
Abstract
Introduction
Breast cancer as a heterogeneous disease
B
ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PR, progesterone receptor.
Current therapies of receptor-positive breast malignancies
Hormone receptor-positive breast cancer accounts for up to 80% of all invasive breast cancers (55). The cornerstone targeted systemic therapy for this subtype of breast cancer is antihormone therapy (55). There are several antihormone therapy options, including gonadotropin-releasing hormone (GnRH) agonists that lead to ovarian function suppression, selective estrogen receptor modulators or downregulators (SERMs or SERDs), and aromatase inhibitors (AIs) (55). Surgical removal of ovaries was the first antihormone therapy used against breast cancers and still remains an option for patients with hormone-positive breast cancer, as is medical ovarian suppression with GnRH agonists (42). The most well-known SERM is tamoxifen (TAM), which selectively blocks ER signaling in the breast, but has agonist activity in other organs such as the uterus (54, 55). TAM can improve survival both in early and advanced breast cancer (20, 55). Fulvestrant, the only SERD approved by the FDA, functions as an ER antagonist by binding to the ER, preventing ER dimerization and facilitating ER degradation. Fulvestrant has been shown to improve survival in patients with advanced hormone-positive breast cancer (15, 16). AIs inhibit the cytochrome P450 component of the aromatase enzyme complex to prevent estrogen biosynthesis and improve survival in postmenopausal women with early and advanced hormone-positive breast cancer (57). Unfortunately, patients with early stage breast cancer may still experience recurrence, and if the disease metastasizes to a distant site, it is not curable and eventually becomes refractory to antihormone therapy, clinically demonstrating that resistance mechanisms can develop. Drugs targeting the mammalian target of rapamycin and cyclin-dependent kinase pathways have already been developed to overcome these resistance mechanisms, but these drugs only provide transient responses in some patients and better therapeutic approaches are urgently needed (67).
Luminal B breast cancers exhibit increased metastatic recurrences
Despite the similarities in the treatments between luminal A and luminal B breast cancer, as well as similar treatment recommendations, patients with the luminal B subtype exhibit significantly worse clinical outcomes than luminal A (31, 64). Pathologically, luminal B breast cancers exhibit higher proliferative indices (i.e., high Ki67), are poorly differentiated, and exhibit a significantly increased risk of recurrence compared with luminal A. Another molecular feature more common in luminal B tumors is a higher p53 mutation. Luminal B breast cancers tend to be less responsive to endocrine therapy and are less likely to benefit from antihormone therapy compared with luminal A (2, 14). The intrinsic antihormone therapy resistance that characterizes luminal B breast cancers leads to increased risk of recurrence. Therefore, additional therapeutics are needed in this particular subtype.
A statistical link between aging, longevity, and breast cancer
One of the fundamental observations in oncology is that the rate of malignancies increases significantly with age (21, 22). In fact, the single strongest prognostic variable that predicts the incidence of cancer is increasing age and this is especially true for breast malignancies (53). As such, breast cancer, together with many other types of cancers, is an aging-related disease (3, 8) and this exponential increase suggests a fundamental link between longevity and carcinogenesis. When the incidence of developing a solid malignancy is analyzed as a function of age, the data clearly show that the risk of developing a solid tumor begins with an early slope that is gradual and flat (Fig. 1). However, as age increases, an inflection point with a steep slope occurs after 40 years (90). Beyond this inflection point, a steep slope is presented in a logarithmic scale, which indicates a late exponential increase of human cancer incidence rate.

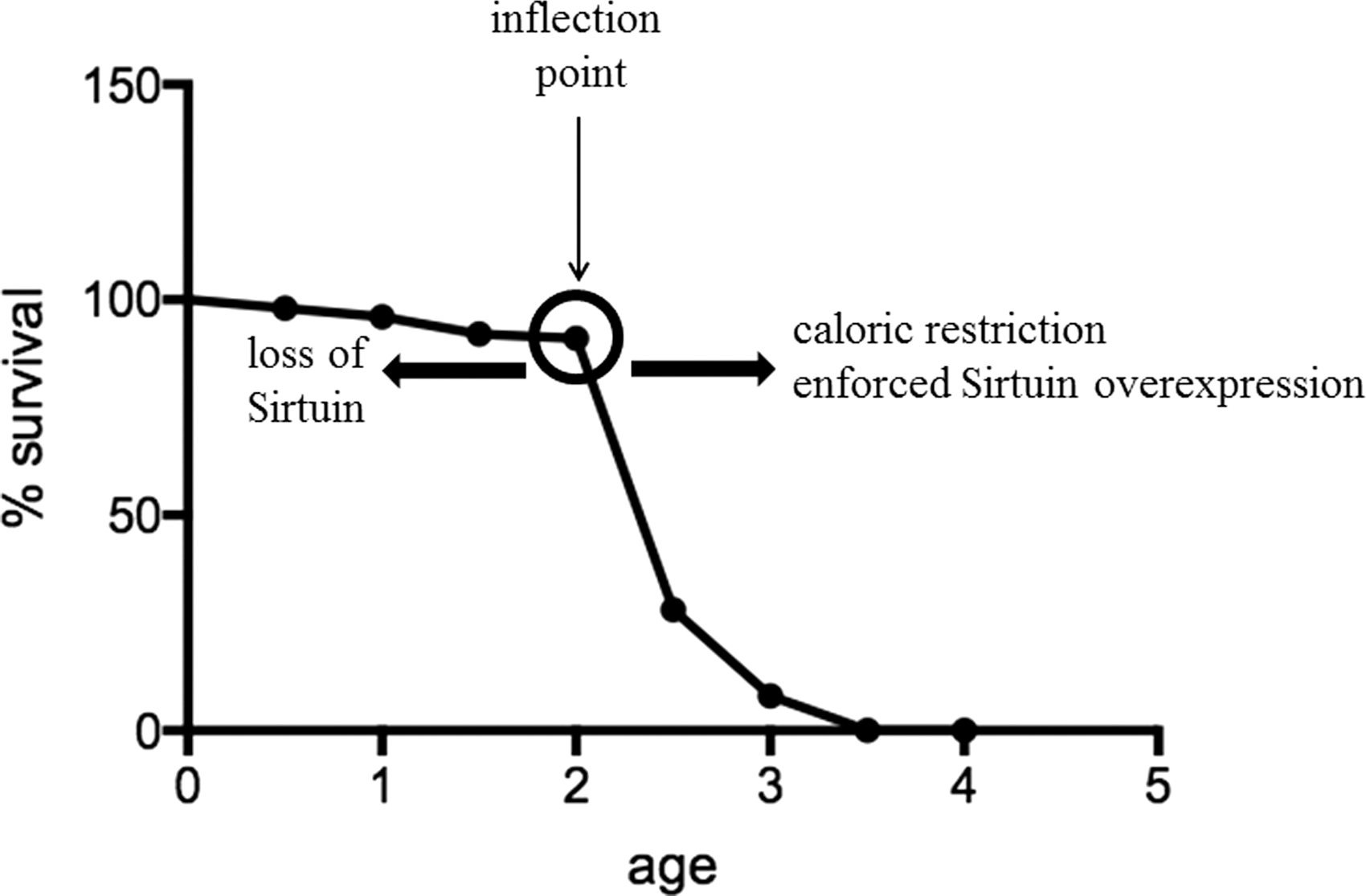
The existence of this inflection point and the steep slope strongly suggests that dysregulation of specific biological processes occurs just before the inflection point. Interestingly, this inflection point is observed not only in human tumors but also in almost all species, including mice and Caenorhabditis elegans (79, 90). Similar to the flat slope of the solid tumor incidence curve, the longevity curve starts with an initial long and flat curve, representing high probability of survival during early life. The flat curve is followed by an inflection point (Fig. 2, presented as a circle) and a steep downward curve, suggesting that the life span has reached its maximum and the probability of survival significantly decreases with age in C. elegans (79, 90). An analysis of these longitudinal data suggests that the curve of the C. elegans longevity is very similar (except that the shape of the curves is inverted) to that observed for the human tumor incidence rate curve (Fig. 1). Interestingly, both curves display a similar inflection point, indicating potential changes in similar or overlapping molecular biological processes. These results strongly suggest that there are specific cellular processes that prevent the damaging effects of increasing age and, at some point, the damage exceeds the cell or organism ability to maintain a reparative state of metabolic homeostatic poise.
The risk of human breast tumor is very similar to what has been observed for the human solid tumor cancer. Approximately half of the newly diagnosed breast cancer patients are older than 65 years (3, 73). In this regard, patients older than 60 years have a twofold increase in luminal B incidence compared with patients between 40 and 59 years old. In contrast, the incidence of luminal A breast cancer remains relatively constant among different age groups (14). Therefore, the incidence of luminal B breast cancer appears to be correlatively linked with aging and, as such, it is hypothesized that there may be a causative relationship between aging-related genes. Based on these observations, including the risk curves for hormone-positive malignancies and how sirtuins direct longevity, it is proposed that sirtuins may function as fidelity to adaptive (reprogramming) proteins that maintain a metabolic homeostatic equilibrium during aging. As such, if the function of sirtuins decreases with increasing age, as has been proposed (6, 66), there may be a mechanistic link between aging, luminal B breast carcinogenesis, and dysregulation and/or decrease in sirtuin enzymatic function.
Sirtuins, aging, and longevity
Sirtuins, which are the murine and human homologs of yeast silent mating-type information regulation 2 (Sir2) gene, were initially discovered in yeast and C. elegans. These genes play a critical role in extending the life cycle by suppressing toxic rDNA formation, suggesting a potential role of sirtuins in the process of longevity and/or organismal aging (32). Studies have shown that overexpression of Sir2 in C. elegans can shift the inflection point on the survival curve to the right (Fig. 2, right arrow), prolonging their life span, whereas knocking out sirtuins in C. elegans can shift the inflection point to the left, reducing their life span (34) (Fig. 2, left arrow). Based on the functions of sirtuins, this observation suggests that sirtuins can either direct the inflection point or direct cellular repair, resulting in a delay of the reflection point occurrence.
Studies have investigated whether the loss of a specific sirtuin gene can affect mice life span. Mice lacking one of the seven sirtuin genes (except Sirt6) do exhibit murine physiological phenotypes similar to that observed in humans for several age-related illnesses, including insulin resistance, cardiovascular disease, neurodegeneration, and most importantly tumorigenesis-permissive phenotypes (17). In this regard, the Gius laboratory showed that mice lacking the Sirt3 gene exhibited dysregulated mitochondrial detoxification pathways as well as contain increased total and mitochondrial reactive oxygen species (ROS). In addition, these mice develop ER-positive mammary tumors that display high Ki67 and are poorly differentiated. These results suggest that the Sirt3 knockout mice may act as murine luminal B-like models that share some biological and histopathological similarities to patients with luminal B breast malignancies (17, 46, 78). Based on these results as well as those published by others (5, 25, 25, 52, 84), it has been proposed that SIRT3 is a bona fide tumor suppressor (TS) protein or gene.
Sirtuins and human breast cancer
Mammalian sirtuins, which are categorized as a class III histone deacetylase (HDAC) family, are different from conventional class I and II HDACs (19, 69). Mammals contain seven sirtuins (SIRT1–SIRT7) that are localized in the nucleus (SIRT1, SIRT6, and SIRT7), cytoplasm (SIRT2), and mitochondria (SIRT3–5) and can direct cellular processes that are related to cellular metabolism (24, 33, 47, 62). Among the mitochondrial sirtuins, SIRT3 appears to be the primary mitochondrial deacetylase (52), which directs mitochondrial energy metabolism (38) and limits the accumulation of mitochondrial ROS (1, 46). In fact, the first publication identifying an SIRT3 downstream deacetylation target was acetyl-coenzyme A synthetase (ACS), providing the first seminal data that SIRT3 plays a critical role in energy homeostasis. Since then, other SIRT3 targets that direct energy metabolism have been characterized, which include long-chain acyl-coenzyme A dehydrogenase (LCAD), 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 (HMGCS2), and all proteins that comprise the adenosine triphosphate (ATP) synthase complex (28, 38, 43, 81, 89).
It has also been shown that critical detoxification enzymes contain SIRT3-targeted reversible acetyl lysines that clearly play a role in the removal of damaging mitochondrial oxidative molecules that are produced by the process of respiration and ATP generation. For example, manganese superoxide dismutase (MnSOD), an SIRT3 deacetylation target (12, 65, 78), directs MnSOD enzymatic activity. The deacetylation of lysines 68 (12) and 122 (78) significantly increases the MnSOD enzymatic activity, thus protecting cells from ROS-induced genomic instability and other deleterious effects or mitochondrial ROS (12, 43, 78). In addition, it is now well documented that MnSOD acetylation is dysregulated in early breast cancer and several groups have demonstrated that MnSOD plays a critical role in breast cancer cell proliferation as well as metastasis. It has been proposed that the ratio of superoxide and hydrogen peroxide (H2O2) production and removal reprograms the mitochondrial as well as other cellular compartments, which subsequently activates redox-sensitive survival and proliferation-related cell signaling pathways (4, 44, 45, 50, 68, 83, 85).
The studies described above also suggest conditions that induce sirtuin deacetylation activity and appear to play a protective role against the development of age-related human pathology, including carcinogenesis (46, 75). Thus, with these results, it seems logical to propose that SIRT3 functions as a sensing or fidelity protein, which can direct downstream targets through post-translational modifications, mainly protein acetylation, to reprogram cellular metabolism and redox balance. These results also raise several important questions regarding the role of SIRT3 in breast cancer: (i) What are the roles of sirtuins, specifically SIRT3, in mammary ductal cell biology and why do deletion and/or dysregulation result in a breast cancer tumorigenic-permissive phenotype? (ii) What is the role of SIRT3 in the direction of mammary ductal metabolic regulation, including cell proliferation and growth?
To understand the role of SIRT3 in breast carcinogenesis, our laboratory has shown that mouse embryonic fibroblasts (MEFs) lacking Sirt3 exhibit significantly higher levels of genomic instability and could be immortalized by the infection of a single oncogene. In addition, these immortalized MEFs were able to grow in low-density soft agar and become tumorigenesis in nude mice (46). More importantly, Sirt3 −/− mice developed mammary gland tumors over 24 months compared with control mice. Interestingly, histological hematoxylin and eosin and immunohistochemistry staining identified these tumors as ER positive and PR positive. These results further suggest that Sirt3 −/− mice developed mammary gland tumors with poorly differentiated, receptor-positive histological characteristics that resemble what is referred to as luminal B malignancies that appear to be more common in older women (46).
When examining SIRT3 protein and RNA expression levels in human breast cancer sample sets, SIRT3 expression was found to be significantly lower in breast cancer samples compared with normal control and it negatively correlates with breast cancer malignancy (46). In addition, low SIRT3 was also correlatively associated with luminal B clinical outcomes (17) as well as other breast cancer subtypes, although to a much smaller degree. Together, results from in vitro, in vivo, and human studies strongly support the hypothesis that SIRT3 is a genomically expressed, mitochondrial localized TS protein. In this regard, results of ER-positive breast cancer from the Sirt3 knockout mice provide a potential argument that SIRT3 may function as a critical regulator at the crossroads between metabolic stress, aging, and aging-related human diseases such as breast cancer. Moreover, the loss of or dysregulation of SIRT3, which is proposed to occur with increasing age, can contribute to a tumor-permissive environment.
In this regard, the increase in cellular ROS observed in tumors lacking Sirt3 is due to, at least in some significant part, dysregulation of mitochondrial energy-generating (i.e., ATP production) pathways as well as mitochondrial proteins involved in the enzymatic detoxification of ROS, such as MnSOD. Most recently, as shown by Dr. Joe Bass's Laboratory (Northwestern University), a mechanistic relationship was discovered between NAD+ metabolism and SIRT3 activity that can direct mitochondrial protein acetylation, including MnSOD activity and ROS levels. Finally, dysregulation of this axis results in an in vitro and in vivo tumor-permissive phenotype that is due to, at least in part, aberrant or increased ROS levels (Fig. 3).
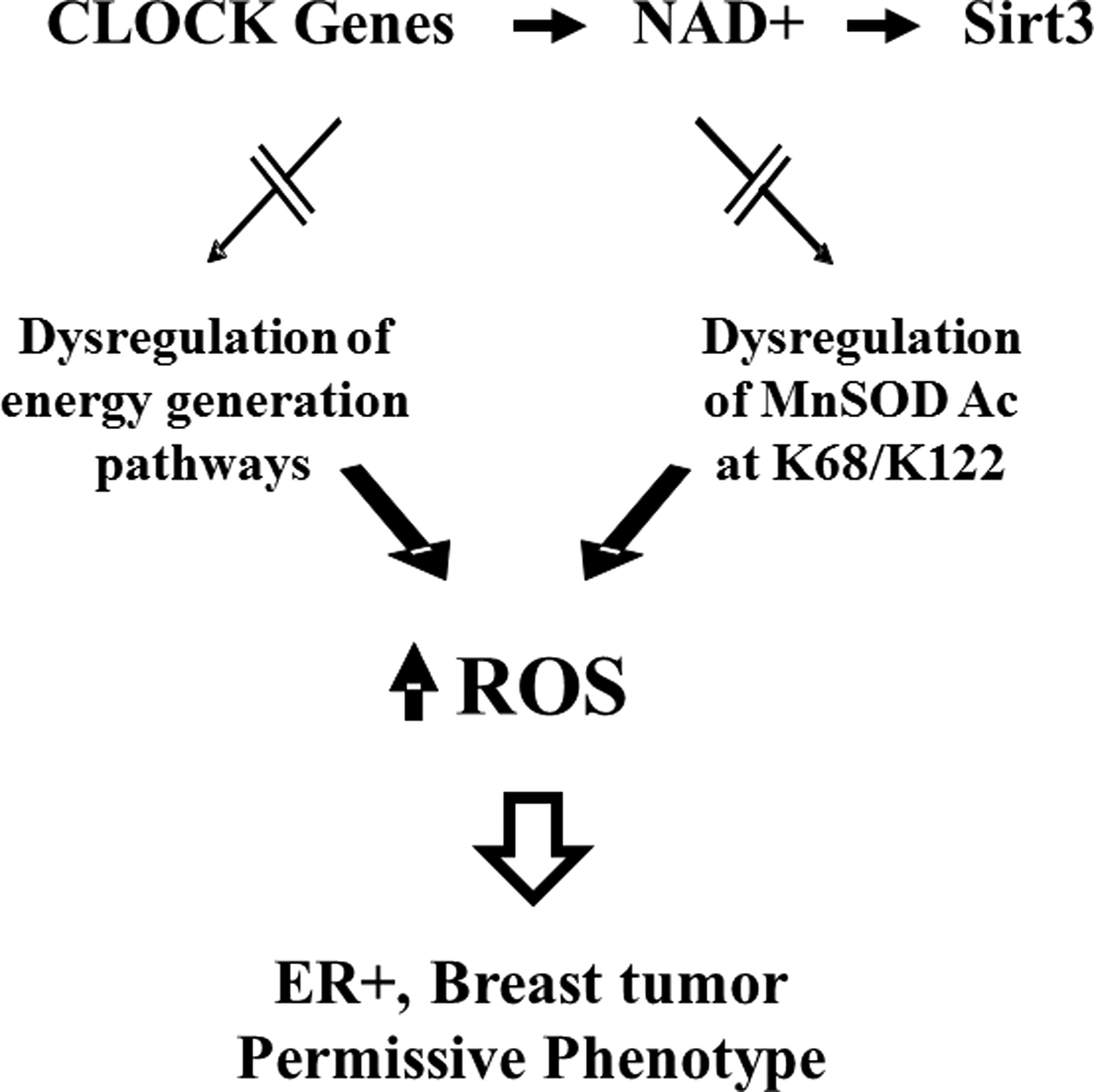
The role of Sirt3 in cellular metabolic stress that directs mitochondrial metabolic reprogramming
Over 60 years ago, Otto Warburg described that tumor cells tend to have aberrant mitochondrial metabolism. Specifically, cancer cells always exhibit higher level of glucose consumption (i.e., glycolysis) when compared with the normal counterparts (86). In this regard, Finley et al. have shown that cells lacking SIRT3 exhibit increased glucose consumption (25). Similarly, Ozden et al. also suggested that loss of SIRT3 could affect the pyruvate dehydrogenase enzymatic activities and promote cells to a more transform phenotype that prefers glycolysis (61). These results suggest that the tumors that develop in mice lacking Sirt3 are a Warburg-like model, providing a potential mechanistic link between mitochondrial acetylome, aging, and carcinogenesis.
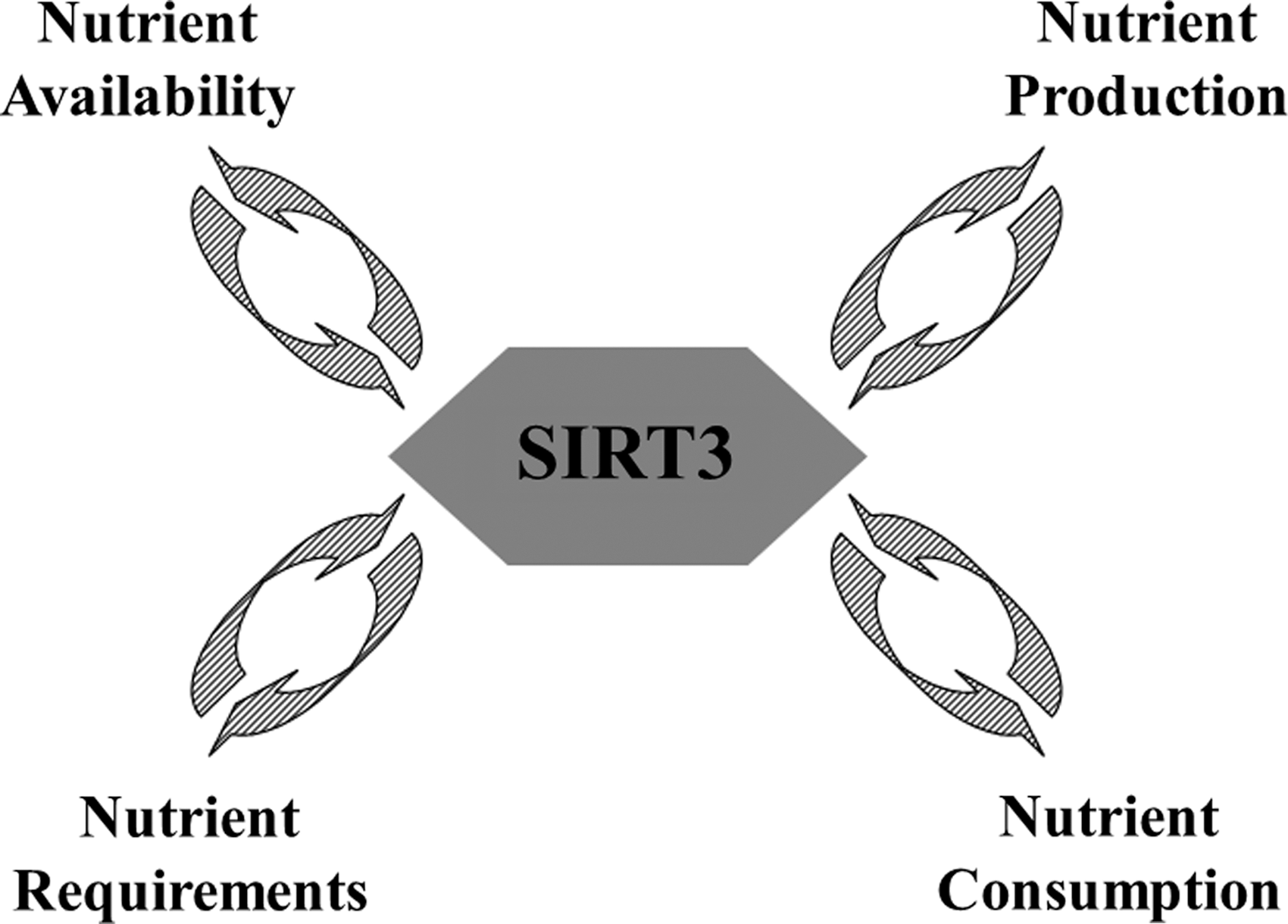
Over the last several years, multiple studies have suggested that SIRT3 plays a critical role in maintaining mitochondrial energy metabolism homeostasis through its deacetylation activity. In this regard, it has been shown that SIRT3 directs the function of tricarboxylic acid cycle activity by deacetylating pyruvate dehydrogenase (61), isocitrate dehydrogenase (IDH) (75), ACS (37, 72), and glutamate dehydrogenase (GDH) (70). It has also been shown that SIRT3 directs ATP production via the deacetylation of electron transport chain subunits (complexes I–III and ATP synthase) (25, 81). SIRT3 also participates in fatty acid metabolism by deacetylating LCAD and HMGCS2 (1, 37, 39). As such, it seems reasonable to propose that SIRT3 and likely other sirtuins sense nutrient status and reprogram mitochondrial metabolism to match energy demand and availability to energy production and consumption (Fig. 4). Thus, loss of Sirt3 results in a mismatch of mitochondrial programming and metabolic stress that favors the accumulation of cell damage. As such, dysregulation of SIRT3 and the subsequent loss of mitochondrial energy coordination result in a cellular environment that is permissive for mammary tissue carcinogenesis (25, 27, 35, 46). Together, these studies strongly support that the deacetylation activity of SIRT3 is an integral part of the signaling pathways in the mitochondria that direct energy metabolic homeostasis.
It is well known that ROS production is closely linked to the mitochondrial energy metabolism. As a result of using oxygen to generate ATP, mitochondria produce ROS as a by-product. Electrons transferred through oxidative phosphorylation (OXPHOS) constitute a major way of ROS production since electrons can leak out of complexes I and III, resulting in one-electron reductions of oxygen to produce the superoxide radical (76). Loss of SIRT3 results in an increase in the acetylation of electron transport chain proteins and induces higher steady-state levels of ROS (25, 81). Thus, it seems reasonable to propose that in response to ROS-induced cellular metabolic stress, SIRT3 is activated and might target the activation of ROS detoxification processes.
In this regard, several studies have shown that MnSOD contains reversible acetyl lysines and that acetylation alters its enzymatic function. These studies also showed that SIRT3 physically interacts with MnSOD and deacetylates MnSOD in cell-free, in vitro, and in vivo (murine) model systems (12, 65, 78). In addition, loss of SIRT3 in different cell lines resulted in increased intracellular and mitochondrial superoxide levels, whereas enforced expression of the wild-type, but not the deacetylation-null, SIRT3 gene decreased cellular ROS and mitochondrial superoxide levels (46, 78). Overall, cells lacking SIRT3 may have dysfunctional coordination of both mitochondrial energy metabolism and detoxification enzymes due to MnSOD hyperacetylation, which can ultimately result in aberrant and potentially damaging ROS production that may have deleterious biological effects.
MnSOD, aging, and cancer
Aberrant and/or prolonged intracellular oxidative stress has been demonstrated to significantly contribute to the aging process and form the central dogma of the Free Radical Theory of Aging. While the targets for this process are likely to be multifactorial, it is well established that increased steady-state levels of ROS damage mitochondrial DNA and proteins that ultimately alter proper function. In this regard, it has been shown that cells lacking ROS scavenging enzymes, such as MnSOD, are more likely subjected to persistent oxidative stress and this has been proposed as one mechanism by which the dysregulation of cellular ROS drives the Free Radical Theory of Aging.
Since MnSOD regulates mitochondrial ROS and increased ROS has been associated with an accelerated aging process, it seems reasonable to propose that changes in MnSOD would connect increased oxidative stress and aging-related diseases. In this regard, loss of MnSOD is embryonically lethal; MnSOD−/− MEFs and MnSOD+/− mice, which have around 60% loss of MnSOD activity, have been used to study the effects caused by loss of MnSOD activity (80). Mice that are monoallelic knockout for MnSOD (MnSOD+/−) mice exhibit elevated superoxide levels as well as aging-related phenotypes, including pathologic heart (88), skin (87), and most importantly carcinogenesis (80, 88). The MnSOD+/− mice were more cancer-prone and exhibited a reduced life span compared with MnSOD+/+ mice, suggesting that the loss of MnSOD activity promotes carcinogenesis. Furthermore, analysis of mitochondrial DNA in breast cancer patients has shown a common deletion of MnSOD in their blood, resulting in an impairment of MnSOD activity and an increase in oxidative damage (58). Overall, a decrease in MnSOD activity can increase oxidative stresses, resulting in aging-related carcinogenesis and promoting breast cancer propagation.
SIRT3 directs MnSOD enzymatic activity via the acetylation lysines 68 and 122
MnSOD was previously regarded as a simple ROS scavenging enzyme and its detoxification activity was stoichiometric depending on the levels of mitochondrial superoxide. However, recent studies have suggested that MnSOD activity can be regulated by several cellular mechanisms, including transcriptional, translational, and, perhaps most importantly, post-translational regulation, depending on the intracellular signals or environmental triggers (18, 40, 41, 51). Furthermore, recognition of specific intracellular physiological conditions and initiation of post-translational signaling cascades by intracellular sensing proteins have been known as a fundamental paradigm in biology (9, 29, 30, 36). Lysine acetylation has recently been regarded as an important post-translational modification mechanism that regulates mitochondrial proteins (13, 48, 49, 52). In this regard, it is logical to hypothesize that MnSOD may contain specific lysine residues, which can be deacetylated by SIRT3.
On this topic, three seminal articles have been published to illustrate how SIRT3 directly affects MnSOD activity through site-specific deacetylation (12, 65, 78). Tao et al. showed that lysine MnSOD K122 is an SIRT3 deacetylation target. When examining the 3D protein structure of MnSOD, lysine 122 is located near the entrance to the MnSOD inner catalytic core. Using site-directed mutagenesis, lysine 122 was mutated to an arginine (positive charge mimicking a deacetylated state, MnSODK122R). This mutation induced a higher level of MnSOD activity and decreased mitochondrial superoxide levels. In contrast, when lysine 122 was mutated to a glutamine (neutral charge mimicking an acetylated state, MnSODK122Q), MnSOD activity decreased and the mitochondrial superoxide level increased. These data validate the electrostatic facilitation model proposed by Dr. Fridovich (7, 78, 89), as published in 1983.
A structural analysis of the MnSOD protein showed that the positively charged lysine residue at position 122 is located very close to the entrance to the catalytic core and ideally oriented to provide superoxide anion attraction. Based on this, we have proposed that the mechanism of increased MnSOD enzymatic activity is due to the attraction of the negatively charged superoxide anion toward the positively charged lysine residues. However, when lysine 122 is acetylated, the electrostatic funnel shows a neutral to negative charge that repels superoxide anion and therefore decreases the possibility of superoxide entering the active site for H2O2 conversion (Fig. 5). In addition, the role of MnSOD lysine 122 acetylation in the process of carcinogenesis has been confirmed using Sirt3 −/− MEFs and in vitro transformation assays (78).

Similarly, deacetylation of lysine 68 was also shown to regulate direct MnSOD enzymatic detoxification activity as a downstream target of SIRT3 (12). In these studies, it was shown that acetylation of MnSOD lysine 68 decreased its detoxification activity, while in contrast, acetylation of MnSOD increased enzymatic activity.
Results showed that SIRT3 was able to deacetylate and further increase the enzymatic activity of MnSOD. It was also shown that when cells were challenged with DMNQ, a reagent that is known to increase mitochondria ROS levels, SIRT3 is induced and leads to MnSOD activation by deacetylation, protecting cells from increased intracellular mitochondrial ROS.
MnSOD acetylation leads to a tumor-permissive phenotype
MnSOD acetylation at lysine 122 results in both a decrease in MnSOD activity and a decrease in ATP production due to impaired OXPHOS (18). Overexpression of MnSOD, on the other hand, increases ATP production and promotes energy generation using OXPHOS (18). Interestingly, it has been previously shown that MnSOD activity is generally lower in cancer cells compared with normal cells (60). One explanation may be provided through the Warburg effect. To provide a rapid ATP supply to cancer cells with defective mitochondria, glucose is converted to lactate through glycolysis and ATP production from glycolysis is more efficient than OXPHOS (86). Therefore, inactivation of MnSOD activity can provide a shift of cellular metabolism from OXPHOS to glycolysis (18, 86).
In addition to its potential role in cellular metabolism, MnSOD can neutralize mitochondrial ROS by converting superoxide to H2O2 and finally to H2O (79) to mediate mitochondrial energetic homeostasis and ROS scavenging (35). A decrease in and/or a loss of SIRT3 enzymatic activity, on the other hand, can result in MnSOD acetylation, which promotes a significant increase in ROS levels and induces aberrant mitochondrial metabolism and a redox imbalance, which may have deleterious biological effects in cells (90). Studies by Spitz and Li have shown that resistance to H2O2 in Chinese hamster fibroblasts results in elevated ROS accumulation as well as genomic instability by nucleic acid peroxidation (77). In addition, high ROS levels can stabilize and activate transcription factors, for example, HIF-1α, which can promote tumor cell proliferation, whereas overexpression of MnSOD can result in a delay in G1-S phase transition, preventing cellular proliferation (5, 35, 71, 82, 89).
Elevated and/or aberrant ROS levels can result in genomic instability, promote transcription of oncogenic genes, and change molecular signaling pathways. These dysregulated cellular processes can promote an oncogenic environment. For example, thymidine phosphorylase, overexpressed in breast cancer, can result in ROS production and accumulation (10). In addition, lactoperoxidase, an enzyme involved in estrogen metabolism, is produced through oxidation of 17-estradiol panoxyl radicals. Overexpression of lactoperoxidase can also result in oxidative stress in the mammary gland, resulting in breast carcinoma (74). Finally, as observed in MCF7 cells, accumulation of ROS can activate mitogen-activated protein kinase pathways, affecting cellular growth and proliferation by promoting many mitogen stimuli-induced progrowth pathways such as ERK and JNK pathways (59).
In addition to the role of elevated ROS in breast carcinogenesis, due to aberrant MnSOD activity, Desouki et al. have found that SIRT3 expression levels are significantly lower in breast cancers. In addition, a decrease in SIRT3 appears to indicate an increased risk for a poor clinical outcome in human breast cancer that is associated with luminal B malignancies (17). These observations suggest that together with SIRT3, MnSOD may protect and/or promote mitochondrial integrity and coordinated energetic homeostasis. On the other hand, acetylation of MnSOD, due to loss of SIRT3 activity, can result in a shift to aerobic glycolysis in cells and ROS accumulation, promoting dysregulation of energy synchrony and tumor-permissive phenotypes. Therefore, this post-translational modification of MnSOD in the form of acetylation can be an indicator for breast cancer tumorigenesis, and more importantly, this observation can provide a potential explanation of how tumors lacking SIRT3 can become resistant to endocrine therapy.
Conclusions
Overall, these results suggest that breast cancer, especially the luminal B subtype, is an aging-related disease. Loss of sirtuins, especially SIRT3, can result in tumor-permissive phenotypes through aberrant regulation of mitochondrial protein acetylation and therefore cells are at risk for oxidative stress (Fig. 6). In this regard, acetylation of MnSOD serves as an integral part in mitochondrial energy homeostasis and aberrant hyperacetylation of MnSOD at lysines 68 and 122, due to loss of SIRT3, can play a role in aging-related cancer, specifically breast cancer. Therefore, detection of MnSOD acetylation at lysines 68 and 122 using lysine-specific antibodies can serve as a potential molecular biomarker for prediction of luminal B breast cancer. In addition, it would be interesting to explore how the transition from luminal A to luminal B breast cancer is connected to loss of SIRT3 activity and hyperacetylation of mitochondrial proteins. Further characterization of mitochondrial proteins that are SIRT3 targets will lead to a better understanding of breast cancer tumorigenesis and may help develop novel therapeutic strategies and improve patient outcomes.

Footnotes
Acknowledgments
D.G. is supported by NCI-2R01CA152601-06A1, NCI-1R01CA152799-01A1, 1R01CA168292-01A1, NCI-1R01CA16383801A1, and the Avon Foundation for Breast Cancer Research and Care Program at the Robert H. Lurie Comprehensive Cancer Center of Northwestern University. We thank Mr. William Nathan for designing and editing the manuscript. Melissa Stauffer, PhD, of Scientific Editing Solutions provided editorial assistance.