
Abstract
Aims:
Internalization of extracellular fluid and its solute by macropinocytosis requires dynamic reorganization of actin cytoskeleton, membrane ruffling, and formation of large endocytic vacuolar compartments, called macropinosomes, inside the cell. Although instigators of macropinocytosis, such as growth factors and phorbol esters, stimulate NADPH oxidase (Nox) activation and signal transduction mediators upstream of Nox assembly, including Rac1 and protein kinase C (PKC), are involved in macropinocytosis, the role of Nox enzymes in macropinocytosis has never been investigated. This study was designed to examine the role of Nox2 and the potential downstream redox signaling involved in macropinocytosis.
Results:
Phorbol myristate acetate activation of human and murine macrophages stimulated membrane ruffling, macropinosome formation, and subsequent uptake of macromolecules by macropinocytosis. Mechanistically, we found that pharmacological blockade of PKC, transcriptional knockdown of Nox2, and scavenging of intracellular superoxide anion abolished phorbol ester-induced macropinocytosis. We observed that Nox2-derived reactive oxygen species via inhibition of phosphatase and tensin homolog and activation of the phosphoinositide-3-kinase (PI3K)/Akt pathway lead to activation of actin-binding protein cofilin, membrane ruffling, and macropinocytosis. Similarly, activation of macropinocytosis by macrophage colony-stimulating factor involves Nox2-mediated cofilin activation. Furthermore, peritoneal chimera experiments indicate that macropinocytotic uptake of lipids in hypercholesterolemic ApoE−/− mice was attenuated in Nox2y/− macrophages compared with wild-type controls.
Innovation and Conclusion:
In summary, these findings demonstrate a novel Nox2-mediated mechanism of solute uptake via macropinocytosis, with broad implications for both general cellular physiology and pathological processes. The redox mechanism described here may also identify new targets in atherosclerosis and other disease conditions involving macropinocytosis. Antioxid. Redox Signal. 26, 902–916.
Introduction
E
The findings of the current study demonstrate that Nox2 signaling via inactivation of phosphatase and tensin homolog and activation of the PI3K/Akt pathway leads to membrane ruffling and macropinocytosis. Activation of this novel signaling pathway stimulates macrophage lipid internalization, leading to foam cell formation in hypercholesterolemic conditions in vivo. The redox mechanism described here may identify new targets in vascular inflammatory disease and other pathological conditions involving macropinocytosis.
The NADPH oxidase (Nox) of “professional” phagocytic cells (a.k.a. phagocyte Nox, Nox2, or CYBB) is the prototype isoform of a family of Nox enzymes (4). Phagocytes, in a process called oxidative burst, increase their oxygen consumption to provide the molecular oxygen (O2) necessary for Nox2-derived superoxide anion (O2 ⋅−) generation and microbicidal activity (54). Nox2 consists of flavocytochrome b558, an integral membrane heterodimer comprising gp91 phox and p22 phox , and four cytoplasmic protein subunits: p47 phox , p67 phox , p40 phox , and the regulatory low-molecular-weight GTPase Rac (4). Nox2 is dormant in resting cells and becomes rapidly activated on stimulation by growth factors, cytokines, and phorbol esters (25, 34). For instance, the 4β stereoisomer of phorbol 12-myristate 13-acetate (4β-PMA) stimulates phosphorylation of p47 phox , followed by translocation of cytosolic subunits to the catalytic core of Nox2 and generation of O2 ⋅−, the precursor free radical of multiple reactive oxygen species (ROS). Nox2 is a target in numerous pathological conditions, as excessive Nox2-derived ROS has been shown to be involved in atherosclerosis, diabetes mellitus, cancer, Alzheimer's disease, and allergic airway inflammation (4).
Interestingly, signal transduction mediators upstream of Nox2 activation, such as Rac1 and protein kinase C (PKC), have been reported to play a role in membrane ruffling and initiation of macropinocytosis (6). In addition, activators of macropinocytosis share the ability to stimulate Nox2 activity and to regulate downstream redox signaling pathways (25, 34). Despite this information, no prior studies have investigated the role of Nox2 in macropinocytosis or elucidated the pathophysiological consequences of Nox-regulated macropinocytosis in a disease setting in vivo.
This study was designed to test the hypothesis that Nox2-derived ROS stimulate macropinocytosis, leading to internalization of macromolecules in phagocytes. The results of the present study provide seminal mechanistic insight into how increased Nox2 activity via inactivation of phosphatase and tensin homolog (PTEN) and activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway leads to cofilin dephosphorylation, membrane ruffling, and macropinocytosis. Moreover, the present study suggests the role of Nox2 in macropinocytotic uptake of lipids by macrophages in an atherosclerotic mouse model in vivo. These findings are the first to demonstrate redox control of macropinocytosis through Nox2 activation.
Results
Stereoisomer 4β-PMA stimulates macropinocytosis
The signaling pathway initiating orchestration of actin reorganization in the submembranous region, leading to membrane ruffling and macropinocytosis is poorly defined. As shown in Figure 1A, high-resolution differential interference contrast (DIC) microscopy indicated that treatment of RAW 264.7 macrophages with 4β-PMA (1 μM, 30 min) induced extensive membrane ruffling, followed by formation of U- and O-shaped ruffles, cup closure, and internalization of macropinosomes, consistent with the ability of phorbol esters to stimulate macropinocytosis (55). As internalization of extracellular fluid and its solute is characteristic of macropinocytosis, we investigated whether 4β-PMA stimulates uptake of native low-density lipoprotein (nLDL) in macrophages. Cells were incubated with nLDL (250 μg/ml), treated with vehicle or 4β-PMA (1 μM, 24 h), fixed; intracellular lipids were stained with the fluorescent lipophilic dye Nile Red; and lipid accumulation was analyzed by confocal laser scanning microscopy and fluorescence-activated cell sorting (FACS). As shown in Figure 1B and Table 1, 4β-PMA induced significant lipid accumulation in macrophages after treatment with nLDL. These results were confirmed by internalization of fluorescently labeled dextran (FITC-dextran, 70,000 MW) after 4β-PMA treatment (Supplementary Fig. S1; Supplementary Data are available online at
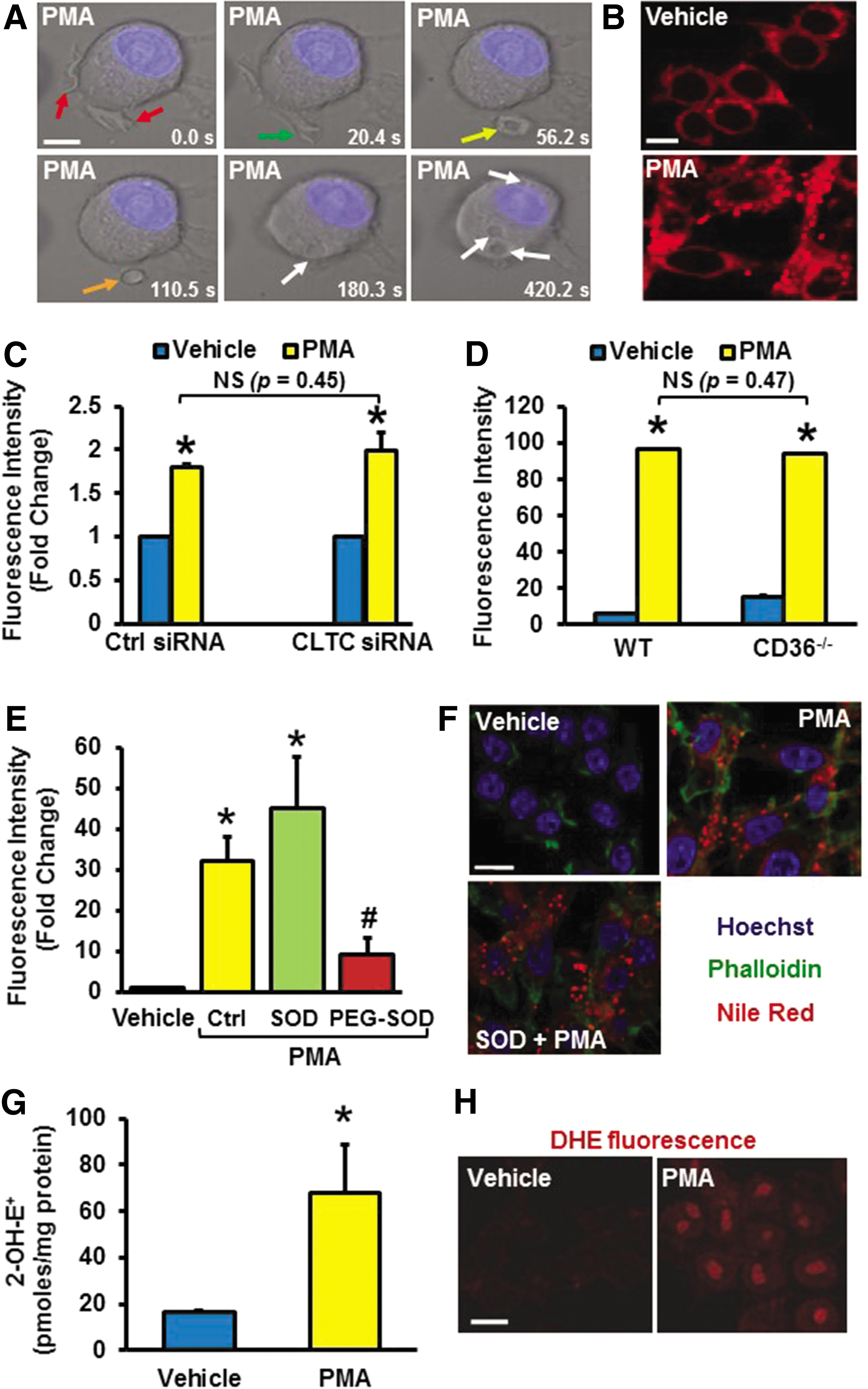
Next, we interrogated whether 4β-PMA stimulates internalization of nLDL via macropinocytosis or other endocytic processes. Macropinocytosis is often characterized by its sensitivity to PI3K inhibitors (32), actin perturbants (1), and Na+/H+ blockers (30). Preincubation of macrophages with the PI3K inhibitor LY294002 (1 μM, 30 min), actin perturbant latrunculin A (1 μM, 30 min), and Na+/H+ blocker amiloride (1 μM, 30 min) significantly attenuated 4β-PMA-induced lipid accumulation after nLDL treatment (Table 1). In contrast, inhibitors of clathrin- [chlorpromazine, (10 μM, 60 min)] (5) and caveolin-mediated endocytosis [nystatin, (10 μM, 60 min)] (61) had no effect on 4β-PMA-induced lipid internalization (Table 1). To complement the pharmacological approach, we silenced the clathrin heavy chain (CLTC) by using siRNA in human macrophages, which did not inhibit FITC-dextran internalization (Fig. 1C). Validation of effective mRNA silencing is shown in Supplementary Figure S2. Finally, Supplementary Figure S3 demonstrating accumulation of 1 μm red (580/605) FluoSpheres after treatment with 4β-PMA is also consistent with 4β-PMA inducing macropinocytosis, as the upper size limits for particles undergoing clathrin- and caveolin-mediated endocytosis are 200 nm and 500 nm in diameter, respectively (52). In summary, these data demonstrate that 4β-PMA stimulates membrane ruffling, macropinosome formation, and macropinocytotic uptake of macromolecules in macrophages.
4β-PMA promotes macropinocytosis via stimulating intracellular superoxide anion generation
4β-PMA mimics endogenously produced diacylglycerol (DAG) and stimulates PKC activation in both macrophages and nonphagocytic cells (29). Interestingly, previous studies demonstrated that phorbol ester-induced macropinocytosis is completely blocked by PKC inhibitors and macropinocytotic uptake of macromolecules is stimulated by activators of PKC (31). As direct stimulation of PKC by 4β-PMA phosphorylates the Nox organizer subunit p47 phox in macrophages, leading to enzymatic assembly of Nox2 and subsequent O2 ⋅− generation, we speculated that (i) O2 ⋅− acts as a signaling molecule in phorbol ester-induced macropinocytosis and (ii) 4β-PMA-induced macropinocytosis involves activation of Nox2.
4β-PMA stimulates extracellular O2 ⋅− generation that may lead to LDL oxidation in the media of cultured macrophages, subsequently stimulating its scavenger receptor (e.g., CD36)-mediated uptake. Consequently, we first tested the role of scavenger receptors in 4β-PMA-induced lipid accumulation. As shown in Figure 1D, 4β-PMA significantly stimulated lipid accumulation in CD36−/− macrophages after nLDL treatment. These data demonstrate that 4β-PMA induces lipid accumulation in macrophages lacking one of the major scavenger receptors, CD36. To confirm these results, we scavenged extracellular O2 ⋅− in the media by the cell-impermeant superoxide dismutase (SOD, 150 U/ml, 1 h), which did not inhibit phorbol ester-induced lipid accumulation in macrophages (Fig. 1E, F). Interestingly, scavenging of intracellular O2 ⋅− generation by the cell-permeant polyethylene glycol–conjugated SOD (PEG-SOD; 50 U/ml, 2 h) significantly attenuated 4β-PMA-induced lipid internalization, suggesting a role for intracellular O2 ⋅− in phorbol ester-induced macropinocytosis (Fig. 1E). To support these findings, HPLC quantification of 2-hydroxyethidium (2-OH-E+), the O2 ⋅− specific oxidation product of dihydroethidium (DHE), indicated that intracellular O2 ⋅− generation was significantly increased in macrophages after 4β-PMA treatment (1 μM, 30 min) (Fig. 1G). Confocal images demonstrating increased DHE fluorescence after 4β-PMA treatment are also shown in Figure 1H. Finally, Electron Paramagnetic Resonance (EPR) using the cell-permeant hydroxylamine spin probe CMH confirmed that 4β-PMA stimulates O2 ⋅− production in macrophages (Supplementary Fig. S4). Taken together, these findings suggest that 4β-PMA stimulates macropinocytosis via a yet undefined mechanism, which involves intracellular O2 ⋅− as a signaling molecule.
Nox2 plays a pivotal role in 4β-PMA-induced macropinocytosis
To investigate the role of the DAG-PKC pathway in phorbol ester-induced macropinocytosis, additional experiments were performed. First, the effect of 1,2-dioctanoyl-sn-glycerol (DOG, 100 μM), a cell-permeant analog of DAG and an activator of PKC (58), was tested on internalization of FITC-dextran. As shown in Figure 2A, DOG significantly stimulated uptake of FITC-dextran in macrophages. Second, the inactive stereoisomer 4α-PMA, which does not activate PKC (44), did not stimulate O2 ⋅− generation and macropinocytosis of FITC-dextran in macrophages (Fig. 2B, C). Third, calphostin c, an inhibitor of conventional and novel isoforms of PKC (33), completely blocked 4β-PMA-induced accumulation of FITC-dextran (Fig. 2D). These data demonstrate that activation of the DAG-PKC pathway is involved in 4β-PMA-induced macropinocytosis.

Because PKC, an upstream activator of Nox2 in macrophages (7), is involved in 4β-PMA-stimulated macropinocytosis, we investigated whether 4β-PMA stimulates macropinocytosis via Nox2 activation. Diphenyleneiodonium (DPI, 10 μM), a widely used inhibitor of flavin-containing oxidases, significantly ameliorated 4β-PMA-induced lipid uptake (Fig. 2E, F), suggesting a possible role of Nox in macropinocytosis. Though multiple isoforms of Nox are expressed in phagocytes, Nox2 is the most highly expressed isoform, and its activation by 4β-PMA leads to a robust generation of O2 ⋅− (Supplementary Fig. S5). Consequently, we silenced Nox2 in THP-1 macrophages using siRNA and tested whether 4β-PMA-induced macropinocytosis is attenuated. A significant decrease of FITC-dextran internalization was observed in macrophages that were transfected with Nox2 siRNA compared with scrambled siRNA-treated cells (Fig. 2G). qPCR data indicated that levels of Nox2 mRNA were decreased by 80% in Nox2 siRNA- versus scrambled siRNA-treated cells (Supplementary Fig. S6). In contrast, we observed no change in Nox1 and Nox5 mRNA levels in Nox2 gene-silenced cells (data not shown). Nox3 mRNA levels are undetectable in macrophages (35) and Nox4 is constitutively active (16). In separate experiments, peritoneal macrophages isolated from respective wild type (WT) and Nox2y/− mice were treated with vehicle or 4β-PMA, and macropinocytosis was investigated. As shown in Figure 2H, 4β-PMA-induced internalization of FITC-dextran was completely blocked in Nox2y/− macrophages compared with WT controls. These results are the first to demonstrate that 4β-PMA stimulates macropinocytosis via Nox2. A distinct role for Nox1 in matricellular protein- and CD47-mediated macropinocytosis adds to the complex role of the Nox in this process and is described in detail elsewhere in this issue.
Nox2 activation is essential for 4β-PMA-induced membrane ruffle formation
The next experiments were designed to investigate which step(s) during 4β-PMA-induced macropinocytosis require(s) Nox2 activation. Scanning Electron Microscopy (SEM) images demonstrated that 4β-PMA stimulates membrane ruffle formation on the dorsal surface of macrophages (Fig. 3A). Preincubation of cells with calphostin c, DPI, and EUK 134 inhibited 4β-PMA-induced membrane ruffling, demonstrating that PKC and O2 ⋅− derived from a DPI-inhibitable enzymatic source are involved in 4β-PMA-induced membrane ruffle formation (Fig. 3A). Next, live cell DIC microscopy was used to investigate the dynamics of 4β-PMA-induced plasma membrane activities (Supplemental Movie S1). 4β-PMA-stimulated plasma membrane extensions dynamically changed their morphology, leading to ruffle bending, cup formation, and closure. DIC imaging confirmed that DPI and EUK 134 block 4β-PMA-induced membrane ruffle formation (Fig. 3B). To investigate whether 4β-PMA stimulates membrane ruffling via Nox2, we silenced Nox2 in macrophages, challenged them with 4β-PMA, and imaged the cell surface by using SEM. As shown in Figure 3C, 4β-PMA stimulated membrane ruffle formation in scrambled but not in Nox2 siRNA-treated cells, demonstrating the role of Nox2 in 4β-PMA-induced membrane ruffling.
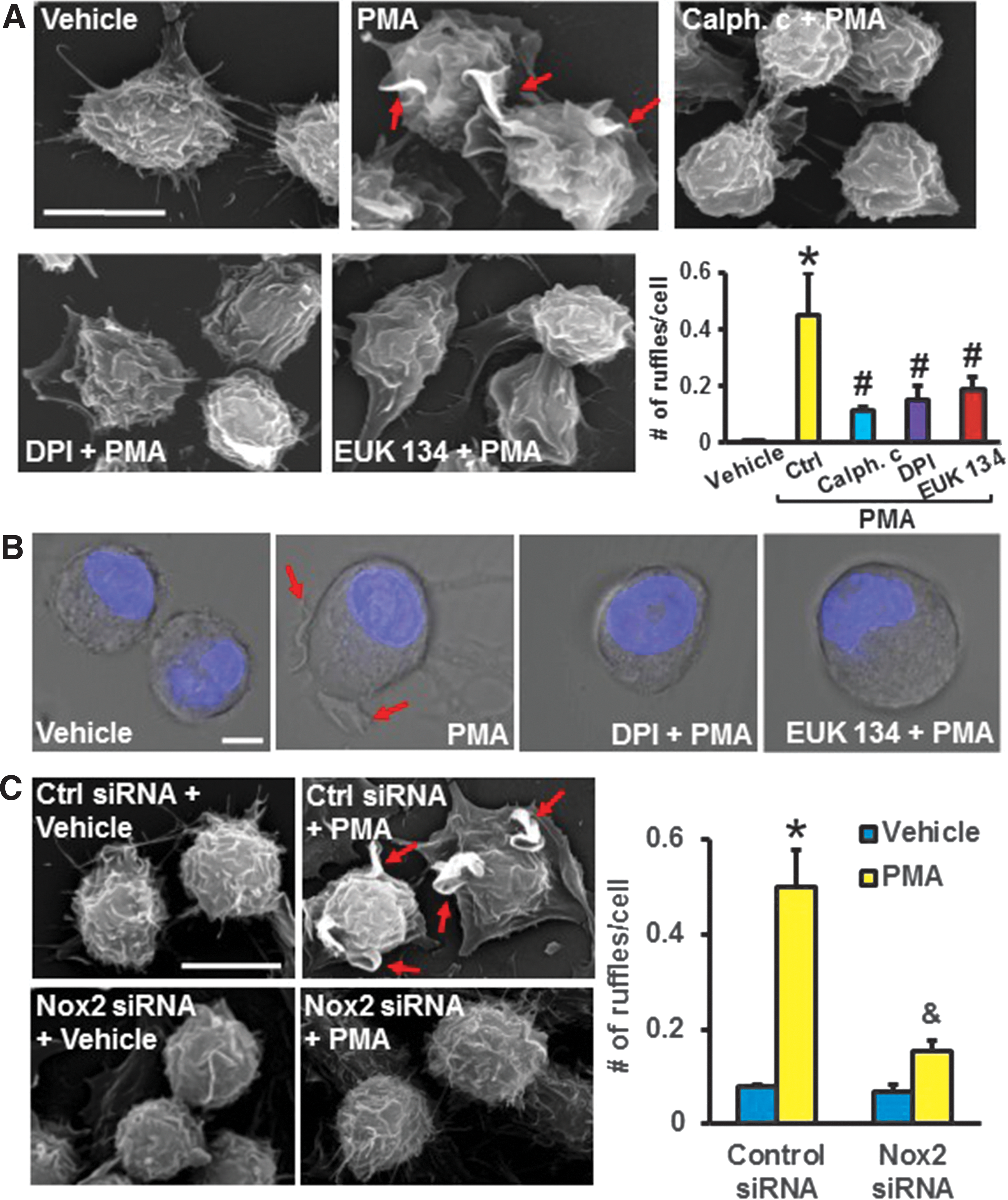
Nox2 induces membrane ruffle formation via dephosphorylation of cofilin
Previous studies demonstrated that binding of epidermal growth factor (EGF) to EGF receptor (EGFR) promotes circular dorsal ruffle formation in epithelial and mesenchymal cells (47). As Nox signaling mediates transactivation of EGFR (13), we tested whether the EGFR tyrosine kinase inhibitor erlotinib inhibits 4β-PMA-induced macropinocytosis. As shown in Supplementary Figure S7, erlotinib did not inhibit macropinocytosis, suggesting that EGFR transactivation is not involved in phorbol ester-induced membrane ruffle formation.
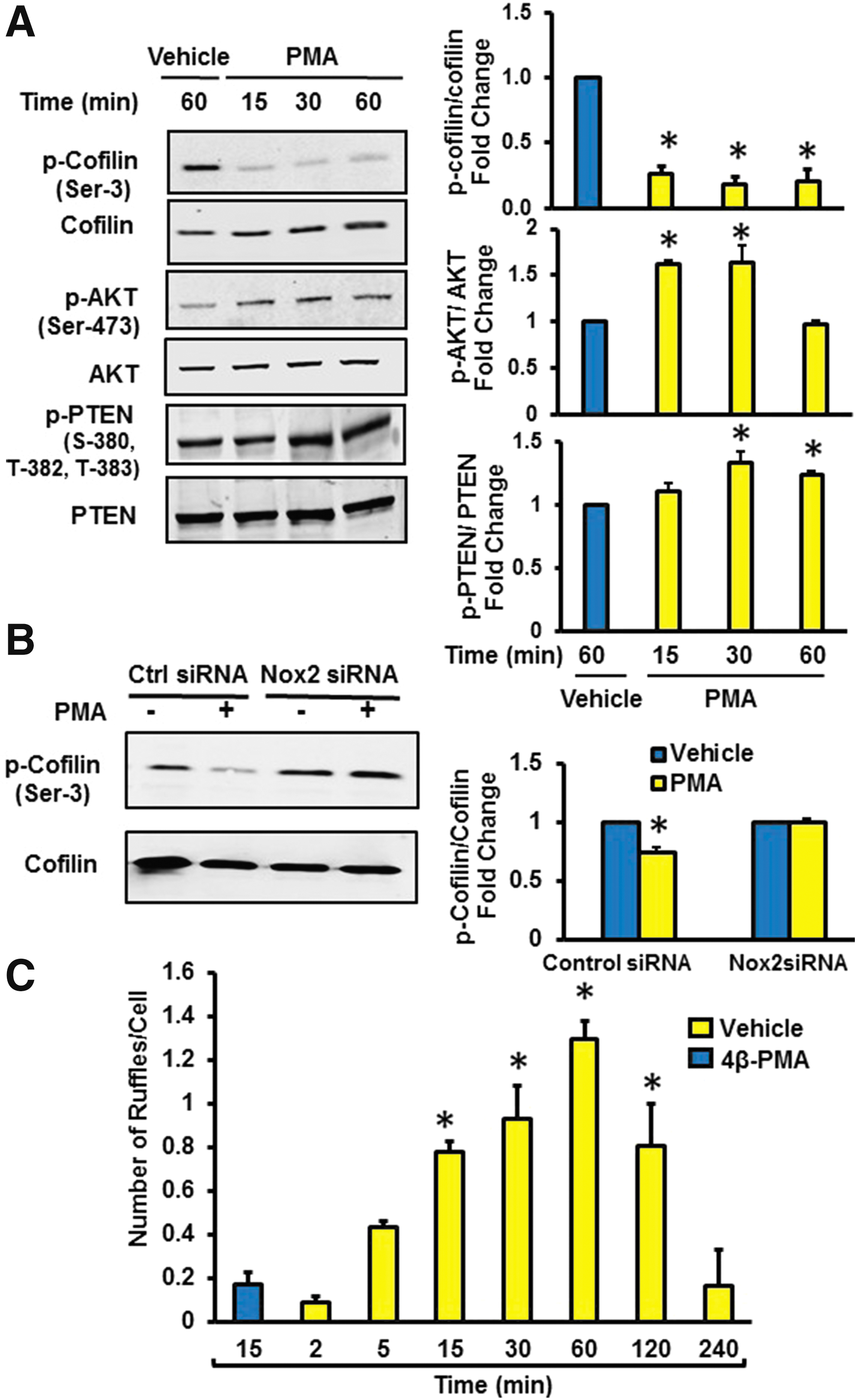
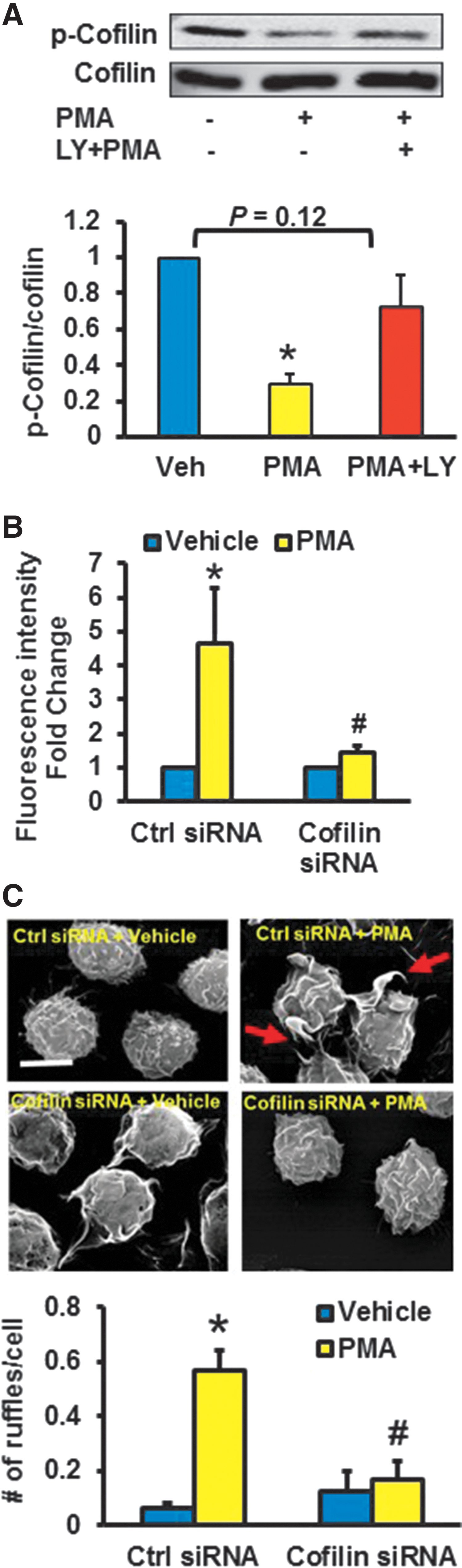
Next, we postulated that actin-binding proteins that regulate rapid actin polymerization are associated with Nox2-mediated membrane ruffling. Accumulated evidence suggests that cofilin, through its actin-severing activity, creates new actin barbed ends for Arp2/3 to rapidly rebuild the actin network near the plasma membrane (19). As the severing activity of cofilin is increased on its dephosphorylation at Ser-3, we first tested whether 4β-PMA dephosphorylates cofilin at Ser-3 in macrophages. Western blot data demonstrated that 4β-PMA activates cofilin within 15 min of incubation (Fig. 4A). This effect was completely inhibited in Nox2 gene-silenced macrophages (Fig. 4B). Time-course experiments indicated that 4β-PMA starts stimulating ruffle formation within the first 15 min of incubation (Fig. 4C). Activated PI3K and its phosphorylation product PI(3,4,5)P3 promote cofilin activation (45); whereas PTEN negatively regulates PI3K/Akt signaling by dephosphorylating PI(3,4,5)P3 and it inactivates cofilin (45). As phosphorylation of PTEN at the serine-threonine cluster (Ser380, Thr382, and Thr383) in the C-terminal tail suppresses its phosphatase activity, we investigated whether 4β-PMA phosphorylates these residues. As shown in Figure 4A, 4β-PMA phosphorylated PTEN at residues Ser380, Thr382, and Thr383 and phosphorylated Akt at Ser473. These data demonstrate decreased phosphatase activity of PTEN and increased PI3K/Akt signaling after phorbol ester stimulation. Consistent with the role of Nox2 in this process, 4β-PMA did not phosphorylate PTEN in Nox2y/− macrophages (Supplementary Fig. S8). To investigate the role of PI3K in 4β-PMA-induced cofilin dephosphorylation, we preincubated macrophages with the PI3K inhibitor LY294002, treated cells with 4β-PMA, and assessed the phosphorylation status of cofilin at Ser-3 via Western blotting. Figure 5A indicates that LY294002 inhibited 4β-PMA-induced cofilin dephosphorylation in macrophages. To provide functional evidence that cofilin is involved in 4β-PMA-stimulated macropinocytosis, we silenced cofilin using siRNA, treated the cells with vehicle or 4β-PMA, and analyzed FITC-dextran uptake using FACS. Figure 5B demonstrates that cofilin siRNA significantly attenuated 4β-PMA-induced FITC-dextran internalization compared with scrambled siRNA-transfected controls. Validation of effective mRNA silencing is shown in Supplementary Figure S9. Finally, SEM data indicated that 4β-PMA-stimulated membrane ruffling was abrogated in cofilin gene-silenced macrophages (Fig. 5C). Taken together, these data demonstrate that 4β-PMA-induced membrane ruffling occurs through Nox2-mediated inactivation of PTEN, stimulation of PI3K/Akt signaling, and cofilin dephosphorylation.
Physiological stimulation of macropinocytosis by M-CSF involves Nox2 activation
To test the physiological importance of this pathway, additional experiments were performed by using M-CSF, a physiologically relevant stimulator of macropinocytosis (60). Importantly, previous data demonstrated that M-CSF stimulates Nox activity in leukocytes (56); however, the role of Nox or ROS in M-CSF-induced macropinocytosis has not been investigated. FACS data indicated that M-CSF treatment (100 ng/ml, 4 h) significantly stimulated internalization of FITC-dextran in WT macrophages (Fig. 6A). The amiloride derivative 5-(N-Ethyl-N-isopropyl)amiloride (EIPA) (10 μM, 30 min) abolished M-CSF-induced FITC-dextran internalization, consistent with previous findings that M-CSF induces macropinocytosis (51). Pretreatment with the cell-permeant antioxidant EUK 134 (10 μM, 1 h) and DPI (10 μM, 1 h) inhibited M-CSF-stimulated macropinocytosis (Fig. 6A). These results suggest that M-CSF-induced macropinocytosis involves O2 ⋅− production that is generated by a DPI-inhibitable enzymatic source. Next, WT and Nox2y/− macrophages were treated with vehicle or M-CSF and macropinocytosis was investigated. As shown in Figure 6B, internalization of FITC-dextran was significantly decreased in Nox2y/− macrophages compared with WT controls.
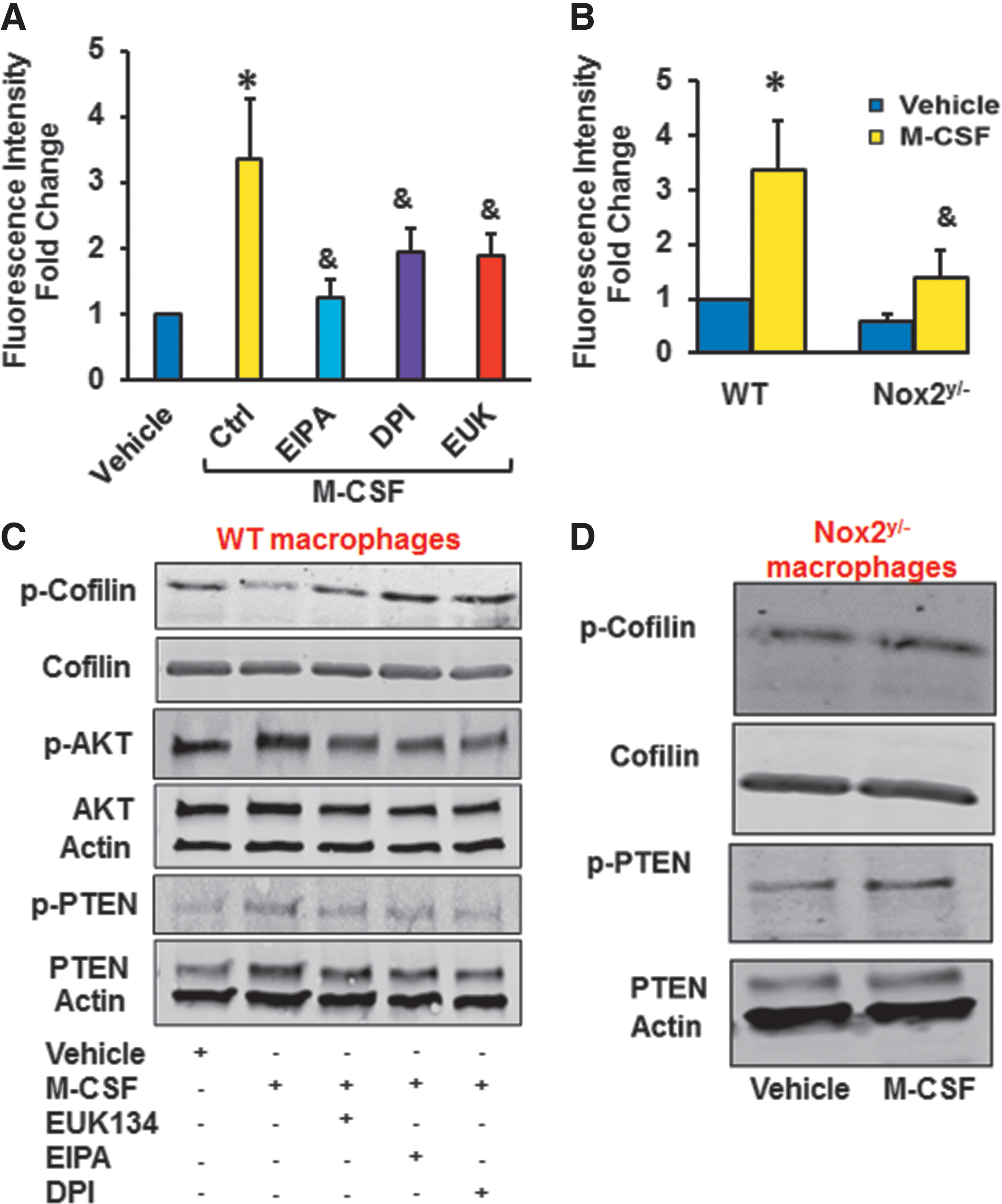
Western blot data indicated that M-CSF rapidly dephosphorylated cofilin at Ser-3 in WT macrophages (Fig. 6C). EUK 134 and DPI inhibited M-CSF-induced cofilin activation (Fig. 6C and Supplementary Fig. S10). The effect of M-CSF on cofilin dephosphorylation was attenuated in macrophages derived from Nox2y/− mice (Fig. 6D). As shown in Figure 6C, M-CSF phosphorylated PTEN and Akt in WT macrophages, demonstrating decreased phosphatase activity of PTEN and increased PI3K/Akt signaling. Incidentally, when cells were pretreated with EUK 134 or DPI, M-CSF-induced PTEN phosphorylation and Akt activation were attenuated. Finally, M-CSF did not phosphorylate PTEN in Nox2y/− cells (Fig. 6D).
Role of Nox2-mediated macropinocytosis in an animal model of atherosclerosis
To determine the role of Nox2-mediated macropinocytosis in vivo, we evaluated the ability of macrophages derived from WT or Nox2y/− mice (donors) to accumulate cholesterol after their adoptive transfer into the peritoneal cavities of high-fat diet-fed ApoE−/− mice (recipients). This approach was validated by previous studies and demonstrated to provide useful information about the role of specific genes in the regulation of foam cell formation in vivo (36). Donor macrophages were fluorescently labeled by using the cell tracker dye CFDA, incubated with vehicle or treated with 4β-PMA (1 μM, 30 min) to stimulate membrane ruffling, and transferred into the peritoneal cavities of ApoE−/− mice (Fig. 7A). Twenty hours later, peritoneal macrophages were isolated, visualized under a fluorescent microscope (Fig. 7A), fixed; lipids accumulated from the peritoneal cavity stained with Nile Red; and CFDA+ (donor) macrophages were selected for FACS analysis (Fig. 7B). 4β-PMA stimulated lipid accumulation in WT-CFDA+ macrophages by approximately four-fold compared with vehicle treatment (Fig. 7D). In contrast, measurement of Nile Red fluorescence by FACS indicated that 4β-PMA-induced lipid accumulation was attenuated in Nox2y/−CFDA+ macrophages (Fig. 7C, D). These data suggest that Nox2-mediated membrane ruffling and subsequent macropinocytosis of cholesterol may lead to significant lipid accumulation in macrophages in hypercholesterolemic conditions.

Discussion
To our knowledge, this is the first study that seeks to examine the role of Nox2 in macropinocytosis. Although our knowledge of the regulatory mechanisms in macropinocytosis has greatly increased in recent years (3, 39, 60), the precise signaling pathway responsible for the initiation of macropinocytosis remains unknown. Results here demonstrate that Nox2 signaling via inactivation of PTEN and stimulation of the PI3K/Akt pathway dephosphorylates cofilin, leading to membrane ruffle formation and macropinocytotic solute internalization. As such, these results may contribute to a better understanding of the underlying mechanisms, regulating membrane ruffling and macropinocytosis.
Although macropinocytosis and phagocytosis share many common features, including a large vacuole size, transient activation of actin cytoskeleton, and other downstream regulatory components, they are fundamentally different processes (42). Phagocytosis is a strictly particle-driven process whereas macropinocytosis is nonspecific internalization of large amounts of fluid and solutes. During phagocytosis, actin modifications remain localized around the particle, whereas macropinocytosis involves global activation of the actin cytoskeleton, resulting in extensive plasma membrane ruffling and fluid internalization. Macropinocytosis is also distinct from receptor-mediated endocytic processes, as it does not require receptor-ligand interaction for internalization. As such, the signaling mechanisms involved in membrane ruffling and subsequent macropinosome formation are different from the mechanisms initiating local activation of actin cytoskeleton during phagocytosis or receptor-mediated endocytosis.
The Nox of “professional” phagocytic cells is a multi-component enzyme that consists of the membrane-bound Nox2 and p22 phox and several cytosolic adaptor proteins (4). The electron transport by phagocyte Nox requires translocation of the cytosolic subunits, p47 phox , p67 phox , and Rac1, to Nox2 and p22 phox (54). In dormant phagocytes, the tandem Src homology 3 (SH3) domain of p47 phox is masked by the auto-inhibitory region (AIR) to keep p47 phox in its inactive conformation (41). Phosphorylation of serine residues within the AIR by DAG-mediated PKC activation exposes the SH3 domain for p22 phox binding and reveals its binding sites for interaction with phosphatidylinositols, such as PI(3,4)P2 (11). During the next steps of Nox assembly, p47 phox brings p67 phox in contact with Nox2, whereas Rac1 induces conformational change of p67 phox , promoting its interaction with the catalytic core of the enzyme, Nox activation, and subsequent O2 ⋅− generation (43). In phagocytes, Nox2 localizes to both intracellular and plasma membranes. This provides the basis for the concept of compartmentalization in ROS-mediated signaling and function (4, 8). ROS generation from intracellular compartments leads to activation of redox-sensitive cell signaling pathways (28), whereas plasma-membrane-bound Nox contributes to phagosomal ROS generation, pathogen killing, and paracrine/autocrine ROS-mediated signaling (54).
Although previous studies provided hints for a possible mechanistic interaction between Nox2 activation and macropinocytosis, this has never been tested experimentally. For example, the classical activators of macropinocytosis, including phorbol esters, growth factors, and M-CSF (3, 32, 51, 59), stimulate Nox activation and ROS generation (24, 34, 56). In addition, the following reports demonstrated that signal transduction mediators upstream of Nox2 activation, such as Rac1, PKC, and phosphatidylinositols, are involved in membrane ruffling and macropinocytosis: (i) Photoactivation of PA-Rac1 in macrophages resulted in plasma membrane ruffling in 1–5 min and subsequent macropinosome formation (23); (ii) M-CSF via stimulation of PI3K and DAG generation leads to PKC activation and cup closure (60); and (iii) sequential dephosphorylation of PI(3,4,5)P3→PI(3,4)P2→PI(3)P→PI is required for EGF-induced macropinocytosis (39).
Previous studies showed that reconstitution of Nox activity in cell-free systems by phorbol esters or anionic amphiphiles is almost instantaneous, peaking within the first 1–2 min of stimulation (15, 43). Results of our O2 ⋅− measurements are consistent with previous reports showing that 4β-PMA stimulates O2 ⋅− generation in 4–5 min in intact cells (24). These data together with our SEM time-course experiments suggest that 4β-PMA-stimulated O2 ⋅− generation precedes or occurs concomitantly with the earliest membrane ruffles in macrophages. To test whether O2 ⋅− is functionally involved in 4β-PMA-induced macropinocytosis, we pretreated macrophages with the cell-impermeant SOD, cell-permeant PEG-SOD, or the synthetic SOD/catalase mimetic EUK 134. FACS data indicated that the cell-permeant O2 ⋅− scavengers, but not the cell-impermeant SOD, attenuated 4β-PMA-stimulated macropinocytosis, suggesting the role of intracellular O2 ⋅− in macropinocytosis. Imaging studies utilizing DHE staining and quantification of the 2-OH-E+ after DHE treatment confirmed that 4β-PMA stimulates intracellular O2 ⋅− generation. These results suggest that 4β-PMA stimulates macropinocytosis via a redox-mediated mechanism, which involves intracellular O2 ⋅− as a signaling molecule.
Earlier studies showed that phorbol ester-stimulated macropinocytosis is inhibited by PKC inhibitors and macropinocytotic uptake of macromolecules is stimulated by activators of PKC (31). We have confirmed this by using the PKC inhibitor calphostin c and demonstrated that the inactive stereoisomer 4α-PMA, which does not activate PKC (44), is unable to stimulate O2 ⋅− generation or induce macropinocytosis. Because analogs of DAG stimulate Nox activity in phagocytes (22), we tested whether incubation of macrophages with the DAG analog DOG stimulates macropinocytosis. DOG increased uptake of FITC-dextran by macrophages, which is consistent with the role of the DAG-PKC pathway in macropinocytosis.
Since Nox2 is the most highly expressed Nox isoform in macrophages and phorbol esters activate Nox2 via the DAG-PKC pathway (35, 50), we tested whether Nox2 is involved in 4β-PMA-induced macropinocytosis. DPI, a small-molecule inhibitor of flavin-containing oxidases, significantly attenuated 4β-PMA-induced macropinocytosis. Although DPI has been widely used to inhibit the oxidative burst by phagocyte Nox, it inhibits all Nox isoforms (26). In addition, as DPI accepts an electron from FAD moieties, followed by a covalent reaction with the flavoenzyme, it inhibits not only Nox enzymes but also xanthine oxidase, mitochondrial complex I, and cytochrome P-450 reductase (4). To further extend our findings and to use a more specific approach, we isolated peritoneal macrophages from Nox2y/− mice, challenged them with 4β-PMA, and investigated whether macropinocytosis is ameliorated. Our data demonstrated that 4β-PMA-induced internalization of fluorescently labeled dextran was significantly blocked in Nox2y/− macrophages compared with WT controls. As compensatory changes in Nox2y/− macrophages in response to the loss of Nox2 function may occur, we silenced Nox2 in macrophages using siRNA and investigated whether it inhibits 4β-PMA-induced macropinocytosis. FACS results indicate that internalization of FITC-dextran was inhibited in Nox2-silenced macrophages after 4β-PMA treatment compared with control siRNA-transfected cells. qPCR demonstrated that Nox2 mRNA levels were decreased in Nox2 siRNA-treated cells, whereas we observed no compensatory changes in Nox1 and Nox5 mRNA levels [Nox3 is undetectable in macrophages (35)]. Importantly, extensive inquiry is required to investigate whether other Nox isoforms under various conditions modulate macropinocytosis in phagocytes and other cell types. Indeed, in an associated paper in this issue, we demonstrate that dynamic Nox1 activation by matrix protein thrombospondin-1 and its cognate receptor CD47 mediates macropinocytosis (15a). On the other hand, as previous studies have reported that Nox4 is constitutively active and it is regulated at the transcriptional level (46), it is likely that Nox4 is not involved in macropinocytosis under acute conditions. However, it is possible that factors that induce Nox4 expression, such as TGFβ1 and HIF-1α (2, 20), may stimulate macropinocytosis under chronic conditions.
The plasma membrane activities leading to macropinosome formation consist of multiple morphologically distinct steps that are each associated with a corresponding sequence of signal transduction mediators. We next used time-lapse DIC and SEM to investigate which morphological step during macropinocytosis is mediated by Nox2. Calphostin c, DPI, and EUK 134 blocked 4β-PMA-induced membrane ruffling, demonstrating that PKC and O2 ⋅− derived from a DPI-inhibitable flavoenzyme are required for the initiation of membrane ruffle formation. In addition, gene silencing Nox2 inhibited 4β-PMA-induced membrane ruffling. Taken together, these findings suggest that phorbol ester PMA stimulates membrane ruffling via Nox2-derived O2 ⋅− generation. In contrast to our observations, a previous study utilizing pharmacological approaches suggested that DAG and PKC are involved in cup closure but not in earlier stages of macropinosome formation (60). In addition, there are inconsistencies between previous results, as some (23), but not all (57), studies demonstrated that Rac is required for membrane ruffling. It is important to note that using different means to activate macropinocytosis, utilizing different cell types that may express different Rac or PKC isoforms, or differences in protein levels of PKC and Nox enzymes may all contribute to these differences.
Cofilin is a key regulator of actin remodeling, lamellipodia formation, and cellular motility (19). The severing and depolymerization activity of cofilin is increased by its dephosphorylation on Ser3, which stimulates its actin-binding activity. Western blot data indicated that 4β-PMA dephosphorylates cofilin at Ser 3 in 15 min, which was inhibited in Nox2-gene silenced as well as in Nox2y/− macrophages. PTEN, a tumor-suppressor protein, inactivates cofilin by antagonizing PI3K signaling and decreasing intracellular levels of PI(3,4,5)P3 (45). On the contrary, PI3K and PI(3,4,5)P3 promote cofilin dephosphorylation and activation. We demonstrated that 4β-PMA decreases the phosphatase activity of PTEN via Nox2 and stimulates Akt signaling. Interestingly, a previous study reported that slingshot phosphatase (SSH) is activated downstream of PI3K/Akt, leading to cofilin dephosphorylation and insulin-induced membrane protrusions (45). Consistent with this study, our data demonstrated that pharmacological inhibition of PI3K inhibits 4β-PMA-induced cofilin dephosphorylation. In addition, we demonstrated that gene silencing cofilin using siRNA inhibited ruffle formation and macropinocytosis after 4β-PMA treatment. When taken together, these data motivate the speculation that redox inactivation of PTEN by Nox-derived ROS leads to activation of PI3K/Akt signaling, cofilin dephosphorylation, and initiation of membrane ruffle formation. Importantly, this signaling pathway was also demonstrated in M-CSF-induced macropinocytosis, demonstrating the physiological relevance of our study.
A large body of literature supports macrophage lipid accumulation as the key initiating event in atherosclerosis (38). After deposition of nLDL in the subendothelial matrix of arteries, monocytes home to the vessel wall, differentiate into macrophages, and imbibe excessive amounts of cholesterol. It is generally accepted that local biological responses to retained nLDL particles lead to their modification, that is, oxidation, and subsequent internalization by macrophage scavenger receptors, leading to foam cell formation and atherosclerosis. In addition to this “classical” mode of lipid uptake, it has been demonstrated in vitro that macropinocytosis may contribute to lipid internalization by macrophages (32). The relative contribution of macrophage nLDL macropinocytosis to atherogenesis is currently unknown, as no pharmacological or genetic tools are available to selectively target this process.
Shifting to an alternative approach, we performed a peritoneal macrophage chimera study to investigate macropinocytic uptake of lipids from the peritoneal cavity of ApoE−/− mice. In this experiment, WT and Nox2y/− macrophages were preincubated with 4β-PMA to stimulate membrane ruffling and injected into the peritoneal cavity of ApoE−/− mice. Consistent with a role of Nox2 in macropinocytosis, our results demonstrated that 4β-PMA stimulated lipid accumulation in WT, but not in Nox2y/−, macrophages. These in vivo data demonstrate for the first time that Nox2-mediated membrane ruffling and subsequent macropinocytosis of cholesterol may lead to significant lipid accumulation in macrophages in hypercholesterolemic conditions. Importanly, recent studies have also demonstrated that macropinocytosis is involved in other pathological processes. For instance, cancer cells utilize macropinocytosis to internalize nutrients and support their metabolic needs, which is critical for their proliferation and tumor growth (14). Macropinocytosis is also a major mechanism by which renal tubular cells endocytose calcium oxalate crystals, leading to kidney stone formation (27). Escherichia coli infection stimulates Shiga toxin 1 macropinocytosis, leading to intestinal and systemic pathologies (40). Thus, a better understanding of the signaling mechanisms regulating macropinocytosis may lead to new pharmacological concepts to target pathophysiological conditions involving macropinocytosis.
To our knowledge, no direct link has ever been made between Nox2 signaling and macrophage macropinocytosis. Importantly, redox regulation of macropinocytosis downstream of Nox2, or any Nox for that matter, had not been previously demonstrated in any cell types or disease model. In addition, the results of the current study provide seminal mechanistic insight into how Nox2 signaling via inactivation of PTEN and activation of the PI3K/Akt pathway leads to cofilin dephosphorylation, extensive membrane ruffling, and macropinocytosis. Peritoneal chimera studies presented are the first that implicate Nox2-mediated macropinocytosis in macrophage lipid accumulation in vivo. These data are expected to reveal an intriguing new line of study in the field of endocytosis, with broad implications for cellular physiology and pathological conditions involving macropinocytosis.
Materials and Methods
Reagents
Cytochrome c, SOD, PEG-SOD, DPI, 4α- and 4β-PMA, Nile Red, LY294002, calphostin c, amiloride, EIPA, latrunculin A, chlorpromazine, nystatin, FITC-Dextran, and thioglycollate medium were purchased from Sigma-Aldrich. DOG and EUK-134 were obtained from Cayman Chemical. Vybrant® CFDA Cell Tracer Kit was purchased from Thermo Fisher. FluoSpheres® were obtained from Life Technologies. Human nLDL was purchased from Kalen Biomedical, LLC. M-CSF was purchased from Miltenyi Biotec, Inc.
Cell culture
RAW 264.7 macrophages (ATCC) were maintained in Dulbecco's modified Eagle's medium (DMEM; Mediatech, Inc.) that was supplemented with 100 IU/ml of penicillin G, 100 μg/ml streptomycin, and 10% (vol/vol) heat-inactivated FBS in a humidified incubator at 37°C and 5% CO2. Thioglycollate-elicited peritoneal macrophages and bone marrow-derived monocytes were cultured in RPMI-1640 medium (without 2-mercaptoethanol) containing 10% FBS. Bone marrow-derived monocytes were differentiated into macrophages using murine M-CSF (20 ng/ml, 6 days) as previously reported (49). THP-1 monocytes (ATCC) were maintained in cell-free conditioned media of RAW 264.7 macrophages and differentiated into macrophages using human M-CSF (20 ng/ml, 9 days). The cells were serum starved (1% FBS, 4 h) before experiments.
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC), Augusta University, Augusta, the United States. Male C57BL/6 (wild type), ApoE−/−, and Nox2y/− mice were purchased from The Jackson Laboratory. CD36−/− mice were kindly provided by Dr. Roy Silverstein (Medical College of Wisconsin).
Imaging experiments
DIC microscopy
DIC microscopy was performed as previously described (48) by using a Zeiss 780 inverted microscope. Cells were imaged by using a 63 × (Alpha Plan-Apo, NA: 1.46) objective. Live images were collected at every second for 2 min. At least four representative fields were chosen for each dish, and the experiment was performed in triplicate.
Confocal microscopy
Macrophages were fixed in 2% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, and stained with Alexa Fluor 488® phalloidin, Hoechst 33342 (Life Technologies), and Nile Red (50 ng/ml; 10 min). Images were taken with a Zeiss 780 inverted confocal microscope.
Scanning electron microscopy
Macrophages were fixed (4% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate solution) overnight at 4°C. Then, the cells were dehydrated through a graded ethanol series (25%–100%) and washed with 100% ethanol before critical point drying (Tousimis Samdri-790). Coverslips were mounted onto aluminum stubs and sputter coated with 3.5 nm of gold/palladium (Anatek USA-Hummer). Cells were imaged at 20 KV by using a Philips XL30 scanning electron microscope (FEI).
Gene silencing
THP-1 macrophages were transfected with siRNA against CLTC, Nox2, and cofilin (OriGene Technologies) by using the transfection reagent TransIT-TKO® (Mirus Bio LLC). To control for possible nonspecific effects of siRNA, a Silencer® negative control siRNA was used. Gene silencing by siRNA was confirmed by qPCR. Cells were used 48 h post transfection.
Real-time PCR analysis
Total RNA was extracted from macrophages by using an RNA extraction kit (IBI Scientific). cDNA was generated by using the TaqMan® Reverse Transcriptase kit (Applied Biosystems). Real-time PCR was carried out by using the SYBR Green Super mix (Applied Biosystems, Inc.). All amplifications were performed in triplicate, and GAPDH was used as the internal control. The primers used for real-time PCR are as follows:
Cofilin: F-TTCAACGACATGAAGGTGCGT, R-TCCTCCAGGATGATGTTCTTCT,
CLTC: F-TGATCGCCATTCTAGCCTTGC, R-CTCCCACCACACGATTTTGCT,
Nox2: F-GTCACACCCTTCGCATCCATTCTCAAGTCAGT, R-CTGAGACTCATCCCAGCCAGTGAGGTAG,
GAPDH: F-CATGTTCGTCATGGGTGTGAACCA,
R-AGTGATGGCATGGACTGTGGTCAT.
Flow cytometry
FACS experiments were performed by using the Becton Dickinson FACSCalibur flow cytometer using a standard protocol. Briefly, macrophages were incubated with nLDL (250 μg/ml) and treated with vehicle or 4β-PMA (1 μM, 24 h). Cells were fixed in 2% PFA and stained with Nile Red (25 ng/ml) for 10 min. In separate experiments, macrophages were incubated with FITC-dextran (70,000 MW, 150 ng/ml) and treated with vehicle, 4β-PMA (1 μM), or M-CSF (100 ng/ml) for 4 h. Fluorosece intensity was measured by using the FL3 channel for Nile Red (Ex: 488 nm, Em: 670 nm) and Fl1 for FITC (Ex: 488 nm, Em: 530 nm). The gating strategy for flow cytometry analysis is shown in Supplementary Figure S11.
Measurement of ROS
Quantification of 2-OH-E ± using HPLC
2-hydroxyethidium (2-OH-E+), the O2 ⋅−-specific oxidation product of DHE, was quantified in RAW 264.7 macrophages as previously described (62). Briefly, an ether-linked phenyl column (100 × 4.6 mm; Phenomenex) was employed by using two mobile phases (Solution A: 50 mM phosphate buffer in 90% water and 10% acetonitrile and Solution B: 50 mM phosphate buffer in 40% water and 60% acetonitrile), and a gradient elution to increase the acetonitrile concentration from 25% to 60% over 10 min at a flow rate of 0.75 ml/min. Forty microliters of sample was injected into the HPLC system (ESA, Inc., with a CoulArray electrochemical detection array system). The areas of the corresponding peaks were measured by using the software provided by ESA, Inc. The limit of detection for this assay was determined from a standard curve and established at 20 fmoles. The concentration of 2-OH-E ± was calculated by using a standard curve and normalized to total protein of the cell lysate.
L-012 chemiluminescence
L-012 chemiluminesce to measure O2 ⋅− generation was previously described (10). Cells (50,000/well) were plated in 96-well plates in sterile phosphate-buffered saline (PBS) containing L-012 (400 μM; Wako Chemicals) and treated with vehicle, 4β-PMA (1 μM), or 4α-PMA (1 μM). Chemiluminescence was measured at 37°C by using a Lumistar Galaxy (BMG) luminometer.
Electron paramagnetic resonance
The spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine hydrochloride (CMH; Alexis Corp.) was used to examine O2 ⋅− production in macrophages by using a Bruker eScan table-top EPR spectrometer (Bruker Biospin). Superoxide production was measured in oxidase assay buffer (65 mM sodium phosphate buffer, pH 7.0, 1 mM EGTA, 10 μM FAD, 1 mM MgCl2, and 2 mM NaN3) that was supplemented with 50 μM CMH as previously reported (17).
Western blot
Western blotting was performed as previously described (18). Briefly, protein lysates were prepared by incubating cells with RIPA lysis buffer (Pierce Biotechnology). The cell lysates were sonicated by using the Sonic Dismembrator, Model 100 (Fisher Scientific) for 2 × 10 s at setting 2 and centrifuged at 14,000 g for 15 min at 4°C to pellet the cell debris. The protein concentration of supernatant was estimated by using the bicinchoninic acid (BCA) protein assay (Pierce Biotechnology), according to the manufacturer's instructions. Equal amounts of protein (30 μg) were heated in Laemmli sample buffer (BioRad Laboratories, Inc.) at 95°C for 5 min, separated on SDS-PAGE gels, transferred onto nitrocellulose membranes (Li-Cor Biosciences), and probed with cofilin, Akt, total PTEN (Cell Signaling), and p-PTEN (Abcam) antibodies. The IRDye-conjugated secondary antibodies (Li-Cor Biosciences) were used to detect the primary antibodies. The internal control β-actin was used to normalize the amount of protein transferred onto the membranes.
Adoptive transfer of macrophages into ApoE−/− mice
The adoptive transfer was performed as previously described, with slight modifications (49). Bone marrow-derived macrophages from WT and Nox2y/− mice (donors) were resuspended in sterile PBS at a concentration of 2 × 106 cells per ml and labeled with 10 μM carboxy fluorescein diacetate-succinimidyl ester (CFDA, Molecular Probes) as per the manufacturer's instructions. Cells were treated with vehicle or 4β-PMA (1 μM, 30 min) and injected into recipient ApoE−/− mice (i.p., 2 × 106 cells/mouse). ApoE−/− mice were fed a high-fat diet for 8 weeks before experiments. Twenty hours later, peritoneal macrophages were isolated, stained with Nile Red, and processed for FACS analysis.
Statistical analysis
The data shown are the means ± SEM. Significance of the differences were assessed by Student's t-test followed by analyzing the confidence interval at 95% or one- or two-way ANOVA, as appropriate for the particular experiment and treatment groups. p values less than 0.05 were considered significant.
Footnotes
Acknowledgments
The authors wish to thank Jeanene Pihkala and Libby Perry (Augusta University) for their help with the FACS analysis and SEM sample preparation, respectively.
This work was supported by National Institutes of Health grants (K99HL114648 and 4R00HL114648-03) awarded to Gábor Csányi.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.