitioned cells were also confirmed through mammosphere formation assays by comparing the mutant cells with their parental controls as well as through tumor onset, survival, and histological analyses of these dedifferentiated cells in the tumors of inducible transgenic mice models under the control of CreERT2.
When Wnts turn Wingmen to EMT
Wnt glycoproteins belong to a highly conserved family of signaling molecules that regulate many aspects of embryonic development in metazoans, such as determining the anterio-posterior axis, cell fate decision, and migration. Additionally, Wnt signaling has a central role in the maintenance of normal multipotent capacity of adult stem cells. Although Wnt signaling is indispensable for orchestrating highly intricate events during embryogenesis, the pioneering member of this pathway, int1, was originally discovered in a mouse model of breast cancer induced by the retroviral mouse mammary tumor virus. Nusse and Varmus made stellar observations herein linking the direct overexpression of the int1 gene to malignant transformation of the mammary tissue (179). Direct links between aberrant Wnt signaling and human cancers only began to emerge when accumulating molecular evidence pinpointed mutations in canonical Wnt signaling pathway components as molecular hallmarks in wide variety of malignancies (85, 106, 223, 266).
In the absence of Wnt signaling, the primal constituent, β-catenin, remains trapped in the cytoplasm and is actively marked for degradation by a β-catenin destruction complex that comprises Axin, CK1α, APC, and GSK3β. Wnt signaling is initiated when Wnt ligands bind to the cysteine-rich domains of Frizzled (Fz) receptors. Ligand binding promotes the formation of a ternary complex between Wnt-Fz and LRP5/6, resulting in a signaling cascade that ultimately disrupts the formation of the destruction complex and leads to the stabilization of cytosolic β-catenin. Free cytosolic β-catenin is subsequently translocated into the nucleus where it binds to the TCF/LEF1 complex and promotes expression of its downstream target genes (Fig. 5). Although the most frequently mutated components in Wnt-driven cancers are adenomatous polyposis coli (APC), Axin, and β-catenin such that Wnt signaling is upregulated in a ligand-independent manner, these mutations have rarely been identified in breast cancer clinically. Truncation mutation in APC, common in colorectal cancer, has only been identified in 1 of 227 breast tumors (141, 235). In addition, no functional mutation in Axin homolog has been detected in breast cancer as well (273). Last, the only clinical evidence implicating aberrant Wnt signaling in breast cancer is the high levels of β-catenin detected in breast tumor samples by immunohistochemistry and Western blotting of tumor lysate (141, 219). However, recent studies have challenged this gray area by highlighting the importance of this signaling pathway in contributing to breast cancer metastases and also in promoting a CSC-like basal cell population in breast cancers (59, 60, 111); employing next-generation sequencing platforms has alluded toward a disclosure of deregulated Wnt signaling through repression of negative regulators of this pathway as well as through direct hyperproduction of Wnts in several cancers proposing a parallel, independent of activating mutations in β-catenin (267).

Based on numerous studies, it was suggested that abnormal autocrine Wnt production indicated by overexpression of Wnt mRNA by breast tumor is the source of constitutive Wnt signaling (51, 117, 124). However, it is important to note that these studies were conducted on very small sample sizes and rigorous studies have not been extended across all Wnt proteins. In addition to excessive Wnt production, expression of physiological Wnt inhibitors was shown to be deregulated. For instance, loss or reduced expression of Wnt-inhibitory factor 1 (WIF-1) and secreted Frizzled-related protein (sFRP) was observed in 60% of breast cancer and 80% of invasive breast cancer, respectively (261, 277). Corroborating studies also identified promoter methylation as the predominant mechanism resulting in loss of expression of both genes (2, 267). Recently, Stewart et al. published a study wherein they discovered a strong association between Her2+ and ER- breast cancer with the Wnt-associated transmembrane protein, wntless, which acts as a carrier for palmitoylated Wnts to be transported to the plasma membrane from the endoplasmic reticulum (242). In recent years, tremendous momentum has been perceived in this scientific arena, thereby suggesting a platform for the development of drugs targeting upstream components, such as wnt receptors and proteins, responsible for efficient processing of Wnts. Indeed, several recent publications targeting the wnt pathway are directed to developing and optimizing inhibitors toward PORCN, which serves to palmitoleate Wnts and is hence necessary for their efficient secretion to the plasma membrane (65, 142, 203).
In vitro, Wnt/β-catenin signaling was reported to upregulate Axin2 expression, which paradoxically results in the cytoplasmic localization of GSK3β, a negative regulator of Snail1 transcription factor, in breast cancer (282). The increase in nuclear Snail1 levels results in initiation of neoplastic EMT programs. In addition, upregulation of Wnt5a was observed in macrophages when cocultured with breast cancer cells and was responsible for the induction of matrix metalloprotease-7, resulting in macrophage-induced invasiveness (204).
Metastatic breast cancers swim in deregulated Hippo pools
The Hippo pathway has recently emerged as a key signaling node central to development and is important in maintaining cellular homeostasis (177). Consequently, its deregulation has been observed in tumors of all origin, making it a critical pathway to elucidate (98, 175, 191, 286). Aberrant overexpression of principal Hippo components, yes-associated protein (YAP) and tafazzin (TAZ), are experimentally defined as sufficient to induce tumorigenesis and inhibit differentiation (31, 96, 210, 239). Oncogenic YAP is a vital effector of downstream mutant KRAS signaling wherein YAP can further potentiate tumor progression and maintenance (103, 227, 287). In 2006, Overholtzer and colleagues presented an interesting corollary by employing both whole genome sequencing analysis and in vivo experimental models, which firmly set YAP in the map of metastasis-promoting factors attributed to mammary neoplasia (188).
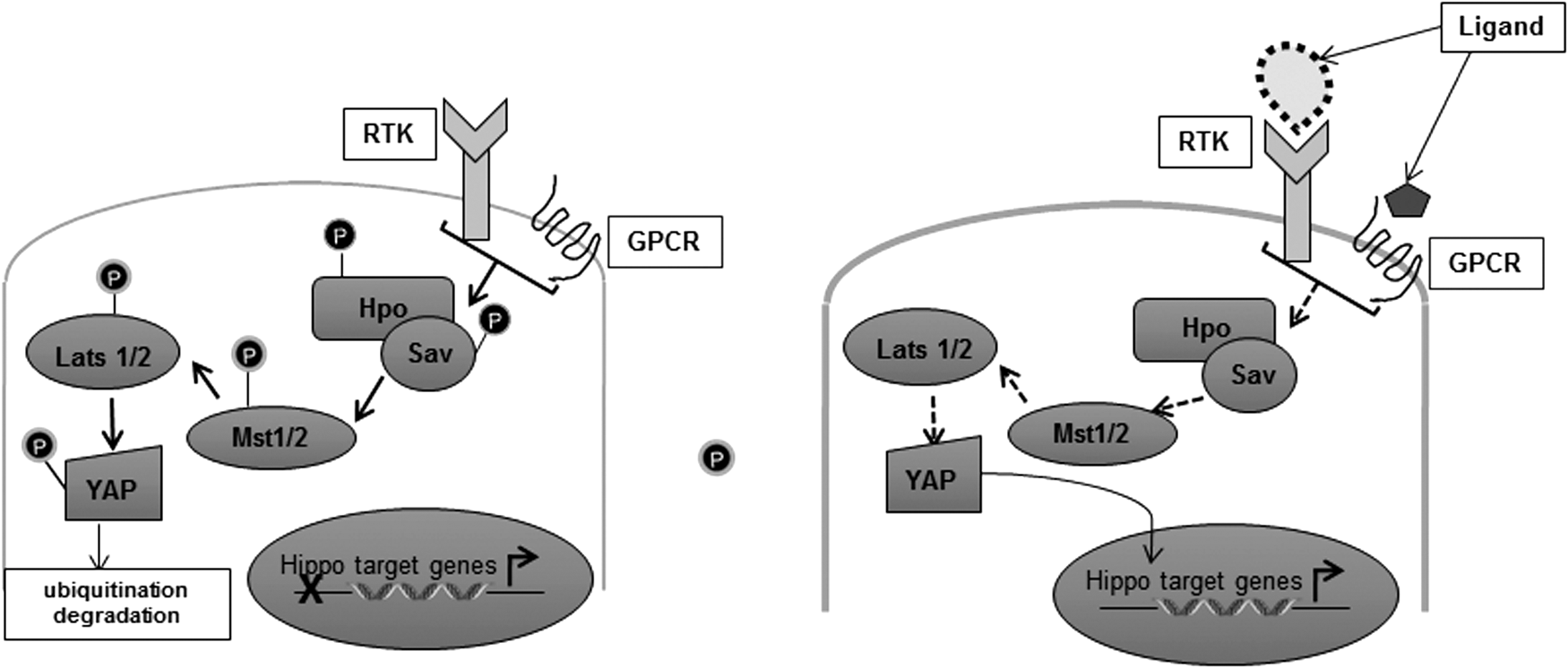
Deregulated Hippo pathway shares features with classical receptor kinase signaling pathways. For one, they are mediated through typical kinase-dependent phosphorylation events that signal to regulate the nuclear localization or cytoplasmic retention of the key terminal effector of this pathway, YAP/TAZ. Second, they signal through serial interaction among their highly conserved motifs known as the atypical WW domains and the PPxF/Y domains. Finally, the temperament of this signaling axis could be canonical or noncanonical depending on the nature of the kinase that contributes to the phosphorylation of the penultimate effector, YAP.
Inhibition of the Hippo pathway results in YAP remaining nonphosphorylated and thus beckons for the nuclear import of YAP tailed by its trussing with the TEAD transcription complex. Nuclear YAP/TEAD complex is subsequently switched on, leading to transcription of genes crucial to fostering proproliferative and survival signals (Fig. 6). Moreover, overexpression of YAP has been profoundly implicated with the gain of metastatic potential and stem cell-associated features in mammary epithelial cells (188). A fascinating new node for mechanosignaling-based activating function is being unraveled in recent years. It is now acknowledged that not only is YAP/TEAD activation a strategic intersection for transcriptional upregulation of actin-associated proteins vital to stemming EMT, but interestingly the noncanonical pathway is also intricately dictated by the cytoskeletal arrangement pattern and cell density, suggestive of a likely feedback loop regulating actin modeling and tumor progression. This novel axis of Hippo-YAP signaling will be discussed in greater detail in the following subsection of mechanosignaling in context with ECM density.
The receptor-independent signaling in breast cancer progression and metastasis
DNA damage and response pathway: failures in repair
Hannahan and Weinberg's, Hallmarks of cancer: the next generation, represent additional aspects that contribute to tumorigenesis and tumor maintenance, which were absent in their original model. One of the principal additions to this model is the role played by genomic instability in leading to carcinogenesis. Cancer cells are exclusively deficient in the crackerjack functioning of their repair pathway enzymes due to the accumulation of mutations in these genes. BRCA1/2, p53, Ku70/80, and FA complex mutations are exclusively acclaimed in regard to breast cancers. BRCA1/2, p53, Ku70/80, and FA complexes are stalwart soldiers in the army of repair enzymes and therefore tumor suppressive in nature. Mutations in the BRCA genes are mostly hereditary and inherited in an autosomal dominant nature. Such mutations acutely raise the risk of developing breast cancer among individuals. It is now well established that cancerous cells present with an increasing number of genetic aberrations, resulting in an overwhelming abundance of errors in DNA replication. This causes strand breaks, which when irreparable, predispose to genomic instability, senescence, or apoptosis-mediated cell death (56, 169). Hence, a continuous loop, in part, may be imagined wherein deficiency of the DNA damage response pathways and carcinogenesis are profoundly linked. Since metabolism is in overdrive, tumor cells accumulate a great majority of mutations due to inappropriate proofreading of the DNA code. Such a situation presents a sui generis therapeutic window to reprogram a cell to commit suicide through targeting its ability to repair damage within itself. Poly (ADP-Ribose) polymerase (PARP) inhibitors deserve exclusive mention in this regard. They have revolutionized this field of research through their ability to contribute to synthetic lethality of cancer cells that are deficient in BRCA1/2, p53, Ku70/80, or FA complex-mediated DNA repair (72, 112, 218, 258, 259).
In cells, PARP functions primarily to identify single-strand DNA breaks (SSBs). (75, 192). Once it detects an SSB, it binds the DNA strand and initiates the synthesis of the poly (ADP ribose) chain in an NAD+-dependent manner, which further provides a site for enhanced protein–protein interactions with several enzymes critical to the DNA repair process. When this step is obstructed through a selective PARP inhibitor, the helix is prone to acquiring double-strand breaks eventually. Double-strand DNA breaks (DSBs) can be repaired only by two mechanisms, namely homologous recombination (HR), which is regarded as an error-free repair process, or its parallel nonhomologous end joining (NHEJ), which is error-prone. BRCA enzymes play an integral role in the repair of these DSBs. While both BRCA1/2 are vital contributors to relieving DSBs via HR, only BRCA1 seems to participate in mobilizing the DSB repair through NHEJ. The FA complex too has a vital, yet poorly understood, role in reinforcing the DSB repair executed by BRCA enzymes. In fact, the FANCD1 gene is now recognized as BRCA2 and is equally crucial in mitigating DNA damage. Resolving DSBs through HR also requires the activation of the MRN complex, which comprises key repair enzymes, including the ATM-Chk2-p53-p21 signaling axis. The Ku70/80 enzymes are mainly involved in augmenting the repair of DSBs through NHEJ (119, 178).
Nevertheless, loss-of-function mutations in aforementioned genes sensitize cancer cell to pharmacological inhibition of PARP (Fig. 8). Several PARP inhibitors have been approved by the FDA, and many are currently being evaluated in clinical settings. Several more are at key stages of development and show promising efficacy in vitro and in vivo (76, 99, 146). Targeting the DNA damage response pathway in this manner promises great opportunities to develop targeted therapies specifically in the case of TNBCs, which are flooded under mutations in the DNA repair machinery (57, 162, 181, 182). Several platforms have now been successfully established to test for BRCA gene mutations at the molecular level. Moreover, BRCA mutations are very frequently associated with additional mutations in the FA complex, ATM/Chk2/p53 pathway, suggesting one strategy to target all. The current pipeline of chemotherapeutics in this avenue therefore represents a novel strategy to selectively treat aggressive breast cancer by striking at its Achilles’ heel.

ECM matters: mammary tissue density serves a prognostic risk factor
Increased breast tissue density was always correlated with greater risk of developing aggressive cancer. However, the explanation for this pathological manifestation remained elusive until recently. Independent researchers have now quoted a host of factors that are strongly associated with increased risk of progressive breast cancer and its correlation with tissue density. Listed below are some of the landmark research articles that provide new insight into this emerging mechanosignaling field and consequently present attractive opportunities for therapeutic intervention.
Mouw and group have reported distinct micro-RNA-dependent regulation in promoting metastasis within luminal breast cancers through sustained activation of the PI3K-AKT as a direct consequence of mammary ECM stiffness (168). They argue that ECM stiffness causes the β1 integrin transmembrane receptors to cluster and thereafter interact with adaptor kinases through their cytoplasmic tails. Such interaction allows for the activation of these adhesion kinase proteins, which subsequently stimulate downstream signaling. One of the primary targets is beta-catenin, which can then transcriptionally upregulate the proto-oncogene c-Myc. Furthermore, activation of miR-18a miRNA through this network, which tightly regulates cellular PTEN and HOXA9, as well as BRCA1 expression, causes their repression and associated augmentation of PI3K–AKT signaling, thus leading to enhanced metastatic potential through a mechanosignaling node.
Another peculiar axis controlled by ECM stiffness brings us to negative regulation of the miR-200c microRNA through leptin-mediated transcriptional repression along an STAT-3-G9a methyltransferase node (39). The authors report that Leptin, a key adipokine that is strongly correlated with obesity, contributes to activating a metastatic and stemness program in mammary epithelial cells. They infer this heightened metastatic program and morphological pathology to be directly governed by activating phosphorylation at the Y-705 residue of STAT-3. The subsequent activation of STAT-3 is accompanied by its association with the G9a histone methyltransferase. This interaction favors the inhibition of the critical miR-200c, which is responsible for repressing metastasis-associated proteins, such as Vimentin and Zeb1. Such repression is mediated through an abundance of H3K9Me2 at the miR-200c promoter region, thereby causing epigenetic silencing through promoter occupancy.
More recently, Seo et al. presented a novel molecular perspective to explaining the association between obesity and tumor advancement (225). Herein, they report obesity-induced ECM stiffness contributing to myofibroblast activation and subsequent interstitial fibrosis in mammary glands in a mouse model. Such fibrosis is strongly connected with augmented YAP/TAZ nuclear localization and transcription of downstream effectors (CTGF and ANKRD). Thus, enhanced YAP/TAZ signaling further promotes tumor invasion and metastasis. Interestingly, deregulation of YAP/TAZ essentially governed through cell morphology and mechanosensitive cues and independent of the canonical signaling components such as LATS/MST1 kinases is a budding concept that we are only beginning to understand. To that end, Cordenonsi's group described a functional role for canonical as well as noncanonical Hippo deregulation in metastatic progression and stemness induction in mammary neoplasia (47). Dupont and colleagues further dissected a critical functional role for oncogenic activation of YAP/TAZ components and contribution to breast cancer progression wherein the potential of YAP/TAZ to act as mechanoresponsive elements in response to Rho GTPase activity and actin cytoskeletal remodeling was elegantly highlighted (64).
Micro-RNA: tiny oligonucleotides make a big difference
Micro-RNAs (miRNAs) constitute small, single-strand, noncoding oligonucleotide sequences roughly measuring about 22 nucleotides, which exert a profound suppressive effect on mRNA during translation. In the early 1990s, miRNAs quite literally punched the clock into modern science as the latest nucleotide members to pave their way into regulating gene expression and protein remodeling. Discovered as a Lilliputian fragment central to controlling larval development in Caenorhabditis elegans, it was the lin-4 miRNA that was determined to wield its effect on the 3′ UTR of lin-14 and execute functional repression of lin-14 protein expression as an aftermath of this sequence of events (6, 10, 73, 133, 276). However, it was not until the let-7 cluster of miRNAs was discovered with homologs in mammalian and other species that the true conservation and extent of regulatory control across species of these Lilliputian nucleotides was realized (193, 212). Presently, miRNAs are convincingly emerging as highly influential mediators of epigenetic silencing events on genes critical to tumor biogenesis and progression (114). It is noteworthy that these noncoding regulatory elements exert their control over routine cellular processes, such as proliferation, apoptosis, angiogenesis, and metabolism, through their tight influence of transcription factors and signal transduction participants (138). Indeed, David Bartel paints a thorough picture on the many physiological factors and cellular events that fall in their jurisdiction in his exhaustive review, which is a must referral for a deeper understanding on this subject (14).
MicroRNAs like other cellular factors represent a double-edged sword in the context of cancer. In most cases, they comprise the pool of tumor suppressors, which function mainly to alleviate neoplastic progression. miR-30 family members represent one such key group of micro-RNAs capable of impairing breast cancer progression, through suppression of genes central to mediating EMT. In an assorted set of breast cancer cell lines, miR-30c expression negatively correlated with chemotherapeutic resistance of these mammary neoplasia as well as with metastatic potential (23, 24). The EMT potential of these cancer cells was greatly diminished primarily through the ability of miR-30c to specifically target cytoskeletal rearrangement-associated proteins, including Focal Adhesion Kinase (FAK), Vimentin, and Twinfilin. Studies from several other research groups further complement this novel regulatory facet of miR-30 family through implicating its function in a myriad of signal transduction pathways central to cancer progression. Some of the integral genes that are controlled through this family include Cels3 (Planar cell polarity), Inhba and Smad1 (BMP/TGF Receptor signaling), Mdm2 (regulator of p53 ubiquitination), Mtdh (EMT transition), and so on (132).
On the contrary, miR-27a has been positively indicated in enhancing the metastatic potential of breast cancer through actively transcribing angiogenic factors of the vascular endothelial growth factor (VEGF) family (159). This axis of miR-27a-mediated oncogenic activation of VEGF and its associated clan is propagated by miR-27a-mediated negative regulation of ZBTB10 (zinc finger and BTB domain-containing protein 10). Normally, ZBTB10 occupies the promoter region of the Sp family of genes, thereby repressing their transcription. Hence, disruption of this axis through deregulated activity of the miR-27a has been associated with active transcription of the Sp-downstream targets that include not only the VEGF family but also the antiapoptotic protein survivin. Furthermore, such activation precipitates the enhanced induction of the G2-M cell cycle phase as a direct result of Myt-1 repression (159). Several other research groups have also implicated miR-27a in breast cancer through its ability to downregulate Foxo1 transcription factor and the RYBP/DEDAF apoptosis-associated protein, thereby leading to the enhanced oncogenic potential of these cells (93, 224).
Thus, one can infer that microRNAs partake in an equally critical manner in surmising the fate of transcription factors and proteins closely associated with maintaining the oncogenic–tumor-suppressive balance and are thus no longer considered redundant, but indeed crucial mediators of signal transduction.
Hedgehog and Lysyl oxidase: predictors of breast cancer metastasis to the bone
It has been clinically observed that of all cancers, breast cancer metastasis is most likely to be slated for camping at the bone tissue matrix. Metastasis of breast cancer to the bone is majorly coupled through two intrinsic events: deregulated hedgehog signaling and lysyl oxidase (LOX) activation (Fig. 9).
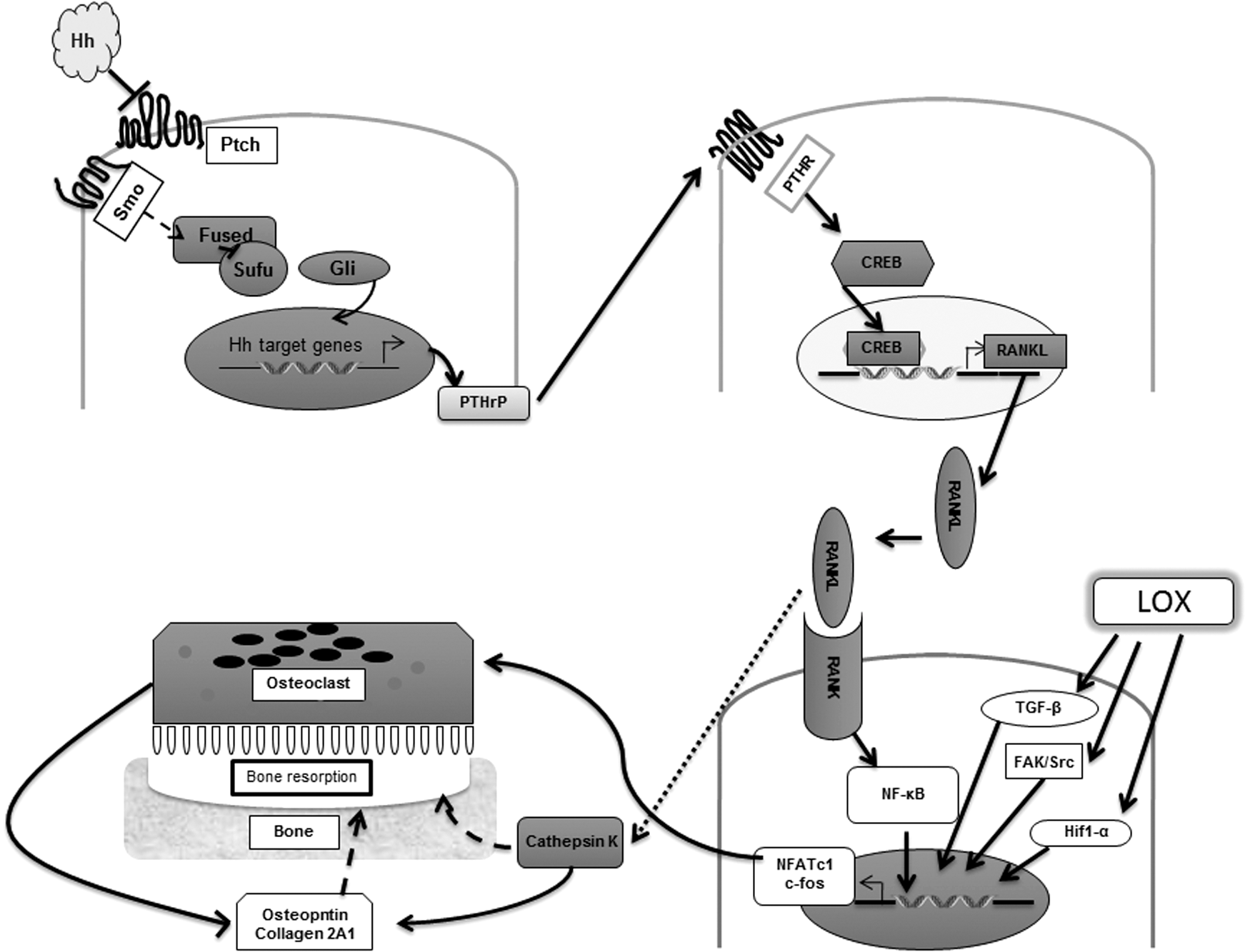
Identification of the hedgehog morphogen (hh) was heralded through a genetic mutation screening in drosophila wherein homozygous deletion of hh was observed to be synonymous with disordered arrangement of the metameric segments in Drosophila melanogaster (180). This led to its discovery as a critical regulator of segment polarity whose loss in the fly led to a lawn-like phenotype characterized by spiky denticles resembling the spines of a hedgehog, hence the distinct name. The hedgehog signaling pathway is essential for several developmental processes during embryogenesis and consequently has been identified as a critical factor in maintenance of stemness. Aberrant activation of hedgehog signaling has been associated with expansion of the mammary stem cell compartment. For instance, it was observed that escalated hedgehog signaling was accompanied by an accumulation in the levels of the polycomb gene family member, Bmi-1, a key factor in regulation of osteoblast and adipocyte differentiation. Breast cancers presenting with overexpression of Bmi-1 levels exhibit enhanced stem-like characteristics apparent through their ability to induce mammary neoplasia in NOD/SCID mice. Furthermore, the mammary stem cell compartment defined by CD44+CD24−/low lin− cells presented with increased expression of the hedgehog signaling members, patched (PTCH1), and the zinc finger proteins, Gli1/Gli2, as well as Bmi-1 (144).
Although aberrant activation of this pathway as a result of mutations of the hedgehog components has been identified in several early breast cancers, it is deregulation of the Indian hedgehog (Ihh) and its role in predisposing to bone metastases of breast cancer cells, which represent the more interesting phenotype. Numerous studies have now pointed toward the ability of Gli2, a transcriptional member of this signaling pathway to favor maturation of osteoclasts and thereby augment bone resorption presenting neoplastic cells with an ideal niche for metastatic colonization (53, 120, 240). Furthermore, paracrine signaling mediated through PTHrP corresponds with elevated levels of Osteopontin (OPN) and consequently cathepsin K and MMP9 that are responsible for increased osteoclastic differentiation and maturation, thus favoring generation of osteolytic lesions and presenting with bone metastasis (52). These studies thus highlight the essential role played by the hedgehog signaling arm in containing breast cancer metastasis and predicting progression-free survival in patients.
Cox et al. identify LOX as a critical factor associated with increasing the metastatic potential of ER-negative breast cancer subtypes exclusively to the bone matrix. Their observation indicates that this spectacle is mediated through the remarkable ability of LOX to generate lesions in bone tissue and provide a unique niche for the recolonization of CTCs detached from primary breast adenocarcinoma (268, 269). LOX was first identified as an amine oxidase, capable of converting the lysine side chain on elastin and collagen proteins in a redox-dependent manner to a peptidyl aldehyde–peptidylα-aminoadipic-δ-semialdehyde, christened allysine for simplicity. This allysine residue was discerned to be essential for the initiation of cross-linking of matrix filaments in bone tissue (201, 234). Despite its early discovery and a primitive knowledge of its functional role, it was not until the scientific infiltration of genomic tools that a positive clinical significance was ascertained for this enzyme in the early 1990s. Gacheru et al. were among the earliest researchers to uncover the structural and catalytic properties of this enzyme, as well as the transcriptional and post-translational mediators of LOX activation in the cellular context (81, 82). The role of LOX in promoting breast cancer metastasis to the bone has been well substantiated by numerous research groups globally (40, 48, 67, 70, 71, 249).
In a decade-old article, Payne and colleagues provided sound evidence through their experiments on Hs578T and MDA-MB-231 invasive breast cancer cell lines that LOX activity was essential for promoting both migration and invasion of these cells in a redox-dependent manner (195). It was discerned that this enhanced metastatic drive of cancer cells was promoted through the activation of the FAK/Src signaling node. Thus, LOX is mainly essential for efficient organization of the bone ECM through generation of hydrogen peroxide (H2O2) upon catalytic activation, which consequently exerts astringent regulatory control of focal adhesion proteins that include the FAK/Src family of proteins.
In an independent study published in 2006, it was elucidated that the hypoxic environment associated with localized tumors provided the ideal milieu for LOX activation. Such activation was further accompanied by the development of pseudopodial attributes, causing these cells to relocate from their primary sites unto new niches and thereby contribute to metastatic progression (70). It is intriguing to speculate herein how both hypoxia and elevated H2O2 concentrations lead to the same regulatory potential on LOX and whether they are related; and indeed if so, which precedes the other, leading us to the chicken and egg problem. Recently, an argument was drawn in an article proposing to partly explain the role of H2O2 in contributing to hypoxia and one could probably extend this argument to the circumstances mentioned above. Qutub and Popel demonstrate through their unique experimental approach that gradient increments in the levels of H2O2 in cancer cells promote neoplastic progression up to a predetermined threshold by favoring hypoxia and thus increasing the angiogenic potential of these tumor cells (206). Thus, it may be derived that LOX catalysis-driven H2O2 production probably contributes to the hypoxic tumor microenvironment, thereby making it more favorable for tumor progression. Moreover, these cells display enhanced metastatic potential not only through their ability to invade the surrounding stroma through the development of motile elements and secretion of matrix metalloproteinases that supplement invasion but also through secretion of proangiogenic factors as a direct consequence of hypoxia-dependent Hif-1α activation.
Recently, Cox et al. provided mechanistic proof for the exact nature of LOX-mediated breast cancer metastasis to the bone matrix in ER−patients (48). They determined that bone tropism of metastatic breast cancers was linked to the ability of LOX to regulate NFATc-1 (nuclear factor of activated T cells, cytoplasmic 1) biology. NFATc-1 has been previously defined as a vital driver of osteoclast formation, the entity that is in tight balance with osteoblast formation enzymes, regulating bone formation (27, 245). A tilt in the balance toward osteoclastogenesis promotes lesions in the bone matrix, typical of elevated NFATc-1 expression. Hence, upregulation of NFATc-1 through enhanced LOX activity and hypoxia in these breast cancer cells promotes lesion formations in the bone, providing these tumor cells with the ideal niche for recolonization, thereby promoting distant metastases.
Last, LOX has also been shown to be an integral component of TGF-β signaling through its ability to control p38-MAPK signaling as a direct consequence of mechanosignaling-mediated activation of the pathway (249). This activity of LOX was also highly dependent on the redox microenvironment within the cancer cells. Moreover, attenuation of the TGF-β signaling node through LOX inactivation further contributes to impaired stem cell potential of these tumors as evident from reduced expression of stem-like markers such as Twist and upregulation of E-cadherin (67). Thus, on the basis of the above arguments, LOX presents an extremely attractive target for therapeutic intervention that is capable of impeding multiple processes critical to metastatic progression in breast cancers. Much research is therefore being carried out in this area to test the potential of LOX inhibitors in a clinical setting.
Potential Role of ROS in Patient Prognosis: A Breast Cancer Perspective
The traditional opinion of ROS causing extensive damage and anarchy within cells has remained a topic of contention among the scientific community. However, mounting evidence seems to suggest a gradient-dependent pathology, wherein there is a customary threshold to good ROS and bad ROS, which diverge on their signaling versus toxicity-inducing functions. The current understanding of ROS biology has come a long way with researchers across the globe beginning to appreciate the positive impact of ROS in modulating signal transduction pathways within the cell. All biological conduits are bound by a tight balance in the various ROS species, a key to maintaining cellular homeostasis. Any skewing of this delicate balance is accompanied by a wave of events that serve to be either favorable or hostile to the cell depending on the nature of the tilt.
Pioneering research in ROS-driven physiology budded with keen observations made by Dr. Otto Heinrich Warburg in his studies on the embryology of sea urchins, wherein he discovered overwhelming changes in respiration/oxygen consumption upon fertilization of the eggs. Warburg's stellar discoveries in the field of oxidative respiration led him to become the recipient of the Nobel Prize in 1931 (272). As early as 1926, Warburg proposed an oxidative and energy model for tumor maintenance in his seminal article titled “The Metabolism of Tumors in the Body.” Presently, we revisit his contributions through digging deeper into the fundamental nature of ROS to exert its profound effect on cellular physiology and metabolism and undertake the lofty mission of curing cancer through manipulating the respiratory potential of the cell, as envisioned by Warburg almost a century ago. In the background of such intricate signaling events being acutely regulated through the redox microenvironment, it is but natural to question the role of these ROS effectors in contributing to metastatic potential and cancer progression.
NOS: the angiogenesis guru
Studies now ascertain that nitric oxide (NO) not only promotes signaling by acting as a post-translational modulator of proteins but also triggers activation of transcription factors that induce angiogenesis and thereby favor the invasion and spread of the primary tumor (79, 251). NO is synthesized by nitric oxide synthase (NOS) through the conjunction of its precursor L-Arginine with univalently reduced molecular oxygen resulting in the formation of L-citrulline as a by-product (243).
Physiologically, there are three isoforms of NOS—the inducible NOS (iNOS) or inducible isoform, the endothelial NOS (eNOS) or endothelial isoform, and the neuronal NOS (nNOS) or neuronal isoform. NO is an extremely stable diatomic radical that is almost incapable of any innate oxidant capacity. The only significant biological activity attributed to NO seems to be its role in binding heme prosthetic groups and serving as a secondary messenger, thereby activating signaling cascades, particularly by stimulating soluble cyclic guanosine monophosphate biosynthesis. In the backdrop of cancer biology, though, NOS is of particular interest to research groups. Lately, a research group reported a significant association between eNOS immunohistochemical staining with augmented activation of metastatic program in murine mammary tumors (109). Alternatively, the role of eNOS in promoting breast cancer metastasis remains poorly understood, although studies indicate that eNOS generation might be important in contributing to angiogenesis and thus increasing the invasive capacity of cancer cells (281). Another study indicated a functional role for nNOS in conjunction with its isoforms in promoting tumor metastasis in a glioblastoma model, wherein they interpret its requirement in the bolstering of angiogenesis and mutilation of the associated vasculature in cancer (115). In contrast, tumor progression in non-neuronal tissues is mainly dictated through NO gradients under the control of the iNOS isoform. Indeed, immunohistochemical analysis of iNOS staining exhibited substantial correlation with the primeval characteristics typical of early metastases as well as overlapped with poor overall patient survival (148). Although reports suggesting a dichotomic role for iNOS in tumor maintenance budded, it was soon apprehended that the apoptosis-promoting function of NO was restricted by its physiological concentration and the time of exposure, as well as tumor or host tissue-mediated secretion, wherein host-derived tumor-associated macrophages, in response to cytokine and T-cell activation, cause NO outburst and are capable of triggering tumor cell death as a function of the intrinsic mitochondrial pathway (279). It prevails that a mild increase in the physiological levels of NO contributes greatly to tumor progression, metastasis, and maintenance both directly through upregulation of antiapoptotic factors and indirectly through the upregulation of proangiogenic factors (79). Treatment with antiangiogenic agents may thus favor inhibition of metastatic and invasive potential of these neoplasia through targeting angiogenesis-promoting factors (30, 32, 62, 252).
Interestingly, there was also a strong association of iNOS expression, loss of WT-p53 tumor suppressor protein, and metastatic potential in a range of tumor cells (7, 25). p53 is a highly durable tumor suppressor that is able to suppress the expression of proangiogenic factors such as VEGF. In a pathology constituted by either loss of p53 or mutant p53 expression, iNOS is able to actively stimulate VEGF generation through its interaction with cyclooxygenase-2 (COX-2), thereby enhancing the metastatic potential of these cells. Moreover, this increased NO-mediated metastatic phenotype was more prominent in cancers that also overexpressed tissue inhibitor matrix metalloproteinase-1 (TIMP-1) in addition to elevated iNOS staining in immunohistochemical samples (214). Herein the authors proposed that TIMP-1 protein nitration as a consequence of increased iNOS expression and NO generation caused binding of the CD63/integrin β1 ligand to the surface of breast epithelial cells, thereby inducing direct activation of the AKT prosurvival pathway. Moreover, inhibiting this post-translational modification on TIMP-1 was sufficient to reestablish chemosensitivity and suppress neoplastic progression through abolishing the FAK/AKT/BAD signaling axis. Nakamura et al. discerned a biochemical signature in patients with invasive breast cancer, wherein they were able to significantly link VEGF-c production and nitrotyrosine expression with augmented lymph node metastases as well as poor survival outcomes, as a direct function of iNOS expression levels in the tumor vasculature (170). Moreover, iNOS-dependent signaling also seems to follow a strict restriction that is dependent on the expression and activity of the different isoforms of the hypoxia-inducible factors in cancer cells, wherein the presence of Hif-1α-associated iNOS activation seems to induce a metastatic phenotype in cancer cells, but expression of Hif-2α mediates an opposing feedback loop (28). Finally, a cohort-based study in patients with breast cancer further complemented the above research studies, providing a strong association between stimulation of proinflammatory cytokines, angiogenic factors, antiapoptotic proteins, and iNOS expression.
In recent years, we and others have demonstrated an association between the levels of nitric oxide in cells and its impact on cellular signaling via peroxynitrite (OONO−; reaction product of NO and O2 .−)-induced post-translational modifications of proteins critical to cell physiology (150, 186). These physiological processes are under strict control of spatial and temporal factors, as well as the concentrations of NO within the cell. As discussed earlier, an abundance of NO in tumor cells is associated with enhanced apoptosis and sensitization of invasive tumor cells to chemotherapy. In particular, Bonavida and Garman discuss the ramifications of such resensitization, which transits these cancer cells onto an alternate pathway through upregulation of apoptosis-associated receptors, such as Fas and DR5, as well as causes suppression of the NF-κB-Snail signaling axis (26). Moreover, NO-inducing prodrugs such as JS-K (O2-(2,4-dinitrophenyl) 1- [(4-ethoxycarbonyl)piperazin-1-yl]-diazen-1-ium-1,2-diolate) can exponentially induce the levels of NO in cells and thus inhibit invasion and metastasis in Matrigel, through mechanistic upregulation of TIMP-2, a metallopeptidase inhibitor protein, across a panel of aggressive breast cancers (230). Ergo, the logical assumption that redox modulation through averting the axis of post-translational modification of proteins favoring cancer progression could mitigate the effects of prosurvival signaling and resensitize aggressive tumors to chemoadjuvant therapy.
The mitochondrial powerhouse leaks irreparably in cancers
Otto Warburg remains the first scientist who conceived the hypothesis that deregulated mitochondria represent a hallmark of cancer cells (272). Presently, it is speculated that mitochondrial deregulation in cancer serves to support the metabolic as well as antioxidant requirements of the highly dynamic tumor cell (104, 271). The mitochondrion represents a distinguished organelle that is indispensable for maintaining redox and metabolic homeostasis, the functional capacity of which is much altered in cancer cells (83, 88, 271). Evidently, it plays an important role in cancer progression and metastasis. In light of breast cancer redox homeostasis and metabolism, it is the manganese-coupled superoxide dismutase (MnSOD), which is mainly implicated.
It is commonly agreed that MnSOD has a tumor-suppressive activity that is strictly regulated through H2O2 flux, associated with the ability of mitochondrial glutathione peroxidase (Gpx) to scavenge ROS or through direct activation of the maspin tumor suppressor protein, thereby alleviating the oxidative stress coupled to the ROS outburst as a consequence of activated signal transduction in nascent tumorigenic cells (63, 139, 154, 163, 183). Finally, epigenetic mechanisms for promoting the tumor-suppressive function were also noted, mostly through hypomethylation and consequent silencing of the promoter region of MnSOD in breast cancers (101, 105).
On the contrary, it is the oncogenic potential of MnSOD that finds greater significance in the establishment of tumors and the setting of metastasis. One propagated theory is that mutational polymorphisms, mutations leading to enhanced promoter occupancy of AP-2 within the MnSOD gene or MnSOD inhibition in response to oxidative DNA damage through DDB2, cause suppression of MnSOD activity in tumors, leading to accumulation of reactive radicals and subsequent carcinogenic progression (18). Several studies have indicated that a marked increase in the expression of MnSOD is observed in the more aggressive breast cancers (46, 69, 118, 174). The role of MnSOD in regulation of breast cancer metastasis is confusing owing to its paradoxical role as a tumor-suppressive antioxidant capable of alleviating oxidative stress in normal cells and as a potential oncogene whose suppression contributes to chemosensitivity.
A positive correlation is observed between the expression of MnSOD and receptor status in breast cancer cells. A refreshing take on this dichotomy was suggested wherein the tumor-suppressive or oncogenic function of MnSOD is attributed to differential expression of the enzyme in different subtypes of breast cancer (69, 118, 128). Particularly, there is a strong implication for the transcription factors, NF-κB and Sp-1, in regulating MnSOD expression in breast cancer subtypes and signaling tumor progression. It is further speculated that based on the subtype-specific expression of MnSOD in basal and luminal subtypes, one could actually program a chemotherapeutic strategy to eliminate these aggressive cancers by exploiting their redox sensitivity.
Kumar et al. portray a unique mechanistic synergism between ROS and chemotherapy, wherein highly aggressive basal breast carcinoma cells, expressing high levels of MnSOD antioxidant levels and refractory to chemotherapy, could be sensitized through MnSOD suppression, providing a more favorable survival outcome in patients (128). Trimmer and colleagues recently provided an alternative take on such redox regulation through overexpressing the tumor-suppressive MnSOD in an orthotopic model of TNBC coupled with the loss of Caveolin 1 in the stroma, causing these cells to undergo oxidative stress-dependent reversion of the protumorigenic phenotype (256). The concept of synthetic lethality was also employed by another research group wherein they employed knockdown of MnSOD in tamoxifen-resistant, highly aggressive, low ROS, and ER-positive breast cancer cells to resensitize these cells to estrogen therapy (211).
In light of proteins innately regulated through ROS, one intriguing effect is also coupled to the capacity of the mitochondrial prosurvival protein, Bcl-2, to regulate the levels of mitochondrial ROS within the cell. While Bcl-2 activation through its serine-70 phosphorylation has already been determined to be an important function of the oxidative milieu within a cell, various research groups, including us, have also pointed toward a more intricate web of feedback signaling loops whereby overexpression of Bcl-2 is associated with augmented mitochondrial O2 .− and redox-dependent survival signaling (43, 149, 150). However, much needs to be uncovered in terms of the mechanism that causes Bcl-2 to mask itself as an antioxidant-like macromolecule in this perspective and restrict the mitochondrial ROS. It is suggested that this restriction is imposed through its influence on complex activity within the electron transport chain and on glutathione activity, as well as the ROS thermostat that signals activation of Bcl-2 in response to oxidative stress. The consequence of this mechanistic control on mitochondrial regulation through the redox-sensing ability of Bcl-2 may hold potential implications in regulation of redox cell homeostasis in MnSOD-dependent manner. Hence, one could extend this redox network model to tailor chemotherapy in cancers besides melanomas where it is already associated with prognostic significance (19).
Warburg's findings dictate that cancer cells are sailing in a sea of ROS metabolites owing to their heightened metabolism and malfunctioning mitochondria. Therefore, they must be more sensitive to elevated ROS insults, as opposed to the normal cells in their vicinity. Thus, if one were to elevate ROS-mediated stress in these cells, they must be more susceptible to programmed cell death than the normal cells that would obviously possess higher tolerance to oxidative stress owing, in part, to their resilient antioxidant defense machinery and slower metabolic rates.
It is therefore stimulating to present some research that has exploited the hypothesis discussed above. In two particularly interesting studies, a novel role for the pentacationic MnTnHex-2-PyP5+ and MnTE-2-PyP5+ MnSOD mimetics as effective strategies to augment apoptotic signaling and induce cell death in lung and skin cancer models, when challenged with oxidative insults initiated through menadione and DMBA (7,12-dimethylbenz[a]-anthracene)/TPA (12-O-tetradecanoylphorbol-13-acetate), respectively, has been demonstrated (3, 288). This potentiates the sensitivity of developing ROS-derived therapeutic approaches to treating cancer, placing special emphasis on the redox microenvironment at the time of treatment. Hence, it presents with an extremely palpable opportunity to target a signaling node that is absolutely vital to cell survival and metabolic homeostasis, yet presents itself with a day and night discrepancy in its exclusive association with tumor cells (199).
ROS topples ligand-stimulated signaling: a domino effect
Over the past several decades, the phenomenon of increased intracellular ROS upon ligand-mediated stimulation of its cognate receptors has been observed in a wide variety of cell types and has inevitably subjected the physiological importance of ROS to intense scrutiny (12, 147, 187, 244). Appreciation for ROS as important cellular entities began to grow with emerging evidence implicating ROS as secondary messengers. One way by which ROS modulate signaling pathways is through the post-translational modifications of signaling components. Sundaresan et al. established strong links between ROS levels and signaling in vascular smooth muscle cells where temporal correlation between ROS levels and global tyrosine phosphorylation levels was observed upon platelet-derived growth factor (PDGF) stimulation (244). The link between ROS and PDGF signaling was further strengthened when it was observed that enzymatic and chemical scavenging of ROS poststimulation abrogated cellular response associated with PDGF stimulation. Similar observations were also made in A431 human epidermoid carcinoma cells in response to epidermal growth factor stimulation (12). Mechanistically, ROS exerts its effect on signaling through reversible oxidation of critical cysteine residues on kinases, phosphatases, and redox-sensitive transcription factors. For instance, disulfide bond formation between key cysteine residues in PKA and PKC as a result of oxidative stress was shown to increase their kinase activity and thus result in enhanced signal transduction (29, 86, 197). Keeping in mind the intricate link between PKA and PKC and the maintenance of stem-like properties in cancer cells, such regulation is particularly noteworthy.
On the other hand, key negative regulators of phosphorylation-dependent signaling, such as protein tyrosine phosphatases and dual-specificity protein phosphatases, have particularly been shown to be inactivated by growth factor exposure-mediated oxidative modification of cysteine residues within their catalytic cores (89, 129, 158). Given the redox sensitivity of these protein kinases and phosphatases, and their importance in regulating protein kinase cascades such as MAPK, PI3K-AKT, and TGF-β-SMAD pathways, it is plausible that the intracellular redox microenvironment could modulate the magnitude and duration of cell signaling activity to elicit specific cellular responses in cancer cells.
Using U-87 MG glioblastoma cells, Leslie et al. (134 –136) reported the increased levels of PIP3 and AKT signaling through the oxidative inactivation of PTEN upon exogenous addition of H2O2, and ROS-induced activation of AKT signaling is inhibited in PTEN-null cells (136). Interestingly, Luo et al. recently demonstrated a paradoxical role for superoxide to inactivate the PTEN phosphatase through S-nitrosylation. However, they also discern a remarkable role for superoxide anions to hyperphosphorylate AKT even in the absence of PTEN in this study (151). Beside the amenability of AKT toward redox regulation, Iskandar et al. have demonstrated the active role of AKT in redox modulation. A concomitant increase in O2 − and H2O2 levels was observed upon activation of AKT through a chemotherapeutic compound targeted specifically against mutant K-Ras. Herein, the increased ROS levels generated through activation of AKT were deemed responsible for the cytotoxicity of this compound in the tumor cell model (108). It is conceivable that cross talk between the PI3K/AKT pathway and cellular redox milieu creates a positive feedback loop to amplify PI3K/AKT signaling in the presence of antagonistic PTEN in breast cancer cells. Of similar note, the redox milieu may serve as a floodgate for AKT signaling.
Furthermore, oxidative stress plays a vital role in activation of the DNA damage response pathway that we have alluded to earlier in this review. Particularly, the BRCA1 and ATM proteins deserve special mention herein as they are vital tumor suppressors capable of relieving cells from oxidative stress through upregulation of the cellular antioxidant machinery (11, 15, 91). While BRCA1 regulates the physiological oxidative stress through direct activation of the antioxidant, Nrf2 (90), the exact nature of ATM-mediated relief from ROS-induced damage is still elusive.
Apart from kinases and phosphatases, ROS has also been observed to regulate protumorigenic transcription factors. One well-characterized, redox-sensitive transcription factor is NF-κB. An extensive review on the redox modulation of NF-κB is presented by Morgan and Liu and is a definite read to understand the intricacy of this signaling axis (167). Briefly, the DNA binding activity and transcriptional activation of NF-κB are critically regulated through a cysteine-62 residue within its p50 subunit (155, 255). Numerous studies have implicated this cysteine residue to be amenable to oxidative modifications (156, 160, 176). In addition, downstream of PKAc activation, ROS has been shown to indirectly phosphorylate Ser-276 of RelA subunit, which is required for expression of a subset of NF-κB response genes (110). Moreover, ROS-mediated upregulation of EMT is also shown to be associated with MMP-3 and NF-κB-mediated upregulation of Snail transcription (45).
Based on the prevailing evidence, we believe that the specific intracellular redox milieu could activate specific subset of signaling pathways resulting in a spectrum of malignant phenotypes. To that end, a whole repertoire of transcription factors that include the bulk of oncogenes, tumor suppressors, and EMT mediators are amenable to oxidative modifications and highly sensitive to redox regulation (5, 33, 41, 45, 61, 78, 87, 91, 121, 126, 140, 160, 222, 231, 264, 285). Although we have tried to list some of the seminal articles discussing redox control of transcription factors in cancer cells, surely we may have missed some key contributions by researchers in this aspect (Table 2). However, the exact mechanism of how ROS specifies cell fate remains foggy and warrants further investigation.
Snapshot of redox-sensitive transcription factors and proteins that critically impact invasive mechanisms in breast cancer progression.
Genomic Portraits of Invasive Breast Cancers: Screens in Diagnostics
It is absolutely essential to understand the innate molecular identity of the cancer to determine treatment strategies that will prove beneficial to the patient. Hence, clinicians rely upon unique molecular expression patterns or biomarkers that comprise a diverse set of factors, prognostic, predictive, or pharmacodynamic in nature. While prognostic biomarkers comprise the factions that determine progression-free survival outcomes, predictive biomarkers consist of those that predict response to the therapeutic treatment. Pharmacodynamic biomarkers on the other hand are a distinctive category of these molecular markers that determine the optimal dosage and therapeutic indices of the speculated treatment (20). Currently, advanced molecular genomic platforms, such as Mammaprint®, Oncotype DX®, Rotterdam signature, genomic grade index, and invasiveness gene signature, to list a few, are often employed to aid in predicting patient prognosis and select suitable adjuvant chemotherapy to treat these metastatic cancers (94).
The Mammaprint is a 70-gene inkjet-synthesized oligonucleotide microarray platform, which is able to foresee prognosis independent of hormone receptor and axillary lymph node status. It also possesses a strong algorithm parameter to predict distant metastases in these models. In an interesting study on this platform, a bioinformatic-based functional annotation of the genes unique to this platform was carried out. Herein, it was observed that the predictive signature converged on the basis of biological function networks to a handful of genes, central to tumorigenic establishment and maintenance, and that the biological significance of these genes spanned all innate features contributing to neoplasia as evident from the six hallmarks of cancer. Moreover, enhanced metastatic potential and relapse-associated functions matched the timbre of this gene signature (254). One shortcoming of the Mammaprint platform is that it needs fresh frozen tissue unlike the Oncotype DX, which is able to analyze formaldehyde-fixed paraffin-embedded tissue (FFPE) samples. The Oncotype DX platform developed by Piek et al. generates the trichotomic Recurrence score based on a 21-gene RT-PCR assay. This platform predicts the risk of relapse over a 10-year period among patients who are ER positive, but lymph node negative. The Rotterdam signature, also developed by a group in The Netherlands, comprises a distinct 76-gene assay signature that provides a prognostic significance to antiestrogen therapy. Calculated by means of an AffymetrixU-133 GeneChip™ System, it is mainly designed to monitor the expression of genes associated with cell proliferation and metastasis, independent of hormone receptor status among patients where the cancer has not spread to the axillary lymph nodes. The genomic grade index is a 97-gene microarray assay developed by MapQuantDx, France, which reads ER+ FFPE samples and is able to redesignate moderately aggressive tumors into low and high molecular grade with significant differences in their prognostic outcomes. The invasiveness gene signature is also developed using the AffymetrixU-133 GeneChip platform that employs a 186-gene signature microarray to predict metastatic potential of tumors. The oligonucleotide platform comprises stemness-associated genes that aid in the prediction of distant metastases to lung, liver, and brain as well as estimate patient prognosis independent of hormone receptor and lymph node status (143). Finally, a gene expression microarray platform from Agilent was recently employed to assess factors leading to distant metastases in breast cancers characterized by ER+ and Her2- receptor status. The platform concluded that late metastases within this subcategory of patients could be associated only with the lymph node status histologically and independently with the expression of CH25H, a gene that codes for cholesterol-25-hydroxylase and is integral to the metabolism of lipids and cholesterol in the human body (165). A novel supplement to this array of platforms is the launch of the METABRIC study platform (Molecular Taxonomy of Breast Cancer International Consortium), which proposed to amalgamate the clinical landscape of breast cancer subtypes with next-generation sequencing methods to identify driver mutations in integrative clusters that exhibited vastly different clinical outcomes (50). A platform such as this is extremely valuable in assigning tumors a cluster group rather than current grade-based classifications, which would provide more insight into the pathways and driver mutations through copy number variation assessment.
The research studies mentioned herein thus present the enormous potential of these platforms in subscribing genes worth targeting to try and diminish the risk of relapses. Drawing out the molecular portraits of these cancers sustains a more confident approach to patient treatment. Knowledge of mutational events in the patient's tumor sample can provide useful insight into the most likely to succeed targeted therapeutics. For instance, the presence of BRCA1/2 mutations suggests more effective treatments with PARP inhibitors, owing to the enhanced sensitivity of these cancers due to their deficiency in the robustness of their repair proteins. Similarly, it is only logical to treat patients harboring anaplastic lymphoma kinase (ALK) amplifications or gain-of-function mutations with ALK inhibitors since they are slated to augment patient response and interpret a better prognosis. Such platforms inclusive of those mentioned above therefore serve greater sensitivity in predicting chemotherapeutic outcomes, thereby offering a more comprehensive risk assessment and therapeutic strategy in treatment of cancers presented with heterogeneous tumor populations.
Current Challenges in Treatment: Where Lay the Bottlenecks?
Since 1989, there is a drastic and steady drop in breast cancer-related deaths among women, attributed mainly to the revolutionary improvements in detection and treatment of the pathology. Early detection of breast cancer through routine breast examination screens and mammograms has proved highly effective in boosting patient survivorship as well as lowered treatment costs. Timely detection is recognized as a significant factor in predicting the outcome of breast cancer treatment and survival.
Breast cancer treatment regimens are subdivided into different stages dependent on their invasive nature, localization, and size. The primordial stage of breast cancer is characterized by carcinoma in situ, which may either be ductal or lobular. Routine mammograms and clinical breast exams, often supplemented with tamoxifen hormone therapy, embrace the current treatment regimen for early stage breast cancers. Additionally, surgery and radiation are recommended to immediately contain and eradicate the cancer cells. Although periodic screening and early diagnosis of breast cancer have been successful in developed countries where awareness campaigns have especially proved highly beneficial in promoting screening, thereby augmenting diagnosis and treatment opportunities, the same is not true for the third-world countries.
Survival rate is directly proportional to early detection before any spread of cancer to other locations such as lymph nodes or distant organs. A 5-year survival rate for localized breast cancer has been estimated to be as high as 99%. Upon metastasis and spread of the neoplasm, the survival rates can drastically drop to a mere 25%. Brain metastases correspond with particularly grim survival rates, accounting for mortalities within 6–9 months of diagnosis. Even in this regard, while macrotumors can be removed to a considerable extent through surgery or radiation, micrometastases within the brain pertain to greater complications and a dismal therapeutic window restricted to chemotherapeutic interventions, with most drugs unable to efficiently access the tumor on account of the impenetrable blood–brain barrier (274). Low and middle-income countries particularly reel under poor clinical outcomes consistent with long delays between diagnosis, medical consultations, and starting treatment (263). Aforesaid provider delay can last up to months resulting in neoplastic progression and failure of treatment, resulting from development of advanced-stage carcinoma. Such social disparity calls for a much needed urge to enhance systemic and local therapeutic strategies focused on treatment of advanced-stage breast cancers. Furthermore, such therapeutic approaches must be amenable in terms of access and affordability among the lower socioeconomic states to address the burgeoning issue of class-dictated disparities in healthcare and management of the etiology.
It is also routinely observed that most breast cancer treatment costs are refuted to improper diagnostics due to lack of accurate identification of the genetic background of the etiology. The advent of routine screening biomarker platforms making it to the clinics is a colossal step up in mitigating the effects of improper treatments culminating from poor knowledge of the molecular and genomic background of these cancers (213). Therapeutic approaches are further confounded by the immense intratumoral and intertumoral heterogeneity in breast cancers, as well as the temporal evolution in the genomic landscape associated with the etiology (13). Such complications can hopefully be resolved through introducing high-throughput screening platforms to assess mainly clinically hostile tumors at multiple times through the course of treatment. In fact, Neagu et al. describe exhaustively the different biomarker platforms that are currently patented, some of which have made it into the clinic for routine screenings (173). Harnessing the full potential of such genomic tools presents an attractive therapeutic window in early detection and alleviation of cancers through tailored chemotherapy that places emphasis on their molecular pathology.
Currently, there is also a massive effort launched to use screening platforms to potentially select therapeutic compounds capable of exclusively targeting the CSC populations. Moreover, sustenance of stemness and redox imbalance seem to have an extremely existential relationship, wherein one phenotype seems to feed the other and thus one can further this speculation that if we were to clamp down the regulatory potential of one axis, it would have a profound impact on the other (125). The rationale behind such screening stems from the theory that metastatic CSCs are more chemoresistant and show increased potential of relapse among patients. They also seem to possess a heightened resistance to apoptosis when targeted with drugs that mediate cell death in nonstem cancer cells, suggesting that they have a differential sensitivity to chemotherapeutics. This propagates the idea that high-throughput screening for compounds targeting cancers might fail to prevent CSC-associated relapse in patients as these stem-associated cancer cells may be missed in screens owing to their small populations in cancers. Hence, chemotherapeutic targeting of the CSC population could particularly synchronize with locoregional therapy and mutually provide a better overall survival statistic through prevention of tumor relapse and progression-free survival in patients. In fact, one such screen led to the identification of the drugs, Salinomycin, Etoposide, and Abamectin, as strong repressors of EMT-transited mammary cancer cells in vivo (92). A remarkable review on the potential of redox chemotherapeutic targeting of stem cells was presented by Ogasawara and Zhang (185). The authors of this review were light-years ahead of their time to speculate and present stunning details of possible mechanisms of redox-mediated targeting of the CSC population. Many of their suggestions warrant experimental validation and may provide clues to targeting these cells and exhausting the possibility of sustenance of the cancer population permanently.
Finally, harnessing tools to detect and sort CTCs presents with a potential axis to determine the molecular subtypes and biomarkers displayed on these CTCs in a noninvasive manner, especially in cancers emanating from breast where clinical heterogeneity is manifest (49, 54). Moreover, since most CTCs are cells dislodged from their primary site harboring metastatic and stem-like potentials, analyses of such CTCs present with opening up the black box in cancers. Such clinical knowledge could predispose to better therapeutic design strategies that could subsequently aid in improving the quality of life for the patient. They may thus contribute to better survival outcomes especially for patients with cancers that comprise satellite tumor populations, each conforming to a distinct chemotherapeutic outcome cluster. Thankfully, next-generation sequencing tools are here to stay and can potentially serve as the tools to provide detailed and quick assessment of the molecular characteristics of the cancer (17, 262). However, there is still much discrepancy in their sensitivities and false-positive outcomes coupled with high costs of assessments, urging for considerable effort to upgrade these platforms so as to bring them from the bench to the clinic.
Summary
To summarize, the review set out to provide a clearer understanding of the cross talk among different pathways intrinsic to aggressive breast cancer as well as the clinical heterogeneity within the different tumor subtypes. We established hitherto the prevalence of shared characteristics among the EMT-bound and CSC populations and established a deeper understanding of the biology of these cancers and the challenges they pose to treatment. The purpose of this review was to comb through the current literature available on breast cancer etiology to identify the bottlenecks hindering breast cancer management owing to their heterogeneity and complex biology. We hope that the in-depth collation of these research articles might provide some refreshment through realizing latent ideas and propositions that we have suggested herein or that may be revealed simply through refamiliarizing oneself with the complex biology and network of factors culminating in aggressive breast cancer.
Footnotes
Acknowledgments
The authors wish to acknowledge all authors whose contributions might have been inadvertently overlooked in this review. S.P. is supported by grants from the National Medical Research Council (NMRC) and the Ministry of Education (MOE), Singapore.