
Abstract
Aims:
The present study was designed to investigate a possible interaction between vascular smooth muscle cell (SMC)-derived proprotein convertase subtilisin/kexin type 9 (PCSK9) and mitochondrial DNA (mtDNA) damage.
Results:
Treatment of cultured SMCs with the proinflammatory stimulus lipopolysaccharide (LPS) stimulated PCSK9 release and induced mtDNA damage. PCSK9 inhibition by its siRNA reduced, and its enhancement increased, mtDNA damage. Induction of mitochondria-derived reactive oxygen species (mtROS) (by rotenone, thenoyltrifluoroacetone, or antimycin A) enhanced mtDNA damage as well as PCSK9 release, suggesting a role of mtROS in PCSK9-mtDNA damage interplay. Induction of mtDNA damage (with the autophagy inhibitor, 3-methyladenine, or DNase II inhibition) enhanced PCSK9 expression, and inhibition of mtDNA damage (with the autophagy inducer, rapamycin) reduced PCSK9 expression, indicating bidirectional interplay between PCSK9 and mtDNA damage. Other studies showed that p38 MAPK is involved in PCSK9-induced mtDNA damage, and mammalian target of rapamycin activation plays a role in mtDNA damage-induced PCSK9 release. Functional impact of PCSK9-mtDNA damage cross-talk was evident in the form of SMC apoptosis, which was enhanced in cells treated with recombinant human PCSK9, but inhibited in cells treated with PCSK9 siRNA. Last, LPS administration in wild-type mice resulted in simultaneous PCSK9 release and mtDNA damage, but mtDNA damage was minimal in PCSK9-null mice given LPS.
Innovation:
Vascular SMC-derived PCSK9 induces mtDNA damage, and damaged mtDNA fragments stimulate PCSK9 release mediated, at least in part, by mtROS.
Conclusions:
These observations suggest positive feedback interplay between SMC-derived PCSK9 and mtDNA damage in the proinflammatory milieu involving mtROS. This interaction results in cellular injury, characterized by apoptosis—a hallmark of atherosclerosis. Antioxid. Redox Signal. 25, 997–1008.
Introduction
M
Proprotein convertase subtilisin/kexin type 9 (PCSK9), an enzyme encoded by the PCSK9 gene in humans, is produced mainly in the liver, kidney, and small intestine (3, 12). This protein plays a regulatory role in cholesterol homeostasis by binding the epidermal growth factor-like repeat A (EGF-A) domain of the low-density lipoprotein receptor (LDLR), inducing LDLR degradation (26). Inhibition of PCSK9 drastically reduces serum LDL levels (11), presumably by stimulating its uptake. Thus, PCSK9 inhibition has emerged as a potential novel drug therapy to treat hypercholesterolemia and associated disease states (19).
Proprotein convertase subtilisin/kexin type 9 (PCSK9) has been identified as a novel target for the development of lipid-lowering therapies for treatment of atherosclerosis and related diseases. However, the role of damaged mitochondrial DNA (mtDNA) in PCSK9 expression remains unknown. In this study, we describe a yet unrecognized phenomenon of cross-talk between PCSK9 and mtDNA damage mediated, at least in part, by mitochondria-derived reactive oxygen species.
Our recent studies showed that PCSK9 is expressed in vascular smooth muscle cells (SMCs) in significant amounts (6). PCSK9 release appears to be stimulated upon exposure of SMCs to stressful stimuli such as lipopolysaccharide (LPS) and oxidized-LDL (ox-LDL), and cellular ROS play a significant intermediary role in this process. We have also shown that damaged mtDNA that escapes autophagy induces intense inflammatory response (4). Since ROS can regulate mtDNA damage and PCSK9 expression, we posited that there might be a mechanistic link between mtDNA damage and PCSK9, which would participate in cellular injury during an inflammatory state.
Materials and Methods
Vascular SMC culture
Human primary aortic SMCs were purchased from ATCC. SMCs were maintained in vascular cell basal medium (ATCC) augmented with SMC growth factors. To elicit an inflammatory state, SMCs were incubated for 24 h with LPS (10–100 ng/ml) (Sigma). The concentration of LPS that induced the most consistent inflammatory reaction (20 ng/ml) was used in subsequent experiments.
PCSK9 inhibition and enhancement in vascular SMCs
To knockdown PCSK9, cells were transfected with 20 nM of siRNA for 24 h with siRNA transfection reagent (Santa Cruz). Briefly, cells were treated with the siRNA duplex solution for 24 h. The medium was then replaced with normal culture medium, and cells were treated with 10 ng/ml LPS for another 24 h. As control, cells were transfected with scrambled sequence control (SSC; Santa Cruz). The cell lysates were utilized for Western blot analysis to verify efficacy of protein knockdown by siRNA.
To mimic PCSK9 overexpression and release, SMCs were incubated with recombinant human PCSK9 (hPCSK9) protein (Life Technologies) in different concentrations (0.5, 2.5, and 5 μg/ml) for 24 h in the presence of LPS.
Treatment of vascular SMCs with inhibitors and inducers
Vascular SMCs were treated with 20 ng/ml LPS for 24 h in the presence or absence of mtROS inducers, rotenone, thenoyltrifluoroacetone (TTFA), or the complex-III inhibitor, antimycin A, p38 MAPK inhibitors (SB202190 or SB203580), autophagy inhibitor (3-methyladenine), or autophagy inducer (rapamycin), and PCSK9 inhibitor, Pep2-8. All chemicals were obtained from Sigma.
Mouse strains and inflammation model
Wild-type (WT, C57BL/6) and PCSK9 knockout (KO) (on C57BL/6 background) mice were purchased from Jackson Laboratory. All animals were housed in the institutional Animal Care Facility. All experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Usage Committee and conformed to the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Only male mice ∼10 weeks of age were used in this study.
To create an inflammatory state, similar weight WT and PCSK9-KO mice were administered LPS (1 mg/kg body weight, given intravenously) (Sigma) or saline as control. Mice were euthanized 24 h later with sodium pentobarbital (80 mg/kg, i.p.), and aortas were collected.
To investigate the role of p38 MAPK in mtDNA damage-PCSK9 interplay, WT mice were administered two different p38 MAPK inhibitors, SB202190 and SB203580 (5 mg/kg) (Sigma). To study the role of mammalian target of rapamycin (mTOR) in the regulation of mtDNA damage by PCSK9, mice were administered rapamycin (2 mg/kg body weight). Two hours later, mice were given LPS.
To study the effect of PCSK9 on mtDNA damage, WT mice were administered hPCSK9 protein (Life Technologies) (2 μg per mouse, intravenously) or PCSK9 inhibitor Pep2-8 (10 μg per mouse, intravenously) or saline (control). Two hours later, mice were given LPS.
At the indicated time points, blood was collected from the tail vein. Four hours after administration of various agents, mice were euthanized with pentobarbital sodium. Then, aorta and other tissues were collected.
ELISA for interleukin-1β and PCSK9
Interleukin (IL)-1β and PCSK9 were measured in mice sera samples by IL-1β ELISA kit (BD Biosciences) or PCSK9 ELISA kit (mouse/human; MBL International) as per manufacturer's instructions.
Western blot
Proteins from SMCs and aortas were purified in the RIPA Lysis Buffer System (Santa Cruz) and loaded onto 12% Mini-PROTEAN® TGX™ Precast Gels (Bio-Rad) for electrophoresis. Size-resolved proteins were transferred to Hybond ECL Nitrocellulose membranes (GE Healthcare, Ho Ho Kus, NJ). After blocking in 5% bovine serum albumin buffer for 1 h, membranes were incubated overnight with primary antibody (1:1000 dilution) at 4°C. After washing with phosphate-buffered saline containing 0.1% Tween-20, membranes were incubated for 1 h with biotin-tagged secondary antibody, which was detected with Pierce ECL Western Blotting Substrate (Thermo Scientific). Band intensities were quantified with ImageJ software and normalized to β-actin.
Antibodies directed at PCSK9, p38 MAPK, mTOR, p-mTOR, and anti-cleaved caspase-3 (17 kDa, ab2302) were purchased from Abcam. Antibody directed at LC3 was purchased from Cell Signaling.
Real-time quantitative polymerase chain reaction
Total RNA was reverse transcribed at 42°C with SuperScript II (Life Technologies). Gene expression was measured using SYBR Green PCR core reagents (Applied Biosystems). Relative mRNA levels for each sample were quantified based on Ct (the amplification cycle threshold) normalized to GAPDH as an endogenous mRNA standard and expressed relative to the control (without LPS), set to a value of 1. Primers used were as follows: PCSK9 forward primer, 5′-TTGCAGCAGCTGGGAACTT-3′; PCSK9 reverse primer, 5′-CCGACTGATGACCTCT GGA-3′; DNase II forward primer, 5′-TTCCTGCTCTACAATGACCAAC-3′; DNase II reverse primer, 5′-GGAAGTTAGGTACACTGTGGACC-3′; and GAPDH forward primer, 5′-GGGTCTTTGCAGCGTAT GG-3′; GAPDH reverse primer, 5′-ACCTCCTGTTTCTGGGGACT-3′.
mtDNA damage analysis
Real-time quantitative polymerase chain reaction (q-PCR) assay was used to assess mtDNA damage as described by Yu et al. (25) and by us (5, 7). Briefly, mtDNA and nuclear DNA (nDNA) were isolated using the genomic DNA extraction kit (Qiagen), and specific primers were used to amplify mtDNA and nDNA fragments. Using comparative Ct method, DNA damage was quantified by comparing the relative amplification of large fragments (∼10 kb) of DNA from treated samples with controls and normalizing this to the amplification of smaller (about 110 bp) fragments. Primers used were as follows: Long primers: producing a 10,235 bp product, FWD: ACATACCCATGGCCAACCT, REV: TATGTTTGCGGTTTCGATGA. Short primers: producing a 113 bp product, FWD: ACATACCCATGGCCAACCT, REV: GGGCCTTTGCGTAGTTGTAT. Cycling parameters for the short reaction were 95°C for 5 min, followed by 45 cycles at 94°C for 30 s, and 60°C for 30 s. Conditions for the long amplification were 94°C for 2 min, followed by 45 cycles at 92°C for 30 s, 71°C for 30 s, and 68°C for 5 min. DNA lesion frequencies were calculated using Poisson transformation.
mtROS measurements
MitoSOX™ Red (Invitrogen), a mitochondrial superoxide indicator, was used to assess mtROS levels by flow cytometry (FACS Vantage SE, BD) according to the supplied protocol. Data were recorded as the M2 percentage fluorescence variation (mean fluorescence intensity), which indicates the fraction of cells with enhanced production of mtROS. In some experiments, mtROS was induced by blocking three key steps in the mtROS respiratory chain (complexes I to III incorporation) by treating SMCs with 20 μM TTFA, 10 μM rotenone, and 40 μg/ml antimycin for 24 h.
Apoptosis analysis
Apoptosis was analyzed by FAM FLICA® Poly Caspase Assay specific for caspase-3 (ImmunoChemistry Technologies) and by TUNEL and Annexin V-FITC staining (Life Technologies) according to the manufacturer-supplied protocol. Apoptosis activity was assessed by flow cytometry, and data were recorded with the use of Flowing Software 2.0 as the M2 percentage fluorescence variation, which indicates the percentage of cells with apoptosis.
Statistical analysis
Data analysis was based on five independent experiments with results expressed as mean ± SD. Groups were compared pairwise by Fisher t tests or by ANOVA with Tukey's post hoc correction when >2 groups were compared. p-Values <0.05 were considered nominally significant, without adjustment for multiple measures, to avoid inflating type-II errors.
Results
LPS induces mtDNA damage, mtROS, and PCSK9 expression
LPS, the major structural element of gram-negative bacterial outer membrane, is known to trigger strong inflammatory responses and cellular injury. It has been used as a proinflammatory trigger by us (4 –6) and others (2). In the present study, treatment of cultured vascular SMCs with LPS resulted in several-fold increase in mtROS (Fig. 1A) as well as in mtDNA damage (Fig. 1B). Both mtROS and mtDNA damage increased in proportion to the amount of LPS used (10, 20, and 100 ng/ml), and the generation of mtROS and the degree of mtDNA damage were similar in magnitude. Simultaneously, there was an increase in PCSK9 expression, which followed the same pattern as mtROS generation and mtDNA damage (Fig. 1C).
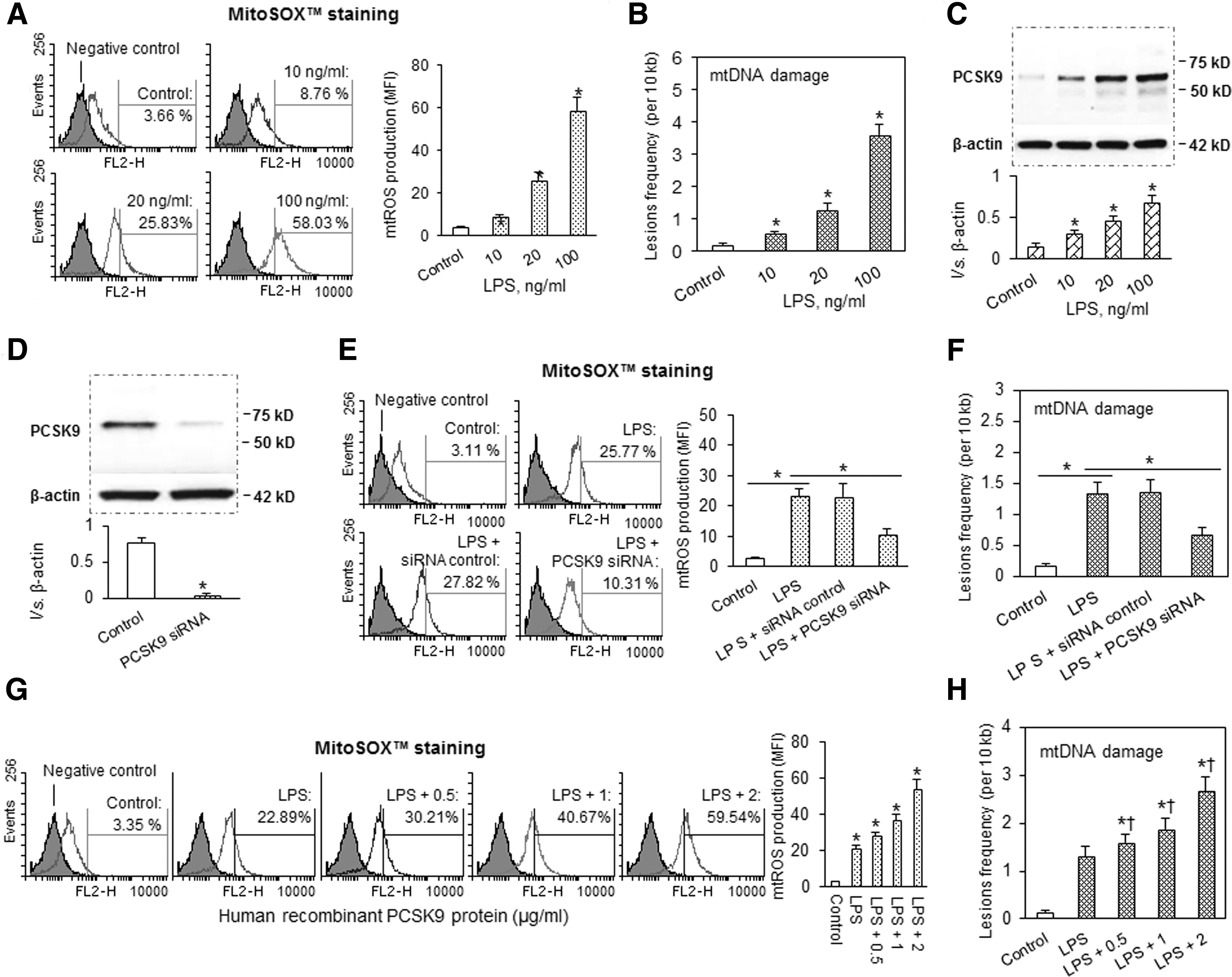
Since 20 ng/ml of LPS gave consistent and stable responses, this concentration was used in subsequent experiments.
PCSK9 regulates mtDNA damage
Next, we inquired whether vascular SMC-derived PCSK9 is involved in the regulation of mtDNA damage. To test this possibility, we inhibited PCSK9 by transfecting SMCs with its specific siRNA. By Western blotting, we confirmed the efficacy of siRNA (Fig. 1D). As shown in Figure 1E and F, PCSK9 inhibition markedly reduced mtROS generation and mtDNA damage. Note that scrambled RNA (SSC) did not affect mtROS generation or the extent of mtDNA damage.
In other experiments, we incubated SMCs with hPCSK9 to mimic its autocrine or paracrine stimulation. We observed that hPCSK9 evoked a near-linear dose-dependent increase in mtROS production (Fig. 1G) and mtDNA damage (Fig. 1H).
mtROS induce mtDNA damage and PCSK9
Because the main cellular source of ROS is mitochondria, we reasoned that mtROS may be the link between PCSK9 and mtDNA damage. mtROS generation was induced by blocking three key steps (complexes I to III incorporation) in the mtROS respiratory chain. As expected (16), mtROS increased markedly after treatment of SMCs with the complex-I inhibitor, rotenone, the complex-II inhibitor, TTFA, or the complex-III inhibitor, antimycin A (Fig. 2A). Concomitant with mtROS production, there was evidence for mtDNA damage and enhanced PCSK9 expression (Fig. 2B, C).
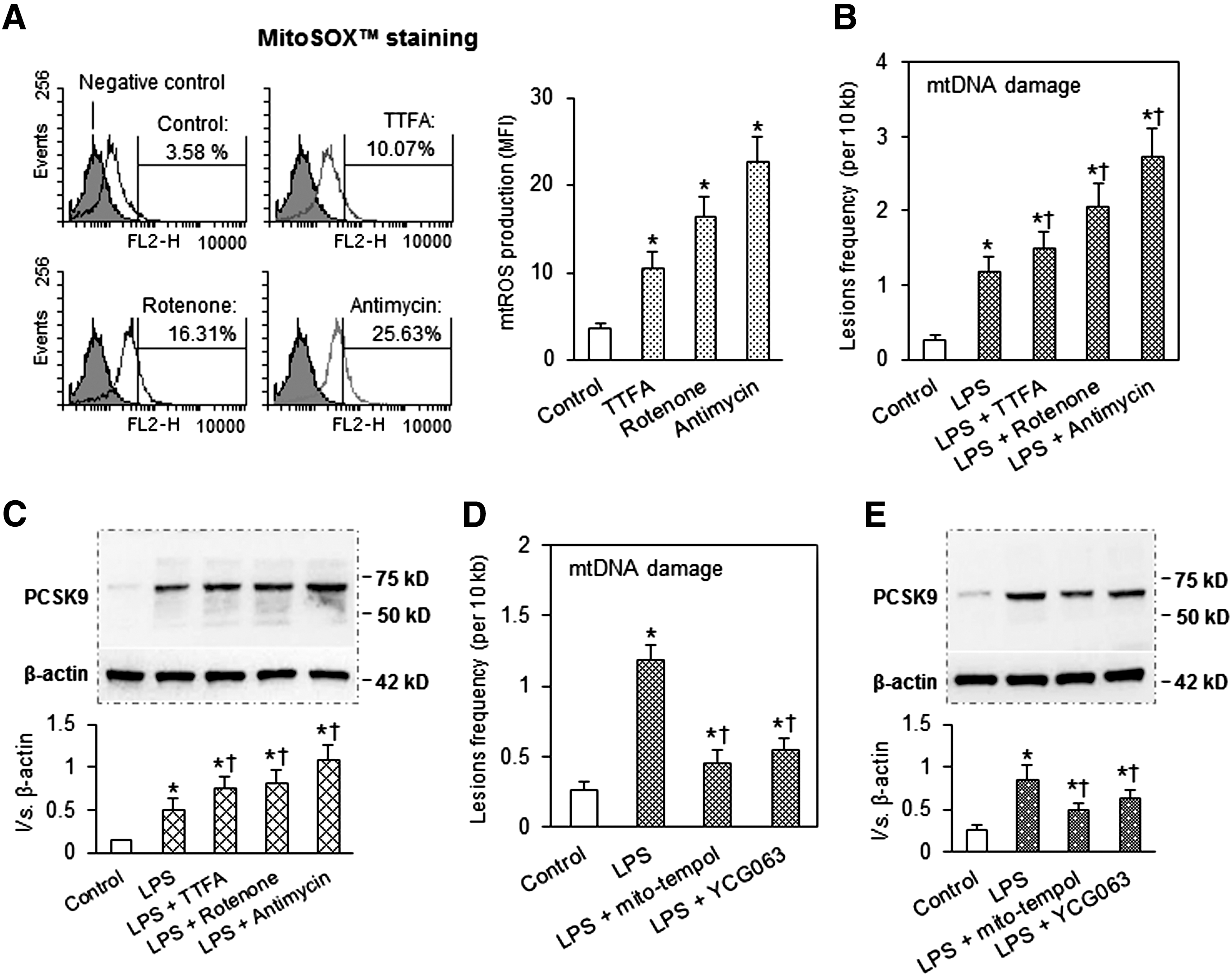
To further investigate the role of mtROS in PCSK9 expression, we used two different mitochondrial-specific antioxidants: mito-tempol and YCG063 (6). As shown in Figure 2D, pretreatment of cells with either of these two different mtROS inhibitors decreased LPS-induced mtDNA damage as well as PCSK9 expression (Fig. 2E), suggesting that mtROS regulates mtDNA damage and PCSK9 expression.
Role of autophagy and DNase II in PCSK9-mtDNA damage interplay
Autophagy is an evolutionarily conserved process for lysosomal recycling of cytoplasmic material, which degrades damaged mtDNA (10). 3-Methyladenine inhibits autophagy by blocking autophagosome formation via the inhibition of type III phosphatidylinositol 3-kinases (PI-3K), while rapamycin induces autophagy by inhibiting mTOR.
To examine the role of autophagy in PCSK9-mtDNA damage interplay, we used several approaches; autophagy inhibitor, 3-methyladenine, autophagy inducer, rapamycin, and DNase II. As shown in Figure 3A–D, 3-methyladenine inhibited autophagy signal LC3-II and induced mtROS generation and mtDNA damage and simultaneously increased PCSK9 expression. On the other hand, rapamycin enhanced autophagy signal and reduced mtROS generation, mtDNA damage, and PCSK9 expression.
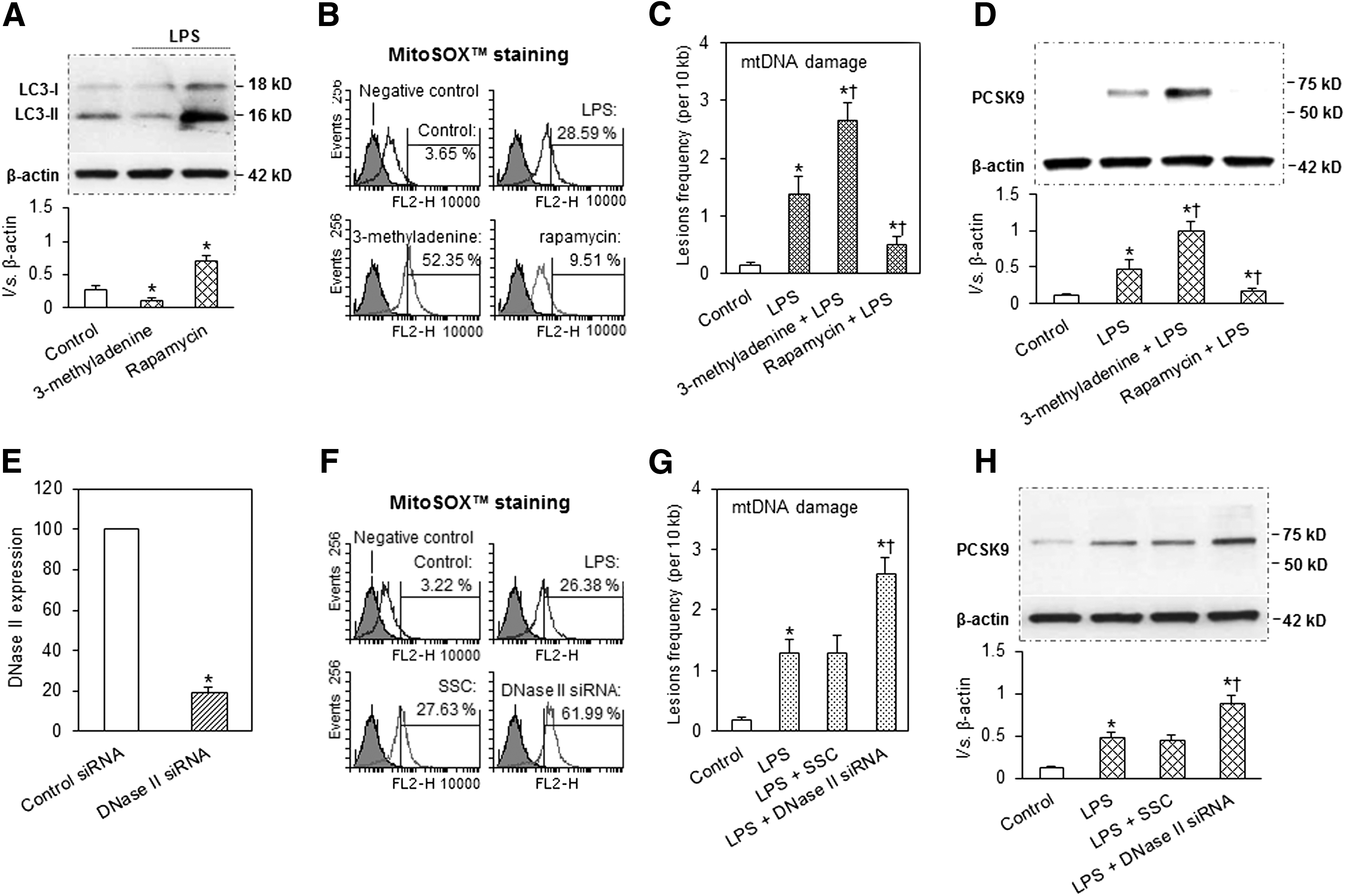
DNase II, a lysosomal enzyme, is expressed in various types of cells, such as macrophages, endothelial cells, and SMCs (16). Similar to the autophagy process, DNase II can digest damaged mtDNA (16). We used siRNA directed at DNase II to inhibit its expression (confirmed by q-PCR, Fig. 3E). As postulated, DNase II inhibition increased mtROS production and mtDNA damage and enhanced PCSK9 expression (Fig. 3F–H). Note that scrambled RNA (SSC) had no effect.
p38 MAPK and mTOR participate in mtDNA damage-PCSK9 interplay
Based on the above results, we concluded that there is bidirectional interaction between mtDNA damage and PCSK9. Next, we attempted to establish the possible mechanism of mtDNA damage-PCSK9 interplay.
First, we investigated the mechanism of damaged mtDNA inducing PCSK9 expression. p38 MAPK is a kinase critically important in the expression of proinflammatory signals (17). To explore whether p38 MAPK is involved in the interplay between damaged mtDNA and PCSK9 expression, we employed SB202190 and SB203580, two relatively specific inhibitors of p38 MAPK activation. We confirmed that treatment of cells with SB202190 and SB203580 inhibited activation of p38 MAPK (measured by Western blot analysis) (Fig. 4A). Treatment with SB202190 or SB203580 resulted in a significant reduction in LPS-induced expression of PCSK9 (Fig. 4B). To further confirm the role of p38 MAPK in mtDNA damage-mediated PCSK9 expression, we first treated SMCs with antimycin (mtDNA damage inducer), followed by SB202190 and SB203580 (p38 MAPK activation inhibitors). As shown in Figure 4C, antimycin markedly enhanced PCSK9 expression and both SB202190 and SB203580 blocked this effect of antimycin, indicating that p38 MAPK activation plays an important role in mtDNA damage-mediated PCSK9 expression.
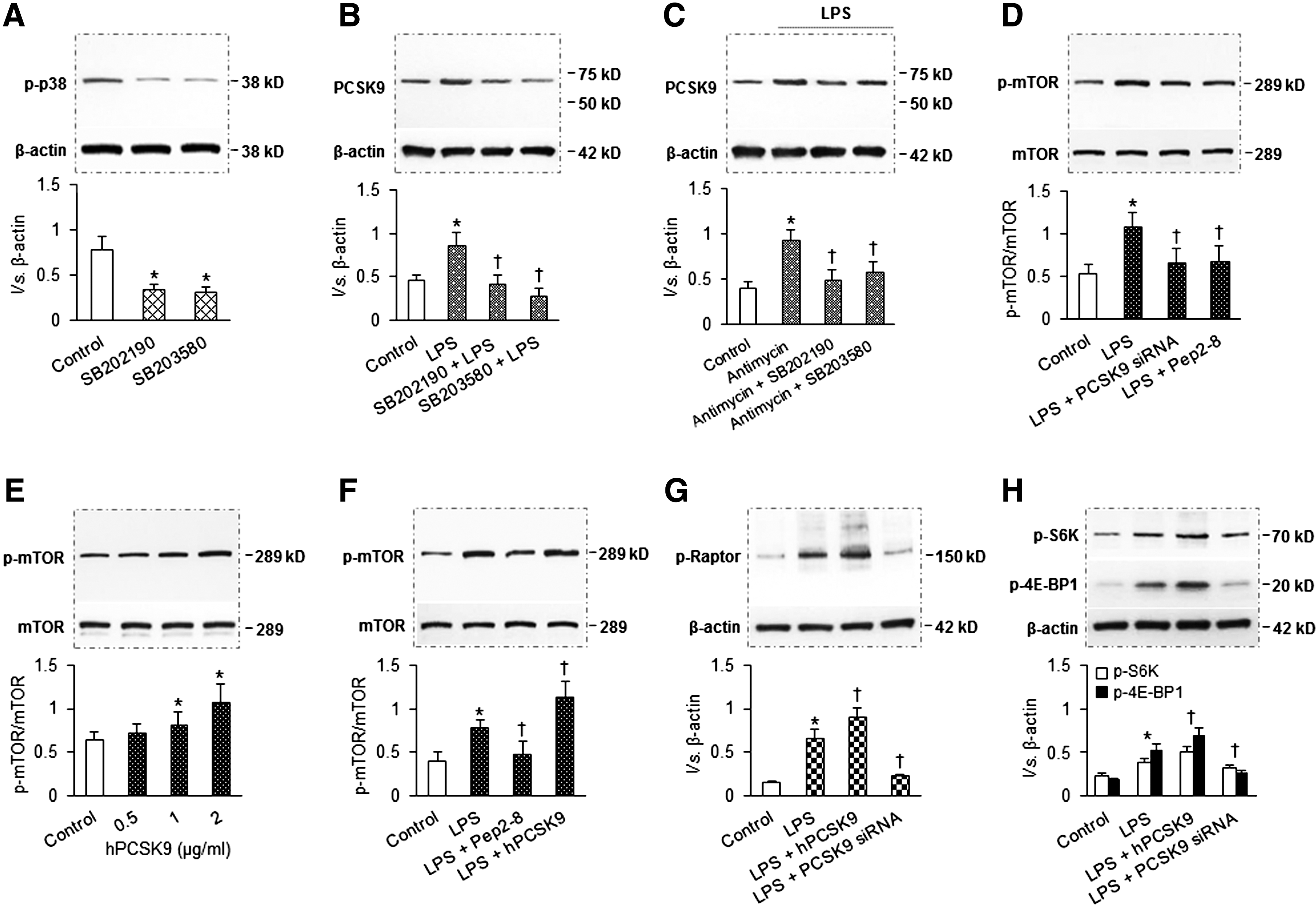
Second, we investigated the mechanism of PCSK9 stimulating mtDNA damage. As shown in Figure 3C, autophagy inducer rapamycin reduced mtDNA damage induced by LPS. Rapamycin is also a specific inhibitor for mTOR, a protein kinase that controls cell growth, proliferation, and survival. Rapamycin treatment also inhibits mTORC1, which contains mTOR, raptor (regulatory associated protein of mTOR), and three other components. The ribosomal S6 kinases and the 4E-BPs (eukaryotic translation initiation factor-binding protein) are two classes of mTORC1 substrates. Recently, Wang et al. (21) found that mTOR inhibition attenuates DNA damage through autophagy-mediated suppression of cAMP response element-binding protein 1 (CREB1). We therefore reasoned that mTOR might play a key role in PCSK9-induced mtDNA damage. To test this hypothesis, we employed both PCSK9 downregulation and upregulation strategies. As shown in Figure 4D, PCSK9 downregulation (with PCSK9 siRNA transfection or treatment of SMCs with PCSK9 inhibitor Pep2-8) significantly decreased phosphorylation of mTOR (Ser2448, p-mTOR). In contrast, PCSK9 upregulation (treatment of cells with hPCSK9) induced p-mTOR expression in a dose-dependent manner (Fig. 4E). We also confirmed that Pep2-8 inhibited, and hPCSK9 enhanced, p-mTOR expression (Fig. 4F). To further determine the role of PCSK9 in mTOR expression, we investigated the effect of PCSK9 upregulation (hPCSK9 treatment) and PCSK9 inhibition (by siRNA transfection) on the expression of p-Raptor, p-S6K, and p-4E-BP1. As shown in Figure 4G and H, hPCSK9 treatment enhanced, while PCSK9 inhibition reduced, the expression of p-Raptor, p-S6K, and p-4E-BP.
Functional significance of mtDNA-PCSK9 interplay
It has been reported that damaged mtDNA induces inflammation and cellular apoptosis (21). We postulated a role for PCSK9-mtDNA damage interplay in the development of apoptosis, measured as proapoptotic protein, Bax, antiapoptotic protein, Bcl-2, and cleaved caspase-3. Western blotting (Fig. 5A–C) showed that PCSK9 upregulation (hPCSK9 treatment) induced, and PCSK9 inhibition (by siRNA) reduced, the expression of Bax and cleaved caspase-3. On the other hand, PCSK9 upregulation reduced, and PCSK9 inhibition enhanced, the expression levels of Bcl-2. Results from flow cytometry (Fig. 5D–F) with Poly Caspase kit specific for caspase-3, TUNEL, and Annexin V-FITC staining confirmed the results of Bax, Bcl-2, and cleaved caspase-3 measurement. Collectively, these observations suggest that PCSK9-mtDNA damage interplay induces SMC apoptosis.

Evidence for mtDNA-PCSK9 interplay in vivo
Next, we studied mtDNA-PCSK9 interplay in vivo using WT and PCSK9 KO mice. Of note, our previous studies showed that compared with SMCs, PCSK9 is expressed in very small amounts in endothelial cells, fibroblasts, and macrophages (4, 6), which are main components of the aorta. Hence, we believe that PCSK9 measurement in the arterial tissue reflects primarily its expression in SMCs.
As expected, LPS administration elicited a marked increase in serum IL-1β (measurement by ELISA) levels in WT mice (Fig. 6A), indicating induction of a systemic proinflammatory state. LPS administration also markedly induced PCSK9 secretion in the plasma of WT mice (Fig. 6B). In keeping with in vitro studies, administration (i.p.) of p38 MAPK inhibitors, SB202190 and SB203580, reduced the expression of PCSK9 in the arterial tissues (Fig. 6C). In concordance with the in vitro observations, rapamycin administration inhibited LPS-induced PCSK9 expression (Fig. 6D). Last, Pep2-8 administration inhibited, while hPCSK9 administration induced, p-mTOR expression (Fig. 6E).
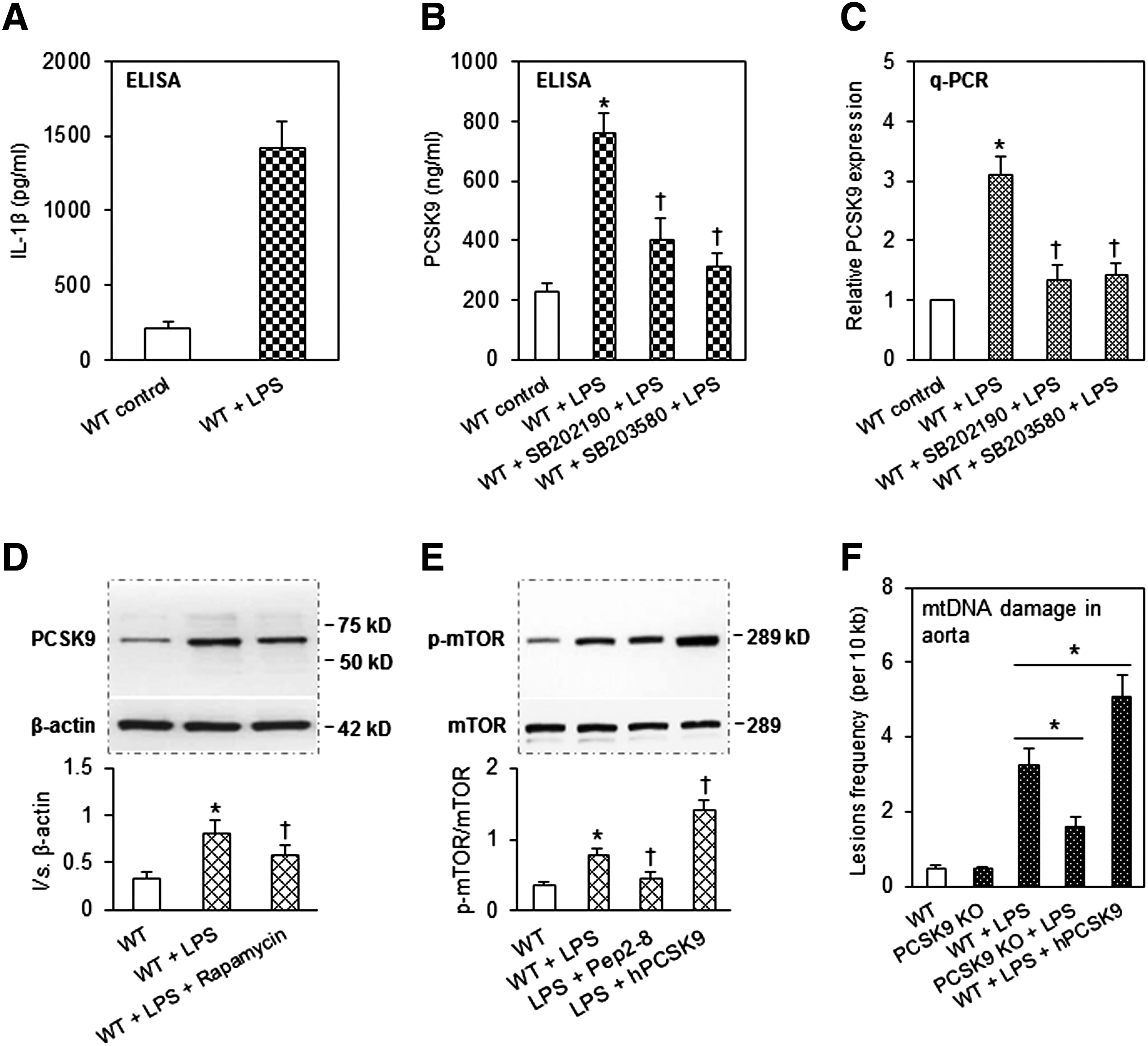
We investigated the impact of PCSK9 in the induction of mtDNA damage in aortic tissues. As shown in Figure 6F, LPS administration in the WT mice induced mtDNA damage (almost threefold increase in lesion density); this increase in lesion density was markedly attenuated in the PCSK9 KO mice (only one to twofold increase). Administration of hPCSK9, on the other hand, resulted in evidence of marked mtDNA damage in the WT mice (almost fivefold increase in lesion density).
We also investigated the appearance of apoptosis, measured as Bax, Bcl-2, and cleaved caspase-3 by Western blot, in the aortas of WT and PCSK9 KO mice given LPS. Apoptosis was quite pronounced in the WT mice, but much less in the WT mice treated with Pep2-8 or in PCSK9 KO mice (Fig. 7A–C). We also noted that systemic administration of hPCSK9 enhanced the expression of Bax and cleaved caspase-3 above and beyond that caused by LPS administration (Fig. 7A, D).

Discussion
By targeting LDLR, PCSK9 can lead to an increase in circulating LDL-cholesterol levels. Although PCSK9 is mainly expressed in the liver and the intestine, many investigators, including our group, have reported that PCSK9 is also expressed in vascular SMCs in significant amounts, especially when the cells are exposed to proinflammatory stimuli, LPS or ox-LDL (6, 9, 24). PCSK9 via increasing serum LDL levels is thought to be involved in atherogenesis, a process that is enhanced in the proinflammatory milieu. LPS is widely used to study the role of the inflammatory state in the pathogenesis of cardiovascular diseases, such as atherosclerosis, hypertension, and myocardial infarction (5, 6). Previously, we showed that LPS induces mtDNA damage in macrophages (5) and enhances PCSK9 secretion in SMCs (6). Based on the results of previous studies, we posited that there might be a link between PCSK9 expression/release and mtDNA damage, which results in vascular injury. The present study was designed to test this postulate.
Indeed, our in vitro studies on cultured vascular SMCs showed that LPS treatment simultaneously induced PCSK9 release and mtDNA damage. Inhibition of PCSK9 by treatment of SMCs with its siRNA inhibited mtDNA damage, and treatment of cells with hPCSK9 (to mimic its autocrine or paracrine release) enhanced mtDNA damage. We also observed that mtDNA damage itself induced PCSK9 expression, suggesting bidirectional interplay between PCSK9 and mtDNA damage. Next, we studied several intermediary signals that might regulate the interplay between PCSK9 and mtDNA damage.
Oxidant damage contributes to retrograde redox signaling from the organelle to the cytosol and the nucleus. A previous study showed an association of cellular ROS and PCSK9 expression (4, 6). Mitochondria are the major source of ROS production in most mammalian cells (8, 13), and generation of ROS by mitochondria is several-fold greater than that from other cellular sources, especially in inflammatory states (22). We studied the role of mtROS as a link between vascular SMC-derived PCSK9 release and mtDNA damage. We employed the same strategy as Oka et al. (16) to induce mtROS production, that is, the use of complex-I/II/III inhibitors. The complex-I inhibitor, rotenone, the complex-II inhibitor, TTFA, and the complex-III inhibitor, antimycin A, each significantly induced mtROS generation, mtDNA damage, and PCSK9 expression. Thus, it appears that mtROS generated in response to the inflammatory stimulus LPS might be a link in PCSK9-mtDNA interplay.
Inflammatory states are often associated with autophagy, a phenomenon which is thought to be protective during stress states, but can result in cellular injury if it proceeds unabated (14). Autophagy removes damaged mitochondria, but the mtDNA fragments that escape autophagy can induce cellular injury (2, 16). We studied the role of autophagy in PCSK9-mtDNA interplay with the use of autophagy inhibitors and inducers, as well as DNase II inhibition by siRNA. We observed that both the autophagy inhibitor 3-methyladenine and DNase II inhibition markedly enhanced mtDNA damage and PCSK9 expression, whereas the autophagy inducer, rapamycin, had the opposite effect on mtDNA damage and PCSK9 expression. These observations collectively suggest a potent, yet undescribed, role of autophagy in PCSK9-mtDNA interplay in vascular SMCs.
Involvement of the proinflammatory kinase p38 MAPK in the induction of mtDNA damage has been previously suggested (27). We questioned if the inhibition of p38 MAPK activation would reduce PCSK9 expression while limiting mtDNA damage. Indeed, we observed that inhibition of p38 MAPK by two different inhibitors reduced PCSK9 expression in vascular SMCs.
The stimulation of mtDNA damage by PCSK9 observed in the present study is particularly intriguing. To examine the potential mechanism of this phenomenon, we inhibited PCSK9 in SMCs (with PCSK9 siRNA and its chemical inhibitor Pep2-8) and observed that PCSK9 inhibition significantly reduced activation of p-mTOR, a protein kinase that controls cell growth, proliferation, and survival. In contrast, PCSK9 upregulation (treatment of cells with hPCSK9) induced p-mTOR expression. Further work showed that PCSK9 upregulation enhanced, while PCSK9 inhibition inhibited, the expression of mTOR-related proteins, p-Raptor, p-S6K, and p-4E-BP1. Of note, we (5) and others (21) have previously shown that mTOR inhibition by rapamycin (an agent that also induces autophagy) decreases mtDNA damage.
mtDNA damage has been shown to upregulate inflammatory signals, which result in cellular injury (1). We questioned if PCSK9-mtDNA interplay may have implications in cellular injury; we did so by measuring vascular SMC apoptosis. Indeed, an increase in PCSK9 activity (incubation of SMCs with hPCSK9) induced apoptosis, and a reduction in PCSK9 (by siRNA transfection of SMCs or incubation of cells with the chemical inhibitor, Pep2-8) decreased SMC apoptosis. We did not measure SMC apoptosis in response to mtDNA damage inducer, but it is intuitive that this would be the case. SMC apoptosis is a hallmark of atherosclerosis, especially in the rupture-prone regions (20). In this study, we show that PCSK9 release (especially in the proinflammatory milieu) is involved in SMC apoptosis. Apoptosis was determined by multiple approaches, that is, cleaved caspase-3 expression and Poly Caspase, TUNEL, and Annexin V-FITC staining. Of the several protein markers of apoptosis, we studied Bax, Bcl-2, and caspase-3. Results of all these measurements were concordant. The role of other pathways in DNA damage-PCSK9 interplay remains to be defined.
In vivo studies in WT and PCSK9-KO mice confirmed the results of the in vitro observations in vascular SMCs. While WT mice showed dramatic PCSK9 release and mtDNA damage following LPS administration, the PCSK9 KO mice showed only limited PCSK9 release and mtDNA damage. Further evidence for the regulatory effect of PCSK9 on mtDNA integrity came from experiments in WT mice given hPCSK9; these mice showed a marked increase in mtDNA damage beyond that caused by LPS alone. Furthermore, the administration of p38 MAPK inhibitors to the mice reduced PCSK9 secretion, indicating a role for this proinflammatory mediator in PCSK9 expression/release. Last, PCSK9-KO mice given LPS showed very little evidence of apoptosis in arterial tissues, whereas administration of hPCSK9 markedly enhanced apoptosis beyond that caused by LPS alone.
The results of our in vitro and in vivo studies collectively provide a strong evidence for bidirectional cross-talk between mtDNA damage and PCSK9 expression in vascular SMCs. mtROS generation as well as the state of autophagy during an inflammatory state seems to play a critical key role in the interaction between mtDNA damage and PCSK9 expression. These observations provide compelling evidence for the role of the mtROS generation-autophagy-mtDNA damage-PCSK9 release pathway, resulting in SMC apoptosis during inflammatory states. These steps are summarized in Figure 7E.
Footnotes
Acknowledgments
This study was supported by funds from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, Washington, DC. Additional support was provided by grants-in-aid from the National Natural Science Foundation of China (No. 11332003, 11572028).
Author Disclosure Statement
No competing financial interests exist.