
Abstract
Aims:
The mechanisms linking deficits in the phagocytic NADPH oxidase 2 (NOX2) complex to autoimmunity are so far incompletely understood. Deficiency in neutrophil cytosolic factor 1 (NCF1) inactivates the NOX2 complex, leading to a dramatic reduction of intra- and extracellular reactive oxygen species (ROS) and enhanced susceptibility to autoimmune disease. The contribution of intracellular NOX2 activity to autoimmune regulation is, however, unknown. Another component of the NOX2 complex, NCF4, directs the NOX2 complex to phagosomal membranes via binding to phosphatidylinositol 3-phosphate (PtdIns3P) and has been proposed to regulate intracellular ROS levels. To address the impact of NCF4 and selective changes in intracellular ROS production on autoimmune inflammation, we studied collagen-induced arthritis (CIA) and mannan-induced psoriatic arthritis-like disease (MIP) in mice lacking NCF4 and mice with a mutation in the PtdIns3P-binding site of NCF4.
Results:
Targeted deletion of Ncf4 (Ncf4−/− ) led to severe defects in overall ROS production due to concomitant reduction of NCF2 and NCF1. These mice displayed delayed neutrophil apoptosis and enhanced innate immune responses, and they developed aggravated CIA and MIP. Disruption of the PtdIns3P-binding site by targeted mutation (Ncf4*/*) resulted in selective defects in intracellular NOX2 activity, which entailed milder effects on innate immunity and MIP but clearly promoted susceptibility to CIA.
Innovation and Conclusion:
This is, to our knowledge, the first study addressing the development of autoimmunity in an organism with selectively compromised NOX2-dependent intracellular ROS levels. Our data reveal a specific role for NCF4-mediated intracellular ROS production in regulating autoimmunity and chronic inflammation. Antioxid. Redox Signal. 25, 983–996.
Introduction
R
Apart from NCF4 (p40phox), the NOX2 complex comprises three additional cytosolic subunits, namely NCF1 (p47phox), NCF2 (p67phox), and the small GTPase RAC1 or RAC2, as well as two transmembrane subunits (CYBB/gp91phox and CYBA/p22phox) that form the heterodimeric flavocytochrome b558 (21). In resting cells, NCF4 and NCF2 co-exist as a complex and NCF2 may also bind NCF1 to form a heterotrimer (21, 36, 57).
Activation of NOX2 requires conformational changes in the cytoplasmic components to allow assembly of the active enzyme complex at a specified membrane location. The core contacts that facilitate enzyme activity are between cytochrome b558, NCF2, and GTP-RAC1/2 (21, 35). These interactions enable the transfer of electrons across the membrane, from NADPH to molecular oxygen, to generate superoxide anion, which is then converted to other reactive oxygen species (ROS) depending on the location and context of activation.
Deficiency in the NADPH oxidase 2 (NOX2) complex to produce intra- and extracellular reactive oxygen species (ROS) poses a genetic risk for autoimmunity, but the specific role of intracellular NOX2 activity is unknown. This study presents functional evidence that a mutation in the phosphatidylinositol 3-phosphate (PtdIns3P)-binding site of the regulatory NOX2 subunit NCF4/p40phox, which reduced intracellular ROS formation, can enhance autoimmune responses. This is, to our knowledge, the first study investigating the development of autoimmunity in an organism with selectively compromised NOX2-dependent intracellular ROS levels.
NCF4 has been shown to regulate NOX2 activity in murine (10, 14, 16, 54) and human neutrophils (38). In addition to the PB1 domain that mediates binding to NCF2, NCF4 contains a Src homology 3 (SH3) domain, a phox homology (PX) domain, and two conserved phosphorylation sites (4). Phosphorylation of the conserved threonine 154 but not serine 315 is required for NOX2 complex activation in response to soluble and particulate stimuli (10). A functional SH3 domain is not required for oxidase activation (10).
The PX domain of NCF4 was shown to play a pivotal role in oxidase activation through its binding to the phospholipid phosphatidylinositol 3-phosphate (PtdIns3P) (17). Structural data revealed that the evolutionarily conserved arginines 58 and 105 are critical for PtdIns3P binding, whereas arginine 57 is vital to stabilize the unique fold of the PX domain (5). The basal distribution of PtdIns3P is partially cytosolic and partially concentrated on endosomal structures, but a dramatic accumulation occurs on internalized phagosomal membranes (15, 20).
An interaction of the PX domain of NCF4 with PtdIns3P was reported to differentially contribute to ROS responses elicited by different agonists, having a profound impact on ROS responses to IgG-coated targets and a partial effect on intracellular ROS responses on phagocytosis of serum-opsonized Staphylococcus aureus and to phorbol 12-myristate 13-acetate (PMA) (1, 14, 54). Thus, binding of NCF4 to PtdIns3P is a regulatory input for oxidase assembly in certain contexts of cell activation, possibly by retaining NCF4 and its cognate binding partner NCF2 on PtdIns3P-containing membranes (54).
Genetic defects in any of the NOX2 subunits that impair the production of ROS are the cause of an innate immunodeficiency that is known as chronic granulomatous disease (CGD). Patients with CGD suffer from recurrent bacterial and fungal infections as well as from granulomatous inflammation in multiple organs. So far, only one case with defects in the NCF4 gene has been reported. In this patient, heterozygosity for a frameshift mutation with premature stop codon and a missense mutation predicting an R105Q substitution in the PtdIns3P-binding site diminished intracellular ROS production, causing CGD-like colitis (38).
Manifestations of autoimmunity are more prevalent in CGD patients. This has been demonstrated for lupus-like disease in a European CGD cohort (56) and in carriers of X-linked CGD (7), whereas other autoimmune manifestations were sporadic (13). Conversely, SNPs in several of the genes coding for proteins forming the NOX2 complex have been linked to chronic inflammatory and autoimmune diseases in humans. A non-synonymous SNP in the NCF2 gene was found to be associated with both childhood- and adult-onset systemic lupus erythematosus (SLE) and a twofold reduction in Fc gamma receptor (FcγR)-elicited ROS production (28). Genetic variants of NCF4 have been associated with RA (43) and Crohn's disease (46, 47), and an increased copy number of NCF1, originally identified as an arthritis-regulating gene in rats and mice (27, 42), was predicted to protect against RA in humans (44).
In rodents, arthritis-regulating genetic variants of Ncf1 greatly diminished or abrogated intra- and extracellular ROS production by the NOX2 complex (27, 42). The intricate role of NCF1 in the assembly and activation of NOX2 involves both phosphorylation-induced conformational changes and a specific interaction of its PX domain with the plasma membrane-associated phospholipids phosphatidylinositol 3,4-bisphosphate and phosphatidic acid (29). Phosphatidylinositol 3,4-bisphosphate also co-localizes with clathrin-coated pits (45). Due to its different phospholipid-binding specificity, NCF4 has been proposed to function as a second carrier protein that mediates recruitment of NCF2 to PtdIns3P-rich phagosomal membranes instead (55). Thus, NCF1 and NCF4 may direct NOX2 activity to different cellular compartments.
Here, we addressed the role of NCF4-dependent NOX2 activity in regulating inflammatory processes targeting articular joints in two mouse models: collagen-induced arthritis (CIA) and mannan-induced psoriatic arthritis-like disease (MIP). CIA is the most commonly studied autoimmune model of RA, which is largely based on the immunological and pathological similarities between the two diseases. Similar to RA, susceptibility to CIA is linked to the expression of specific major histocompatibility complex (MHC) class II genes and both diseases share pathological features, including synovitis, pannus formation, erosion of cartilage and bone, fibrosis, and joint stiffening (23). MIP resembles human psoriasis and psoriatic arthritis-like disease in many pathological aspects. Innate immune cells and NOX2-derived ROS were shown to regulate the inflammatory processes in MIP, whereas the adaptive immune system was redundant (32).
By studying the CIA model, we hope to elucidate the role of NCF4-dependent NOX2 activity in the regulation of autoimmune arthritis, whereas the MIP model may provide further insights into the redox regulation of innate immunity and inflammation.
Results
Differential expression of cytosolic NOX2 subunits in Ncf4-targeted mice
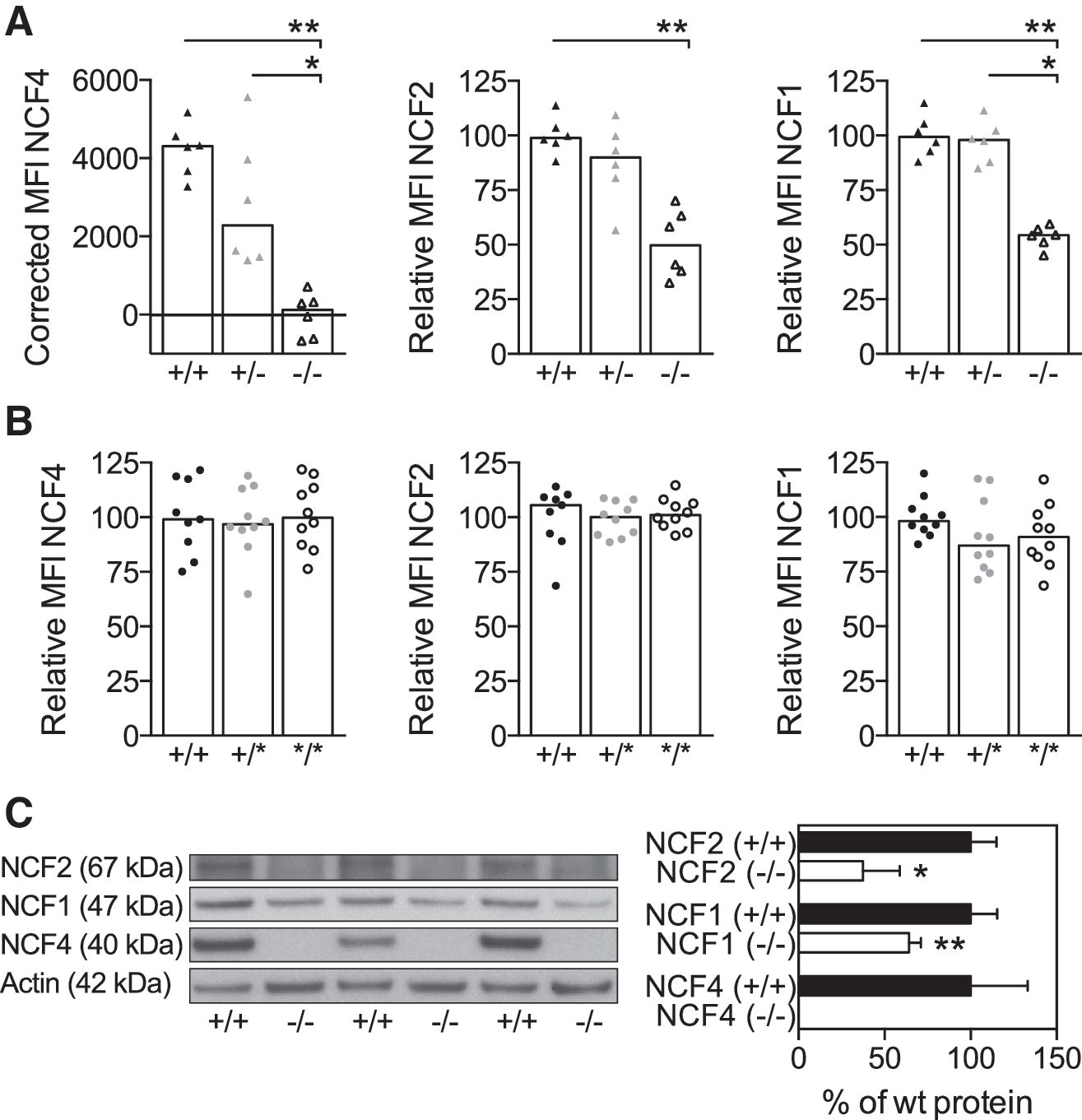
The lack of NCF4 protein in BQ.Ncf4−/− mice has previously been shown to alter the level of NCF2 (16), whereas the R58A mutation in the PtdIns3P-binding site of NCF4 was reported to not affect the expression of cytosolic NOX2 subunits (14). We sought to re-examine the protein expression of intracellular NOX2 subunits in our BQ strains. As expected, NCF4 protein was absent in BQ.Ncf4−/− (Fig. 1A, C) but was normally expressed in BQ.Ncf4*/* and BQ.Ncf4+/* polymorphonuclear neutrophils (PMNs) (Fig. 1B). Expression of NCF2 in BQ.Ncf4−/− PMNs was reduced to 50.6% ± 6.3% (mean ± SEM) of the wild-type (wt) median fluorescence intensity (MFI) (Fig. 1A), which was in accordance with published results (16). Besides NCF2, protein levels of NCF1 were also reduced in BQ.Ncf4−/− PMNs (Fig. 1A, C). Importantly, protein levels of NCF2 and NCF1 were comparable between BQ.Ncf4*/*, BQ.Ncf4+/*, and wt PMNs (Fig. 1B).
NOX2 activity titrates with NCF4 defects and regulates apoptosis
We next characterized ROS production in BQ.Ncf4−/−
and BQ.Ncf4*/* granulocytes on stimulation with the PKC activator PMA or in response to serum-opsonized S. aureus (SOS) particles. Lack of NCF4 in BQ.Ncf4−/−
bone marrow neutrophils (BMNs) dramatically reduced ROS production in response to PMA (Fig. 2A, B), whereas the mutation in the PX domain caused a moderate defect in intracellular PMA-induced ROS levels (58.6% ± 3.0% [SEM] of wt response) and an increase in extracellular PMA-induced ROS levels (Fig. 2A, B). We also detected less intracellular PMA-induced ROS in BQ.Ncf4+/* BMNs (85.7% ± 3.5% [SEM] of wt response), demonstrating that two functional copies of Ncf4 are needed for full NOX2-mediated intracellular ROS responses. After stimulation with SOS particles (Fig. 2C, D), primed BQ.Ncf4−/−
BMNs displayed severe defects in ROS production (22% ± 1% and 12.5% ± 1.2% [SEM] of wt intra- or extracellular ROS levels, respectively), whereas the mutation in the PtdIns3P-binding site in BQ.Ncf4*/* BMNs caused a moderate defect in intracellular ROS formation (73.8% ± 6.2% [SEM] of wt levels) but no defect in extracellular ROS formation. The defect in SOS-triggered intracellular ROS production was also seen in non-primed BQ.Ncf4*/* BMNs (Supplementary Fig. S1A; Supplementary Data are available online at
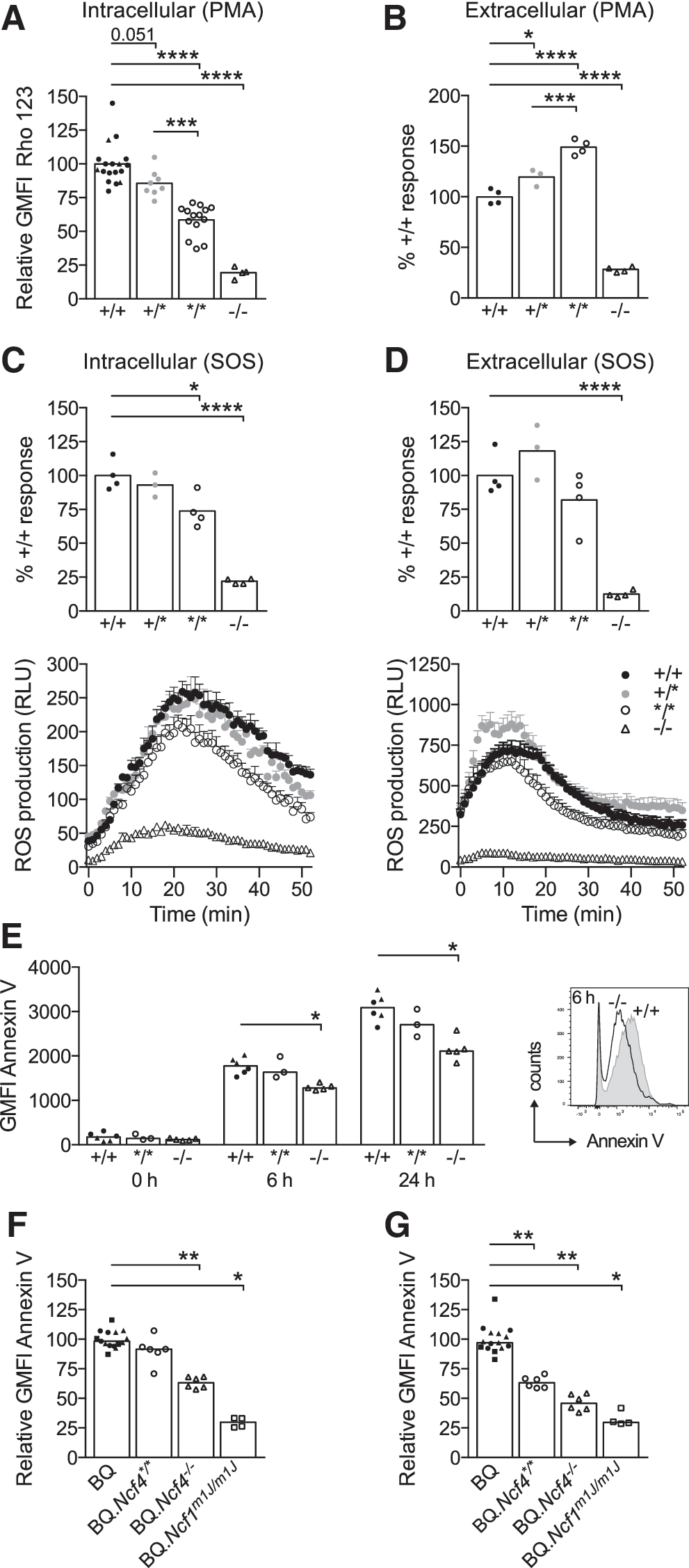
To evaluate whether reduced ROS production in Ncf4-deficient mice alters the induction and progression of apoptosis, we assessed phosphatidylserine exposure by Annexin V staining on purified CD11b+ Gr-1hi/++ BMNs (Fig. 2E) or bone marrow leukocytes gated on CD11b+ Gr-1hi/++ BMNs (Fig. 2F, G) after stimulation with SOS. Dead cells were excluded by staining with a live/dead cell stain. After 6 h, we observed similar apoptosis in BQ.Ncf4*/* and wt BMNs (Fig. 2E, F), suggesting that milder defects in ROS production are tolerated. However, reduced phosphatidylserine exposure on gated CD11b+ Gr-1hi/++ BQ.Ncf4*/* BMNs in comparison to wt cells after a 24 h culture of bone marrow leukocytes with SOS (Fig. 2G) indicated that a reduction in intracellular ROS may impair apoptosis progression to some extent. The severe defects in ROS production in BQ.Ncf4−/− BMNs clearly delayed apoptotic phosphatidylserine exposure at all time points (Fig. 2E–G). Even more dramatic effects were seen in BQ.Ncf1m1J/m1J BMNs, which completely lack ROS production by the NOX2 complex (Fig. 2F, G).
NCF4 defects promote type II CIA
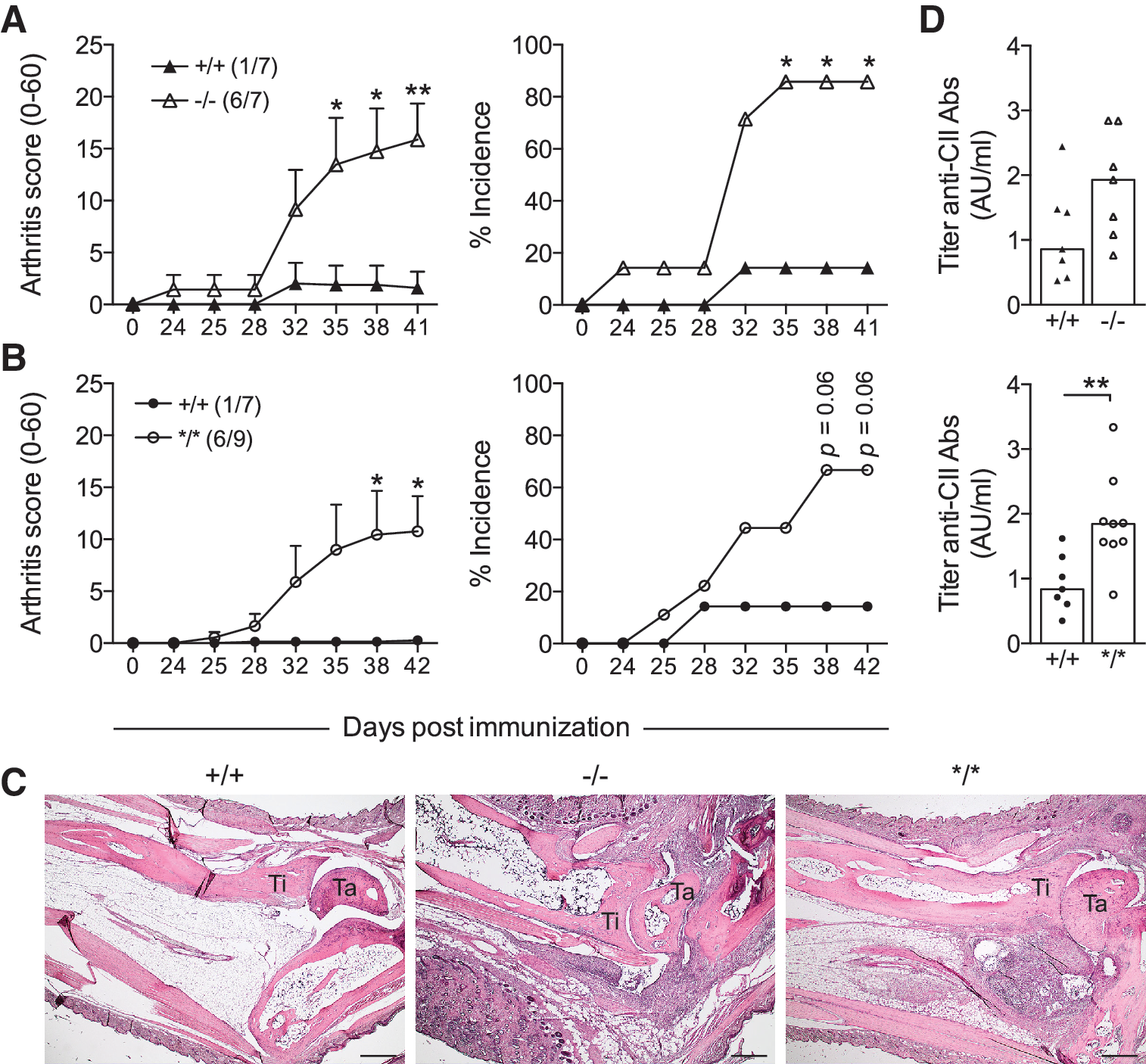
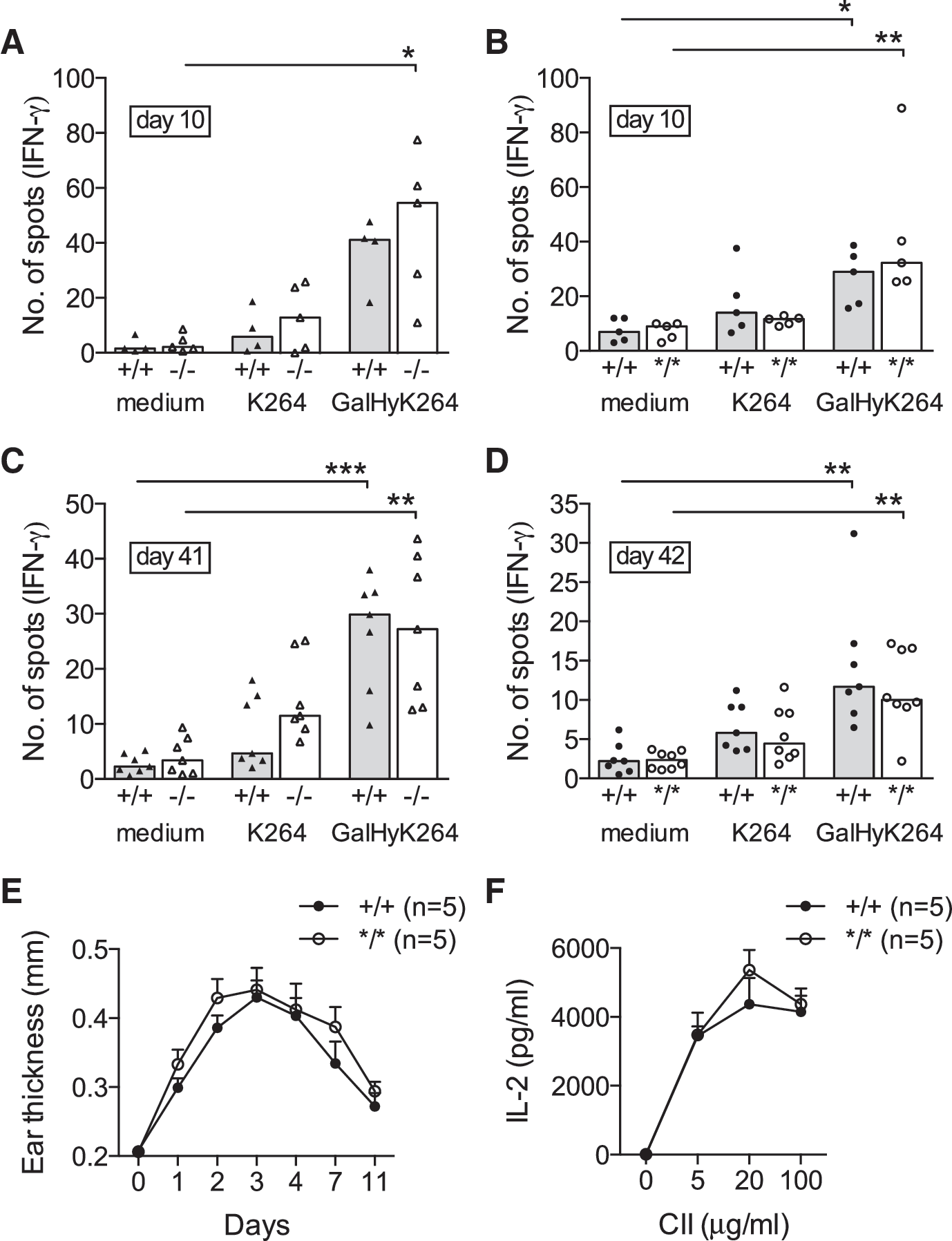
Next, we were interested as to whether the NOX2 defects in Ncf4-targeted mice would affect CIA development. On immunization, BQ.Ncf4−/− (6/7) and BQ.Ncf4*/* mice (6/9) were more prone to develop CIA compared with wt mice (1/7) (Fig. 3A, B and further data summarized in Table 1). Mice with heterozygous expression of the Ncf4 mutation also developed more severe arthritis compared with wt mice (Table 1). Histopathological analysis of ankle joint sections reflected the macroscopic arthritis scores. Arthritic joints of Ncf4-targeted mice displayed synovial infiltration, pannus formation, and incipient cartilage and bone erosion; whereas joints of wt mice (except 1/7) were largely intact with few infiltrates (Fig. 3C). Focal collections of tartrate-resistant acid phosphatase (TRAP+) osteoclasts were readily detectable at sites of bone erosion in ankle joints of Ncf4-targeted mice (Supplementary Fig. S2). BQ.Ncf4−/− mice showed a tendency toward higher serum antibody titers against CII on immunization, and we detected increased titers in BQ.Ncf4*/* compared with wt mice (Fig. 3D). Since the humoral response to CII is known to be T cell-dependent and enhanced activation of arthritogenic CD4+ T cells has been reported in the absence of NOX2-derived ROS production (19, 27), we next investigated the T cell response in Ncf4-targeted mice. However, no difference in the Th1 cell response to the MHC class II-restricted major epitope of heterologous CII (GalHyK264) or the non-modified CII peptide (K264) could be seen in BQ.Ncf4−/− or BQ.Ncf4*/* compared with wt mice either after priming (Fig. 4A, B) or during established disease (Fig. 4C, D). Moreover, we observed no differences in the Th17 cell recall response (Supplementary Fig. S3A, B), the total number of CD4+ and CD8+ T cells (Supplementary Fig. S3D), or CD69 expression (Supplementary Fig. S3E, F). The delayed-type hypersensitivity (DTH) reaction (Fig. 4E) and antigen presentation of CII (Fig. 4F) were also not affected by the NCF4 mutation. Taken together, our data show that defects in NCF4 function, even milder defects caused by the NCF4 mutation, promote CIA.
Booster immunization on day 35. Data represent male mice.
Combined data from four independent experiments (endpoint on day 54–75).
Combined data from three independent experiments (endpoint on day 64–119).
Values represent mean ± SEM of diseased mice and mice receiving a score >1 once.
Values represent mean ± SEM of all mice.
p < 0.01, *** p < 0.001.
NCF4 deficiency affects innate inflammatory pathways in CIA
To assess whether NCF4 affects inflammatory pathways during the development of CIA, we next determined systemic and local cellular and cytokine responses. We observed a dramatic increase in circulating CD11b+ Ly-6G+ neutrophils in BQ.Ncf4−/− mice both before (day 25) and during established arthritis (day 39) (Fig. 5A), but no significant increase before and only a moderate increase during established arthritis were observed in BQ.Ncf4*/* (Fig. 5B) compared with wt mice. This suggested a dose-dependent effect of ROS on peripheral neutrophil counts. The frequencies of peripheral classical and non-classical monocytes were not affected by deficiency in NCF4 (Supplementary Fig. S4A). To explore the underlying cause of peripheral neutrophilia, we determined the percentage of receptor tyrosine kinase c-Kit+ FcγRII/III+ granulocyte-macrophage precursor cells (GMPs) among bone marrow multipotent progenitor cells lacking all lineage markers (Lin − ) and the stem cell antigen Sca-1. A higher frequency of GMPs in BQ.Ncf4−/− and a modest increase in BQ.Ncf4*/* mice indicated that the differentiation of precursor cells sustained peripheral neutrophil numbers (Fig. 5C). The frequency of GMPs in naive mice was comparable between genotypes (data not shown).
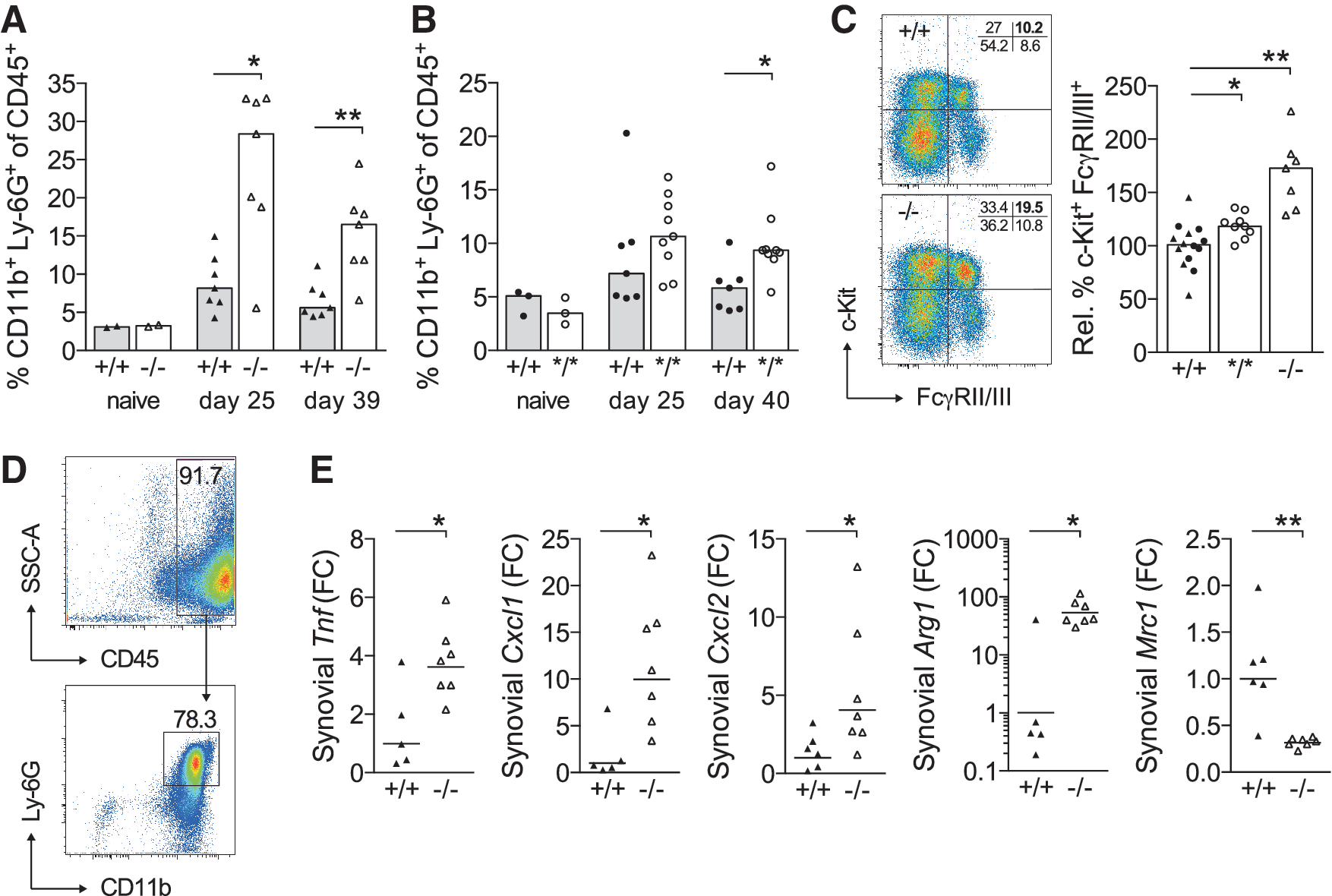
Synovia from joints with active and newly developed arthritis are dominated by infiltrating neutrophils, which is exemplified here in the synovium of an arthritic BQ.Ncf4*/* mouse containing around 80% neutrophils (Fig. 5D). Consequently, synovia from arthritic joints of BQ.Ncf4−/− mice have elevated expression of neutrophil chemoattractants (Cxcl1, Cxcl2) and tumor necrosis factor (Tnf) as a result of acute arthritis (Fig. 5E). Higher expression of arginase 1 (Arg1), an arginine-hydrolyzing enzyme that is inducible in murine neutrophils, macrophage/monocyte lineage cells, and dendritic cells (40, 41), reflected myeloid cell infiltration/activation and could play a role in the modulation of the inflammatory immune response. Expression of mannose receptor, C type 1 (Mrc1), a marker associated with tissue-resident and alternatively activated macrophages (37), was downregulated in BQ.Ncf4−/− synovium. This points to a reduced frequency of Mrc1-expressing macrophages among synovial cells.
Activation of interferon (IFN) signaling with prominent upregulation of the transcription factor signal transducer and activator of transcription 1 (STAT1) in several inflammatory cell types has previously been identified as a pathway downstream of a deficient NOX2 complex in BQ.Ncf1m1J/m1J mice completely lacking ROS production by the NOX2 complex (31). Based on that, we assessed STAT1 levels in myeloid cells in peripheral blood after immunization (day 25). STAT1 levels in circulating neutrophils (Supplementary Fig. S4B) and classical monocytes (data not shown) were comparable between Ncf4-targeted and wt mice. STAT1 levels in CD11b+ Ly-6G − Ly-6C − blood cells (mainly non-classical CD43+ CD62L − monocytes) were elevated in BQ.Ncf4−/− but not BQ.Ncf4*/* mice (Supplementary Fig. S4B). Higher serum concentrations of the proinflammatory cytokines IFN-γ, TNF, and interleukin (IL)-6 in BQ.Ncf4−/− mice reflected an improper regulation of the inflammatory response (Supplementary Fig. S4C). STAT1 levels in peripheral blood myeloid cells of naive BQ.Ncf4−/− but not BQ.Ncf4*/* mice were elevated (Supplementary Fig. S4D), whereas the amount of phosphorylated STAT1 (pY701) in response to IFN-γ stimulation was comparable to wt cells (Supplementary Fig. S4E). In summary, deletion of NCF4, which severely reduced ROS responses and delayed apoptosis induction in neutrophils, led to an increase in the number of circulating neutrophils, STAT1 levels, and proinflammatory cytokines in CIA. On the other hand, the more limited ROS defects created by the NCF4 mutant had a lesser impact on the neutrophil expansion, systemic cytokines and no effect on STAT1 levels.
Acute inflammation titrates with NOX2 deficiency
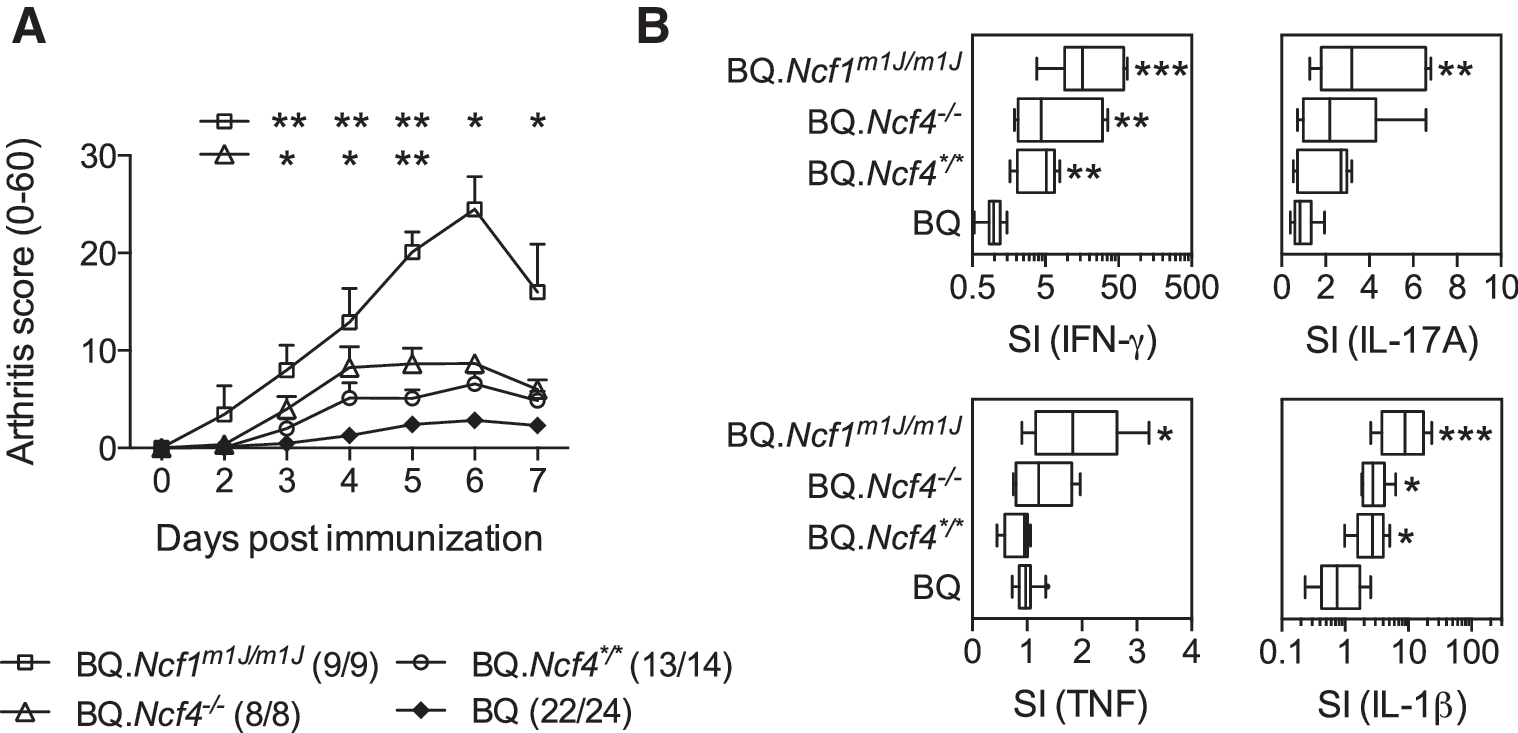
Intraperitoneal injection of yeast mannan causes a ROS-regulated, macrophage- and neutrophil-driven inflammatory response targeting the skin and articular joints, which is dramatically enhanced in BQ.Ncf1m1J/m1J mice lacking ROS production by the NOX2 complex (32). We assessed joint and cellular phenotypes after mannan injection in Ncf4-targeted mice in parallel to BQ.Ncf1m1J/m1J mice and found that joint swelling (Fig. 6A) and production of proinflammatory cytokines (Fig. 6B) titrated with the degree of NOX2 defects (BQ.Ncf1m1J/m1J mice > BQ.Ncf4−/− > BQ.Ncf4*/* > BQ). Remarkably, the disease scores in BQ.Ncf4*/* mice were not different from wt controls; IL-17A and TNF responses, shown to be driving the disease in BQ.Ncf1m1J/m1J mice (32), were not enhanced in BQ.Ncf4*/* mice. Taken together, specific targeting of Ncf4 without affecting other NOX2 subunits led to aggravated autoimmune arthritis but to only a mild inflammatory response in MIP.
Discussion
As the last NOX2 subunit that was identified, NCF4 has since been established as a positive regulator of NOX2 activity in phagocytes. By targeting NCF4 function in BQ.Ncf4−/− and BQ.Ncf4*/* mice, we were able to tune NOX2 activity in vivo and to study the functional impact of distinct changes in NCF4-dependent ROS location and levels. NCF4 defects promoted CIA in mice, demonstrating that Ncf4 alleles associated with lower NOX2 activity decrease the threshold for autoimmunity and regulate chronic inflammation.
Reduced levels of NCF2 and NCF1 in BQ.Ncf4−/− mice highlight the importance of NCF4 as a stabilizing partner for the other regulatory subunits. The discrepancy between our study and the study by Ellson et al. (16) regarding NCF1 levels in BQ.Ncf4−/− mice (detected by Western blot) may be explained by the higher purity of our neutrophil preparation and the normalization of our data to an internal loading control. Reduced levels of NCF2, the primary binding partner of NCF1 in the resting state (36), could curtail NCF1 protein stability in BQ.Ncf4−/− mice. Likewise, loss of NCF1 in BQ.Ncf1m1J/m1J mice affected NCF2 and NCF4 levels (unpublished data).
Lack of positive regulation through NCF4, including domains other than the PtdIns3P-binding site, and reduced levels of NCF2 and NCF1 explain the severe defects in NOX2 activity that were observed in BQ.Ncf4−/− mice. On the other hand, selective disruption of the PtdIns3P-binding site in NCF4 caused a moderate reduction in intracellular ROS levels in response to PMA and bacterial particles, in line with previous reports (1, 14). PtdIns3P binding to NCF4 may, however, not be critical for intracellular ROS production induced by fungal particles in murine neutrophils, as has been suggested from in vitro studies (3). This may explain why BQ.Ncf4*/* mice were not as susceptible to the ROS-regulated MIP model as were BQ.Ncf4−/− and BQ.Ncf1m1J/m1J mice. In summary, NOX2 defects in BQ.Ncf4*/* mice are more restricted both quantitatively and spatially, and they depend on the context of cell activation.
The NOX2 subunits are expressed in phagocytes and antigen-presenting cells (33, 34, 49), and both innate and adaptive immunity contribute to the development of chronic autoimmune inflammation as seen in RA and CIA. Ncf1 alleles that reduce intra- and extracellular ROS production by the NOX2 complex have been shown to enhance several autoimmune and acute inflammatory diseases in both rats and mice, modeling (i) RA (27, 42), (ii) psoriatic arthritis (32), (iii) SLE (8, 31), and (iv) gout (50). The proposed causative mechanisms triggered by a decreased ROS response are divergent and involve (i) increased arthritogenic T cell responses and dysregulated macrophages (19, 26) (although the effect on T cell responses has been variable) (58), (ii) αβ T cell-independent activation of macrophages, leading to IL-17A secretion and neutrophil-driven inflammation (32), (iii) a STAT1-dependent type I IFN response signature (31), or (iv) an inability to form neutrophil extracellular traps (NETosis), giving rise to higher levels of proinflammatory cytokines (50). The different possible disease mechanisms have recently been reviewed (24).
The mechanisms by which NCF4 deficiency promotes CIA are likely to be complex and may involve both innate and adaptive pathways that are associated with CIA pathogenesis. One of the phenotypes that we observed in BQ.Ncf4*/* mice was an increased antibody response to CII, which is known to mediate arthritis and to be T cell-dependent. However, measurements of CII-specific T cell responses were comparable between mice with NCF4 defects and wt mice. It should be kept in mind that the T cell response in CIA involves both an autoreactivity to mouse CII and a heteroreactivity to rat CII used for immunization. Therefore, the autoimmune response remains to be investigated as was done by using mice mutated at the major T cell epitope of CII (26). Our previous results from an investigation of the role of NCF1 in the CIA model point toward a central role of macrophages (19). Macrophages could, however, operate as both antigen-presenting cells and regulatory cells controlling the development of chronic inflammation. Surprisingly, BQ.Ncf4*/* mice lacked immune defects seen in more severely ROS-compromised mice, such as upregulation of STAT1 and strong enhancement of inflammatory cytokine secretion. The mechanisms by which reduced intracellular ROS levels in these mice enhance CIA remain to be investigated in more detail.
Granulopoietic cytokines have been shown to play a critical role in the pathogenesis of CIA (9, 18) and depletion of neutrophils after disease onset arrests CIA progression, demonstrating a requirement for neutrophils in disease perpetuation (18). Interestingly, lack of NCF4 has previously been linked to exacerbated inflammation with enhanced neutrophil recruitment and a deficiency to resolve dextran sodium sulfate-induced colitis in Ncf4−/− mice (11). The authors suggested that an inability to contain gut microbes under inflammatory conditions contributed to excessive inflammation in Ncf4−/− mice. In our setting, it is possible that commensal bacteria and mycobacterial components in the adjuvant change the threshold for autoimmunity in BQ.Ncf4−/− mice. It has, however, been reported that NOX2/NCF1 deficiency promotes arthritis development in germ-free mice (58) and in the absence of adjuvant (22), suggesting that ROS have a regulatory function in autoimmunity that is decoupled from host defense.
Based on our data, a complex set of phenotypes emerges in BQ.Ncf4−/− mice with severe ROS deficiency, and this is likely shared by and further amplified in BQ.Ncf1m1J/m1J mice. It involves delayed apoptosis induction, accelerated myelopoiesis of precursor cells with subsequent neutrophilia and enhanced inflammatory cytokine secretion, which may promote the susceptibility to both autoimmune arthritis and the T cell-independent MIP model (illustrated in Supplementary Fig. S5). Delayed apoptosis induction, as depicted for SOS-induced apoptosis, could prolong the survival of ROS-deficient neutrophils at inflammatory sites. Consequently, the amount of proinflammatory cytokines secreted by these neutrophils increases, hindering the resolution of inflammation and promoting disease progression. Impaired apoptosis has also been observed in neutrophils from X-linked CGD patients (6, 30, 48).
Based on our findings, the functional consequence of the RA-associated SNP in human NCF4 (43) should be evaluated in the future. Our work reflects the complex genetics that underlies RA and autoimmunity, and variants of NCF4 may also pose a genetic risk for the development of other autoimmune diseases. The particular type of autoimmune disease that may manifest in CGD patients and individuals with genetic polymorphisms in NOX2 genes is likely a combination of genotype, interacting genes, and environmental factors, as exemplified by the CGD patient harboring mutations in NCF4 (38). Even synonymous SNPs in NCF4 may be linked to functional consequences (53), demanding a careful analysis of the possible ROS phenotypes that are associated with polymorphisms in NCF4. Thus, identification of additional cases of individuals with genetic variants of NCF4 will help dissect NOX2-regulated innate and adaptive pathways that are involved in the pathogenesis of autoimmune disorders.
Materials and Methods
Mice
Ncf4− /− (16) and Ncf4R58A/R58A mice (14) (abbreviated Ncf4*/*), generously provided by Phillip T. Hawkins (Babraham Institute, Cambridge, UK), were crossed with C57BL/10.Q/rhd mice (originally from Jan Klein mouse colony, Tübingen University, Germany, and kept in our animal facility for more than two decades) to introduce the arthritis-permissive MHC class II H2-Aq haplotype and backcrossed to C57BL/10.Q/C57BL/6N.Q background (in total n ≥ 10, short name BQ). Genotyping of mice was routinely performed by TaqMan SNP assay with a specific primer set (BQ.Ncf4− /− : forward 5′ GCCGCTATCGCCAGTTCTAC 3′, reverse 5′ GGTGAAAGGGCTGTTCTTGCT 3′; BQ.Ncf4*/*: forward 5′ CAAAAGGAGGGTCCAAGTATCTCA 3′, reverse 5′ CAAACCGCTCCTCGAGCTT 3′) and two allele-specific fluorescent dye-tagged TaqMan probes (Ncf4− /− : AGCGGTAAGGGCC [FAM], TGGAGGAGCGGTTTG [VIC]; Ncf4*/*: TACCGCGCCTATC [FAM], TACCGCCGCTATC [VIC]). Age- and sex-matched littermates from heterozygous intercrosses were used in all experiments. The m1J point mutation in Ncf1 (25) was backcrossed onto the C57BL/10.Q/rhd background, designated as BQ.Ncf1m1J/m1J mice. Age- and sex-matched C57BL/10.Q/rhd mice served as wt control for BQ.Ncf1m1J/m1J mice. Mice were housed in the Scheele animal facility (Karolinska Institute, Stockholm) under SPF conditions. The local ethics committee approved all animal experiments that were performed (permits N134/13 and N490/12).
Flow cytometry
Antibodies were purchased from Biolegend, R&D Systems, or BD Biosciences and included anti-CD11b (M1/70), anti-Ly-6G (1A8), anti-Gr1 (RB6-8C5), anti-CCR2 (475301), anti-Ly-6C (HK1.4), anti-CD43 (S11), anti-CD62L (MEL-14), anti-CD45 (30-F11), anti-CD3ɛ (145-2C11), anti-TCRβ (H57-597), anti-CD4 (RM4-5), anti-CD8α (53-6.7), anti-CD45R (RA3-6B2), anti-TER-119 (TER-119), anti-c-Kit (2B8), anti-Sca-1 (D7), and anti-FcγRII/III (2.4G2). Bone marrow cells lacking expression of the lineage markers CD3ɛ, CD4, CD8α, CD11b, CD45R, Gr-1, and TER-119 were defined as Lin − . Intracellular NOX2 subunits were detected after cell fixation and permeabilization (Fixation/Permeabilization solution kit; BD Biosciences): anti-NCF1 (sc-17845; Santa Cruz), anti-NCF2 (07-502; Upstate Biotechnology), and anti-NCF4 (07-501; Upstate Biotechnology). Biotinylated primary or secondary antibodies were detected with Qdot 655- or APC-conjugated streptavidin. PE-conjugated antibodies were used for detection of STAT1 (clone 1/Stat1; BD Biosciences) or pY701 STAT1 (clone 4a; BD Biosciences) after cell fixation and permeabilization (10 min Cytofix/Cytoperm solution [BD Biosciences] at 37°C followed by 30 min on ice with 90% methanol). Specificity of STAT1 staining was verified with an isotype control. For detection of pY701 STAT1, cells were either stimulated with recombinant mouse IFN-γ (1500 U/ml, Biolegend) or left unstimulated for 10 min at 37°C before fixation. Live and dead cells were distinguished by fixable near-IR dead cell stain kit (Molecular Probes). Cells were acquired on an LSR II flow cytometer (BD Biosciences), and data were analyzed by FlowJo 9.8.5 and 10.1 software.
Western blotting
Bone marrow cells were flushed out of femur and tibia bones with 10 ml sterile phosphate-buffered saline (PBS). Untouched BMNs were isolated using a MACS neutrophil isolation kit (Miltenyi). Purity was typically 85–92% CD11b+ Ly-6G+ of live cells. BMNs were lysed in a buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate, and protease inhibitor cocktail (Roche). Cleared lysates (1 × 106 cells) were subjected to denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred, and blotted for NCF1 (07-500), NCF2 (07-502), NCF4 (07-501; all Upstate Biotechnology), and beta-actin (Abcam). Peroxidase-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch) was used for signal detection by enhanced chemiluminescence (Amersham Hyperfilm ECL; GE Healthcare).
Detection of ROS production and apoptosis
For flow-cytometric detection of intracellular ROS, bone marrow cells were incubated with 3 μM dihydrorhodamine (DHR) 123 (Invitrogen) and stimulated (20 min, 37°C) with 200 nM PMA (Sigma-Aldrich). Geometric mean fluorescence intensity of rhodamine 123 was corrected for background fluorescence (dimethyl sulfoxide). ROS production by primed (1000 U/ml TNF, 1 h, 37°C) or non-primed BMNs (4 × 105 or 3 × 105 cells/well, respectively) stimulated with PMA (100 nM) or SOS particles (BMN: particle ratio of 1:20) was measured in duplicate by chemiluminescence assay in HBSS++ containing 150 μM isoluminol/18.75 U/ml HRPII (extracellular ROS) or 150 μM luminol/375 U/ml SOD (intracellular ROS). Extracellular ROS production by bone marrow leukocytes (5 × 105 cells/well) was assessed in response to 5 μM fMLF. S. aureus BioParticles (Molecular Probes) were opsonized by incubation in DPBS with 10% mouse serum (30 min, 37°C), washed, and resuspended in HBSS++. Data output was measured in relative light units (RLU), and total RLU integrated over time (50 min for primed BMNs and fMLF-stimulated bone marrow leukocytes, 90 min for non-primed BMNs) was normalized to wt controls. For apoptosis assessment, 3.2 × 105 purified BMNs or 5 × 105 bone marrow leukocytes were resuspended in RPMI 1640 with 4% or 5% fetal calf serum (FCS) and stimulated with SOS (1:20) for the indicated time. Washed cells were stained with surface markers, fixable near-IR dead cell stain, and FITC-Annexin V (Biolegend) and they were analyzed by flow cytometry.
Induction and evaluation of CIA
Rat type II collagen (CII) was obtained from pepsin-digested SWARM chondrosarcoma (52) and, subsequently, processed as described earlier (2). Mice were immunized with 100 μg CII emulsified 1:1 in 50 μl complete Freund's adjuvant (CFA) containing 25 μg Mycobacterium butyricum (smegmatis) (Difco) intradermally (i.d.) at the base of the tail. When indicated, the mice were boosted with 50 μg CII emulsified 1:1 in 25 μl incomplete Freund's adjuvant (Difco). Arthritis development was evaluated using a macroscopic scoring system as described earlier (27). Mice were bled from the sub-mandibular vein at the indicated time points. Antibodies in serum against CII were detected by ELISA. Briefly, serum dilution series were added to CII-coated plates, and bound Igs were detected with HRP-conjugated rat anti-mouse Ig kappa (Southern Biotech) and ABTS (Roche). The absorbance was read at 405 nm (Synergy-2; BioTek Instruments). Pooled serum from immunized mice was used as a standard. Sera were also assayed for cytokines by LEGEND-plex immunoassay (Biolegend). For histopathological analysis, PFA-fixed, decalcified (0.27 M Titriplex III [Merck] containing 7.5% polyvinylpyrrolidone in 0.1 M Tris, pH 6.95), dehydrated ankle joints were mounted in paraffin and sectioned (6 μm). Deparaffinized and rehydrated slides were then stained with hematoxylin and eosin or TRAP stain and Fast Green counterstain. Images were acquired on a Zeiss Axioplan microscope that was equipped with an Olympus SC30 digital camera.
T cell assays
Peptides spanning the sequence 259–273 of CII with a non-modified lysine at position 264 (K264) or with a β-D-galactopyranosyl residue on L-hydroxylysine at position 264 (GalHyK264) were synthesized as previously described (39). For recall assays, 5 × 105 draining (inguinal, axial) lymph node cells were plated per well (#MSIPS4W10; Merck Millipore); coated with anti-IFN-γ (AN18, 10 μg/ml) or anti-IL-17A (TC11-18H10.1, 5 μg/ml); and stimulated with K264 or GalHyK264 (25 μg/ml) for 24 h. Bound cytokines were detected with biotinylated anti-IFN-γ (R46-A2, 2 μg/ml) or anti-IL-17A (TC11-8H4, 1 μg/ml), followed by alkaline phosphatase-conjugated streptavidin. Spots were developed with BCIP/NBT (Sigma-Aldrich). Scanned wells (ImmunoScan) were analyzed with ImmunoSpot software (Cellular Technology Ltd.).
For antigen presentation assays, 3 × 104 peritoneal macrophages (isolated by adherence) were co-cultured with 1 × 105 HCQ.3 hybridoma cells (12) for 24 h in 96-well plates in the presence of CII. IL-2 in supernatants was detected by ELISA using capture antibody Jes6-1A12 (2 μg/ml), biotinylated detection antibody Jes6-5H4 (1 μg/ml), and Eu-labeled streptavidin (PerkinElmer). The Eu3+ label was measured by dissociation-enhanced time-resolved fluorometry (excitation 360/40 and emission 620/40, Synergy-2; BioTek Instruments).
DTH response
Mice received an i.d. injection containing 10 μg CII in 0.02 M acetic acid in both ears on day 10 after immunization with CII/CFA. Swelling of left and right ears was measured and averaged per mouse. In control experiments, an injection of 0.02 M acetic acid alone did not induce a DTH response.
Synovial cell isolation and quantitative polymerase chain reaction
For flow cytometric analysis, synovial cells were isolated from the synovial membrane by enzymatic digest in medium containing 1 mg/ml collagenase IV (Sigma), 0.2 mg/ml DNase I (Roche), and 10% FCS. For gene expression studies, RNA was isolated from synovial tissue (RNeasy Micro Kit; Qiagen), reverse transcribed to cDNA (iScript cDNA synthesis kit; Bio-Rad), and tested for expression of Tnf, Cxcl1, Cxcl2, Arg1, Mrc1, and Actb using iQ SYBR Green Supermix (Bio-Rad) and gene-specific primers (Tnf: forward 5′ GTCCCCAAAGGGATGAGAAGT 3′, reverse 5′ GTGTGAGGGTCTGGGCCATA 3′; Cxcl1: forward 5′ GCTTGAAGGTGTTGCCCTCAG 3′, reverse 5′ AAGCCTCGCGACCATTCTTG 3′; Cxcl2: forward 5′ GCGCTGTCAATGCCTGAAGA 3′, reverse 5′ TTTGACCGCCCTTGAGAGTG 3′; Arg1: forward 5′ CTCCAAGCCAAAGTCCTTAGAG 3′, reverse 5′ AGGAGCTGTCATTAGGGACATC 3′; Mrc1: forward 5′ CCACAGCATTGAGGAGTTTG 3′, reverse 5′ ACAGCTCATCATTTGGCTCA 3′; Actb: forward 5′ CCACACCCGCCACCAG 3′, reverse 5′ TCTGACCCATTCCCACCATC 3′). Data were normalized to Actb using the ddCt method (51).
Induction and evaluation of mannan-induced inflammation
Mice were injected intraperitoneally with 20 mg mannan from Saccharomyces cerevisiae (Sigma-Aldrich) and scored daily for inflammation in the peripheral joints as previously described (32). On day 4, peritoneal cells were harvested by lavage using 10 ml ice-cold PBS, and 1 × 106 cells were stimulated with PMA (50 ng/ml) and ionomycin (1 μM) for 20 h. Supernatants were assayed for cytokines by LEGEND-plex immunoassay (Biolegend) or CBA Flex Set (BD Biosciences).
Statistical analysis
Data were analyzed using Prism software (Version 6.0f; GraphPad), and information on the specific statistical test performed is included in all figure legends. p-Values smaller than the significance level (set to 0.05) are indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Footnotes
Acknowledgments
The authors thank Vilma Urbonaviciute and Katrin Klocke for their helpful discussion and critical reading of this article. They also thank Emma Mondoc and the staff at the Scheele animal facility for technical assistance. They are grateful to Phillip T. Hawkins for kindly providing Ncf4−/− and Ncf4R58A/R58A mice. This work was supported by grants from the Konung Gustaf V:s 80-Årsfond (SGI2014-0031) (S.W.), the Karolinska Institute Foundation (S.W.), the Swedish Foundation for Strategic Research (SSF), the Swedish Research Council, the Wenner-Gren Foundations (S.W.), the Knut and Alice Wallenberg Foundation, and the European Community's Seventh Framework Programme (HEALTH-2011-278611 [Neurinox]).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.