
Abstract
Introduction
H
Insult or injury resulting in cellular stress destabilizes intracellular heme proteins and results in the generation of reactive oxygen species (ROS) and lipid peroxidation. The kidney has high metabolic activity and maintains a relatively hypoperfused medulla. Therefore, one may conclude it is highly challenged with oxidizing agents. Extensive evidence exists for the importance of HO in maintaining homeostasis in the presence of oxidative stress, and it is involved in the pathophysiology of many organ systems. Due to this, interest in HO continues to grow rather exponentially (Figs. 1 and 2). This review will update the reader on new findings regarding the role of HO in kidney homeostasis and pathology.


Biochemistry of HO
HO is a microsomal cytoprotective enzyme that is induced in response to injury and cellular stress [reviewed in Refs. (47, 48, 99, 146)]. Importantly, HO mitigates toxicity by degrading heme released from heme-containing proteins, including hemoglobin, myoglobin, and cytochromes, utilizing NADPH and molecular oxygen as cofactors (Fig. 3) (150). The enzymatic mechanism has been elucidated (150), and structural studies provide insight into function (12, 42, 120). Bilirubin is a free radical scavenger that blocks lipid peroxidation (3, 126, 139). CO is a physiologically important vasodilator, acting via cyclic guanosine monophosphate (cGMP) (69, 163). Iron induces ferritin (Ft), which is protective (23). While each product exhibits beneficial properties, potential adverse effects may result if they reach high levels (Fig. 3).

Three reported isoforms of HO include HO-1, −2, and −3 (2, 86, 93, 94). HO-1 is the highly characterized, inducible form, and its role in kidney health and disease is a focus of this review. The human HO-1 gene is 13 kb in length and its locus is 22q12 (HGNC:5013). It encodes a 288 amino acid monomer with molecular mass of 32 kDa (UniProtKB P09601). Post-translational modifications affecting the size of HO-1 have been reported and are important for cellular function (82). HO-2 is constitutively expressed at low levels in the nervous system, reproductive system, cardiovascular system, kidney, and liver (63, 87, 156, 163). The human HO-2 gene is 36 kb in length, is located on 16p13 (HGNC: 5014), and encodes a 36 kDa monomer consisting of 315 amino acids (UniProtKB: P30519). Three protein isoforms (a, b, and c) have been reported (Entrez Gene: 3163). Recently, endothelial induction of HO-2 was demonstrated in response to vascular stress in an arteriovenous (AV) fistula model (63). The HO-3 gene was found to produce no functional protein and was subsequently identified as a pseudogene (52).
Induction of HO-1
The kidney encounters toxic oxidants, endogenous and exogenous, as well as bound iron from the circulation. Free heme-induced oxidative stress is an important physiologic mechanism of induction of HO-1 [reviewed in Ref. (125)]. In addition, non-heme molecules such as heavy metals, endotoxin, UV radiation, prostaglandins, H2O2, certain growth factors, cytokines, and other stimuli can induce HO-1 in the kidney (8, 125). Ft is coinduced with HO-1 and both play a key role in iron homeostasis and the oxidative stress response (4, 13, 23, 100, 158).
The classical means of induction of renal HO activity is treatment with the heme analog, hemin (78). Tin protoporphyrin (SnPP) and zinc protoporphyrin (ZnPP) have been used to inhibit HO activity (91, 134). Several protoporphyrins modulate HO activity; however, these molecules have off-target effects and have a narrow therapeutic window. More sophisticated recombinant animal models have been developed to study the role of HO-1 in a variety of biological processes (59, 68). HO-1 is induced in animal models of ischemia, hypertension, glomerulonephritis, AKI, and chronic kidney disease (CKD) (4, 5, 61, 100, 141).
Nuclear factor erythroid 2-related factor 2 (Nrf2) is an important regulator of HO-1 transcription in response to oxidative stress. It binds to stress response elements in the gene promoter (7, 8). In mice, the Bach1 transcription factor represses HO-1 expression by competing with Nrf2 binding sites in the HO-1 promoter region. In steady state, Nrf2 is bound to a cytosolic repressor called Kelch-like ECH-associated protein 1 (Keap1), which aids in ubiquitination and proteolytic degradation of Nrf2. During oxidative stress, Keap1 is covalently modified by electrophiles and ROS at its cysteine residues, causing release of Nrf2 and decreased targeting for proteasomal degradation (128). This permits nuclear translocation of Nrf2 and promotes HO-1 transcription in the setting of increased oxidant burden.
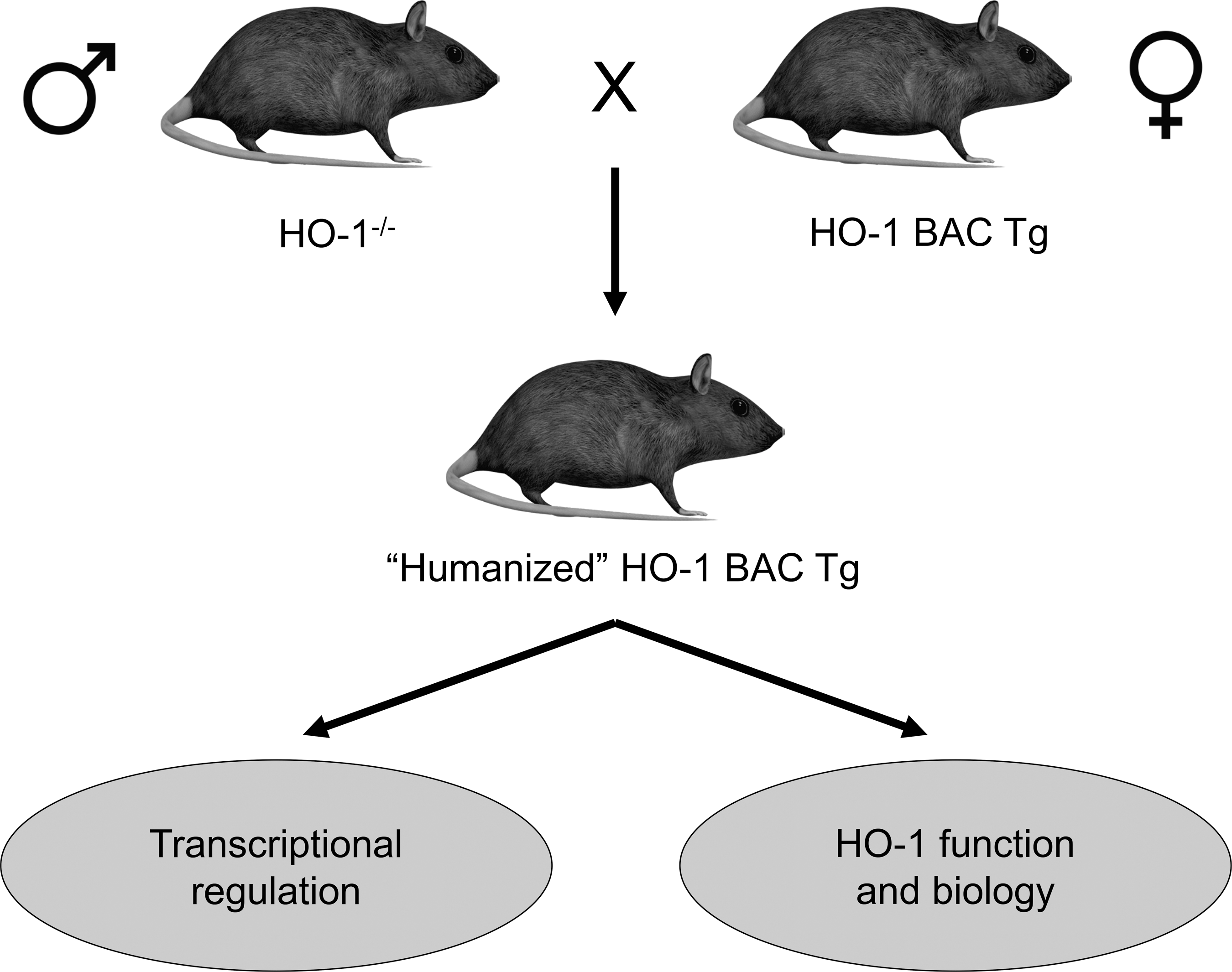
Several reports have elucidated the molecular regulation of HO-1 expression, which is robust and complex [reviewed in Refs. (47, 125)]. HO-1 is tightly regulated at the transcriptional level [reviewed in Ref. (125)]. To study in vivo regulation of HO-1 expression, our laboratory generated a humanized HO-1 transgenic mouse model, utilizing a bacterial artificial chromosome (BAC) containing the entire human HO-1 gene, including its regulatory sequences. When crossed with HO-1-deficient mice, the resultant strain globally expressed human HO-1 (humanized BAC [hBAC] mice), but not endogenous murine HO-1 and was able to rescue the phenotype of the HO-1−/− mouse (Fig. 4) (68). The murine transcription factors specificity protein 1 (Sp1), JunB (activator protein-1 family, AP-1), and upstream stimulatory factor (USF)-1 and −2 regulated human HO-1 gene expression in this model (68).
HO-1 expression is regulated differently in humans and mice (Table 1). For example, heat shock induces HO-1 in mice but not in humans (121, 122). In mice, hypoxia and cytokines such as interferon-γ (IFN-γ) are potent inducers of HO-1, whereas they are repressors in humans (71, 121). In humans, a GT-repeat polymorphism in the HO-1 promoter modulates the level of transcription (27, 70, 148). Long polymorphisms, defined as greater than 25–27 repeats (L allele), in this region are associated with lower inducibility of HO-1 and increased risk of disease (14, 27, 30, 107, 148). Significant correlation with disease was identified in the settings of renal transplantation, diabetic kidney disease, CKD progression, AV fistula failure, and others (30, 76, 81, 107, 148). Recent evidence demonstrated microRNAs are involved in the regulation of human HO-1 expression in vitro (15). The promoter sequence of the human HO-1 gene has binding sites for several transcription factors, including nuclear factor-κB (NF-κB), AP-1 and −2, Sp1, USF-1 and −2, and interleukin (IL)-6. These sites include response elements for STATx, c-Rel, hepatocyte factor-1 and −4, and GATA-X (75, 98, 119, 129, 130, 138). In human renal epithelial cells, an intronic enhancer controls HO-1 expression via chromatin looping and Sp1-dependent interaction with the −4.0 kb HO-1 promoter region (36).
Reproduced with permission from Sikorski et al. (125).
E1 and E2 = −4.0 and −10 kb distal promoter elements upstream of the HO-1 gene in mice.
A and B regions = analogous to E1 and E2 in mice. −4.0 and −9 kb distal promoter elements for human HO-1 gene containing stress and cadmium response elements.
HO, heme oxygenase.
Cellular Localization of HO-1
While HO-1 localizes predominantly to the endoplasmic reticulum, expression in the nuclear (18, 82), mitochondrial (17, 21), and secretory (83, 151) subcellular compartments has been reported. In the setting of hypoxia- or heme-mediated oxidative stress in vitro, HO-1 localized to the nucleus and modulated transcription of oxidative stress response genes (82). Cleavage of a hydrophobic C-terminal domain caused HO-1 to localize to the nucleus in cell culture. There, nuclear HO-1 protected the Nrf2 transcription factor from glycogen synthase kinase 3β-mediated phosphorylation and subsequent degradation (18). HO-1 targeted to mitochondria was protective against hypoxia-induced oxidative damage in human embryonic kidney cells. Protection was superior in mitochondrial-targeted HO-1 overexpressing cells compared with cytoplasmic overexpression (21). Bindu et al. found mitochondrial expression of HO-1 in a rat model of gastric mucosal injury, which protected from mitochondrial stress, improved cellular respiratory function, and prevented apoptosis (17). Zager et al. showed that plasma and urinary HO-1 levels positively correlated with renal cortical expression in several models of AKI, suggesting HO-1 as a candidate AKI biomarker (155).
Localization of HO-1 in the Kidney
HO-1 is induced in a variety of kidney substructures in response to injury, including proximal tubules, glomeruli, and renal interstitium, as well as in renal mononuclear phagocytes (RMPs) (4 –6, 59, 65, 100). We define RMPs here as tissue-resident and infiltrating macrophages, dendritic cells, and monocytes. In most models of AKI, HO-1 is induced predominantly in renal proximal tubules. HO-1 expression is seen in glomeruli in models of diabetes (6) and in infiltrating macrophages in acute renal transplant rejection (5). Understanding limitations in our approach, we categorized renal pathologies by kidney substructure (Table 2), and implications of expression of HO in each are discussed.
HO-2: The Constitutive Isoform of HO
The promoter region of the HO-2 gene contains a glucocorticoid response element, which regulates HO-2 expression in the brain (88, 111). In addition, cellular oxygen tension regulates HO-2 expression in endothelial cells (46, 53). Post-translational modifications, such as casein kinase 2-dependent phosphorylation, also affect HO-2 activity in vitro in hippocampal cells (20).
In 1997, Maines described heme regulatory motifs (HRMs) in HO-2 (87), proposing that the protein might serve as a cellular redox sensor, in addition to catalyzing a regulated step in redox signaling. These HRMs are independent of the enzymatic active site and bind heme with higher affinity (87). Yuan et al. recently substantiated the role of HO-2 and HRMs in redox signaling and demonstrated a role in control of respiratory rate (152). Biochemical and structural NMR studies further supported the conclusion that HRMs in HO-2 allow cellular sensing and response to redox changes (12, 42).
Changes in HO-2 induction occur with age. Increasing age results in a decreased ability to induce renal HO-2 (102). HO-2 knockout mice challenged with heme proteins demonstrated worse renal function, increased histologic damage, and greatly increased elaboration of pro-inflammatory IL-6 compared with wild-type controls (102). Thus, a decreased induction of HO-2 may be related to increased sensitivity to heme protein toxicity in aging mice.
HO-1 and Autophagy
Autophagy is defined as a lysosome-mediated degradation of cellular material, such as proteins and organelles [reviewed in Ref. (25)], a process critical for maintaining cellular homeostasis. Dysregulated, prolonged, or incomplete autophagy results in accumulation of damaged cellular material, eventually leading to cell death. It is a highly complex process starting with the formation of autophagosomes and terminating with fusion to lysosomes (79).
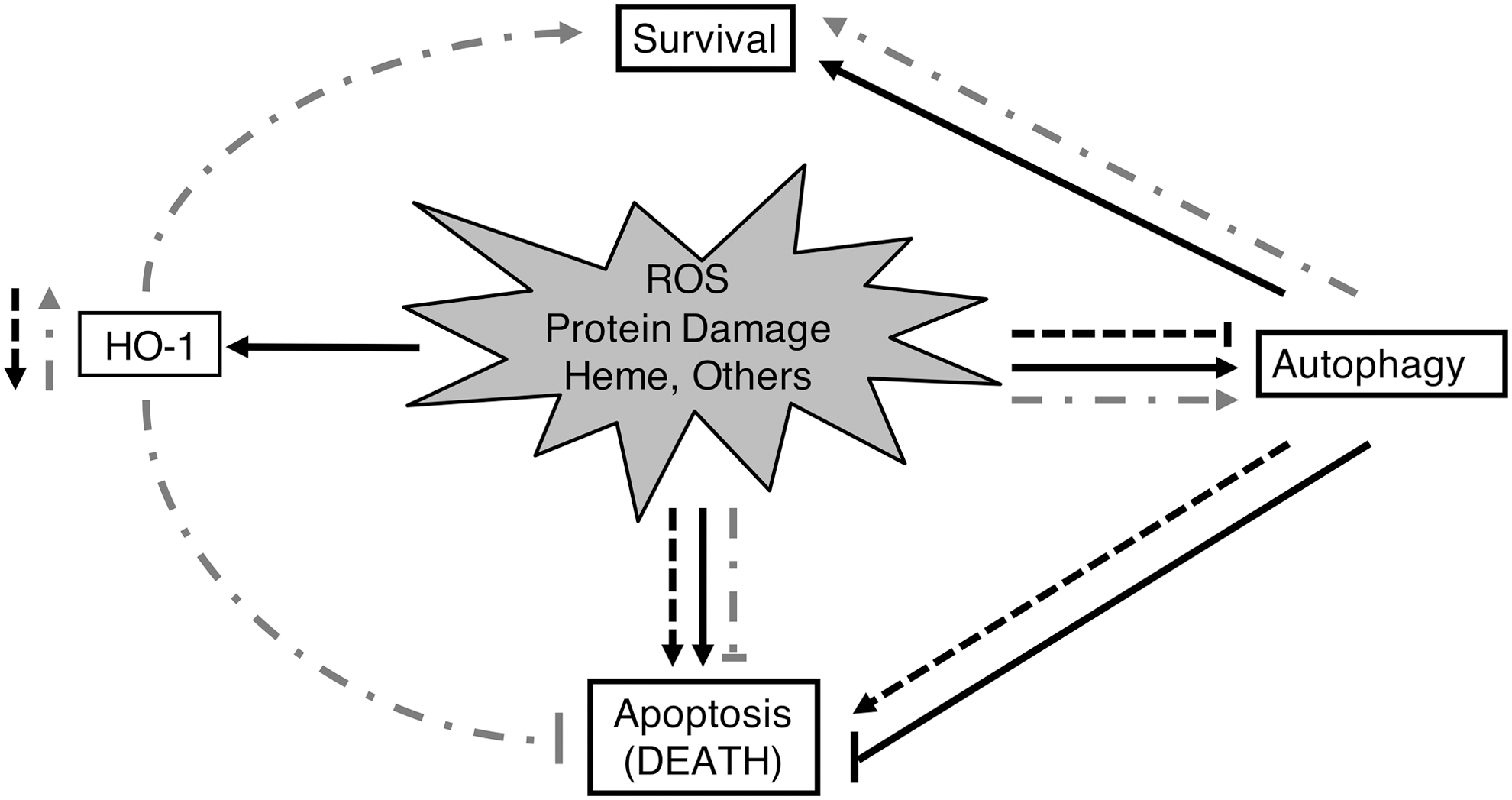
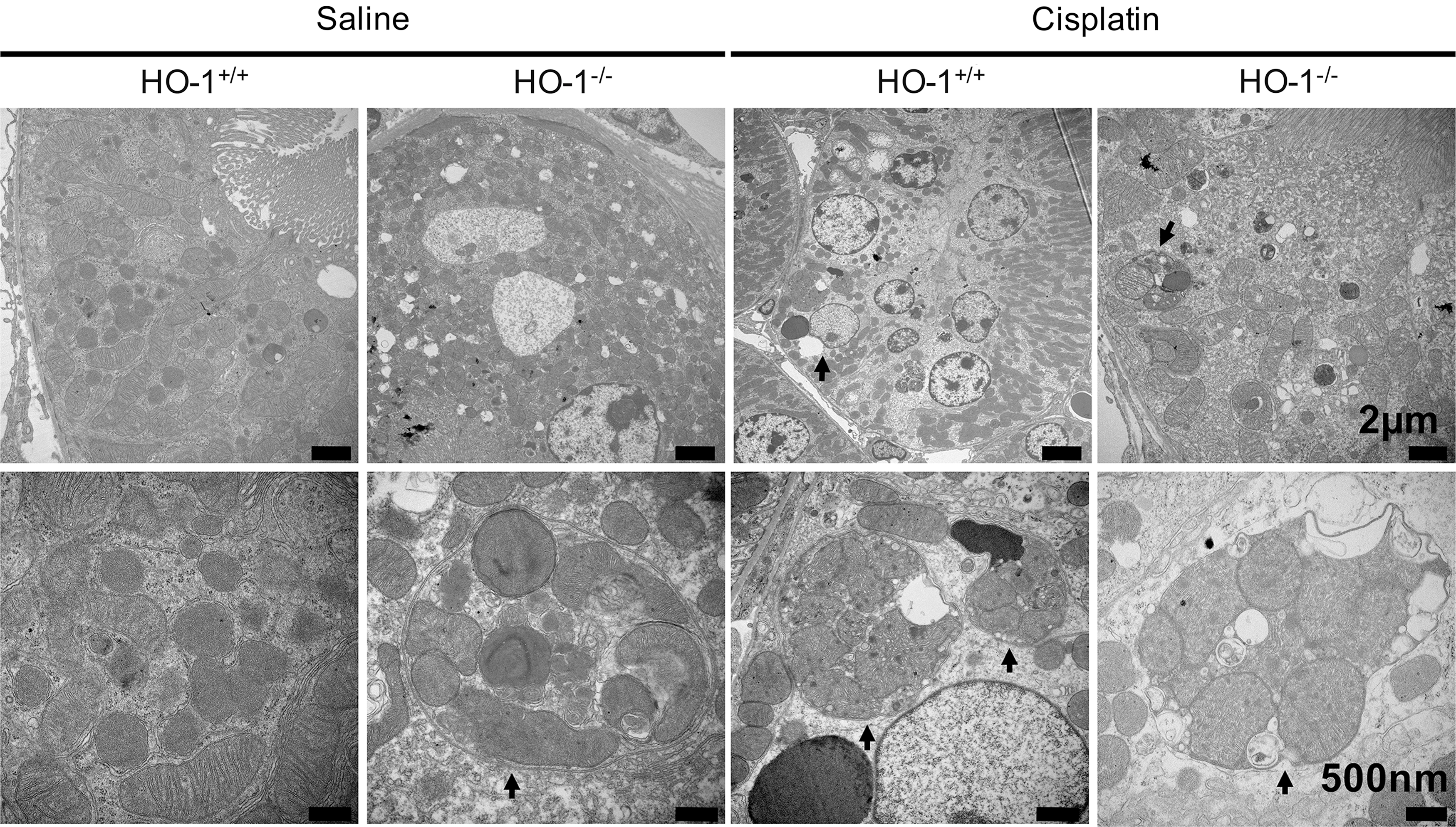
Mammalian markers of autophagy include autophagy proteins Atg5, Atg6 (beclin1, Becn1), Atg7, and Atg8 (LC3, Map1lc3). Oxidative stress can trigger autophagy and lead to cell survival or death [reviewed in Ref. (117)]. A role for HO-1 mediating autophagy in response to oxidative stress has been proposed (Fig. 5). Autophagy and HO-1 induction are protective against injury to renal proximal tubular cells (RPTCs) mediated by cisplatin (CP) (22, 64, 108). Kidneys of HO-1 knockout mice show increased numbers of autophagic vesicles both basally and after CP administration (Fig. 6) (22). In CP-treated primary RPTCs from wild-type mice, Atg5, −6, −7, and −8 were elevated. However, RPTCs from CP-treated HO-1 knockout mice did not exhibit upregulation of these autophagic markers and instead showed increased apoptosis. Overexpression of human HO-1 in hBAC mice reversed maladaptive changes, including low levels of autophagic activity (22). These data collectively suggest that HO-1 induction promotes autophagy and cell survival in CP nephrotoxicity.
HO Mediation of Vascular Tone: A Role for CO
Early studies demonstrated that heme-induced expression of HO in spontaneously hypertensive rats decreases blood pressure, an effect that is reversed with HO inhibition (90). CO, one of the products of the HO reaction, regulates vascular tone (69). The contribution of CO to relaxation of vascular tone was particularly important in a diabetic kidney disease model, characterized by low renal NO bioavailability (24). CO mediates vasoactive effects through synthesis of cGMP, activation of K+ channels, and blockade of vasoconstrictive endothelin-1. CO production by HO stimulates angiogenesis. Also, it is anti-inflammatory, antiapoptotic, antithrombotic, and exhibits antioxidant properties. Dulak et al. have extensively reviewed the diverse functions of CO in this context (38).
HO in Renal Inflammation
Early studies demonstrated that inflammation in glomeruli induced HO-1 in the renal tubules (96, 141). Hemin treatment reduced glomerular inflammation in a model of nephrotoxic nephritis (NTN) (96). The role of specific cytokines and molecular signals in modulating inflammation will be discussed in a disease-specific context. Recently, an important role for HO activity in the RMP compartment has been demonstrated.
HO in mononuclear phagocyte-mediated renal inflammation
HO-1 is induced in RMPs, in addition to renal parenchymal cells, in the setting of injury (59, 100). RMPs include tissue-resident leukocytes that perform homeostatic functions and those that transmigrate during inflammation or injury. The predominant functions of RMPs are phagocytosis (66), coordination of the immune response, and production of cytokines (58).
In kidney injury, oxidative stress results in destabilization of intracellular heme proteins, organelle damage, and cell death. HO activity is beneficial in oxidative stress due to production of antioxidant bilirubin (57) and vasodilatory CO (55, 69), both of which are anti-inflammatory. In addition, induction of HO-1 is associated with increased Ft levels, which exhibits anti-inflammatory effects as well (23).
RMP trafficking and antigen presentation may be related to HO activity. Global HO-1 knockout animals demonstrated greater homing of tissue-resident RMPs to primary lymphoid organs after injury (59). Also, HO-1 inhibited autophagy, a process necessarily preceding antigen presentation (22). It may be that tissue-resident RMPs contribute to immune tolerance when appropriate, as has been suggested for tissue-resident mononuclear phagocytes (MPs) in the liver (37). MPs support integration of the innate and adaptive immune systems; they contribute to differentiation and activation of lymphocyte subsets, including T lymphocytes and innate lymphoid cells (11). This may be due to effects beyond the elaboration of enzymatic products. Putative signaling mechanisms are linked to redox status (58, 144). Further studies are needed to obtain a full understanding of the role HO plays in kidney inflammation and homeostasis.
HO-1 in Kidney Disease
Glomerular diseases
Diabetic kidney disease
ROS are important in multiple pathways in the pathogenesis of diabetes and its complications (26, 106). On one hand, induction of antioxidant proteins is protective, while blockade or decreased activity worsens damage in diabetic kidneys in animal models and humans (33, 77, 95). In humans, serum bilirubin levels were inversely correlated with progression of kidney disease in type II diabetics in two large-scale clinical trials (115), suggesting bilirubin is a target biomarker for progression of diabetic kidney disease (57).
Streptozotocin (STZ) is a pancreatic islet toxin that is used to induce a model of type I diabetes in animals (136). Within the kidney, HO-1 is induced specifically in the glomeruli in this model (6, 77). Lee et al. provided evidence for functional importance of renal induction of HO-1 in diabetes (77). The authors showed that the enzymatic activity was increased in diabetic rat kidney glomeruli and cultured podocytes stimulated with high glucose. ZnPP, an inhibitor of HO activity, worsened apoptosis of podocytes in vivo and in vitro (77). Inhibition of HO activity also increased albuminuria (77). Induction of HO-1 with hemin abrogated podocyte apoptosis (77). They suggested hypoxia inducible factor (HIF)-1 as an upstream signaling molecule for induction of HO-1 and demonstrated a positive correlation between HIF-1 and HO-1 in diabetic rat glomeruli and glucose-stimulated podocytes (77). In STZ diabetic mice, hemin treatment induced HO-1. In addition to increasing atrial natriuretic peptide (ANP) and adiponectin, treatment caused a decrease in the pro-inflammatory cytokines tumor necrosis factor-α (TNF-α), IL-1β, IL-6, redox response transcription factor NF-κB, and markers of fibrosis (105).
Mouse mesangial cells treated with high glucose demonstrated increased ROS levels, transforming growth factor-β1 (TGF-β1) expression and proliferation (80). Inducing expression of Nrf2 caused a decrease in markers of ROS and oxidant-mediated damage in the presence of high glucose. Upon Nrf2 induction, increased levels of HO-1 and glutamate cysteine ligase, an enzyme involved in antioxidant glutathione synthesis, were observed (80). Upregulation of Nrf2 may provide an avenue for therapeutics for diabetic kidney disease. In a large multicenter trial, patients with type II diabetes and CKD were treated with a potent Nrf2 activator, bardoxolone methyl, or placebo. Although initial results were encouraging, the trial was terminated before completion due to increased risk of cardiovascular events in the treatment group (35).
Oxidative stress plays an important role in early pathological changes in the kidney in STZ diabetic rats (116). Rodriguez et al. demonstrated that the HO system regulates hemodynamics in diabetic rats with hyperglycemia. Treatment with the antioxidant tempol showed similar responses as seen with HO-1 induction (116). Thus, HO-1 may be beneficial in diabetic kidney disease due to its ability to restore the redox health of the tissue. The authors demonstrated low renal cortical NO, vasoconstriction, and hypoperfusion in diseased animals. Induction of HO or treatment with CO reversed these adverse changes by attenuating detrimental vasoconstriction (116).
Hayashi et al. used STZ-induced diabetes in rats and evaluated a number of antioxidant enzymes, including HO-1, catalase, glutathione peroxidase, and zinc–copper superoxide dismutase (51). They showed that HO-1 expression was specifically increased. Insulin and vitamin E treatment reversed induction of HO-1, suggesting that hyperglycemia and oxidative stress were involved. Immunohistochemistry studies provided evidence that podocytes and mesangial cells expressed HO-1 in this model (51).
Akita hyperglycemic mice develop diabetic kidney disease (1). These mice have a heterozygous deletion for the insulin gene and demonstrate induction of renal HO-1. Abdo et al. overexpressed catalase in Akita transgenic mice, decreasing oxidative stress, and found lower levels of Nrf2 expression. Catalase overexpression coincided with lower levels of HO-1 induction, providing evidence that oxidative stress contributes to renal induction of HO-1 in the Akita model of diabetes (1).
These studies collectively demonstrate a protective role for HO in diabetic kidney disease. HO provides this protection via diverse mechanisms. HO-1 induction mitigates oxidative stress-mediated damage, inflammation, vasoconstriction, glomerular histopathologic changes, and decreased kidney function, all of which are important in the pathogenesis of diabetic kidney disease.
Lupus nephritis
Tissue-resident and monocyte-derived MPs are important in the pathogenesis of renal disease in systemic lupus erythematosus (SLE). Significant MP infiltration occurs in lupus nephritis (43, 73), but MPs appear to function poorly because they fail to clear apoptotic fragments. Poor phagocytic function is thought to drive the development of autoreactive anti-double-stranded DNA (anti-dsDNA) antibodies. Patients with SLE show reduced expression of HO-1 in circulating monocytes (54), raising the possibility of a connection between myeloid cell HO-1 expression and lupus nephritis.
Recently, Mackern-Oberti et al. have demonstrated a beneficial role for HO activity in FcγRIIb−/− mice, a model for SLE (84). The FcγR is found on MPs and is important for their phagocytic function. These mice develop proteinuria and renal inflammation that is improved with HO-1 induction or CO treatment (84). The authors showed that HO-1 mRNA transcription was decreased in CD11b+, CD11c+, and CD4+ inflammatory cells in the spleens of FcγRIIb−/− mice relative to controls. CD11b+ cells expanded in the spleens of FcγRIIb−/−, and CO treatment decreased CD11b+ cell numbers and CD11b+ Gr-1+ neutrophils. FoxP3 transcription was reduced and the proportion of CD4+ FoxP3+ Treg cells was reduced in the spleen and inguinal nodes of FcγRIIb−/− mice. This provided evidence for an anti-inflammatory and renoprotective role for HO-1 in a mouse model of lupus nephritis (84).
Takeda et al. used MRL/lpr mice to model SLE. These mice develop impaired kidney function and glomerular injury. Induction of HO-1 with hemin resulted in decreased proteinuria, reduced glomerular immune complex deposition, and increased expression of inducible nitric oxide synthase (iNOS) in the spleen and kidney of diseased mice. Serology showed decreased levels of anti-dsDNA immunoglobulin (132), providing evidence that HO-1 induction changes the immune status and thus the pathogenesis of lupus nephritis.
Nephrotoxic nephritis
NTN is a model of acute glomerulonephritis achieved by intravenous administration of antiglomerular basement membrane antibodies. Vogt et al. used subnephritogenic levels of nephrotoxic serum (NTS) to precondition Sprague Dawley rats before glycerol-induced rhabdomyolysis. NTS pretreatment abrogated the increase in serum creatinine and morphologic damage normally associated with rhabdomyolysis (141). Surprisingly, HO-1 was upregulated in tubules but not glomeruli in this model of glomerulonephritis. NTS treatment also increased Ft levels but failed to increase other antioxidants such as catalase or glutathione peroxidase. Treatment with the HO inhibitor SnPP abolished the beneficial preconditioning effect of NTN (141). The authors suggested elaboration of TNF-α by glomeruli, released into the urinary space or delivered to tubules via peritubular capillaries, as the mediator of HO-1 induction. They therefore postulated a novel mechanism for glomerular–tubulointerstitial communication, such that glomerular inflammation induced an acquired, protective, HO-1-mediated response in the tubules.
Glomerular induction of HO-1 has been shown in acute NTN in rats (96). Positively stained cells appeared to be mononuclear in morphology and coexpressed proinflammatory macrophage markers. In acute NTN, pretreatment with hemin decreased acute inflammation, proteinuria, glomerular microthrombi, and ED1 (macrophage marker)-positive cells (96).
Datta et al. provided evidence for an intersection of iNOS and HO-1 in NTN rats (34). HO-1 and iNOS mRNA were increased in glomeruli of nephritic animals relative to control. Nephritic animals demonstrated a significant increase in urine protein to creatinine ratio that was attenuated by pretreatment with hemin. Induction of HO-1 coincided with decreased iNOS expression and activity (34). The authors suggested that inhibition of iNOS could be attributed to (i) degradation of the heme prosthetic group required by iNOS or (ii) CO product binding to heme iron and interference with iNOS activity. Also, iron, which is liberated by HO-1, is known to downregulate iNOS, and hemin treatment decreased transcription and activity of iNOS in nephritic animals.
Tubular Disease
Polycystic kidney disease
Data suggest polycystic kidneys are in a state of chronic oxidative stress with altered levels of antioxidant enzymes (92). HO-1 mRNA levels were elevated in diseased kidneys from the Han: SPRD-Cy rat and C57BL/6J-cpk mouse indicating the presence of oxidative stress. Also, polycystic kidneys demonstrated a decrease in expression and activity levels of antioxidant enzymes, including extracellular glutathione peroxidase, mitochondrial superoxide dismutase, peroxisomal catalase, and cytosolic glutathione S-transferase (92).
AKI caused by ischemia–reperfusion (IR) or nephrotoxic agents accelerated the progression of cyst formation in mouse models of polycystic kidney disease (PKD) (50, 131). Zhou et al. showed induction of HO using cobalt protoporphyrin (CoPP) reduced cytogenesis in cpk mice, a model of autosomal recessive PKD. There were fewer cysts and smaller kidneys in treated mice compared to controls. In contrast, HO inhibition with SnPP produced the opposite effect, resulting in increased cystogenesis and larger kidneys (162). These data provide evidence that HO-1 modulates disease progression in PDK, possibly by altering the redox balance.
Acute kidney injury
IR injury
Bilateral renal ischemia for 30 min followed by reperfusion increased renal heme content and induced HO-1 expression and HO activity in mice (89). In a similar, uninephrectomized rat model, increased renal heme load appeared to induce HO-1 expression. Rats treated with the HO inhibitor SnPP increased heme levels in the kidney, with worsening renal function and widespread tubular epithelial damage (123).
NF-κB and monocyte chemoattractant protein-1 (MCP-1) are upregulated in oxidative stress that occurs in renal IR injury (IRI). These signaling molecules facilitate recruitment of monocytes and macrophages to the site of ischemic injury. HO-1-deficient mice, when subjected to 10-min bilateral ischemia, exhibited more kidney damage and significantly increased MCP-1 and NF-κB levels, compared with wild-type mice (109, 127). Bone marrow-derived macrophages overexpressing HO-1 localized to the injured kidney when administered intravenously in mice undergoing IRI. Mice adoptively transferred with HO-1 overexpressing MP exhibited improved renal function 24 h after injury compared with controls (41).
Cheng et al. recently showed that adiponectin could protect against IRI-induced renal damage (31). Adiponectin upregulated HO-1 expression via the peroxisome proliferator-activated receptor α (PPARα) pathway. Upregulation of cyclooxygenase 2 was required for adiponectin-mediated induction of HO-1 (31).
Ischemic postconditioning (IPo) is an IR model where the animal is subjected to intervals of ischemia after the primary insult. Guo et al. subjected rats to a bilateral 45-min IRI and showed that the beneficial effect of IPo was due to upregulation of heat shock proteins (HSPs), including HO-1 (49). As expected, IR rats demonstrated severe kidney damage and impaired renal function. There was increased lipid peroxidation, inflammation, and tubular epithelial apoptosis in the kidneys of the IR rats and these were markedly reduced in the IPo group after IRI. Quercetin, a nonspecific HSP inhibitor, significantly repressed the renoprotective effects of IPo (49). In addition, remote ischemic preconditioning has demonstrated HO-1-mediated cardioprotection (160, 161) and improved outcomes in liver IRI (62, 143).
More recently, Hull et al. demonstrated that HO-1-deficient mice are highly susceptible to bilateral ischemia for 10 min when compared to wild type, confirming previous findings by Pittock et al. (109). They used Cre-lox recombinant mice, in which a lysozyme M promoter was utilized to drive the Cre gene, allowing flox-mediated deletion of HO-1 expression in myeloid lineage cells (macrophages, dendritic cells, monocytes, and neutrophils) (HO-1LysM−/−). When these mice were subjected to 25-min bilateral IRI, they exhibited a higher number of renal tubular casts, persistent loss of proximal tubule brush border, increased collagen deposition and fibrosis compared to controls (59). These studies underscore the critical role of HO-1 in the kidney after IRI.
Rhabdomyolysis
Rhabdomyolysis is characterized by widespread necrosis of striated muscle, leading to a massive release of myoglobin into the blood. Exposure of the kidney tubules to heme and heme proteins contributes to AKI in this setting. Nath et al. demonstrated, for the first time, the functional role of HO-1 in a rat model of glycerol-induced rhabdomyolysis. A few hours after glycerol, HO-1 mRNA was increased in the kidneys, accompanied by increased HO activity. SnPP treatment resulted in significantly worse kidney function and kidney damage (100). Prior conditioning of the animals with hemoglobin resulted in induction of HO-1, and these animals were protected from AKI (100). These findings were further confirmed in HO-1 knockout mice that demonstrate 100% mortality following glycerol-induced rhabdomyolysis (68, 104). In contrast, mice that genetically overexpressed HO-1 were highly protected from rhabdomyolysis (68).
In the setting of kidney injury and iron accumulation, investigators have demonstrated subsequent upregulation of Ft coupled to HO-1 induction (4, 100). The importance of Ft in the protective effects of HO-1 was highlighted in recent studies using proximal tubule-specific Ft knockout mice. These mice exhibit significant increase in mortality and worsening of renal injury when compared to control mice subjected to rhabdomylosis and CP-induced AKI (158). More recently, Boddu et al. characterized a leucine-rich repeat kinase 2 (Lrrk2) deletion model of Parkinson's disease in rats, wherein hemoglobin accumulated in the kidney with concomitant induction of HO-1. The Lrrk2 knockout rat model was protected against glycerol-induced rhabdomyolysis. Preconditioning of these rats with endogenous hemoglobin conferred protection against AKI (19).
Sepsis and endotoxin-induced injury
The pathophysiology of AKI associated with sepsis is poorly understood; however, decreased renal blood flow, inflammation and injury of the renal tubular epithelium, and oxidative stress have been implicated in sepsis-associated AKI (9). Endotoxin treatment modeled sepsis in rats and was protective against rhabdomyolysis-induced AKI when given before glycerol (140). Rats treated with endotoxin 24 h before glycerol exhibited HO-1 and Ft induction and improved renal function compared with rats that received glycerol and endotoxin simultaneously. Inhibition of HO resulted in worse renal function in this model (140).
Free heme and HO are important in the pathogenesis of septic shock in a cecal ligation and puncture model of sepsis. Heme levels increased as a result of polymicrobial infection and were associated with tissue damage and reduced immune tolerance (74). Wegiel et al. demonstrated the significance of MP HO-1 in a tissue-specific knockout mouse. They showed beneficial effects of administration of CO on bacterial killing in an animal model of sepsis and primary human macrophages in culture. The Nacht, LRR, and PYD domains-containing protein 3 (NALP3) inflammasome was important for CO-mediated improved bacterial clearance by macrophages (145).
CP nephrotoxicity
Nephrotoxicity secondary to CP treatment is well-established (114). In animals, CP accumulates in the proximal tubules where it causes renal tubular necrosis, cast formation, and loss of the brush border, secondary to oxidative stress (124). In rats, increased kidney heme content resulted from CP nephrotoxicity, and consequently, HO-1 was induced in this model (4). Treatment with the HO inhibitor SnPP resulted in kidney damage (4). Overexpression of HO-1 is protective against CP-induced toxicity (68, 124, 159) [reviewed in Ref. (99)]. Further, CP causes more severe functional and structural renal damage in HO-1 knockout mice compared to wild-type mice (124).
Contrast-induced nephropathy
Contrast-induced nephropathy (CIN) is a common cause of AKI, especially in patients with pre-existing renal disease. Several mechanisms of damage from iodinated contrast administration have been proposed, including (i) decreased renal blood flow, (ii) direct toxicity to renal tubular epithelial cells, and (iii) increased oxidative stress.
Goodman et al. demonstrated that HO-1 induction mitigates AKI from radiocontrast media (45). In this study, uninephrectomized, salt-depleted, and indomethacin-treated, male Sabra rats, a model with pre-existing kidney dysfunction, were given radiocontrast. HO-1 was induced in the renal cortex in this model. Induction of HO-1 with CoPP improved renal function after CIN in rats, whereas treatment with HO inhibitor tin mesoporphyrin (SnMP) led to worsening of renal function. Upregulation of HO-1 was also associated with an increase in antiapoptotic proteins Bcl-2 and Bcl-X in the kidney. The expression of proapoptotic proteins caspase-3 and caspase-9 was reduced (45). These findings illustrated a protective role for HO-1 in CIN. A caveat to these studies is that the model of CIN involved multiple insults, such as reduction of nephron mass, salt-depletion, and indomethacin treatment. Compound insults and radiocontrast cause injury, whereas contrast administration alone fails to cause AKI in rodent models. The requirement for multiple insults may confound results in animal models of CIN.
Gentamicin nephrotoxicity
Gentamicin is an antibiotic that can be nephrotoxic by causing renal tubular epithelial necrosis, primarily in the cortical proximal tubules. Administration of gentamicin in rats caused induction of HO-1 within 6 h, remaining elevated for several days (4). Kidney dysfunction was not abrogated by SnPP inhibition of HO (4). Taye et al. obtained contrary results, demonstrating that ZnPP abolished the renoprotective effect of HO-1 in this model. They found HO induction decreased inflammatory markers in the kidney, conferring protection (133). These studies indicate heterogeneity in the gentamycin model of AKI.
Mercuric chloride nephrotoxicity
Mercuric chloride (HgCl2) causes nephrotoxic AKI in experimental models. HgCl2 increases oxidative stress, depletes antioxidant glutathione, and increases lipid peroxidation in the kidney. Similar to other nephrotoxins, such as CP and gentamicin, HgCl2 caused robust renal induction of HO-1 in rats (101, 149). Preconditioning of rats with hemin resulted in HO-1-mediated protection against HgCl2-induced nephrotoxicity, however, HO activity inhibition with SnPP did not worsen outcomes relative to control (101).
Cyclosporine nephrotoxicity
In cyclosporine (CsA) nephrotoxicity, tubulointerstitial fibrosis occurs secondary to increased production of free radicals, lipid peroxidation, and tissue hypoperfusion. Several reports have demonstrated a protective role for HO-1 in CsA-induced nephrotoxicity (112, 113). CsA nephrotoxicity alone reduced HO-1 levels and HO activity. Rats treated with CsA and the HO inhibitor SnPP exhibited severe renal injury characterized by tubulointerstitial scarring and fibrosis. However, rats treated with CoPP in the setting of CsA nephrotoxicity, showed normal kidney morphology coinciding with HO-1 induction. These data suggested that HO-1 was not induced in response to CsA, but its induction with CoPP was protective in this model of AKI.
In vitro studies using LLC-PK1 cells demonstrated that preincubation with bilirubin, ANP, or cGMP analog, reduced CsA-induced toxicity in a dose-dependent manner and increased cell survival (110). ANP and 8-bromo cGMP induced HO-1 expression and enzyme activity. RPTCs treated with the HO inhibitor SnPP demonstrated heightened sensitivity to CsA-induced cellular toxicity (110).
More recently, mice treated with oleanolic acid (OA), a Nrf2 pathway activator, and CsA were protected from tubulointerstitial fibrosis (56). The CsA + OA-treated mice demonstrated increased nuclear translocation of Nrf2 and HO-1 expression (56). Taken together, these data highlight the beneficial role of HO-1 induction in CsA nephrotoxicity.
Interstitial Renal Disease
Renal transplant rejection
HO-1 is induced in infiltrating MPs in acute renal allograft rejection in rats (5). In this study, the authors demonstrated infiltration of inflammatory cells 5 days post-transplant that costained for ED1, a marker for rat MPs, and HO-1 or iNOS. Infiltration occurred in the peritubular interstitium and glomeruli. This study provided the first description of induction of HO-1 and iNOS, both gas generating systems, in renal transplant rejection.
Magee et al. studied the effects of a peptide inducer of HO-1, RDP1258, in rat renal allograft rejection (85). This peptide was modeled after a region in the HLA-B molecule that was shown to be protective in other models of transplant rejection. Administration of this molecule demonstrated an increased splenic HO activity and reduced activation of inflammatory cells. In rat kidney transplantation, administration of RDP1258 increased survival of transplant recipients (85). This study was limited by a lack of investigation for off-target effects of RDP1258. Therefore, one cannot conclude that improved survival was solely due to HO-1 induction.
In 2002, Tullius et al. found that induction of HO-1 using CoPP improved survival, kidney function, and morphologic characteristics of grafts in a rat renal transplantation model (137). They showed that kidneys from CoPP-treated donor animals could be stored under ischemic conditions for up to 32 h with 60% of recipients surviving 24 weeks. Comparing this to recipients of untreated donors, 40% survival was observed after only 4 h of ischemia and storage. Morphologically, HO-1 induction protected the grafts. The authors demonstrated HO-1 induction increased Bcl-X and decreased TNF-α (137).
Heat preconditioning or CoPP induced expression of HO-1, HSP70, HSP90, and Bcl-X in a rat kidney transplant model. Both treatments improved graft survival and function. Markers of apoptosis and morphologic signs of injury were decreased in animals where HO-1 was induced. These beneficial effects were reversed with SnPP inhibition of HO activity in both heat preconditioned and CoPP-treated animals (142).
GT-repeat length polymorphisms in the HO-1 promoter correlate with renal function post-transplantation in humans (40). Kidney function from donors with at least one short GT-repeat (S allele) was improved compared with donors with long GT-repeats. Results from multivariate analysis, controlling for a variety of parameters, showed that serum creatinine of recipients, who received a graft from S allele carriers, was 0.81 times that of noncarriers (95% confidence interval [CI]: 0.70–0.95, p = 0.01) (40). No difference was found between the two groups for rejection or delayed graft function. This study demonstrated human relevance for HO-1 expression in renal graft function in the setting of transplantation.
Ozaki et al. further demonstrated the importance of HO-1 inducibility in renal allograft function in humans. They examined the HO-1 promoter GT-repeat length of both donors and recipients. Donor GT-repeat length was more important for renal function. Donors containing at least one S allele demonstrated better function over a 3-year follow-up period (107). HO is important for renal allograft function and survival, representing an avenue for therapeutic intervention.
Obstructive uropathy
HO-1 is induced in renal interstitial cells in animal models of obstructive uropathy, for example, in unilateral ureteral obstruction (UUO). Such expression was observed in periglomerular and peritubular interstitial cells (65). Increased α-smooth muscle actin (αSMA) expression and F4/80+ MP infiltration were observed in the interstitium. HO-1 levels peaked 12 h after obstruction and decreased over 7 days (65). These findings indicate damage secondary to UUO in animals is mediated by oxidative stress and is characterized by inflammation and fibrosis. HO-1 is induced in the interstitium presumably in response to increased oxidative stress.
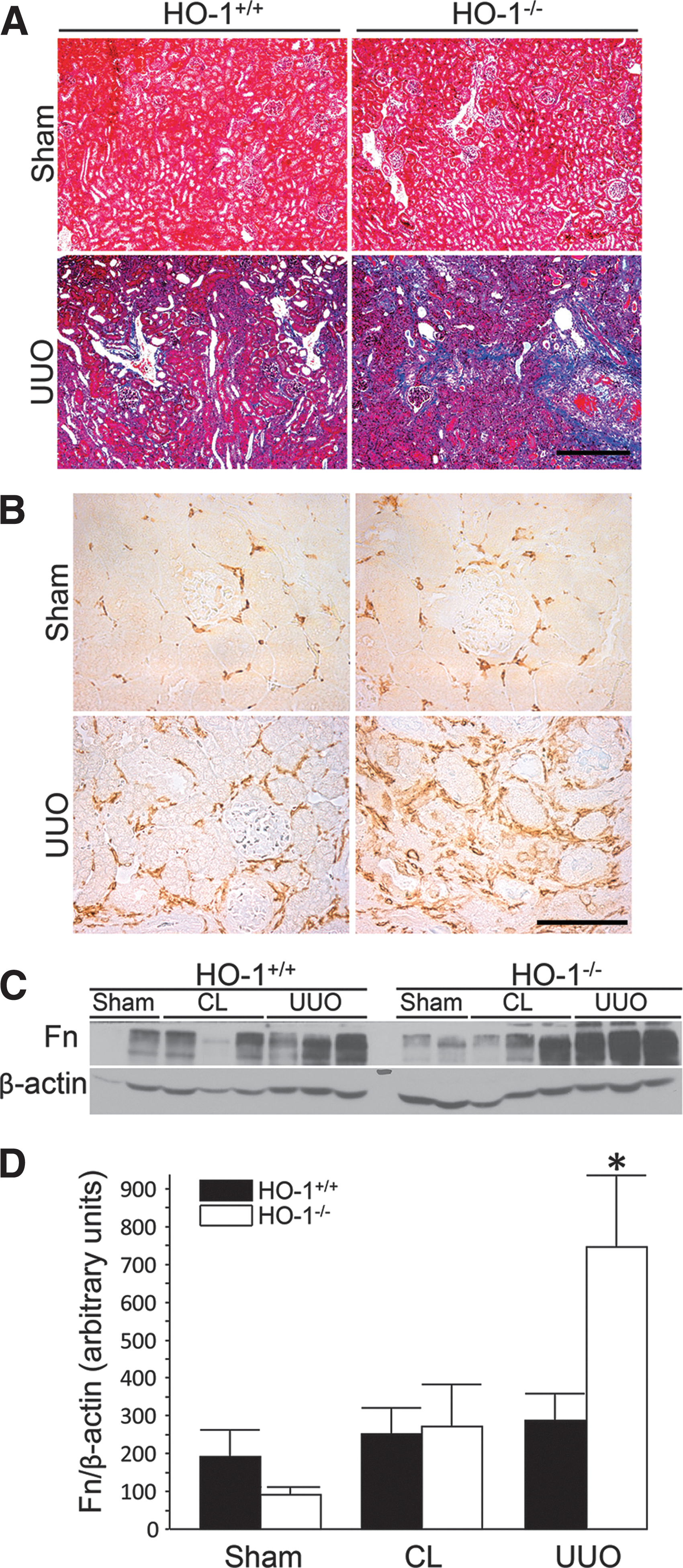
UUO in HO-1 knockout mice confirmed a protective role in renal damage and fibrosis in the setting of obstruction (67). Hydronephrosis was similar in HO-1−/− and control mice, but inflammatory infiltration and cast formation were increased in the knockout mice. Interestingly, the contralateral, unobstructed kidney appeared to display greater inflammatory infiltrate and expressed greater levels of TGF-β compared to sham control in HO-1 deficiency. This observation indicates the possibility of inflammatory crosstalk between the kidneys in this model. UUO caused more severe fibrosis in HO-1 knockout kidneys, and fibrosis was positively correlated with TGF-β and αSMA expression. MP infiltration, defined by F4/80 expression, was greater in HO-1-deficient mice, coinciding with elaboration of profibrotic TGF-β (Fig. 7) (67).
Correa-Costa et al. performed functional studies and investigated the ability of HO activity to reverse fibrosis in UUO (32). HO-1 was induced in rats by pretreatment with hemin. UUO caused proteinuria and increased protein to creatinine ratio that was reduced with HO-1 induction. UUO did not alter glomerular filtration rate, however, it increased mean arterial pressure and kidney hypertrophy. These pathologic changes were reversed with hemin treatment. Morphologically, hemin treatment decreased fibroblast proliferation and inhibited fibrosis at 7 and 14 days postobstruction. HO-1 induction was associated with decreased levels of TGF-β. Also, HO-1 induction decreased expression of IL-1β and IL-6, lymphocyte activation marker IFN-γ, and increased anti-inflammatory IL-10 (32). These investigators also performed experiments where HO-1 was induced 1 week after obstruction to study the ability of HO-1 to reverse functional decline. While not as effective as pretreatment, hemin administration 7 days after UUO improved function and decreased inflammation and fibrosis. Also, kidney hypertrophy was reduced (32). This study provides evidence of functional and morphologic improvement with HO-1 induction in the UUO model of obstructive uropathy.
Chronic kidney disease
AKI is associated with increased risk of CKD (29), however, a causal relationship has not been demonstrated. Poor response to injury may contribute to consequent progression to CKD. Inflammation and repair pathways are dysregulated following unilateral renal IR (154). Several factors implicated in the pathogenesis of CKD (e.g., TGF-β) are activators of HO-1 expression. TGF-β is a cytokine important in kidney tissue repair, regeneration, and fibrosis. TGF-β may beneficially induce HO-1 and conversely, induction of HO-1 has been proposed as a therapeutic approach in mitigating TGF-β-mediated renal disease (157). It is possible that repeated bouts of AKI with poor healing predisposes to the development of CKD. This was best demonstrated in patients with sickle cell disease (SCD) who experience acute renal ischemia during vaso-occlusive events, wherein an association between polymorphisms in the HO-1 gene and the development of CKD has been reported (118).
Zager et al. developed a model of IR-induced AKI that progresses to CKD in mice (154). The protocol for this model involves unilateral IRI and leaving the contralateral kidney intact. Upon infliction of relatively mild IRI, the injured kidney develops progressive histologic damage, inflammation, fibrosis, and functional decline over a 3-week period (148). TNF-α, MCP-1, TGF-β1, and collagen III were upregulated in injured kidneys in these animals. Cortical expression of anti-inflammatory IL-10 spiked postischemia, then dropped to levels below baseline at 1 and 3 weeks. Gene activating histone acetylation was observed at the MCP-1, TGF-β1, and collagen III gene loci (153, 154). As expected, cortical HO-1 was induced in the acute phase and dropped to near baseline levels at 3 weeks (154).
Incorporating toxic levels of adenine into the diet of rats caused tubulointerstitial nephropathy and CKD (10). These animals demonstrated impaired Nrf2 activation and decreased elaboration of its downstream targets, including HO-1, glutamate-cysteine ligase, and catalase. Low Nrf2 activity resulted in increased oxidative stress and activation of the transcription factor NF-κB (10).
Ishima et al. performed a biochemical and in vitro study to learn the effects of uremic toxin indoxyl sulfate (IS) on ROS formation and the role HO-1 plays in this system (60). IS and other uremic toxins are elevated in serum of patients with CKD, and IS has been proposed to play a pathological prooxidant role (39). Induction of HO-1 in HK-2 cells with hemin resulted in an increase in ROS formation that was reversed by inhibition with SnPP (60), suggesting HO-1 induction is a maladaptive response in the progression of CKD. However, this study is limited by lack of experiments in animal models.
In contrast, Chen et al. provided evidence that HO-1 may protect against progression to CKD in patients with coronary artery disease. In a prospective clinical study consisting of a cohort of 386 patients, the authors found that S allele carrier status was associated with a decreased risk of progression (30). Genotypic data were collected from these patients and analyzed for GT-repeat length in the proximal HO-1 promoter. Primary renal endpoints were focused on kidney function, doubling of serum creatinine and end-stage renal disease (ESRD) requiring hemodialysis. Mortality was a secondary endpoint. Adjusted hazard ratios and associated 95% CIs for homozygous L allele individuals (long GT repeats, >27) were 1.99 (1.27–3.14; p = 0.003) for renal endpoints and 1.36 (1.04–1.79; p = 0.03) for mortality (30). This study provides associative evidence that inducibility of HO-1 is protective from progression and beneficial in patients with CKD.
Vascular Disease
Hypertension
A role for HO-1 in the development of hypertensive kidney disease was first shown in spontaneously hypertensive rats (78, 90). Using Doppler flowmetry, Zou et al. found HO-1 modulated renal medullary blood flow (MBF) (163). Treatment with a zinc porphyrin HO inhibitor caused a selective drop in MBF, decreased cGMP concentration from medullary dialysate, and coincided with vasoconstriction and hypoperfusion (163). These findings indicate HO-1 is important in maintaining medullary perfusion under stress, effects that are mediated, at least, in part, via CO derived from the HO-1 catalyzed reaction.
Levere et al. used heme arginate to induce HO-1 in spontaneously hypertensive rats. Systolic blood pressure was decreased with heme arginate treatment, a change that was abrogated following treatment with a zinc porphyrin inhibitor of HO (78). Martasek et al. found that such protection against rising blood pressure was due to elaboration of bilirubin and decreased vasoconstrictive arachidonate metabolites (90).
Use of a subpressor dose of angiotensin II (Ang II) in a model of hypertension showed HO-1 induction protected kidney function and prevented inflammation and fibrosis, but did not change urinary neutrophil gelatinase-associated lipocalin (NGAL) levels (28). SnPP inhibition of HO enzyme activity worsened renal dysfunction, inflammation, injury, and fibrosis. Since CoPP treatment protected against renal injury and kidney dysfunction, but did not reverse hypertension, the authors suggested that HO-1 may mediate renoprotective effects independent of decreasing blood pressure in this model (28).
Recently, Konvalinka et al. used bioinformatics and proteomics in cell culture models to identify, and animal models to verify, candidate biomarkers of renin–angiotensin–aldosterone system (RAAS) activity in the kidney (72). Ang II induced expression of HO-1 in renal cortices. Nrf2, an important transcription factor involved in HO-1 regulation, was increased (72). HO-1 was identified as the top candidate regulated by Ang II activity in the kidney, and it also provided evidence that urinary HO-1 could serve as a marker for renal RAAS-mediated activity.
HO-1 induction is not limited to renal tissue in hypertension (61, 97). In fact, HO-1 may be induced in the vasculature systemically in response to chronic high blood pressure (61). Ishizaka et al. demonstrated increased HO-1 levels in rat aortas of animals infused with Ang II. Whereas basal expression was found in adventitial and medial smooth muscle cells, induction occured in adventitial and endothelial cells (61).
A direct role for HO-1 in the regulation of vascular tone and health was recently demonstrated in a combined animal and human study (147). Compared to wild-type mice, HO-1-deficient mice exhibited increased vascular dysfunction in multiple models, including Ang II-infusion, STZ-induced diabetes, and aging. The authors also evaluated length polymorphisms of the HO-1 promoter region in a cohort of 4937 individuals. HO-1 expression levels in monocytes correlated with flow-mediated dilation and inversely with CD14 in a subset of hypertensive subjects. Importantly, L allele homozygosity in the HO-1 promoter correlated with an increased prevalence of hypertension and reduced survival during a long follow-up period (>7 years) (147).
Sickle cell nephropathy
Cell-free hemoglobin and heme levels are elevated in sickle cell nephropathy (118). During the course of renal vaso-occlusion, intravascular hemolysis occurs and the kidney is exposed to massive amounts of toxic heme. Not surprisingly, Nath et al. found HO-1 was induced in the kidney in mouse models of SCD (103). Human transgenic sickle mice demonstrated increased body weight and kidney hypertrophy relative to control mice. HO activity was increased in the sickle mouse kidney compared with controls. Glutathione peroxidase was decreased and catalase was unchanged, indicating a specific role for HO-1. Use of a prooxidant glutathione synthesis inhibitor caused diffuse sickling throughout the kidney, supporting a pathologic role of redox dysregulation in sickle cell nephropathy. In a patient with SCD, HO-1 was expressed in the tubular epithelium, interstitium, endothelium, and smooth muscle cells of the renal vasculature. Glomerular staining for HO-1 was also observed, however, the authors were uncertain whether the expression was in mesangial or infiltrating inflammatory mononuclear cells (103).
Belcher et al. also found HO-1 was induced systemically in transgenic sickle mice. Pharmacologic HO-1 induction with hemin reduced IR-induced hemostasis in the lungs, liver, spleen, and skin (16). An associated decrease was observed in endothelial activation. In addition, treatment with HO products CO and biliverdin inhibited stasis and endothelial activation (16). Ghosh et al. investigated differential expression of genes in multiple organs of mouse models of SCD. There was selective HO-1 induction in the kidney and liver in both Berkeley and Townes mouse models of SCD (44).
In patients with SCD, Saraf et al. found that L allele homozygosity in the HO-1 promoter was associated with decreased glomerular filtration rate (118). Based on in vitro human RPTC gene expression, the authors found enrichment of single nucleotide polymorphism downstream of HO-1 in sickle patients that was associated with CKD stage (OR = 2.8, p = 0.0003) or ESRD (OR = 9.8, p = 0.0004) (118).
Conclusions
During the past few decades, an important role for the HO-1 enzyme system in the pathophysiology of kidney diseases has been supported by several lines of evidence. (i) HO-1 is induced in the kidney in both animal models and humans with a variety of kidney diseases; (ii) HO-1 expression levels determine the course of kidney disease—deficiency or inhibition of HO-1 in animal models worsens renal structure and function, and increased expression is protective; (iii) plasma and urine levels of HO-1 have been implicated as biomarkers for AKI in both animal models and humans; (iv) polymorphisms in the human HO-1 promoter correlate with HO-1 expression and outcomes in several kidney diseases; (v) many pharmacologic interventions used in preclinical models of kidney disease, including α-melanocyte-stimulating hormone, erythropoietin, IL-10, and NGAL, are potent inducers of HO-1 and mediate their effects, at least, in part, through HO-1 induction; and (vi) several clinical trials targeting the HO-1 pathway in the kidney, heart, and other organ systems are ongoing or in various phases of completion (
Footnotes
Acknowledgments
The authors acknowledge support from NIH grants R01 DK59600 (to A.A.), the core resource of the UAB-UCSD O'Brien Center (P30 DK079337; to A.A.), and partial support from NIGMS MSTP T32GM008361 (to J.M.L.).