
Abstract
Aims:
Reactive oxygen species (ROS) play a pivotal role in different pathologic conditions, including ischemia, diabetes, and aging. We previously showed that ROS enhance miR-200c expression, causing endothelial cell (EC) apoptosis and senescence. Herein, we dissect the interaction among miR-200c and three strictly related proteins that modulate EC function and ROS production: sirtuin 1 (SIRT1), endothelial nitric oxide synthase (eNOS), and forkhead box O1 (FOXO1). Moreover, the role of miR-200c on ROS modulation was also investigated.
Results:
We demonstrated that miR-200c directly targets SIRT1, eNOS, and FOXO1; via this mechanism, miR-200c decreased NO and increased the acetylation of SIRT1 targets, that is, FOXO1 and p53. FOXO1 acetylation inhibited its transcriptional activity on target genes, that is, SIRT1 and the ROS scavengers, catalase and manganese superoxide dismutase. In keeping, miR-200c increased ROS production and induced p66Shc protein phosphorylation in Ser-36; this mechanism upregulated ROS and inhibited FOXO1 transcription, reinforcing this molecular circuitry.
These in vitro results were validated in three in vivo models of oxidative stress, that is, human skin fibroblasts from old donors, femoral arteries from old mice, and a murine model of hindlimb ischemia. In all cases, miR-200c was higher versus control and its targets, that is, SIRT1, eNOS, and FOXO1, were downmodulated. In the mouse hindlimb ischemia model, anti-miR-200c treatment rescued these targets and improved limb perfusion.
Innovation and Conclusion:
miR-200c disrupts SIRT1/FOXO1/eNOS regulatory loop. This event promotes ROS production and decreases NO, contributing to endothelial dysfunction under conditions of increased oxidative stress such as aging and ischemia. Antioxid. Redox Signal. 27, 328–344.
Introduction
H
We previously showed that reactive oxygen species (ROS) increase miR-200c in endothelial cells, causing apoptosis and senescence. The SIRT1/eNOS/FOXO1 regulatory loop plays a key role in endothelial function and the miR-200c role on this loop was unknown. Herein, we demonstrated that miR-200c directly targets SIRT1, eNOS, and forkhead box O1 (FOXO1); consequently, miR-200c decreased nitric oxide (NO) and increased ROS production. In human cutaneous old fibroblasts, in murine femoral arteries of old mice, and in mouse hindlimb ischemia, an increase in miR-200c expression and a decrease in zinc finger E-box binding homeobox 1 (ZEB1), SIRT1, endothelial nitric oxide synthase (eNOS), and FOXO1 were found. These data suggest the possibility that miR-200c may contribute to endothelial dysfunction in aging and in ischemia.
miRNAs are 21–23 nucleotide-long small noncoding RNAs that act predominantly as inhibitor of protein translation in mammals; as an additional effect in most of the cases, they can affect mRNA stability of their gene targets (5). The miRNA/mRNA target recognition is complex and still incompletely understood (5). For miRNA-mRNA-mediated inhibition, a complete pairing between the 3′ untranslated regions (UTRs) of the mRNA target and the seed sequence of the miRNA, a region centered on nucleotides 2–7, is required. Thus, seed pairing is a necessary requirement for most target prediction algorithms.
We previously reported that miR-200 family was upregulated by ROS in ECs (27). The miR-200 gene family consists of five members: miR-200c and miR-141 are clustered on chromosome 12, whereas miR-200a, miR-200b, and miR-429 are clustered on chromosome 1. Moreover, miR-200 family can be divided into two subgroups according to their seed sequences—subgroup I: miR-141 and miR-200a; and subgroup II: miR-200b, miR-200c, and miR-429.
miR-200c was the most upregulated family member and the induction upon in vitro exposure to hydrogen peroxide (H2O2) was not only restricted to ECs but occurred also in other cell types, including murine myoblasts and myotubes, and human cutaneous fibroblasts. An increase in miR-200c in response to enhanced oxidative stress was also found in vivo in mouse skeletal muscles following hindlimb ischemia (27). Finally, we demonstrated that miR-200c overexpression downregulated its target protein zinc finger E-box binding homeobox 1 (ZEB1) and recapitulates oxidative stress-induced phenotype, causing cell senescence and apoptosis (27). Subsequent studies have confirmed that miR-200c is strongly modulated by ROS also in different tumor cells (29, 30).
In the present study, we aimed at dissecting the molecular mechanisms leading to endothelial dysfunction and senescence in response to an increase in miR-200c and we examined three functionally related proteins, which have a major role in vascular homeostasis: silent mating type information regulation 2 homolog (sirtuin 1 or SIRT1), endothelial nitric oxide synthase (eNOS), and forkhead box O1 (FOXO1) transcription factor. SIRT1 is an NAD-dependent deacetylase known to play a pivotal role in cell senescence. SIRT1 has antioxidative and anti-inflammatory effects and has a major role in EC biology (6). Notably, excessive ROS levels or aging decrease SIRT1 expression, causing endothelial dysfunction and senescence (37); furthermore, in ECs, SIRT1 activation ameliorates oxidative stress response, prevents endothelial senescence, and promotes eNOS-derived nitric oxide (NO) bioavailability and mitochondrial biogenesis (6, 37). In turn, NO enhances SIRT1 mRNA and protein stability; therefore, a positive loop exists between eNOS activity and SIRT1 expression level (37).
NO is a key regulatory molecule that controls EC function and vascular tone relaxation (13, 32). Moreover, reduced eNOS expression and/or NO bioavailability are associated with decreased EC survival, vascular smooth muscle cell proliferation, and migration from the media to the intima leading to enhanced neointima accumulation and failure of the arterial system to relax in response to a variety of stimuli, such as an increase in shear stress, inhibition of the angiogenic response to ischemia, and increased thrombogenicity due to platelet aggregation and adhesion. In addition, a decrease in NO has multiple proatherogenic actions, including (i) increased leukocyte adhesion to the vascular endothelium and migration through the capillary wall, (ii) increased influx of lipoproteins into the vascular wall, and (iii) induction of low-density lipoprotein oxidation (25, 26).
A strict link exists between NOS activity and ROS production since NOS uncoupling leads to the production of superoxide anion rather than NO (2, 29, 46). In the eNOS/SIRT1 molecular circuitry, an important role is also played by FOXO1 transcription; FOXO1 is a direct target of SIRT1 deacetylation, which in turn induces its transcriptional activity on SIRT1 promoter as well as on ROS scavenger promoters, that is, catalase (CAT) and manganese superoxide dismutase (MnSOD or SOD2) (12).
Another important player in oxidative stress modulation and response is the p66 isoform of ShcA protein (p66Shc), a fundamental regulator of mitochondrial ROS production by a variety of different stimuli (19).
P66Shc is phosphorylated at the Ser-36 site in response to several stress stimuli, including UV irradiation and H2O2, and this modification is crucial for oxidative stress response and modulation of ROS production (28) by three mechanisms restricted to the nucleus, the plasma membrane, and the mitochondria, respectively: 1. The nuclear mechanism involves a p66Shc-mediated FOXO transcription factor inhibition, leading to the decreased expression of ROS-scavenging enzymes, CAT and MnSOD (33). 2. At the plasma membrane, p66Shc promotes RAC1 activation and triggers NADPH membrane oxidase-ROS production (23). 3. p66Shc acts also in the mitochondrial intermembrane space where it binds to cytochrome c, acting as an oxidoreductase and generating ROS. These ROS, in turn, activate the permeability transition pore, triggering organelle dysfunction, massive release of mitochondrial apoptotic factors, and ROS and eventually inducing cell apoptosis (18).
Given the importance of p66Sch in ROS modulation, we also investigated the role of miR-200c on p66Shc phosphorylation and ROS production.
Results
SIRT1, eNOS, and FOXO1 are modulated by H2O2
We initially established the effect of ROS on SIRT1, eNOS, and FOXO1 mRNA and protein expression in our experimental conditions. To this aim, human umbilical vein ECs (HUVECs) were treated with 200 μM H2O2 for 8, 16, and 24 h. We first evaluated miR-200c expression and confirmed the upregulation at each time point, in keeping with our previous results (27), and an miR-200c peak expression (∼30-fold) at 16-h treatment (Supplementary Fig. S1a; Supplementary Data are available online at

At 8-h treatment, there is a discrepancy between mRNA induction and protein inhibition of SIRT1 and FOXO1, but miRNA action is the inhibition of translation, and in some cases, the decrease in protein is associated with a compensatory increase in mRNA levels.
SIRT1, eNOS, and FOXO1 are direct targets of miR-200c
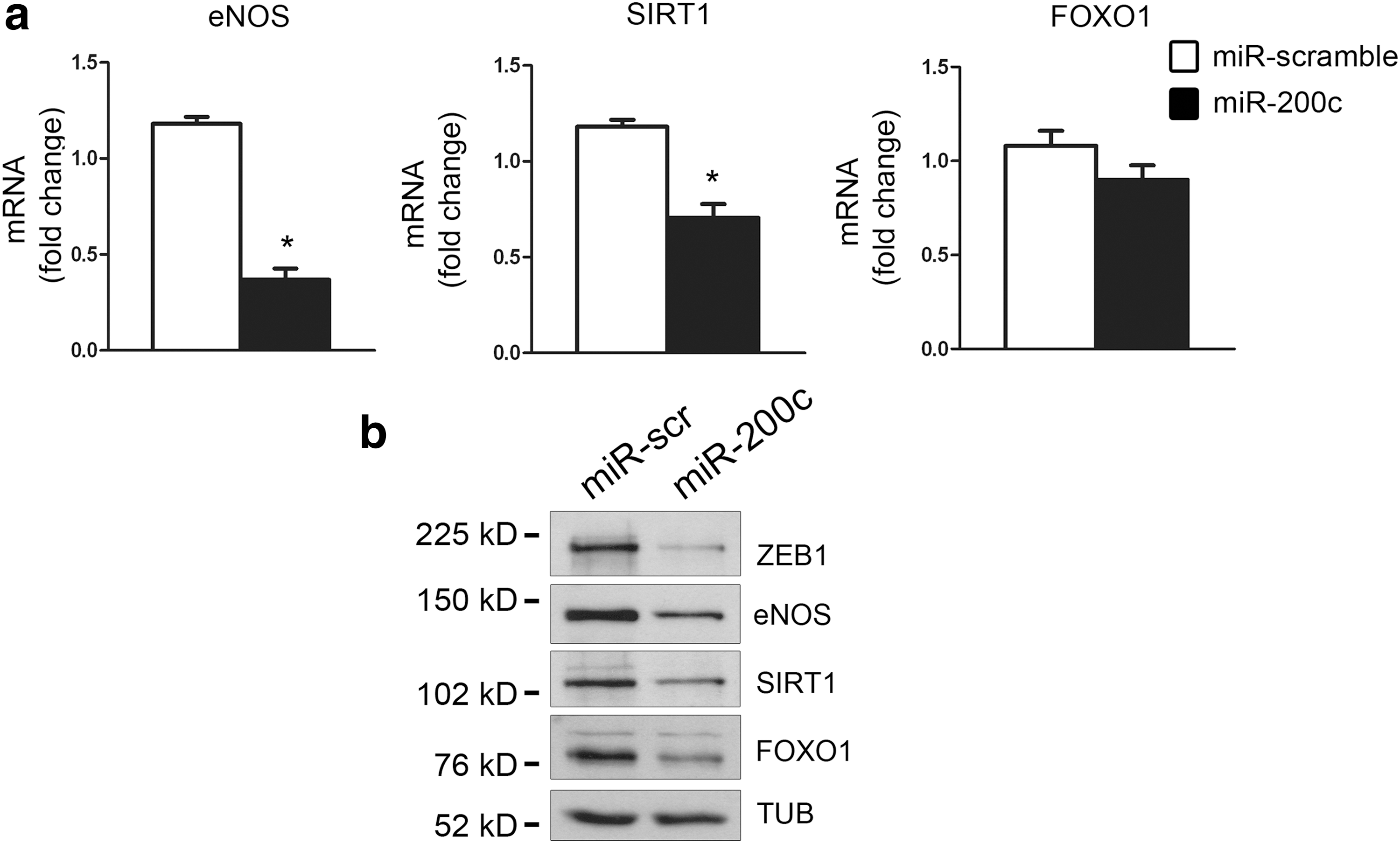
We previously demonstrated that H2O2 treatment in HUVECs induces cellular senescence and apoptosis, at least in part, through a mechanism involving miR-200c upregulation (27). Given the importance of SIRT1, eNOS, and FOXO1 in EC function, we established whether oxidative stress-dependent increase in miR-200c modulates these targets. HUVECs were transduced with lentiviruses expressing either pre-miR-200c or a control sequence. We observed a significant downmodulation of SIRT1 and eNOS mRNA (Fig. 2a) and protein (Fig. 2b and Supplementary Fig. S2a for quantification) upon miR-200c overexpression; in contrast, FOXO1 mRNA was not affected (Fig. 2a), whereas FOXO1 protein levels exhibited a significant decrease (Fig. 2b and Supplementary Fig. S2a for quantification). Similar results were also observed in human embryonic kidney 293 (HEK293) cells (Supplementary Fig. S2b, c) and C2C12 murine myoblasts (Supplementary Fig. S2d, e). Furthermore, in all cell types, ZEB1 protein decreased, as expected, upon miR-200c forced expression (Fig. 2b and Supplementary Figs. S1b and S2b–e).
In silico analyses indicated that SIRT1, eNOS, and FOXO1 are all potential targets of miR-200c. Specifically, computational miRNA target analysis revealed that SIRT1 has two predicted seed sequences for miR-200c in its 3′UTR (seed a and seed b) (Supplementary Fig. S3). In contrast to SIRT1, eNOS and FOXO1 have only one seed sequence for miR-200c in their 3′UTRs (Supplementary Fig. S3). We cloned each miR-200c seed complementary sequence and the surrounding nucleotides of SIRT1, eNOS, and FOXO1 3′UTRs downstream of a luciferase open reading frame (pLUC-miR-200c seed wt); additionally, we also generated the luciferase constructs deleted of the miR-200c binding site (pLUC-miR-200c seed del) to define their effective targeting by miR-200c.
We found that miR-200c inhibited all the pLUC-miR-200c seed wt constructs, but not the pLUC-miR-200c seed del constructs (Fig. 3a).
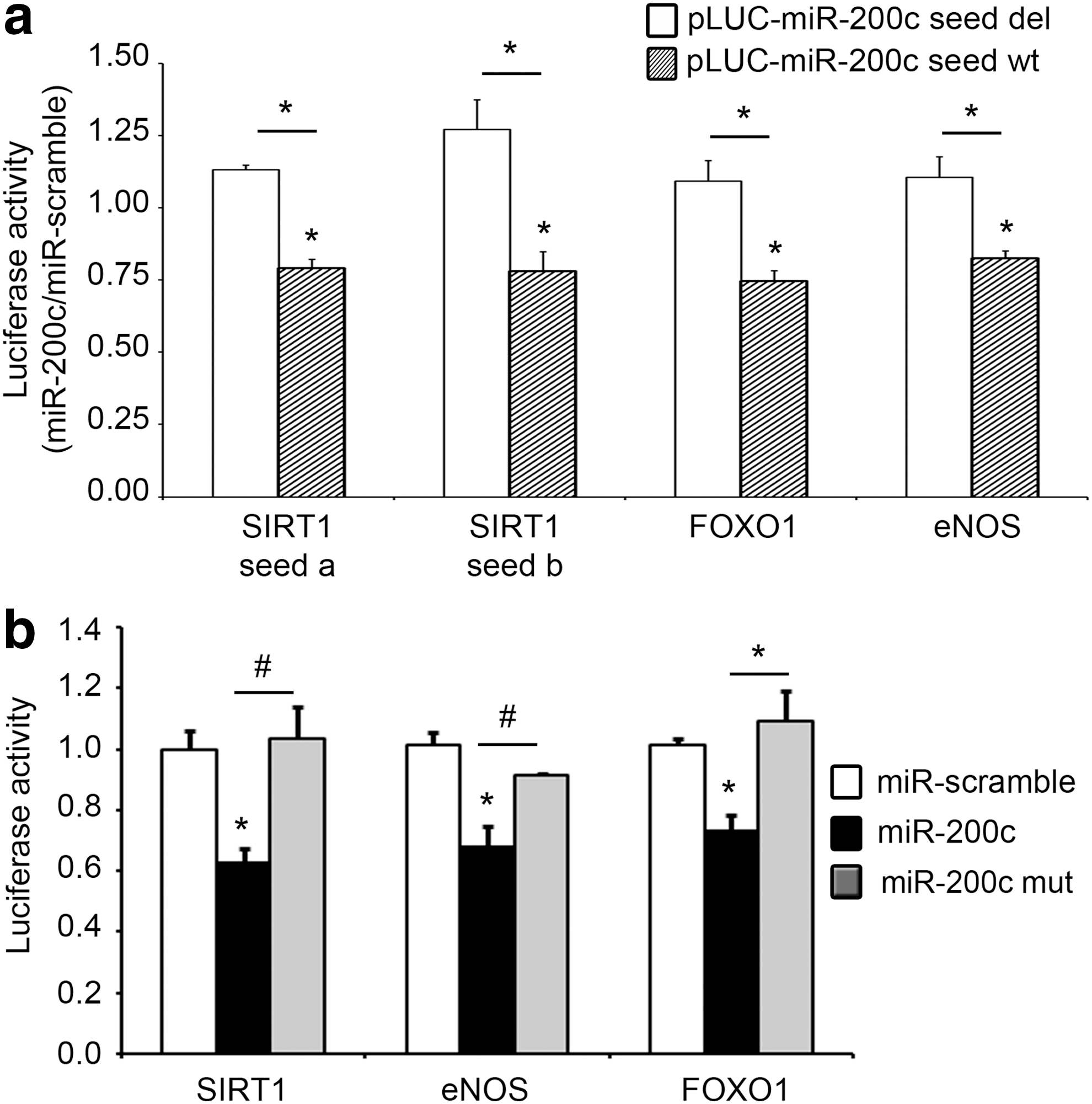
We also performed these experiments with the complete 3′UTRs of SIRT1, eNOS, and FOXO1, and we observed the downmodulation of the luciferase activity for all constructs upon miR-200c overexpression (Fig. 3b). To gain specificity, we used miR-200c mutated in the seed sequence (miR-200c mut) as a negative control and we did not observe any decrease of luciferase activity with any of the 3′UTRs (Fig. 3b). As expected, the complete 3′UTR of SIRT1, which contains both seed a and b sequences, was more efficiently downmodulated by miR-200c overexpression than the reporters containing only a single binding site (∼40% reduction compared with ∼20%) (Fig. 3a, b); in contrast, the constructs with the complete 3′UTRs of eNOS and FOXO1 exhibited a downmodulation of luciferase activity comparable with the constructs containing only the seed sequence, that is, ∼20% (Fig. 3a, b).
Taken together, these results show that SIRT1, eNOS, and FOXO1 are all direct miR-200c targets.
miR-200c upregulation is required for SIRT1, eNOS, and FOXO1 downmodulation by H2O2
We investigated whether miR-200c upregulation was an integral part of H2O2-induced downmodulation of SIRT1, eNOS, and FOXO1. To this end, ROS-induced miR-200c was antagonized with anti-miR-200c and the protein levels of its targets were quantified. HUVECs transfected either with anti-miR-200c or with scramble oligonucleotides were exposed to H2O2 for 8 h (Fig. 4a, b). miR-200c inhibition caused, per se, a slight increase of SIRT1, eNOS, and FOXO1 protein levels, although not significant, while upon H2O2 exposure, SIRT1, eNOS, and FOXO1 protein decrease was significantly attenuated in anti-miR-200c-transfected cells versus control cells (Fig. 4a, b). As expected, we also observed a comparable attenuation of ZEB1 protein downmodulation in cells treated with anti-miR-200c upon H2O2 exposure versus control cells (Fig. 4a, b).

The same results were obtained using another source of oxidative stress, that is, the alkylating agent 1,3-bis(2 chloroethyl)-1-nitrosourea (BCNU), a glutathione reductase inhibitor that blocks the conversion of oxidized to reduced glutathione (17).
We have already shown that 2-h treatment of BCNU upregulates miR-200c in HUVECs and downregulates ZEB1 protein (27). Using the same experimental conditions, we found that BCNU downregulated ZEB1, eNOS, and SIRT1 proteins and that anti-miR-200c treatment partially prevented their expression decrease (Supplementary Fig. S4a, b). FOXO1 protein upon BCNU exposure, differently from H2O2 treatment, was also post-translationally modified, as detectable by the slower mobility band in BCNU-positive lanes. Anyhow, also in this case, there was a decrease in protein levels that was reverted by anti-miR-200c treatment (Supplementary Fig. S4a, b).
miR-200c overexpression inhibits NO production
Since we showed that miR-200c downmodulates eNOS protein levels, we then asked whether miR-200c also inhibits NO production in HUVECs.
In view of the fact that HUVECs produce very low amounts of NO in vitro in basal conditions, that is, in the absence of shear stress forces (35), we used cilostazol, a drug that stimulates eNOS activity, and hence NO production, via a cyclic AMP/PKA- and phosphatidylinositol 3-kinase/Akt-dependent mechanism (21), to appreciate differences in NO modulation upon miR-200c overexpression.
Moreover, we used two different methods to determine NO production, an indirect one, measuring nitrite formation, and a direct one, DAF2 fluorescence.
In both cases, we found that miR-200c overexpression inhibited NO levels both under baseline conditions and in response to 30 μM cilostazol (Fig. 5a, b).
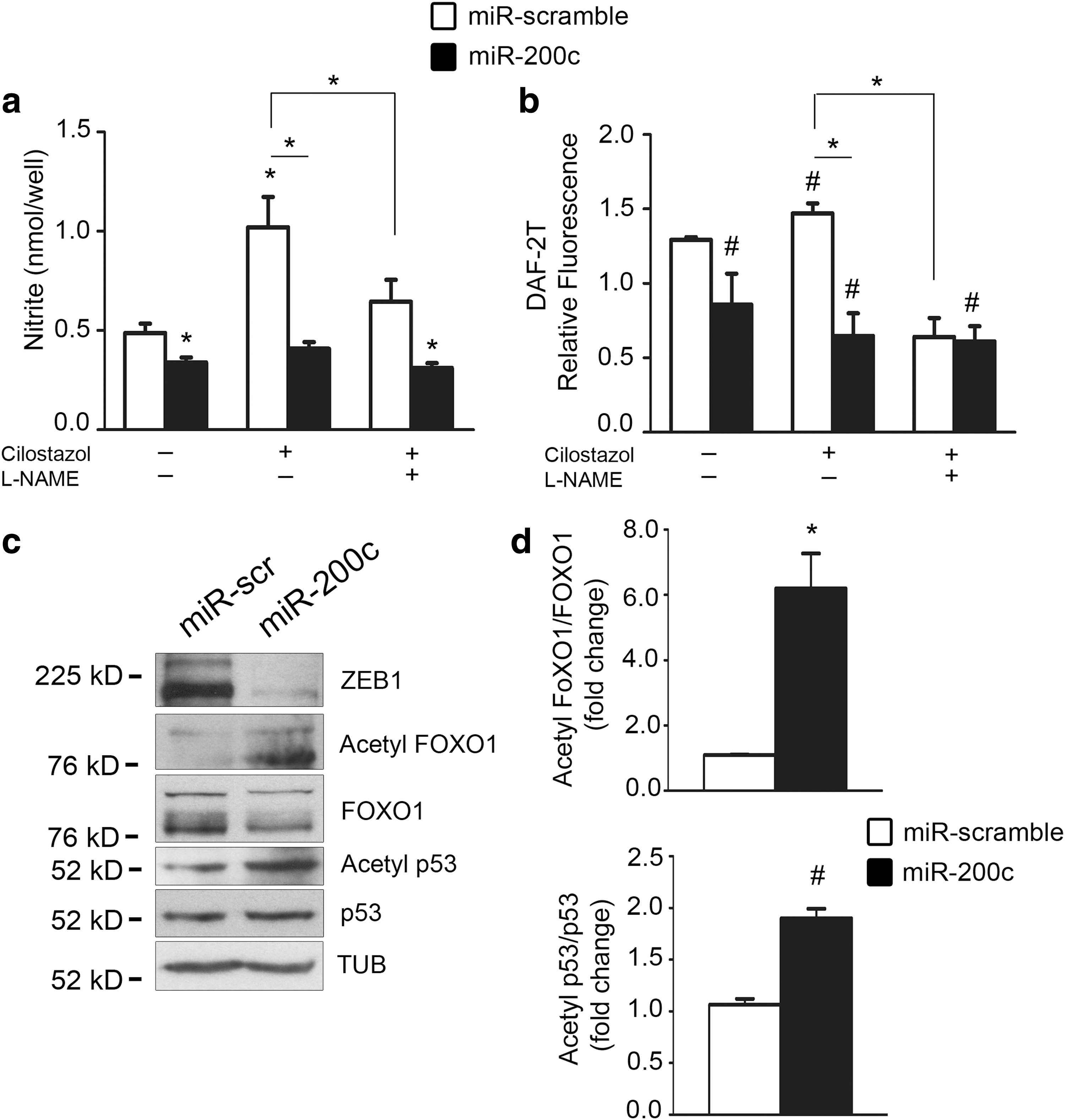
Furthermore, to assess whether NO formation was eNOS specific, we used an NOS inhibitor, NG-nitro-L-arginine methyl ester (L-NAME). We found that NO production upon cilostazol decreased both in scramble and in miR-200c-overexpressing HUVECs (Fig. 5a, b).
miR-200c modulates SIRT1 target acetylation, further enhancing ROS production
Since SIRT1 is a deacetylase, SIRT1 protein downregulation, caused by miR-200c, should be associated with enhanced acetylation on SIRT1 targets.
Among SIRT1 targets, we first analyzed FOXO1 and found that FOXO1 lysine acetylation increased in miR-200c-overexpressing HUVECs (Fig. 5c, d). Moreover, miR-200c overexpression in HUVECs induced also the acetylation of p53, another important SIRT1 target (Fig. 5c, d).
Since FOXO1 acetylation causes detachment of this transcription factor from MnSOD and CAT promoters (12), via this mechanism, the production of these ROS scavengers is expected to diminish and oxidative stress is expected to increase.
We therefore examined whether miR-200c, regulating SIRT1 and FOXO1 protein expression, can induce oxidative stress. To this aim, HUVECs were transduced with miR-200c or miR-scramble and oxidative stress was evaluated by a dihydroethidium (DHE) red/ox-sensitive fluorescent probe. We found that miR-200c overexpression enhanced oxidative stress (Fig. 6a, b). To understand whether miR-200c elicits peroxide species production, we used a specific H2O2 assay that contains a substrate capable of reacting with H2O2, generating a measurable luciferin product. As shown in Figure 6c, we observed a significant increase in H2O2 production upon miR-200c overexpression in HUVECs compared with scramble control cells.

In light of these results, we examined whether miR-200c modulated the mRNA expression of different ROS scavengers: superoxide dismutase 1 (SOD1) and MnSOD (SOD2), CAT, and peroxiredoxin 2 (PRDX2) (Fig. 6d). We found that all ROS scavengers, but SOD1, were significantly downmodulated by miR-200c (Fig. 6d). Interestingly, PRDX2 is a known miR-200c direct target (9), and SOD2 and CAT are both transcribed by FOXO1 (12). Moreover, PRDX2 and CAT proteins were downmodulated in miR-200c-overexpressing HUVECs (Supplementary Fig. S5a, b).
As a further confirmation that miR-200c plays a major role in ROS production, we found that miR-200c overexpression in HUVECs induced p66Shc phosphorylation in Ser-36 (Fig. 6e, f).
We also asked whether protein target downregulation by miR-200c was, at least in part, caused by ROS formation induced by miR-200c. To this aim, we treated HUVECs overexpressing miR-200c with 10 mM N-acetyl-L cysteine for 8 h and, in these experimental conditions, we did not find any increase of protein targets (data not shown).
miR-200c is upregulated with aging
Increased oxidative stress has been associated with the aging process and with a decrease of SIRT1 protein levels (37). Since oxidative stress enhances miR-200c, which in turns decreases SIRT1 protein expression, we established whether the levels of miR-200c increased with aging. Specifically, we assayed skin fibroblasts from five healthy young donors (4–6 years old) and six old subjects (65–80 years old) at low culture passage (p4-p5).
We found that miR-200c was significantly induced in skin fibroblasts from old versus young donors (Fig. 7a). Moreover, we assayed the mRNA expression of miR-200c target genes ZEB1, SIRT1, FOXO1, and eNOS (Fig. 7b) and found that all of them were significantly downmodulated in fibroblasts of old compared with young subjects; it is noteworthy that eNOS was very low expressed and notwithstanding this, eNOS mRNA decrease was still present (Fig. 7b).
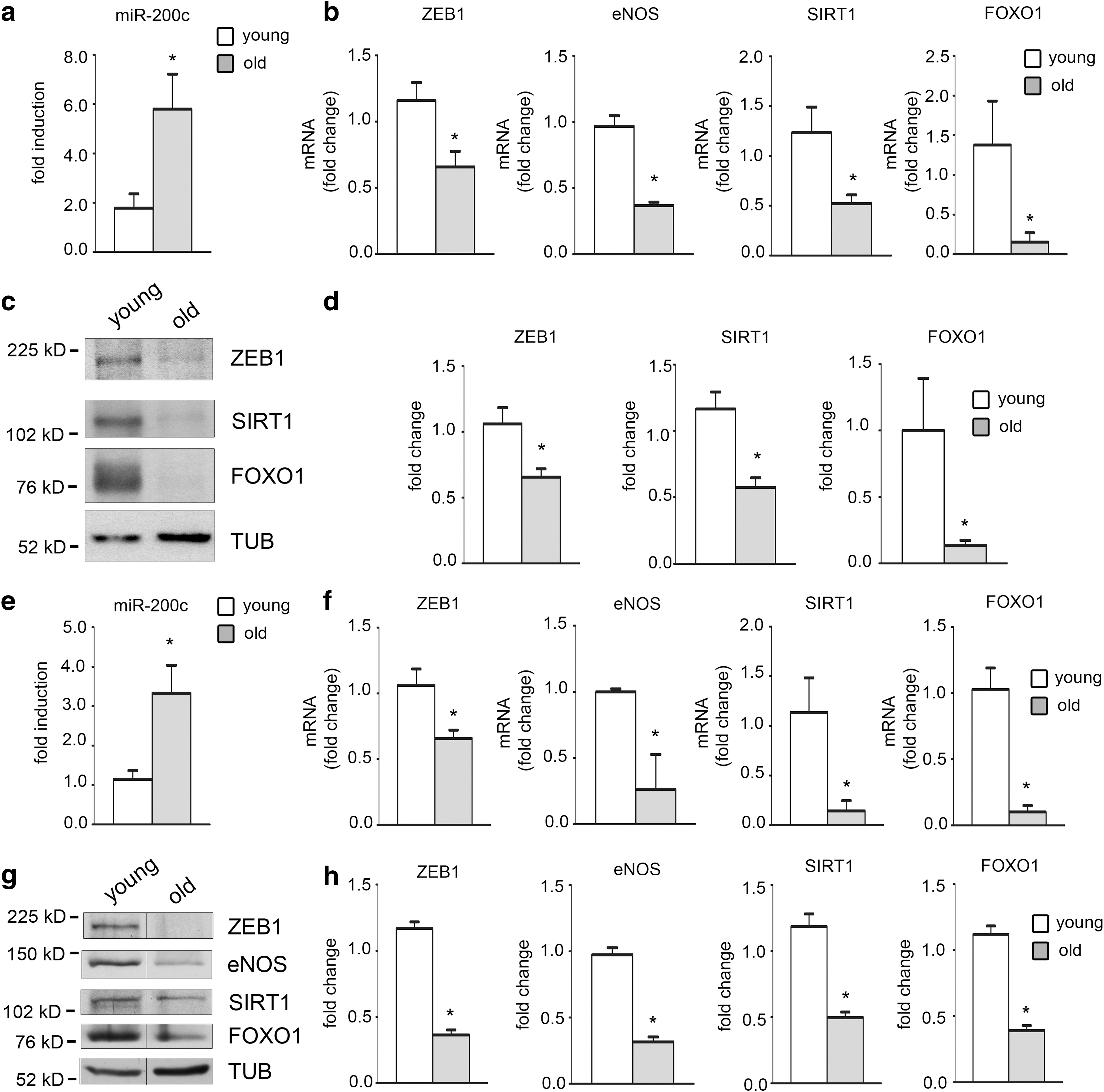
We also confirmed these results by protein expression analysis (Fig. 7c, d). We found that ZEB1, SIRT1, and FOXO1 were all downmodulated in fibroblasts from old versus young donors (Fig. 7c, d); eNOS could not be assessed since the protein was undetectable in these cells.
Interestingly, we also found a decrease of PRDX2 and CAT proteins in fibroblasts from old compared with young donors (Supplementary Fig. S5c, d). These results are in agreement with the age-dependent increase of oxidative stress (11).
We confirmed these data also in femoral arteries of 8 young (2–3 months) versus 7 old mice (18–21 months). We found an upregulation of miR-200c and a downregulation of mRNA and proteins of ZEB1, SIRT1, FOXO1, and eNOS in old femoral arteries compared with young (Fig. 7e–h).
miR-200c and its targets are modulated in ischemia
We have previously shown that miR-200 family is induced in the skeletal muscle of a mouse model of acute hindlimb ischemia and that this induction was dependent on ROS production caused by ischemia (27). Thus, in the present work, we established whether in the same model of hindlimb ischemia, miR-200c induction was associated with a decrease of its target genes.
In agreement with our prior study (27), we confirmed that miR-200c was upregulated at 1, 2, and 5 days after acute hindlimb ischemia (Fig. 8a). We then assayed the mRNA and protein expression levels of its targets and found that ZEB1, eNOS, SIRT1, FOXO1, and ZEB1 were all significantly downregulated 5 days after surgery (Fig. 8b–d).

Interestingly, we also found a decrease of PRDX2, CAT, and SOD2 proteins upon 5 days of hindlimb ischemia (Supplementary Fig. S5e, f). These results are in agreement with the increase of oxidative stress upon ischemia (47).
Anti-miR-200c treatment in mice rescues hindlimb ischemia effects on its targets and improves perfusion
To better characterize the role of miR-200c in ischemia and to give a stronger cause–effect role on its targets, we performed an anti-miR-200c treatment in mice upon hindlimb ischemia.
We first assessed whether miR-200c levels were decreased upon anti-miR-200c treatment. We found a significant downregulation of miR-200c upon ischemia (Supplementary Fig. S6a). We found that anti-miR-200c treatment was able to prevent the decrease of mRNA (Fig. 9a) and protein levels of miR-200c targets upon ischemia (Fig. 9b and Supplementary Fig. S6b for quantification).
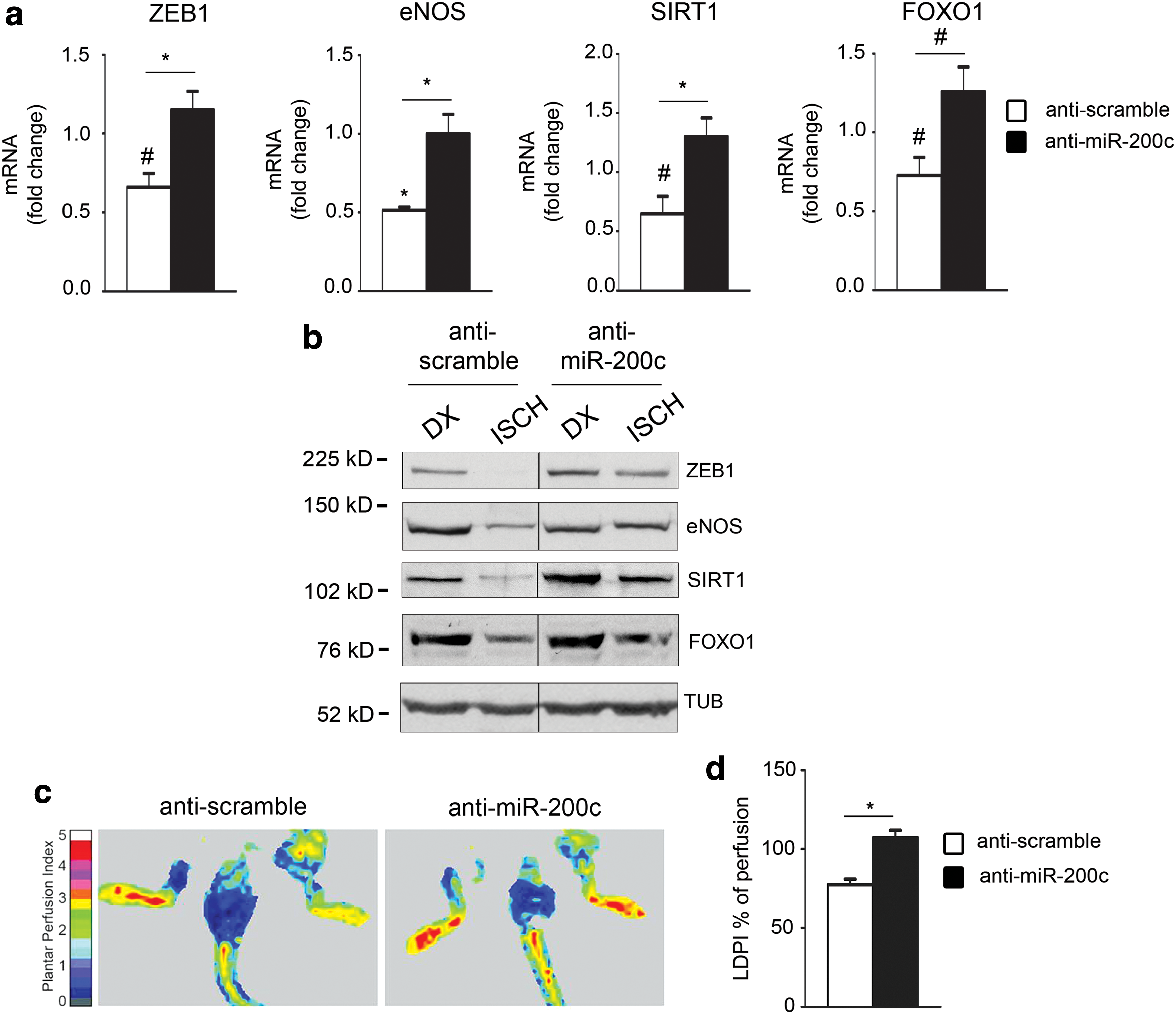
We also performed a laser Doppler perfusion imaging (Lisca) analysis and we found that anti-miR-200c treatment was able to revert completely the decrease of perfusion ratio (ischemic limb/normoperfused limb) 5 days after surgery compared with antiscramble control mice (Fig. 9c, d).
These data clearly demonstrated that antimiR-200c administration ameliorates perfusion of the ischemic limb and the expression of ZEB1, eNOS, SIRT1, and FOXO1 mRNA and proteins.
Discussion
Oxidative stress plays a causal role in aging and in a variety of age-associated diseases, including atherosclerosis, ischemia and ischemia/reperfusion of multiple tissues and organs, Alzheimer's disease, diabetes, and age-related macular degeneration (20). ROS have been shown to affect cell signaling, triggering apoptosis, cell senescence, and endothelial dysfunction (31).
In the present study, we aimed at elucidating the role played by oxidative stress-induced miR-200c on the important regulatory loops among SIRT1, eNOS, and FOXO1. We have focused on these proteins because of their key role in endothelial function, cell senescence, and oxidative stress endurance (28, 37). In addition, we examined whether miR-200c, which is induced by ROS, modulated oxidative stress.
In this study, we demonstrated that SIRT1, FOXO1, and eNOS are direct targets of miR-200c. Moreover, we found that miR-200c overexpression downmodulates different ROS scavengers and also induces p66Shc protein phosphorylation in Ser-36 residue, enhancing oxidative stress.
A complex regulatory loop is involved in the response to miR-200c. The miR-200c-dependent downmodulation of SIRT1 and FOXO1 protein levels is also associated with FOXO1 hyperacetylation, an SIRT1 target. This leads to FOXO1 detachment from the promoter sequences of SIRT1, further decreasing SIRT1 expression. Moreover, FOXO1 transcribes the ROS scavengers, CAT and MnSOD (12), and FOXO1 acetylation and diminished expression also lead to a decrease in CAT and MnSOD mRNA expression.
It is important to underline that p66Shc phosphorylation in Ser-36 induces its translocation in the nucleus, where it inhibits FOXO1 transcriptional activity (33); therefore, miR-200c phosphorylating p66Shc inhibits FOXO1 transcription also via a different mechanism, further decreasing ROS scavenger transcription (33). Moreover, miR-200c-dependent phosphorylation in Ser 36 of p66Shc elicits oxidative stress production by two other mechanisms: (i) activating RAC1 and triggering NADPH membrane oxidase-ROS production (23) and (ii) in the mitochondria by p66Shc binding to cytochrome c, acting as an oxidoreductase and generating ROS (18).
Another mechanism by which miR-200c enhances ROS production is by directly targeting PRDX2 (9).
PRDX2 is one of the six members of the mammalian peroxiredoxin family and acts as a selective scavenger for H2O2 having an efficiency similar to CAT or glutathione peroxidase for this reactive species (36).
PRDX2 is also known to regulate several signaling pathways in different cell types (24, 42, 43).
Interestingly, PRDX2 positively regulates the VEGFR activation state in vascular ECs and its downregulation has been shown to suppress tumor angiogenesis in vivo (22). Finally, recent molecular evidence suggests that PRDX2 may have an important role in the crosstalk between oxidative metabolism and cellular molecular clock, probably by modulating the activity of Sirt1 in the nucleus (3, 4, 38).
Our results showed that PRDX2 mRNA and protein were downmodulated by miR-200c overexpression in HUVECs, corroborating the already demonstrated role of miR-200c in angiogenesis inhibition (7, 41) and on SIRT1 activity via a PRDX2-dependent mechanism.
In summary, our results show that miR-200c enhances oxidative stress and this effect is associated with downmodulation of ROS scavengers and p66Shc phosphorylation in Ser-36.
Our data, obtained in primary ECs, are in agreement with previous studies that linked miR-200 family increase with ROS production in tumor cells (9, 30).
Another consequence of miR-200c-induced SIRT1 downmodulation is the increased acetylation of p53, another important SIRT1 target (40, 45). An increase in p53 acetylation has been demonstrated to play a positive role for its transcriptional activity and also in apoptosis induction via a p53 transcription-independent pathway and to provoke an increase in ROS (45). Moreover, p53 acetylation has been also shown to be important in DNA damage responses (40). In keeping, we previously showed an increase in apoptosis upon miR-200c overexpression and also a p53 transcriptional role for miR-200c induction upon H2O2 treatment (27). Therefore, our results might suggest a positive feedback loop of miR-200c in sustaining its induction by H2O2 via a p53 hyperacetylation mechanism.
Interestingly, it had already been shown that miR-200a, another miR-200 family member, directly targets SIRT1, and by this pathway, miR-200a modulates epithelial-to-mesenchymal transition in cancer development (14). However, there were no prior studies on miR-200 family and SIRT1 interaction in nontransformed cells. It is important to underline that miR-200a displays a different seed sequence from miR-200c. Our results demonstrate that miR-200c also targets SIRT1. We previously showed that the entire miR-200 family is induced by oxidative stress (27); consequently, since both miR-200 family seed sequences (subgroup I and II; see Introduction) are present in SIRT1 3′UTR, the entire miR-200 family targets SIRT1 upon oxidative stress.
Indeed, miR-200a seed sequence is conserved in humans and mice, whereas human miR-200c seed sequences are not conserved in mice. Notwithstanding this, two very close seed sequences of subgroup II are present in the 3′UTR of the murine SIRT1 gene. Therefore, SIRT1 is a likely miR-200c target also in mice. As a further confirmation, we observed downmodulation of SIRT1 also in murine myoblasts upon miR-200c overexpression.
Our previous results showed that miR-200c induced a strong increase in HUVEC senescence (27). It is well established that aging is associated with increased oxidative stress (11), SIRT1 expression levels decrease with oxidative stress and aging, and that SIRT1 decrease is associated with cell senescence in vitro (37). Therefore, we examined whether miR-200c increased with aging. Interestingly, we found that miR-200c levels increase in human fibroblasts (HFs) of old versus young donors and also in femoral arteries of old mice compared with young mice. In keeping with the results obtained in HUVECs upon miR-200c overexpression, we found a decrease in PRDX2 and CAT protein levels in old fibroblasts compared with young fibroblasts, raising the possibility that the age-dependent oxidative stress increase may be due, at least in part, to higher H2O2 levels being PRDX2 and CAT H2O2 scavengers.
Moreover, the results of the present study indicated that also in a physiological model of oxidative stress, such as aged fibroblasts, there is upregulation of miR-200c expression and decrease of SIRT1, FOXO1, and ZEB1.
Since in fibroblasts eNOS protein was not detectable, we confirmed these data also in femoral arteries derived from young and old fibroblasts, showing that in this case also, miR-200c was upregulated and ZEB1, SIRT1, eNOS, and FOXO1 were inhibited.
Further experiments are needed to demonstrate a clear role of miR-200c not only in aging, but it is interesting to note that also in another condition associated with an increase of ROS, such as aging, miR-200c and its targets are modulated.
FOXO1 had been shown to negatively regulate eNOS expression (1), therefore miR-200c targeting FOXO1 should induce eNOS transcription; in contrast, we found that miR-200c targets eNOS directly, with the ultimate effect of inhibiting NO production both under baseline conditions and upon eNOS activation.
Indeed, our results are in agreement with a previous study that showed that SIRT1 activation with resveratrol induces eNOS transcription via FOXO1 and forkhead box O3 (FOXO3) transcription factors (44). In that article, the importance of FOXO transcription factors in the positive transcriptional regulation of eNOS in ECs has been underlined (44).
We also examined the effect of ischemia on miR-200c targets. In keeping with our previous study (27), we confirmed an miR-200c increase in skeletal muscles of mice with acute hindlimb ischemia. We also found that ZEB1, SIRT1, FOXO1, and eNOS were downmodulated upon ischemia as well as PRDX2, CAT, and SOD2.
The increase of miR-200c has been recently related to the increase in oxidative stress in diabetes and has been recently demonstrated to play a major role in vasoconstriction via a ZEB1-dependent pathway finally leading to the increase in inflammation (48); moreover, a role in angiogenesis inhibition and EC migration inhibition was also described (7, 41) since miR-200c inhibits the VEGF pathway targeting its receptor VEGFR2 (41).
In keeping, we found that miR-200c-overexpressing HUVECs do not migrate in response to VEGF (data not shown). Moreover, we found that in ischemic mice treated with anti-miR-200c, the inhibition of miR-200c ameliorated perfusion in the ischemic limb, at least in part, because of the restoration of regulatory loop among eNOS/SIRT1/FOXO1. This loop, in fact, is deeply implicated in the modulation of ROS and in the production of NO, which play a major role in vascular tone relaxation.
As summarized in Supplementary Figure S7, we show that the oxidative stress-induced miR-200c inhibits SIRT1, FOXO1, and eNOS expression in ECs. Furthermore, miR-200c directly targets PRDX2 and induces p66Shc phosphorylation in Ser36 that enhances ROS production in different ways, one of which is by inhibiting FOXO1 transcriptional activity. FOXO1 inhibition, in turn, downmodulates CAT and MnSOD expression; via all these mechanisms, miR-200c sustains ROS production (Supplementary Fig. S7). These findings raise the possibility that miR-200c and possibly also other miR-200 family members, by disrupting the SIRT1/FOXO1/eNOS loop, contribute to endothelial dysfunction found in conditions associated with enhanced oxidative stress, such as aging, ischemia, reperfusion, and diabetes.
Materials and Methods
Cell culture
Cells were cultured in a humidified atmosphere of 5% CO2, 95% air. HUVECs (Clonetics) were grown in EGM-2 (Cambrex). HEK293 cells (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Euroclone). C2C12 myoblasts were cultured in DMEM supplemented with 20% FBS and 100 U/ml penicillin and 0.1 mg/ml streptomycin.
Fibroblast isolation and culture
Cultures of HFs were established from foreskin tissue of five healthy young donors (<10 years) and from abdominal or breast plastic surgery of six healthy old donors (>65 years) without any comorbidities (such as diabetes and ischemia). Procedures were performed in accordance with the ethical standards of the Committee on Human Experimentation of IDI-IRCCS. The study was conducted according to the Declaration of Helsinki principles.
Briefly, full-thickness skin biopsies were cleaned with surgical scissors to remove most of the subcutaneous tissue and the remaining skin was finely minced into small pieces. These pieces were placed in a 25-cm2 culture flask and cultured in fibroblast growth medium (DMEM containing 10% FBS, 50 IU-50 μg/ml penicillin–streptomycin and 4 mM glutamine) to obtain fibroblast cultures. The subconfluent primary HF cultures were amplified twice in a 1:6–1:7 split ratio. Subconfluent tertiary HF cultures were used for further molecular experiments.
miRNA target prediction
Computational prediction of miR-200c target genes was done using published algorithms: TargetScan (
Western blot analysis
Cells were lysed in a buffer containing 100 mM Tris (pH 6.8), 20% glycerol, and 4% sodium dodecyl sulfate (SDS). Protein concentration was determined by BCA protein assay kit (Pierce). Then, dithiothreitol 200 mM was added and lysates were boiled for 5 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membrane by standard procedures. The membranes were blocked with 5% nonfat dry milk powder in 0.05% Tween 20 phosphate-buffered saline (PBS-T) for 1 h. Immunodetection was performed by incubating the membranes with different primary antibodies for 2 h at room temperature or overnight at 4°C. After four washes with PBS-T, the membranes were incubated with secondary antibody conjugated with horseradish peroxidase for 1 h. After four washes, blots were developed with Amersham ECL Plus Western Blotting Detection Reagents and membranes were exposed to Amersham Hyperfilm ECL (GE Healthcare Life Sciences). Protein levels were evaluated by densitometric analysis using a GS-710 scanner (Bio-Rad) and Quantity One software (Bio-Rad). Protein expression was normalized for α-tubulin protein levels.
The following primary antibodies were used to detect the proteins of interest: anti-ZEB1 (H-102), anti-SIRT1 (H-300), anti-FOXO1 (H-128), and anti-acetyl-FOXO1 (D-19) (Santa Cruz Biotechnology); anti-eNOS, anti-CAT (Calbiochem), and anti-PRDX2 (EPR5154) (Abcam); anti-acetyl-p53 (Lys382) (Cell Signaling Technology); anti-p53 (DO1) and anti-α tubulin (Ab-1) (Oncogene Research Products); anti-SHC (Transduction Laboratories); and antiphopsho p66-pSer-36 (6E10) (Abcam).
Drug treatments
Cells were treated with the following drugs: H2O2 Perdrogen (30% wt/wt solution; Sigma) was administered to the cells as a 100 mM solution in PBS; cilostazol 30 μM in dimethyl sulfoxide (Sigma). N-acetyl-L-cysteine (Sigma) and BCNU (Sigma) were dissolved in water.
miRNA overexpression
Stable expression of miR-200c or miR-scramble in HUVECs, HEK293, and C2C12 cells was generated by viral infection using lentiviral supernatants. These viruses were produced as previously described (16). In summary, cells were infected with lentiviral virus for 2 h and then were recovered in complete fresh medium for 24 h. Afterward, infected cells were selected by puromycin-containing medium (Sigma) for 72 h. miR-200c overexpression was controlled by quantitative real-time PCR (RT-qPCR) (see methods below).
Anti-miRNAs
Locked nucleic acid oligonucleotides against miR-200c or a control sequence (Exiqon) were transfected by siRNA transfection reagent (Santa Cruz Biotechnology) in 40% confluent HUVECs (5000/cm2) at the final concentration of 40 nM. After 16 h, cells were incubated with fresh medium, and then 200 μM H2O2 was added to the cells for additional 8 h. The expression level of miR-200c was evaluated by qRT-PCR in HUVECs transfected with anti-miR-200c and compared with control sequence transfected cells. The decrease obtained was ∼30%.
miRNA and mRNA quantification
Total RNA was extracted using QIAzol (Qiagen). miRNA levels were analyzed using the TaqMan RT-qPCR and quantified with the ABI Prism 7000 SDS (Applied Biosystems).
Primers for miR-200c, miR-16, U6, and reagents for reverse transcriptase and RT-qPCRs were all obtained from Applied Biosystems.
miRNA expression levels in each sample were normalized to miR-16 and to U6 small RNA expression. As previously found (27) and in the experimental conditions of the present study, these two RNAs were not modulated by H2O2 treatment.
mRNA complementary DNA (cDNA) was generated by the SuperScript First-Strand Synthesis System (Invitrogen) and real-time PCR was performed with the SYBR GREEN RT-qPCR method (Qiagen) using ABI Prism 7000 SDS (Applied Biosystems).
mRNA expression was normalized for beta-2-microglobulin (B2 M) levels or ribosomal protein L13 (RPL13).
The following primers were used for SYBR GREEN real-time PCR:
B2 M human: forward: 5′-TTCTGGCCTGGAGGCTATC-3′; reverse: 5′-TCAGGAAATTTGACTTTCCATTC-3′.
ZEB1 human: forward: 5′-GGGAGGAGCAGTGAAAGAGA-3′; reverse: 5′-TTTCTTGCCCTTCCTTTCTG-3′.
eNOS human: forward: 5′-GCATCCCTACTCCCACCAG-3′; reverse: 5′-TTCTTCACACGAGGGAACTTG-3′.
SIRT1 human: forward: 5′-AAATGCTGGCCTAATAGAGTGG-3′; reverse: 5′-TGGCAAAAACAGATACTGATTACC-3′.
FOXO1 human: forward: 5′-AAGGGTGACAGCAACAGCTC-3′; reverse: 5′-TTCCTTCATTCTGCACACGA-3′.
RPL13 human: forward: 5′-GGAGTACCGCTCCAAACTCA-3′; reverse: 5′-GGTGGCCAGTTTCAGTTCTT-3′.
CAT human: forward: 5′-GGAGCAGGGGCCTTTGGCTACTT-3′; reverse: 5′-TCCAGCAACAGTGGAGAACCGAACT-3′.
SOD1 human: forward: 5′-GGCCAAAGGATGAAGAGAGGCATGTT-3′; reverse: 5′-GACCACCAGTGTGCGGCCAA-3′.
SOD2 human: forward: 5′-GGTGGAGAACCCAAAGGGGAGTTG-3′; reverse: 5′-TTATTGAAACCAAGCCAACCCCAACCT-3′.
Relative RNA expression was calculated using the comparative Ct method (2−ΔΔCt) (26a).
NO assay
Indirect method
The total nitrite/nitrate of the cell supernatant was measured by the nitric oxide assay kit (Abcam), as previously described (34). Briefly, the reaction consists of a two-step process: in the first step, nitrate was converted to nitrite by a nitrate reductase; in the second step, nitrite was converted into deep purple azo compound by the Griess reagent. The amount of this purple compound can be measured at 540 nm absorbance and reflects the NO levels. The detection limit of this assay is ∼0.1 nmol/well or 1 μM.
Direct method
NO production was evaluated by adding 4,5-diaminofluorescein 10 μM (DAF2 DA) (Alexis) to HUVEC medium (endothelial growth basal medium without phenol red) for 30 min. Afterward, the fluorescent product was detected by a fluorescence microplate reader, Victor 3 1420 Multilabel Counter (Perkin Elmer).
Oxidative stress
DHE staining is specific for superoxide anions only when used in HPLC, otherwise it is used to measure ROS increase in both superoxide and peroxide.
Intracellular ROS production was evaluated by incubating the cells with 2 μM DHE (Sigma-Aldrich) at 37°C for 15 min. Then, cells were fixed with 4% paraformaldehyde, nuclei were stained with Hoechst 33342, and fluorescence was revealed by fluorescence microscopy, using the same exposure conditions in each experiment and quantified using Scion Image software as described previously (15).
ROS-Glo™ H2O2 Assay (Promega) was used to measure H2O2 levels in HUVECs. This assay measures the levels of H2O2 directly in cell culture. The derivatized luciferin substrate is incubated with samples and reacts directly with H2O2 to generate a luciferin precursor, therefore generating a light signal proportional to the level of H2O2 present in the sample.
Briefly, cells were seeded on a microplate of 96 wells at the density of 4.000 cells per well. The day after ROS-Glo H2O2 substrate was added to the wells of cultured cells, they were incubated for 6 h under normal mammalian cell culture conditions. After incubation, ROS-Glo detection solution was added and the plate was incubated for 20 min before reading luminescence using a Victor 3 1420 Multilabel Counter (Perkin Elmer).
Animal model and surgical procedures
All experimental procedures complied with the Guidelines of the Italian National Institutes of Health and with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Bethesda, MD) and were approved by the institutional Animal Care and Use Committee. Hindlimb ischemia was induced in 2- to 3-month-old C57BL/6 N male mice. Before all surgical procedures, mice were anesthetized with an intraperitoneal injection of 10 mg/kg xilazine (Intervet Farmaceutici) and 100 mg/kg ketamine (Ketavet 100; Intervet Farmaceutici). Hindlimb ischemia was induced by dissection of the left femoral artery, as previously described (47). At various times after surgery, gastrocnemius and tibialis anterior muscles were collected for RNA and protein extraction.
Anti-miR-200c delivery
For anti-miR-200c treatment in mice, acute hindlimb ischemia was induced in 2- to 3-month-old C57BL/6 N male mice; 2 days before ischemia and 2 days after ischemia, lentiviruses encoding either anti-miR-200c sequence or a scramble sequence were administered into mice through tail intravenous injection (106 TU in total; divided in the two administrations).
Blood flow measurement by Laser Doppler Perfusion Imaging (Lisca) was performed as previously described (10).
Plasmids
Plasmids used in luciferase assays were generated by cloning oligonucleotides bearing the wt or deleted miR-200c pairing site of SIRT1, FOXO1, and eNOS gene +/−20 bases, in pMIR-REPORT-Luciferase (pLUC; Ambion) downstream of the stop codon, between SpeI and HindIII restriction sites. A BamHI restriction site was added as control of correct insertion. The positions of different seed sequences are depicted in Supplementary Figure S2. The following oligonucleotides were used (5′-3′, miR-200c seed sequence is underlined):
SIRT1_seed a_forward:
CTAGTGGATCCACTGTGGTAGAGCTTGCATTGATCTTTTCCACA
SIRT1_seed a_reverse:
AGCTTTTCACATTTTGGCAGTT
SIRT1_seed a_del_forward:
CTAGTGGATCCTAAATACCTATCACTGTGGTAGAGCTTGCAACTGCCAAAATGTGAATATGCAAAGCCTA
SIRT1_seed a_del_reverse:
AGCTTAGGCTTTGCATATTCACATTTTGGCAGTTGCAAGCTCTACCACAGTGATAGGTATTTAGGATCCA
SIRT1_seed b_forward:
CTAGTGGATCCGGATTTGGTGTTAAATACCAAACTGCTAGCCCT
SIRT1_seed b_reverse:
AGCTTCATCATGTTCATCTCCAT
SIRT1_seed b_del_forward:
CTAGTGGATCCCTCCCATTGGGAGGATTTGGTGTTAAATATGGAGATGAACATGATGATGTAACTTGTAA
SIRT1_seed b_del_reverse:
AGCTTTACAAGTTACATCATCATGTTCATCTCCATATTTAACACCAAATCCTCCCAATGGGAGGGATCCA
eNOS_forward:
CTAGTGGATCCGTATCTTACCTGTAAAGTCTAATCTCTAAATCA
eNOS_reverse:
AGCTTGGTAAATCTTCAATAATA
eNOS_del_forward:
CTAGTGGATCCCCTCTCTCAGGAGTATCTTACCTGTAAAGATTATTGAAGATTTACCATAAGGGACTGTA
eNOS_del_reverse:
AGCTTACAGTCCCTTATGGTAAATCTTCAATAATCTTTACAGGTAAGATACTCCTGAGAGAGGGGATCCA
FOXO1_forward:
CTAGTGGATCCAATTAATTGGCTTTGTGTCCTATTTAGTCCAT
FOXO1_reverse:
AGCTTCTTTCCACATGACTTGAA
FOXO1_del_forward:
CTAGTGGATCCCCCAAAGTCATCAATTAATTGGCTTTGTGTCAAGTCATGTGGAAAGCCCAAAGTCATCA
FOXO1_del_reverse:
AGCTTGATGACTTTGGGCTTTCCACATGACTTGACACAAAGCCAATTAATTGATGACTTTGGGGGATCCA
Plasmids for the full-length 3′-UTR luciferase assay were purchased from Genecopoeia and were all cloned in pEZX-MT01 vector system. The following plasmids were used:
pEZX-MT01-ZEB1 3′UTR (NM_001128128.2),
pEZX-MT01-SIRT1 3′UTR (NM_012238.3),
pEZX-MT01-NOS3 3′UTR (NM_0000603.3), and
pEZX-MT01-FOXO1 3′UTR (NM_002015.3).
Anti-miR-200c construct was generated cloning the oligonucleotides against the mature sequence of miR-200c:
5′CCGGGCCTCCATCATTACCCGGCAGTATTATTTTTG 3′ oligo (forward) and 5′ AATTCAAAAATAATACTGCCGGGTAATGATGGAGGC 3′ oligo reverse were annealed and cloned into EcoRI and AgeI restriction sites in plKo.1 vector purchased from Sigma.
miR-200c mutant construct was derived by plko.1-miR-200c previously described (27) by site-directed mutagenesis in the seed sequence region. The mutant miR-200c sequence was modified from the original sequence TAATACTGCCGGGTAATGATGGA to
Luciferase assay
The rationale of this assay is based on translational inhibition of the firefly luciferase gene by the cloning of miRNA-targeted sequences in its 3′UTR.
For the site-specific luciferase assay, HEK293 cells were plated in 12-well plates and transfected either with 30 ng of pLUC or different pLUC-miR-200c seed wt or pLUC-miR-200c seed del (pLUC-Sirt1-seed a-wt, pLUC-Sirt1-seed a-del, pLUC-Sirt1-seed b-wt, pLUC-Sirt1-seed b-del, pLUC-eNOS-wt, pLUC-eNOS-del, pLUC-FOXO1-wt, pLUC-FOXO1-del; see Plasmid section), together with 100 ng of plKO.1-pre-miR-200c or plKO.1-scramble, and 25 ng of pRL-null renilla luciferase.
HEK293 cells were plated in 12-well plates and were transfected with 0.4 μg of the full-length 3′-UTR luciferase contructs or empty vector (pEZX-MT01, pEZX-MT01-ZEB1, pEZX-MT01-SIRT1, pEZX-MT01-FOXO1, pEZX-MT01-NOS3) together with 0.5 μg of plKO.1-pre-miR-200c, plKO.1-pre-miR-200c-mut, or plKO.1-scramble.
Cellular extracts were tested with dual-luciferase assay (Promega) according to the manufacturer's instructions, using a Victor 3 1420 Multilabel Counter (Perkin Elmer). Values were normalized according to renilla luciferase activity.
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM) analyzed by Mann–Whitney nonparametric test using GraphPad Prism software (Version 5.0). p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
This study was supported by Ministero della Salute GR-2010-2309531 to A.M.; by Ministero della Salute RF-2010-2318330, RC-2015, AIRC-IG2011-ID11793 to M.C.C.; by Ministero della Salute, Fondazione Cariplo grant No. 2013-0887, Telethon-Italy grant No. GGP14092, and AFM Telethon grant No. 1847 to F.M.; by FP7-PEOPLE-2011-CIG, N.294176, to D.A.; and by AFM Telethon grant No. 16772, Duchenne Parent Project-Netherlands#DeSanta-DPPNL, to F.D.S.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.