
Abstract
Definition and Characteristics of Cardiorenal Syndrome
S
CRS, cardiorenal syndrome.
Type 1 CRS, in which acute kidney injury (AKI) occurs as a complication of acute decompensated heart failure, is also recognized as worsening renal function (WRF) in the field of cardiology. WRF predicts much higher rates of mortality and hospitalization in heart failure (19, 20). In addition to the acute decline of cardiac output (low cardiac output syndrome), renal congestion caused by fluid overload has recently been suggested as a possible mechanism for AKI in heart failure (51). Type 2 CRS is characterized by progressive WRF along with chronic heart failure, including several different conditions such as atrial fibrillation, congenital heart disease, constrictive pericarditis, and chronic ischemic heart disease (22, 25, 31, 32). However, type 4 CRS is defined as chronic heart disease occurring with chronic kidney diseases (CKD). It should be noted that ∼70%–80% of patients with end-stage renal disease have complications of cardiac dysfunction (18). Although the definition of type 2 and type 4 CRS seems to differentiate the two different disease conditions clearly, previous epidemiological studies have not shown a clear distinction between type 2 and type 4 CRS (6). This is because both CKD and chronic heart failure are strong risk factors for each other and clinical observational studies could not demonstrate the causal relationships between the kidney and heart diseases. Type 3 CRS is characterized by acute reduction of renal function that leads to acute cardiac injury and/or dysfunction, including acute coronary syndrome, congestive heart failure, and arrhythmias. Any cause of AKI may contribute to acute cardiac dysfunction, possibly by retention of uremic solutes and/or volume overload. In addition, activation of systemic immunologic reactions, sympathetic nervous and renin–angiotensin–aldosterone system, and increased oxidative stress are considered as contributing factors to cardiac injury in AKI (8, 56).
Type 5 CRS is characterized by acute or chronic systemic illnesses that concurrently induce both cardiac and kidney dysfunction. Many contributing factors for acute and chronic systemic conditions for type 5 CRS have been suggested. Among them, sepsis is a prototypical condition that leads to acute type 5 CRS. Reportedly, 45%–70% of all AKIs are associated with sepsis (9, 47, 61). The prognoses of patients with both sepsis and AKI are extremely poor and their mortality rate is unacceptably high (7, 47). There are several pathophysiological mechanisms relatively specific for sepsis-induced AKI, that is, a proinflammatory state caused by complement and coagulation activation, protease activation (heparan sulfate, elastase), free radical formation, cytokine production (interleukin [IL]-1, IL-6, IL-18, tumor necrosis factor [TNF]-α), and cell activation (neutrophil, macrophage, platelet, endothelial cell). In parallel, an anti-inflammatory state will also be induced by cytokine (IL-10) production, reduced phagocytosis and chemotaxis, and rearranged immune function (lymphocyte apoptosis). Dysregulation of microcirculation is often seen in septic AKI such as vasodilation-induced glomerular hypoperfusion and abnormal blood flow within the peritubular capillary network in the kidney. However, ∼30%–80% of patients with sepsis show cardiac-specific troponin elevation and reduced cardiac function (2, 3, 5, 41, 59). Cardiac dysfunction that occurs in sepsis is now called septic cardiomyopathy. In addition to changes of circulating volume and peripheral vessel dilation, mitochondrial dysfunction, downregulation of beta-adrenergic receptors and postreceptor signaling pathways, impairment of calcium release from the sarcoplasmic reticulum, and impairment of electromechanical coupling at the myofibrillar level are suggested as pathophysiological mechanisms in septic cardiomyopathy (57). As seen in septic AKI, in septic cardiomyopathy, there is a role for inflammatory cytokines.
Mitochondrial Dysfunction in Acute Kidney Injury
Tubular epithelial cell injury plays a central role in the pathophysiology of AKI. Although acute tubular necrosis was considered as the dominant cause of cell death of tubular epithelium in AKI, experimental studies have clarified that apoptosis also contributes to the pathogenesis of AKI. The renal ischemia–reperfusion model shows many apoptotic cells in injured tubules with activation of caspases (36, 38, 39). Upregulation of Bcl-2 expression was observed in ischemic AKI models (12, 30). Another AKI model of cisplatin injection also shows renal tubular cell apoptosis in vivo as well as in vitro in a dose-dependent manner. Cisplatin induces apoptosis in renal tubular epithelial cells via three major pathways: (i) the extrinsic pathway through death receptors, (ii) the intrinsic pathway on mitochondria, and (iii) the endoplasmic reticulum stress pathway (49).
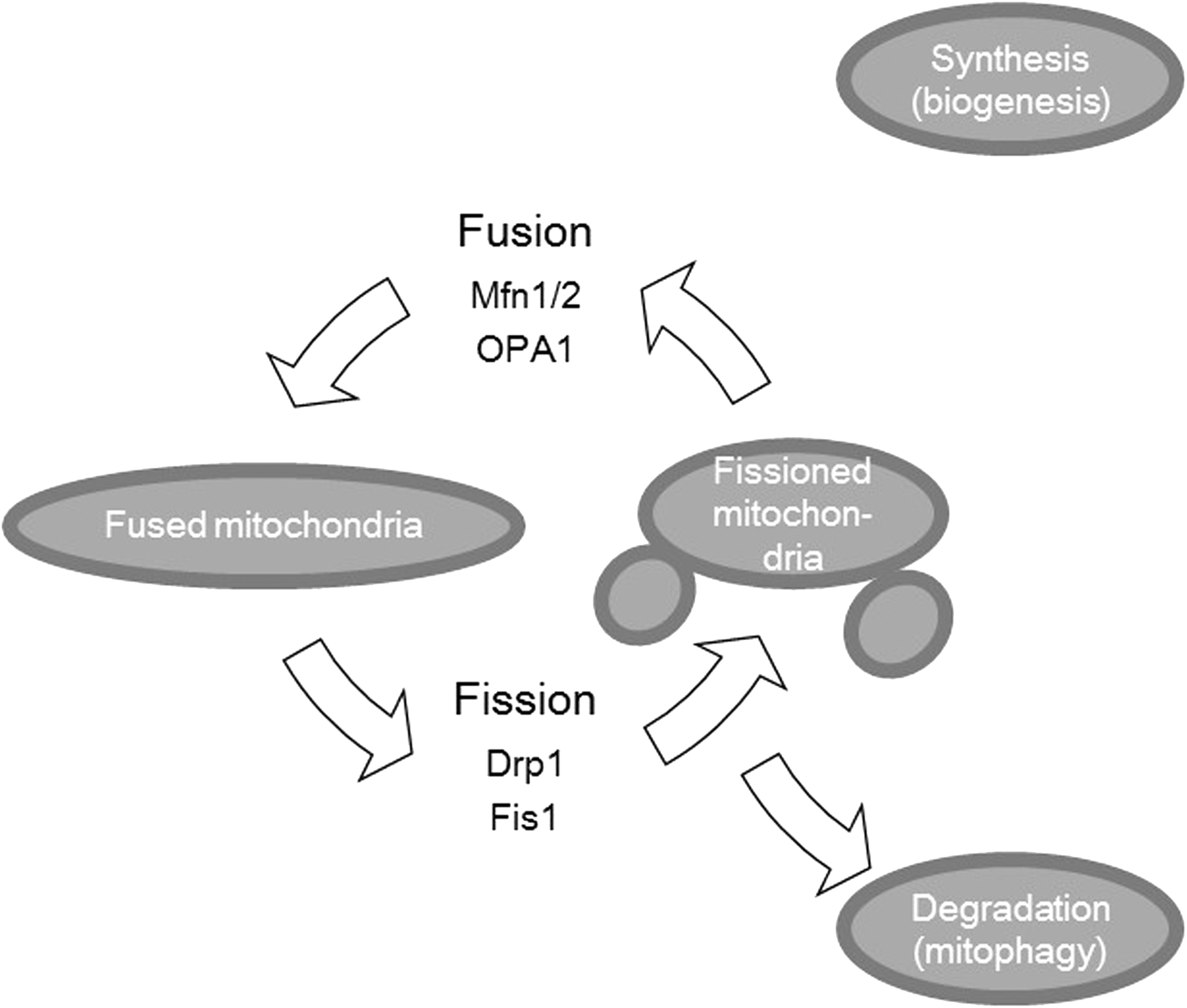
Mitochondria continually divide and fuse under normal conditions and mitochondrial dynamics are involved in many biological processes, including apoptosis. Although guanosine triphosphatases (GTPases) mediate fusion and fission, fission is mainly regulated by dynamin-related protein 1 (Drp1). Drp1 is a cytosolic protein that moves to the outer mitochondrial membrane by several different activations. Fusion of the inner membrane involves optic atrophy 1 (OPA1) (1), whereas the outer membranes are regulated by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) (43, 58) (Fig. 1). Mitochondrial fission is also an early event during apoptosis, which occurs before caspase activation and membrane blebbing. Cytochrome c release through mitochondria is considered the primary trigger of the caspase cascade. Cytochrome c is released from the outer mitochondrial membrane during apoptosis simultaneously with mitochondrial fragmentation (35). Inhibition of Drp1 can occur through a dominant-negative inhibitor or by using RNAi, which reduces mitochondrial fragmentation during apoptosis and cytochrome c release (13, 28, 29, 42). Germain et al. reported that Drp1-dependent crista remodeling triggered cytochrome c release from mitochondria (29). Ban-Ishihara reported that Drp1-dependent mitochondrial fission regulates cytochrome c release by mitochondrial DNA distribution and crista reformation (11).
Brooks and colleagues reported a remarkable morphologic change of mitochondria in AKI. First, they observed mitochondrial fragmentation preceding apoptosis in cultured rat proximal tubular cells subjected to azide-induced adenosine triphosphate (ATP) depletion or cisplatin exposure. Dominant-negative and small interfering RNA knockdown experiments showed successful blockade of mitochondrial fragmentation, cytochrome c release, and finally apoptosis. In vivo analysis with three-dimensional image reconstruction from serial electron microscopy sections showed mitochondrial fragmentation in murine proximal tubular cells during renal ischemia–reperfusion injury and cisplatin-induced nephrotoxicity. In addition, treatment by mdivi-1, a pharmacological inhibitor of Drp1, attenuated apoptosis of tubular epithelial cells and renal dysfunction in these in vivo models. The efficacy of mdivi-1 in renal ischemia–reperfusion was also confirmed by our recent report (Fig. 2). Mdivi-1 has been identified as an inhibitor of mitochondrial division by yeast screening of chemical libraries and found to inhibit Drp1 assembly and GTPase activity (16). Tang et al. evaluated the protective effects of mdivi-1 on rat rhabdomyolysis-induced AKI developing after intramuscular glycerol injection. They observed that mdivi-1 reduced Drp1 accumulation and Bax expression in mitochondria and inhibited the release of cytochrome c and cell apoptosis (65).

Recent studies have clarified a possible pathway by which mitochondrial fragmentation is induced by cisplatin exposure to renal tubular epithelial cells. Sphingosine-1-phosphate (S1P) is the natural sphingolipid ligand for a family of five G protein-coupled receptors (S1P1 to S1P5) and regulates diverse cellular signaling responses. S1P binds extracellularly to those receptors for regulating cell movement (62). Bajwa et al. found that overexpression of sphingosine-1-phosphate receptor-1 in cultured renal tubular epithelial cells made them resistant to cisplatin exposure through prevention of cell apoptosis and mitochondrial fragmentation (10).
Sirtuins are nicotinamide adenine dinucleotide (NAD)-dependent deacetylases that are highly conserved from bacteria to human. Sirtuin 3 (SIRT3) is mainly localized in the mitochondrion. SIRT3 is the major mitochondrial deacetylase that maintains basal ATP levels as well as reactive oxygen species (ROS) homeostasis through the regulation of detoxifying enzymes (45). Morigi et al. reported that SIRT3 restoration, by adenosine monophosphate (AMP)-activated protein kinase agonist 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) or the antioxidant agent acetyl-
Mitochondrial Dysfunction in Heart Disease
The kidney and heart, rich in mitochondria, require large amounts of energy production. In the heart, mitochondrial function is assumed as an important determinant of myocardial contractility. Fusion of mitochondria in the heart was evaluated with simultaneous conditional knockout of Mfn1 and Mfn2. Embryonic cardiac Mfn1/Mfn2 ablation was lethal, whereas conditional combined cardiac Mfn1/Mfn2 ablation in adults induced mitochondrial fragmentation along with progressive and lethal dilated cardiomyopathy (17). Another group developed genetically Mfn1/Mfn2 inactivated mice using different promotor systems (50). The former study used Nkx-2.5cre that is active in cardiac progenitor cells and therefore has the potential to recombine loxP sites in all cardiac cells, including cardiomyocytes and vascular smooth muscle, and mesenchymal cells. The latter study used the Myh6-cre that is restricted only to cardiac myocytes. In this study, cardiac morphology and function of knockout mice were normal at birth. However, abnormal mitochondrial morphology, manifesting as unpacking of cristae and loss of matrix density, developed at postnatal day 7 together with the symptoms of cardiomyopathy. Mitochondria biogenesis genes were downregulated and the amount of mitochondrial DNA was reduced.
Recently, mitochondrial structural changes have been evaluated in ischemia–reperfusion-induced cardiac dysfunction. Ong et al. demonstrated that reducing mitochondrial fission by transfecting a dominant-negative mutant of Drp1, or by mdivi-1 treatment, reduced mitochondrial permeability transition pore sensitivity and cell death after simulated ischemia–reperfusion injury. In vivo treatment by mdivi-1 reduced infarct size in a coronary artery occlusion model of mice (48). Sharp et al. examined the role of mitochondrial fission in ischemic cardiac injury with isolated neonatal murine cardiomyocytes and Langendorff isolated perfused hearts of rats. In addition to the finding that Drp1 inhibition preserved mitochondrial structure after ischemia–reperfusion, they demonstrated that ischemia–reperfusion induced Drp1 activation via calcineurin-dependent Drp1 S637 dephosphorylation. A calcineurin inhibitor FK506 and therapeutic hypothermia inhibited Drp1 S637 dephosphorylation and attenuated mitochondrial morphological changes and myocardial dysfunction (60). Taken together, Drp1 inhibition seems to be a promising strategy for preserving myocardial function after ischemia–reperfusion.
Role of Mitochondrial Dysregulation in Cardiorenal Syndrome
As discussed earlier, sepsis causes type 5 CRS, that is, simultaneous complication of septic AKI and septic cardiomyopathy. Regarding these two sepsis-induced organ dysfunctions, emerging evidence indicates the role of mitochondrial dysregulation. Mitochondrial biogenesis (new mitochondria generation by mitochondrial DNA replication) is essential for maintaining the energy-generating capacity of mitochondria. Peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) is known to be a positive regulator of mitochondrial biogenesis (69). A number of different signals deriving from normal metabolic fluctuations, as well as exaggerated signaling induced by an injury control with PGC-1α, indicate its role in mitochondrial biogenesis within normal metabolic adaptation or in response to pathologic conditions. For instance, ATP depletion and increased ROS induce PGC-1α (54, 63). Tran et al. examined the role of PGC-1α in septic AKI by using animal models of cecal ligation and puncture (CLP) and lipopolysaccharide injection (67). Sepsis reduced PGC-1α expression in the renal tubular epithelial cells. Fluid resuscitation as a treatment of septic shock preserved PGC-1α expression to the same degree as that seen in sham-operated animals. PGC-1α knockout mice, which show normal renal function and mitochondrial structure under stable conditions, showed persistent AKI, whereas their wild-type littermates had recovered function after sepsis exposure. Proximal tubular cell-specific PGC-1α knockout mice showed similar results, suggesting the PGC-1α expression is critical for recovery from septic AKI.
Septic cardiomyopathy is a well-recognized complication of severe sepsis and septic shock with a number of potential mechanisms, including cytokines, nitric oxide, complement activation, apoptosis, and energy metabolic abnormalities suggested (27). However, it should be noted that many controversies exist in understanding the pathophysiology of septic cardiomyopathy. Sepsis is generally characterized as an overwhelming cascade of inflammatory reactions, especially at its early stage, with significant amounts of ROS and NO produced systemically in addition to cytokines. Since mitochondria are one of the main field of ROS and NO, production of NO and ROS, protein nitration, and lipid peroxidation are increased in mitochondria of the heart in septic animal models (15, 34, 68). Peroxynitrite (ONOO−) is a noxious oxidant that is produced by a reaction of NO with O2
•− in about diffusion limit. Sepsis induced by CLP showed increased activity of the inducible mitochondrial nitric oxide synthase (i-mt NOS) and mitochondrial ONOO− levels (26). Increased oxidative stress and decreased ATP production were improved in inducible NOS (iNOS) knockout mice. Moreover, treatment with melatonin, an inhibitor of iNOS, improved mitochondrial homeostasis after sepsis, restored ATP level, and improved survival. Another study in sepsis evaluated several different pharmacological inhibitors of NOS (71); nonselective NOS inhibitor N-nitro-
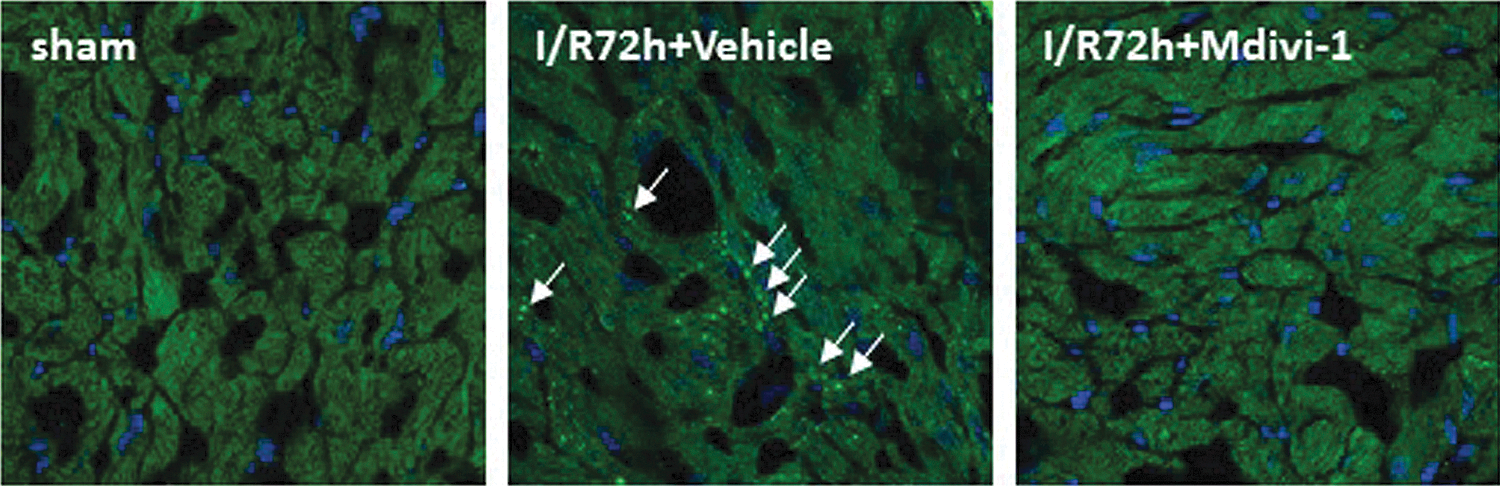
Reach out and touch of kidney suffered with AKI on the heart (i.e., Type 3 CRS, acute renocardiac syndrome) have not been clarified sufficiently to date. Only a few animal studies have conducted and found that cellular apoptosis and capillary vascular congestion were observed in the heart after renal ischemia–reperfusion or glycerol injection-induced rhabdomyolysis (37, 46, 53). AKI has been shown to lead to cardiac hypertrophy (14) and increased cardiac infiltrating macrophages (66). Recently, changes in mitochondrial dynamics have been reported to play a role in cardiac apoptosis induced by AKI (64). Fragmented mitochondria in cardiomyocytes were observed after renal ischemia–reperfusion (Fig. 3). In this renal ischemia–reperfusion model, the M mode echocardiogram revealed a statistically significant reduction of fractional shortening l (Fig. 4). Western blot analysis demonstrated that Drp1 was increased not in cytosolic, but in the mitochondrial, fraction of the heart tissue after renal ischemia–reperfusion. A Drp1 inhibitor mdivi-1 reduced mitochondrial fragmentation (Fig. 3) and cardiac apoptosis (Fig. 5) induced by AKI. The mechanisms by which AKIs enhance mitochondrial fragmentation in the heart are still unclear. Several possible pathways can be considered, by and large related to unknown humoral mediator accumulation in the blood. Increased blood levels of high-mobility group box 1 protein and IL-6 were reported in mouse AKI models (23, 40, 52). Chemokine (C-X-C motif) ligand 1, IL-1β, and TNF-α were increased not only in the kidney but also in the spleen and liver in renal ischemia–reperfusion injury (4). One basic study demonstrated that mitochondrial dysfunction in the brain of Caenorhabditis elegans induced the mitochondrial unfolded protein response (UPRmt) in a distant organ of intestine, although no secreted factor was defined yet (24). Further investigations are indispensable to ascertain the responsible pathways in cardiac injury caused by AKI.


Perspectives
The transmission electron microscopic image in Figure 6 shows the proximal and medullary thick ascending limb of Henle; both are vulnerable to ischemic and toxic renal injuries. The amount of mitochondria can account for the differences in vulnerability. They are rich in mitochondria; its cellular occupancies can be more than 30%. Admittedly, the kidney is the organ that is second-most rich in mitochondria. The shape of organ mitochondria varies based on the different ATP demands of the organ that induce different surface areas between the external and internal membranes unique to the organ. The mitochondrial inner membrane is the reaction field that transforms the energy from oxidative reactions into cellular ATP. ATP depletion in mitochondria is induced by many factors related to ROS and intracellular calcium concentrations. ATP depletion can trigger mitochondrial fragmentation even in distant organs.

Persistent reduction of mitochondrial function and suppression of biogenesis would finally be detrimental to highly differentiated organs. Direct application of antioxidant will result in organ protection, including mitochondrial function in either primary or distant organs, although the effectiveness of this scavenging strategy almost always declined with time. Oversuppression of ROS will easily disrupt mitochondrial hormesis, orchestrating oxidant traffic as essential intracellular signaling molecules for differentiation, growth, cell death, and senescence (33, 72). Recently, long-term impacts of AKI on the heart have been suggested based on several clinical epidemiology data. One clinical observational study reported a significant association of AKI with higher risk of coronary events and all-cause mortality independently from subsequent progression to CKD (70). This observation may suggest that long-term dysregulation of mitochondrial hormesis by AKI could contribute to heart disease. The restoration of mitochondrial biogenesis within mitochondrial hormesis would serve the improvement of chronic CRS. So, AKI transmits its injury to the heart, or the heart might draw a lesson from the kidney injury. Further studies are certainly necessary to obtain a more detailed picture of organ cross talk in AKI.
Footnotes
Author Disclosure Statement
No competing financial interests exist.