
Abstract
Introduction
M
By providing ∼90% of cellular ATP through oxidative phosphorylation (6), mitochondria are one of the main sources of reactive oxygen species (ROS) (83), which can act as signaling molecules in healthy cells, regulating a wide range of physiological processes (40, 55). Produced in excessive amounts, however, ROS can lead to mitochondrial dysfunction and disruption and ultimately to cell death (92). The central role of these organelles in glucose, lipid, and amino acid metabolism positions them as key players in global energy modulation (21).
Over the past decade, growing evidence has suggested a causative link between mitochondrial dysfunction and major phenotypes associated with aging. Given the intimate relationship between aging and energy homeostasis, manipulations aimed at preventing or delaying metabolic changes, such as restriction of caloric intake, have been effective in extending lifespan and conferring a longevity phenotype.
Besides its implication for the aging process, mitochondrial dysfunction and metabolic impairment have been identified as hallmarks in the initiation and progression of end-organ injury in a wide range of diseases, such as cardiac dysfunction, cancer, and neurodegenerative disorders. Along with the heart and brain, the kidneys are one of the organs that are highly enriched in mitochondria, which provide the energy supply necessary for their function. Thus, it is not surprising that mitochondrial dysfunction can lead to kidney failure. In humans, inherited mitochondrial cytopathy is frequently associated with a renal phenotype that manifests as tubular disorders, but also as focal segmental glomerular sclerosis and cystic kidney disease (19, 30, 105). Acquired mitochondrial dysfunction is also emerging as a player in the pathogenesis of kidney injury and poor outcome (19, 25, 106, 147).
Here we review current knowledge of mitochondrial dysfunction in the pathogenesis of aging, and, in particular, of how mitochondrial regulation can affect disease outcome. In this context, we focus on the silent information regulator 3 (Sirt3), the primary mitochondrial deacetylase, and highlight its role as the master regulator of mitochondrial integrity during the aging process, while discussing its potential implication as a molecular target for mitochondrial protection in chronic diseases focusing on renal injury as a meaningful example.
Mitochondrial Function
Mitochondrial function: what we already know
Mitochondria are complex organelles containing two different membranes that allow for peculiar metabolism compartmentalization (32). The outer mitochondrial membrane (OMM) acts as a permeability barrier to the cytosol and has a composition similar to the plasma membrane. It contains porins that make mitochondria permeable to metabolites and small peptides (71). Conversely, the inner mitochondrial membrane (IMM) serves as an electrical insulator through the electron transport chain (ETC) proteins anchored on the typical invaginations (cristae) responsible for ATP synthesis. Impairment of cellular metabolic function is often accompanied by exuberant ROS accumulation, and oxidative damage has been associated with multiple pathologies (101), given that mitochondria are particularly susceptible to injury.
It is also widely acknowledged that mitochondria play a key role in cellular calcium homeostasis and are intimately involved in the regulation of apoptosis (29, 38). Intrinsic apoptosis is primarily, through not exclusively (70), dependent on the mitochondrial permeability transition pore (mPTP) opening that leads to mitochondrial membrane potential (ΔΨ) dissipation, cytochrome c release, and the activation of proapoptotic pathways (38).
Mitochondrial function: recent advances in mitochondrial dynamics
In the past two decades, extensive research in the field has provided compelling evidence that mitochondria are unlikely static organelles, but their size, number, and location may vary significantly depending on cell-specific energy demands. The remodeling of mitochondrial structure and function in response to metabolic changes is controlled by a perpetual process of fusion, fission, motility, and morphological changes, defined as mitochondrial dynamics, that also help maintain mitochondrial functional integrity when cells experience environmental stress (108, 120, 140).
Fusion is mediated by optic atrophy protein 1 (OPA1), an IMM dynamin-related GTPase, as well as by the OMM dynamin-related GTPases Mitofusins (Mfn) 1 and 2. It acts by mixing mitochondrial contents, thus partially buffering mitochondrial defects and mitigating transient stressors (33, 157). Fission depends on cytosolic dynamin-related protein (Drp) 1 (59) and its OMM receptors, including mitochondrial fission factor (93), fission 1 (Fis1) (145), and the 49 and 51 kDa mitochondrial dynamics proteins (MiD49-51) (95). Fission creates new mitochondria in growing cells and also contributes to quality control by enabling the removal of damaged mitochondria by mitophagy and facilitating apoptosis during high levels of cellular stress (2, 75, 88).
Mitophagy is mainly regulated by the Ser/Thr kinase PTEN-induced putative kinase 1 (PINK1), which under physiological conditions is imported to mitochondria, where it is steadily cleaved and degraded. Upon loss of ΔΨ, PINK1 accumulates on the OMM, where it recruits Parkin, a cytoplasmic E3 ubiquitin ligase, to target the damaged organelles for degradation (146).
Despite the impressive progress in our understanding of mitochondrial biology in recent years, many important aspects remain unresolved, and the multiple mechanisms through which mitochondrial structure and function can be modulated in physiological and pathological conditions remain to be fully elucidated.
Role of Sirtuin 3 in Regulating Mitochondrial Function
Post-translational modifications of mitochondrial proteins are deeply involved in the regulation of mitochondrial function to maintain normal cellular homeostasis, and lysine acetylation—the covalent addition of an acetyl group from acetyl CoA to the ɛ-amino group of lysine residues—is the most important form. Acetylated residues are observed in all major metabolic pathways in the mitochondria (151), and reversible lysine acetylation affects enzymatic activity, protein stability, protein–protein interaction, and subcellular localization of target proteins (42).
The enzymes that regulate deacetylation include the Sirtuins family of NAD+-dependent deacetylases that consume NAD+ during the catalytic reaction, yielding to O-acetyl ADP ribose, the deacetylated protein substrate, and nicotinamide (NAM). Sirtuins are evolutionarily conserved from bacteria to humans (35). In mammals, seven family members (Sirt1-7) that differ with regard to catalytic activities, subcellular localization, protein targets, and biological functions have been described (45, 57).
Among Sirtuins located within the mitochondrial matrix, Sirt3 is the major regulator of global protein acetylation in mitochondria, as documented by findings of high amounts of hyperacetylated mitochondrial proteins in Sirt3 knockout mice compared with Sirt4 and Sirt5-deficient animals (53, 79). Since its initial discovery, extensive research has identified several substrates for Sirt3 deacetylase activity that can affect different mitochondrial processes, underlining the key role of Sirt3 in maintaining mitochondrial vitality.
Regulation of energy, metabolism, and redox homeostasis
Sirt3 functions to maintain basal ATP levels by physically interacting with and deacetylating components of the ETC and F1F0 ATPase complexes (1, 131, 141). As a metabolic sensor to changes in the energy status of the cell, it modulates the activity of key metabolic enzymes involved in fatty acid oxidation (52), amino acid and acetate catabolism (46, 113, 114), ketogenesis (118), and the urea cycle (47).
Sirt3 maintains redox homeostasis through a dual mechanism. It prevents excessive electron leakage and a consequent increase in ROS generation by regulating the function of ETC complexes I and III. However, it contributes to ROS detoxification by activating mitochondrial antioxidant enzymes such as manganese superoxide dismutase (MnSOD/SOD2), isocitrate dehydrogenase, and thioredoxin 2 involved in the clearance of superoxide and hydrogen peroxide (4). Besides regulating the activity of antioxidant enzymes, Sirt3 can also induce the transcription of MnSOD/SOD2 and catalase through the deacetylation of the transcription factor Forkhead box O3A (FOXO3A) (60, 125).
Moreover, Sirt3 inhibits the activity of cyclophilin D, the regulatory component of the mPTP complex, thus preventing mPTP opening and loss of ΔΨ. Mitochondrial swelling due to mPTP opening has been observed in cardiac myocytes from Sirt3 knockout mice, which develop accelerated cardiac hypertrophy and fibrosis with age and show reduced survival to heart stressors (44).
Regulation of mitochondrial quality control and DNA repair
Oxidative damage to proteins leads to their misfolding and accumulation within mitochondria, engaging the unfolded protein response (UPRmt) that limits the proteotoxic stress and restores protein homeostasis within the organelles. The UPRmt relies on the activation of C/EBP homologous protein and estrogen receptor α, and Sirt3, very recently, has been added to the list. It has been reported that proteotoxic stress induces Sirt3, concomitant with the upregulation of FOXO3A, MnSOD/SOD2, catalase, and the autophagy marker LC3BII. In contrast, Sirt3 silencing decreases the antioxidant defense and cell viability under proteotoxic stress, highlighting the role of Sirt3 in regulating mitochondrial quality control (97).
Furthermore, Sirt3 has a role in mitochondrial DNA repair. The most frequent mutagenic lesion induced by oxidative stress is the generation of 8-oxoG, an oxidative form of guanine. By deacetylating 8-oxoguanine-DNA glycosilase 1 (OGG1), a DNA repair enzyme that excises 8-oxoG from damaged genome, Sirt3 protects mitochondrial DNA from oxidative damage and prevents apoptotic cell death under oxidative stress (20).
Regulation of mitochondrial dynamics and biogenesis
Sirt3 has recently been disclosed as a new regulator of mitochondrial dynamics by targeting OPA1, which displays profusion activity and antiapoptotic action by maintaining the tightness of cristae junctions and preventing the release of cytochrome c (34). Sirt3 directly binds to and deacetylates OPA1, thus enhancing its GTPase activity. This contributes to the preservation of mitochondrial networking and protection from mitochondrial fragmentation and cell death (111).
Mitochondrial biogenesis relies on joint action between the nucleus and mitochondria. PPARγ coactivator-1α (PGC-1α), a major regulator of mitochondrial biogenesis (112, 134), activates Sirt3 transcription through the orphan nuclear receptor Err (estrogen-related receptor)-α, which binds to the Sirt3 gene promoter (72). In turn, Sirt3 can directly activate the PGC-1α promoter via cAMP response element binding protein phosphorylation, suggesting a feed forward loop (94, 117). Overexpression of Sirt3, as well as PGC-1α, leads to higher mitochondrial DNA (mtDNA) content in cultured skeletal muscle cells, whereas Sirt3 knockdown inhibits mitochondrial biogenesis and induction of mitochondrial-related genes (72). Regulation of mitochondrial biogenesis is part of the mechanism through which Sirt3 protects cells from oxidative injury (27).
Mitochondrial Dysfunction in Aging
A decline in bioenergetics underlies the general frailty of old age and a broad spectrum of metabolic and degenerative diseases. The elderly are more prone to developing diabetes (119) and chronic kidney disease (CKD) than young adults because of progressive structural and functional alteration in the kidney (41, 107) and constitute a growing proportion of end stage renal disease (ESRD) patients (
During the aging process, mitochondrial biogenesis, function, and dynamics decline (10, 22), with the net result of dysfunctional mitochondria favoring oxidative stress-induced NLR pyrin domain-containing protein 3 (NLRP3) inflammasome activation, a family member of nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs) (110). Inflammation associated with aging (98) contributes to sarcopenia (61), which is linked to a reduction in energy expenditure and insulin resistance (23, 67).
In rodents and in nonhuman primates, calorie restriction (CR) is the most effective nutritional intervention for slowing aging and decreasing the incidence of age-related diseases (62, 91). Mechanisms underlying protection from age-associated organ damage include PGC-1α-dependent mitochondrial biogenesis (80) and Sirt3 upregulation (94, 128).
Due to its profound impact on diet adaptation via deacetylation in proteins involved in metabolic pathways, Sirt3 was first indicated as a central agent of adaptation to CR, capable of prolonging lifespan and delaying age-associated disorders (50, 104).
Of note, Sirt3 maps on human chromosome 11p15.5 in a large cluster of prosurvival genes (76), and polymorphisms in the Sirt3 gene linked with increased Sirt3 expression are associated with longevity, providing evidence of a possible Sirt3-dependent regulation of human aging (5). To this end, we have recently documented that mice with targeted disruption of the Agtr1a gene encoding for angiotensin type 1A receptor develop fewer aortic atherosclerotic lesions and less cardiac hypertrophy and fibrosis than wild type mice, which is likely caused by the reduced generation of peroxynitrite in the aorta and heart (7). Importantly, multiple organ protection from age-induced oxidative damage is accompanied by the preservation of the number of mitochondria in renal proximal tubules, linked to Sirt3 upregulation, which translates into a prolonged lifespan (7).
Accordingly, Sirt3 expression is regulated with age and it has been identified as a key regulator of several age-associated diseases including cancer, cardiac hypertrophy, and neurodegenerative and metabolic diseases [see review (85)]. In particular, Sirt3 has been associated with metabolic age-associated disorders such as metabolic syndrome and insulin resistance in peripheral tissues, which are primary risk factors for type 2 diabetes. Skeletal muscles of elderly individuals have reduced Sirt3 and PGC-1α expression than those of younger subjects (66), as occurs for Sirt3 mRNA in aged hematopoietic stem cells that show impaired self-renewal potential and differentiation capacity (14).
Sirt3 is crucial for regulating metabolic flexibility, that is, the ability to adapt fuel oxidation in response to fuel availability. When nutrients are abundant, enhanced muscle Sirt3 activity fosters glucose metabolism by deacetylating pyruvate dehydrogenase (PDH). Conversely, during fasting, muscle Sirt3 decreases, leading to impairment of PDH activity and to the switch of substrate utilization from glucose to fatty acid oxidation for energy production (65). Unlike skeletal muscle, during fasting liver Sirt3 expression is induced, favoring fatty acid oxidation through the deacetylation of long-chain acyl coenzyme A dehydrogenase (52), suggesting that Sirt3 regulates different enzymes involved in substrate utilization in different organs based on nutrient availability.
Nutrient excess, as in the case of a high-fat diet (HFD), invariably downregulates Sirt3 both in skeletal muscles and in the liver (54, 64). The functional relevance of loss of Sirt3 and consequent increase in mitochondrial protein acetylation on metabolic phenotype have been addressed in Sirt3 knockout mice. Except for one study showing that these animals display glucose intolerance associated with impaired insulin signaling in skeletal muscles (64), all other investigations reported that lean Sirt3 knockout mice have no overt metabolic dysfunction (74), whereas glucose intolerance and insulin resistance can only manifest in aged animals (54). When Sirt3 knockout animals are fed HFD, metabolic abnormalities occur earlier and are exacerbated (54, 74).
Consistent with the development of a metabolic syndrome phenotype in Sirt3 knockout mice, human subjects carrying the Sirt3 rs11246020 single nucleotide polymorphism—which induces a mutation within the catalytic domain of Sirt3 (V208I) affecting its enzymatic activity—display increased susceptibility to metabolic syndrome (54).
Another trigger responsible for insulin resistance is angiotensin II (Ang II), the main effector of the renin–angiotensin system (51, 90, 130). Ang II promotes insulin resistance in rat skeletal muscle cells via mitochondrial abnormalities (69), including excessive ROS production and Sirt3 dysregulation (82). Enhanced superoxide generation, by opening mPTP, induces mitochondrial depolarization and lowers Sirt3 protein expression and activity, impairing antioxidant defense. Ang II-induced Sirt3 dysfunction involves a reduction in AMP-activated protein kinase (AMPK) signaling and the downregulation of nicotinamide phosphoribosyltransferase, the rate-limiting enzyme in the biosynthesis of NAD+ (58).
Skeletal muscle cells are rescued from Ang II-induced insulin resistance upon treatment with acetyl-
Besides its role in maintaining a proper metabolic control during aging, Sirt3 has been linked to protection from pathogenic mechanisms, leading to aged phenotype. Indeed, it has recently been reported that Sirt3 negatively regulates the aging-associated tissue fibrosis of multiple organs by activating glycogen synthase kinase 3β, which inhibits transforming growth factor beta (TGF-β1) signaling (124).
The antiaging effect of Sirt3 has been attributed to its ability to control redox homeostasis through the activation and induction of ROS detoxifying enzymes, although this is part of a wider protective effect of Sirt3 on mitochondrial integrity (85). Altogether, these results indicate that Sirt3 plays a role in promoting longevity and attenuating age-related disease, such as metabolic syndrome and fibrosis, thus suggesting that Sirt3-dependent pathways could be targeted to influence lifespan in mammals.
Mitochondria Dysfunction in Organ Injury, with Special Emphasis on the Kidney
The main manifestation of the aging process is a progressive decline in the functional maintenance of tissue homeostasis and an increased susceptibility to degenerative diseases and death (49).
Many theories have been proposed to explain the process of aging (63), but the most widely accepted is free radical theory, according to which aging is the result of the accumulation of deleterious effects induced by free radicals and the inability of an organism to manage ROS-induced cellular damage, thus determining organ dysfunction and affecting lifespan (48). As both the major producer and primary target of ROS, mitochondria are thought to play an important role in mediating end-organ injury (138). Although mitochondrial structure and function can be different depending on the tissue of origin, or cell type within the tissue of origin, efficient energy supply is an essential feature for most of the highly differentiated functions of mammalian cells.
Disruption of mitochondrial integrity is a unifying common mechanism of cell functional loss and death in several organs, and a heterogeneous class of disorders with a broad spectrum of complex clinical phenotypes has been linked to mitochondrial defects and oxidative stress (135). In particular, mitochondria are thought to play an important role in the pathogenesis of several neurodegenerative diseases and cardiac dysfunction (101). This is not surprising, as neurons and cardiomyocytes (24, 28) are particularly sensitive and vulnerable to any abnormalities in mitochondrial function because of their high energy demand. This applies to the kidney too, in which alterations in mitochondria have been recognized as a hallmark of the initiation and progression of acute and chronic renal diseases (19).
In the kidney, tubular cells are extremely rich in mitochondria because of their high energy request for solute and metabolite reabsorption, and mitochondrial dysfunction in these cells contributes to renal failure in acute kidney injury (AKI). In experimental sepsis-dependent AKI, the suppression of PGC-1α results in time-dependent inactivation of renal mitochondrial respiratory ETC complexes I and II/III, and sustained inhibition of the antioxidant activity of MnSOD associated with increased superoxide generation, all of which indicate early events in renal function decline (99) (Fig. 1A). Notably, persistent depression of mitochondrial homeostasis may be the functional link between AKI and the development of early fibrosis (122).

Mitochondrial fragmentation is central to renal tubular cell injury and death, and is the result of the imbalance between fission and fusion. Intracellular signaling pathways involved in mitochondrial dynamics perturbation during renal cell injury have recently been reviewed (148). It is reported that mitochondria from cultured renal proximal tubular epithelial cells (RPTECs) exposed to ATP-depleting agents such as azide, or chemotherapics as cisplatin, undergo rapid fragmentation that is inhibited by B-cell lymphoma (Bcl) 2 and precede cytochrome c release and cell apoptosis (11). Mechanistically, the profission protein Drp1 translocates from the cytoplasm to mitochondria, where it interacts with Fis1, activating fission (Fig. 1B).
The inhibition of Drp1 by siRNA knockdown of Drp1, or the expression of a dominant negative Drp1, significantly reduces mitochondrial fragmentation, cytochrome release, and apoptosis in injured RPTECs, supporting a functional role of Drp1-mediated mitochondrial fragmentation in tubular cell apoptosis. To this end, pharmacological blockade of mitochondrial fission by mdivi-1, a Drp1 inhibitor, improves renal function and ameliorates renal tubular damage and apoptosis in mice with ischemia/reperfusion or cisplatin-induced AKI (11).
Mitochondrial fragmentation can also derive from the inhibition of fusion. Excessive proteolytic cleavage of the OPA-1 by the zinc metalloprotease OMA1 (3) results in mitochondrial fragmentation in ischemic AKI, with consequent tubular damage/apoptosis and renal failure (142) (Fig. 1B). In addition, Mfn1 and Mfn2 interact with Bak—a member of the Bcl-2 protein family with multi-Bcl-2 homology domains—contributing to the maintenance of a filamentous mitochondrial network in healthy cells. Upon exposure to azide or cisplatin, Bak dissociates from Mfn2 and interacts with Mfn1, leading to mitochondrial fragmentation and permeabilization, resulting in apoptosis (12) (Fig. 1B).
In vivo studies demonstrated that Bak knockout protects against ischemic AKI through a mechanism involving the maintenance of mitochondrial dynamics and integrity. At variance with wild type mice, germline Bak knockout mice show improved renal function and attenuation of kidney injury in terms of reduced mitochondrial fragmentation and apoptosis in renal tubular cells after AKI induction. Similar results were observed in mice with a conditional deletion of the proapoptotic Bax in proximal tubular cells (139). Evidence of Bak/Bax having a pathogenic role in mitochondrial fragmentation and renal tubular cell death in experimental ischemic AKI may have important implications for humans, as the mitochondrial apoptotic pathway is activated in human kidney allografts after ischemia/reperfusion injury (17).
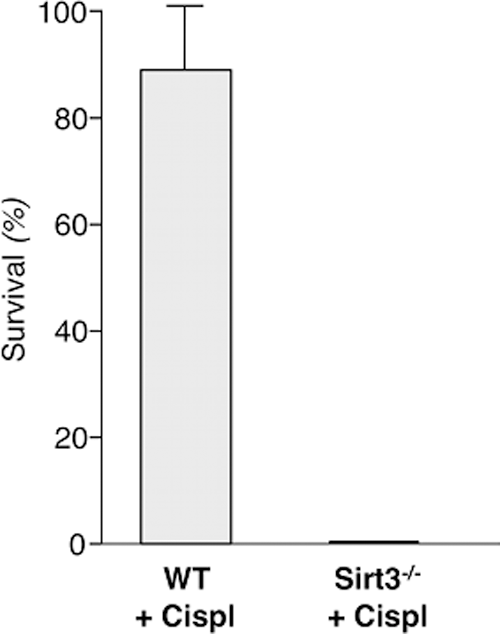
A direct role of Sirt3 in regulating mitochondrial dynamics was recently provided by experimental evidence showing that Sirt3 is a master regulator of injury and repair in AKI. Mice with cisplatin-induced AKI developed renal tubular damage related to oxidative stress and mitochondrial abnormalities associated with reduced expression of renal Sirt3 (87). Administration of the AMPK agonist AICAR or the antioxidant and mitochondrial protective agent acetyl-
The biological relevance of Sirt3 was further confirmed by findings that both AICAR and acetyl-
Taken together, the above evidence strongly supports the concept that mitochondrial dysfunction plays a key role in AKI, and suggests that restoring mitochondrial function within the nephron may be critical to functional recovery. Despite extensive research and scientific advances in the pathophysiology of AKI, the molecular target for early diagnosis and timely intervention for improving survival after an AKI episode has not been identified so far, thus slowing the development of effective therapeutic tools.
To achieve renoprotection, molecules that selectively target and concentrate in the mitochondria have been developed to improve efficacy, limiting adverse effects. They range from triphenyl alkyl phosphonium cation (TPP+)-conjugated antioxidants (i.e., the coenzyme Q10 mimetic MitoQ and SOD mimetics Mito-CP, MitoTEMPO), which accumulate in the mitochondrial matrix, and Szeto-Sciller (SS), small peptides (i.e., SS-31/Bendavia) that selectively partition to the IMM binding to cardiolipin, to mitochondrial division inhibitors (11).
The efficacy of mitochondria-targeted molecules in inducing mitoprotection has been documented in several experimental models of AKI [see review (126)]. Importantly, MitoTEMPO has been shown to improve renal function and the survival rate of septic mice, even when given as delayed therapy (99). Moreover, by protecting endothelial cell mitochondria, SS-31/Bendavia reduces microvascular rarefaction and the associated interstitial inflammation and fibrosis 4 weeks after acute ischemia/reperfusion injury (78). Clinically, a phase 2a trial is ongoing to evaluate the impact of intravenous Bendavia (MTP-131) on ischemia reperfusion injury in patients undergoing percutaneous transluminal angioplasty of the renal artery (126).
The recent realization that the disruption of mitochondrial dynamics is a key event in renal tubular cell damage in AKI and that Sirt3 is a target for renal protection offers new proof that boosting Sirt3-dependent biological pathways might represent a new important clue for obtaining organ protection in acute settings.
More recently, the role of mitochondria dysfunction is emerging as a new possible pathogenic mechanism in podocytes too, which require a high energy supply to maintain their complex cytoskeletal network to sustain the permselective function of the glomerular filtration barrier (115). There is evidence that mitochondrial dysfunction contributes to several intracellular processes involved in CKD progression, including glomerular and tubular epithelium injury, renal inflammation, and fibrosis.
One of the primary cellular sites of damage in CKD are podocytes, which are key regulators of the glomerular filtration barrier, and their injury and loss contribute to the development of proteinuria and ultimately to glomerulosclerosis (37, 81). These cells are the targets of the noxious effects of hormones and nephrotoxic drugs that promote cell damage through a mechanism that involves mitochondrial dysfunction.
Podocytes exposed to aldosterone generate excess ROS and show reduced expression of PGC-1α and its downstream mitochondrial transcription factor A, which results in lower levels of mtDNA copy number and impairment of ETC complex activity (153) (Fig. 3). Similar mitochondrial abnormalities can be observed in podocyte of aldosterone-infused mice (123) and of aristolochic acid-induced nephropathy (152), leading to the development of proteinuria (Fig. 3). Furthermore, puromycin and adriamycin induce podocyte apoptosis associated with a reduction in Mfn1 expression and induction of mitochondrial fission. Recovery of Mfn1 expression, as well as inhibition of Drp1, promotes mitochondrial fusion and prevents podocyte damage (77) (Fig. 3).

The functional and structural properties of glomerular filtration rely on finely tuned paracrine crosstalk between podocytes and endothelial cells (26). Dysfunctional podocytes release endothelin-1 (ET) that, by binding to its type A receptor on glomerular endothelial cells, induces mitochondrial oxidative stress, which is in turn responsible for endothelial dysfunction and podocyte apoptosis. In keeping with this, kidney biopsies of patients with focal segmental glomerulosclerosis show increased endothelial mtDNA damage associated with ET type A receptor overexpression in segmental sclerosis lesions (26).
Impairment of glomerular podocytes leads to protein ultrafiltration that contributes to the progression of CKD through its well-recognized toxic effect on renal tubular epithelium (156). Albumin (Alb), the main component of protein ultrafiltrate, induces apoptosis in RPTECs by triggering Bax protein upregulation and translocation to mitochondria, ultimately leading to ΔΨ alterations and cytochrome c release (31) (Fig. 4). Mitochondrial abnormalities characterized by swollen mitochondria with disorganized and fragmented cristae, associated with an increased release of cytochrome c into the cytosol, a reduced mtDNA copy number and expression of NADH dehydrogenase-1, as well as ATP synthase, are also observed in the renal tubules of Alb-overloaded mice (155).
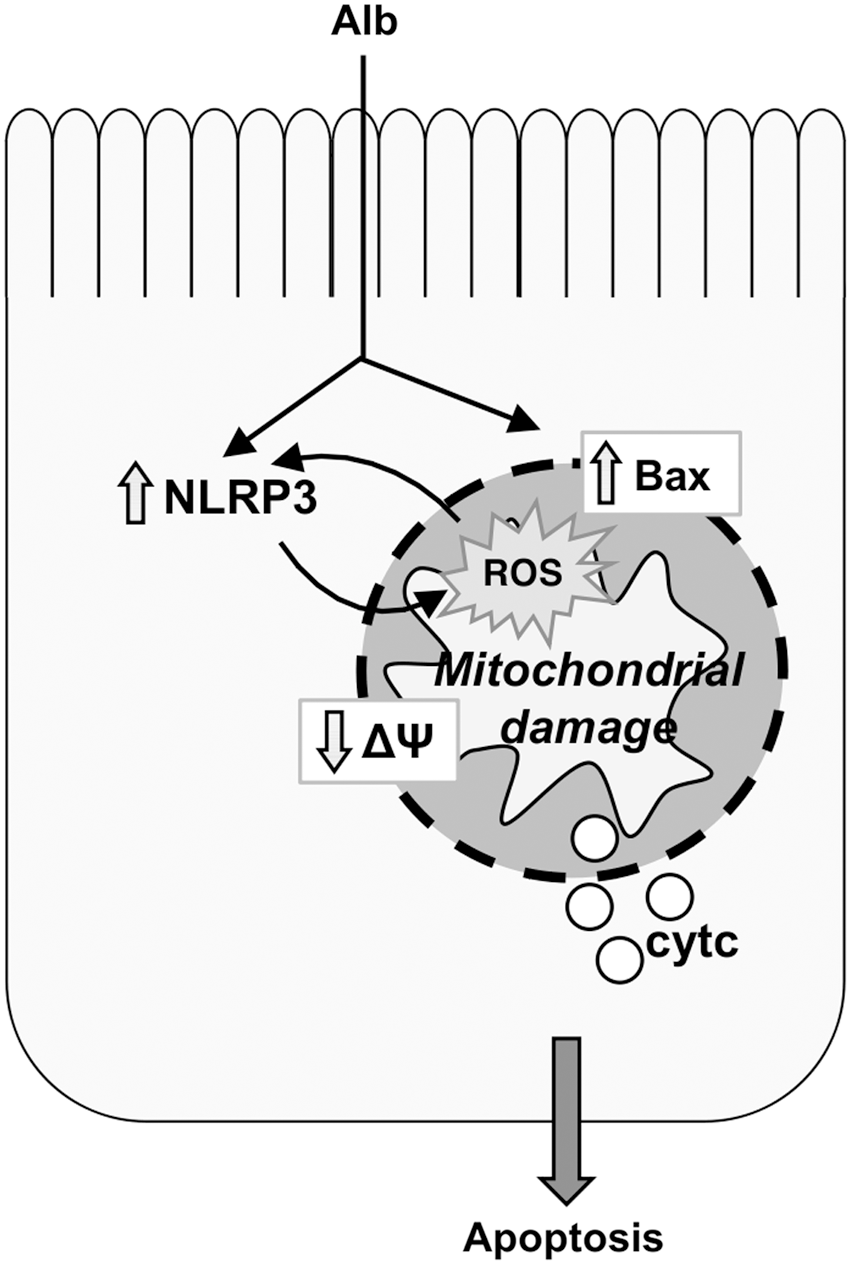
Moreover, tubule apoptosis can sustain renal inflammation through a mechanism that involves intracellular danger recognition pathways. Apoptosis-associated proteins interact with NLRs and with procaspase-1, forming the inflammasome that cleaves proinflammatory cytokines to their mature form. NLRP3, a member of the NLR subfamily, is upregulated in kidney biopsies from nondiabetic patients with CKD and correlates positively with serum creatinine (132). Moreover, higher NLRP3 protein expression is found in kidneys from proteinuric patients and parallels the severity of proteinuria (154). There is a feed forward loop between mitochondrial dysfunction and NLRP3 inflammasome activation that mediates Alb-induced renal tubular injury (154, 155) (Fig. 4).
Perturbation in mitochondrial dynamics is emerging as an important contributor to glomerular and tubular epithelial cell injury in diabetic nephropathy (DN). Decreased PGC-1α expression in the renal cortex of diabetic rats is associated with increased glomerular mitochondrial ROS generation, hypertrophy, and proteinuria. As the in vitro counterpart to these abnormalities in DN, exposure of mesangial cells to high glucose (HG) downregulates PGC-1α, promoting cell hypertrophy through a mechanism involving Drp1-mediated mitochondrial fragmentation and ROS generation (Fig. 5). Overexpression of PGC-1α by downregulating Drp1 rescues mesangial cells from hyperglycemia-induced hypertrophy (43).
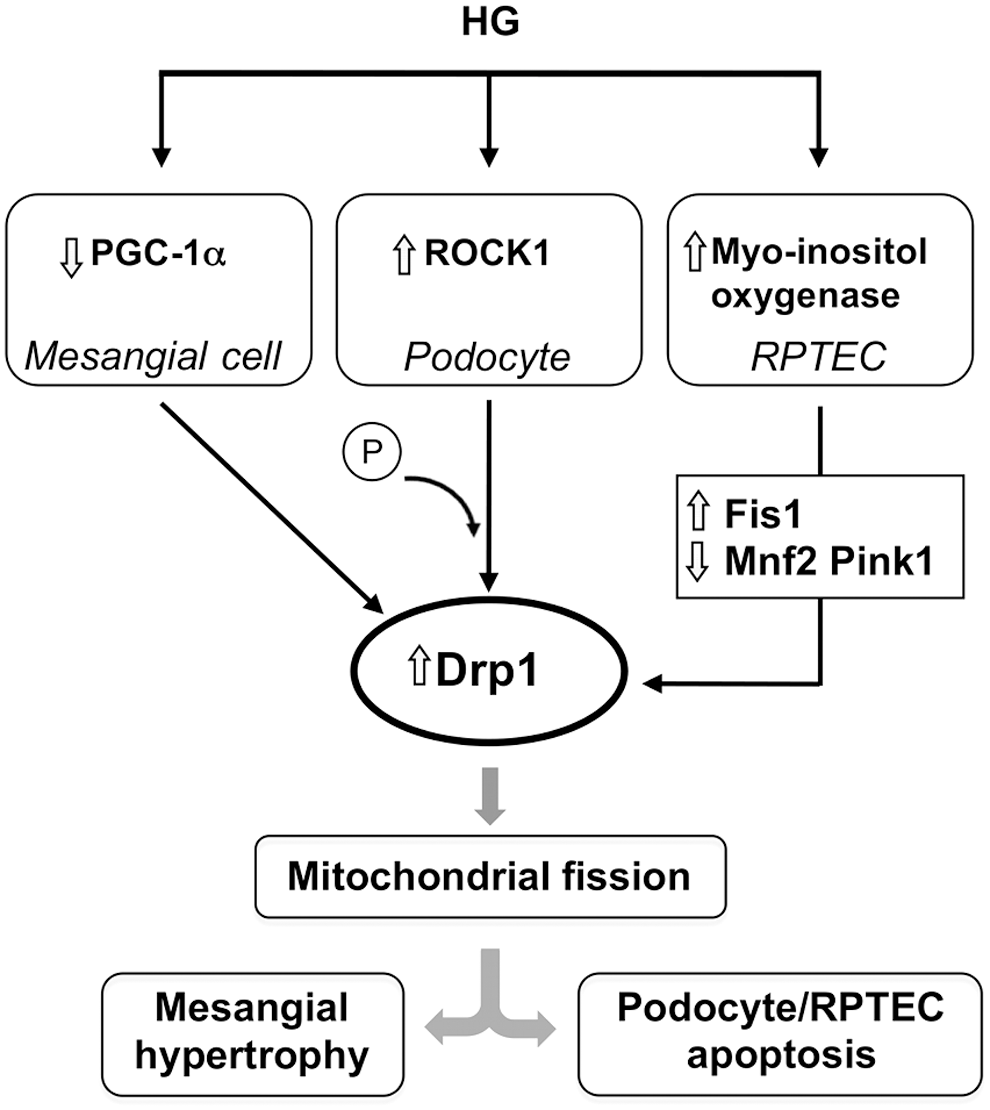
In addition, data from genetically engineered mouse models unraveled Rho-associated, coiled-coil-containing protein kinase 1 (ROCK1), a regulator of podocyte actomyosin cytoskeleton, as a key regulator of mitochondrial dynamics in DN (136). The deletion of ROCK1 in both type 1 and type 2 models of diabetes prevents an increase in mitochondrial Drp1 levels, fragmentation, ROS production, and the associated apoptosis in diabetic glomeruli, mainly in podocytes. This translates into reduced albuminuria and preservation of podocyte number. Conversely, the inducible expression of constitutively active mutant of ROCK1 in podocytes increases mitochondrial fission and exacerbates albuminuria. The mechanisms underlying the detrimental effect of ROCK1 activation in podocytes seem to be related to phosphorylation of Drp1 at Ser600 and the recruitment of the activated fission protein to mitochondria (153) (Fig. 5).
Disruption of mitochondrial homeostasis contributes to tubulopathy in DN. HG upregulates myo-inositol oxygenase, which regulates glucose metabolism, in cultured RPTECs (149). This effect is associated with the increase in mitochondrial profission proteins and the downregulation of profusion and mitophagy-related proteins (Fig. 5). The net result is mitochondrial fragmentation and defective autophagic removal of damaged mitochondria that eventuates in mitochondrial oxidative stress and tubular cell apoptosis. Myo-inositol oxygenase inhibition restores mitochondrial quality control, thus attenuating diabetes-induced renal tubular damage (149).
In addition to experimental studies showing mitochondrial dysfunction in DN, a quantitative urine metabolomics approach in diabetic patients has recently revealed a characteristic signature of suppressed mitochondrial function in patients with DN that has also been validated in independent studies (116).
There is little data describing the role of Sirt3 in the pathophysiology of CKD and it is limited to renal dysfunction associated with lipotoxicity, obesity, and diabetes. It has recently been documented that Sirt3 has a role in modulating lipotoxicity-related renal inflammation in free fatty acid-bound bovine serum albumin (BSA)-overloaded mice that display reduced renal Sirt3 gene expression and increased monocyte chemoattractant protein-1 (73) (Fig. 6).

Moreover, Sirt3 mRNA expression is downregulated in kidney biopsies from patients with obesity-related glomerulopathy and DN concomitant with a reduction in TGR5 (137), a G protein-coupled receptor that regulates glucose and lipid homeostasis (129). Reduced renal expression of TGR5 correlates with the progression of kidney disease in patients and is associated with nephropathy in diet-induced obesity and diabetic mice. The finding that TGR5 knockout mice have decreased PGC-1α, ERRα, and Sirt3 renal expression suggests that the receptor may act as an endogenous modulator of Sirt3 via PGC-1α-ERRα signaling. In keeping with this, TGR5 activation by increasing Sirt3 protein expression and activity improves mitochondrial biogenesis, inhibits renal oxidative stress, and prevents obesity and diabetes-associated kidney disease (137) (Fig. 6).
Studies with antioxidant molecule targeted to mitochondria in CKD are very few and limited to MitoQ or coenzyme Q10 treatment. Oral administration of MitoQ to Akita mice, a model of type 1 DN, reduces albuminuria and interstitial fibrosis associated with the activation of the TGF-β and β-catenin signaling pathways, and prevents glomerular basement membrane thickening (18). Similarly, coenzyme Q10 prevents renal mitochondrial dysfunction and ameliorates DN in db/db mice (100, 121).
The overexpression of mitochondrial proteins by gene transfer is another means to ameliorate renal dysfunction. Restoring Mfn2 expression in streptozotocin-induced DN exerts a renoprotective effect at an early stage of the disease by reducing oxidative stress and preventing mitochondrial dysfunction (127). Gene delivery of translocase of IMM 44 (TIMM44), which is involved in importing mitochondria-targeted preproteins into the mitochondrial matrix, attenuates proteinuria and oxidative stress and inhibits renal cell proliferation and tubular apoptosis in accelerated DN. Consistently, overexpression of TIMM44 inhibits HG-induced cell proliferation and apoptosis of cultured RPTECs and corrects mitochondrial abnormalities (150).
To date, no experimental data have been provided that elucidate what role Sirt3-targeted therapies could have in CKD, thus encouraging further research that will test whether stimulation of Sirt3-dependent biological pathways could be a valuable tool for offering organ protection in chronic settings too.
Sirtuin Modulators
So far research and development on sirtuin activators have mainly focused on Sirt1, the prototype of the sirtuin family, as a target. The first discovered sirtuin activator is resveratrol, a natural polyphenolic molecule found in grapes and red wine, which was originally described as a direct activator of Sirt1, although this evidence has been challenged by more recent findings showing that AMPK is required for the activation of Sirt1 by resveratrol (8).
Resveratrol alone does not affect Sirt3 expression levels (128) or activity—likely because of its reduced bioavailability—whereas combined with β-hydroxymethylbutirate, a breakdown product of the amino acid leucine, it can increase Sirt3 activity and stimulate fatty acid oxidation under hyperglycemia (86). Nutraceutical formulations of resveratrol with improved bioavailability have been developed (133), and naturally occurring resveratrol derivatives have recently been characterized chemically (68). Among them, trans(−)-ɛ-viniferin is able to increase Sirt3 expression and provides protection in cell models of Huntington's disease (36). Other natural polyphenols found in green tea have been described as attenuating HFD-induced renal oxidative stress through Sirt3-dependent deacetylation of MnSOD (143).
An additional pharmacological manipulation able to modulate sirtuins is represented by the direct activation of AMPK by AICAR, in particular Sirt3, as we documented in experimental AKI (87). The clinical relevance of this therapeutic intervention has been further supported by finding that intravenous administration of AICAR to type 2 diabetic patients reduces hepatic glucose output while stimulating fatty acid oxidation and lowers plasma glucose (9).
Another possible way to activate sirtuins is by promoting NAD+ biosynthesis. Although NAD+ can be synthesized de novo from the essential amino acid tryptophan via the kynurenine pathway, the main source of NAD+ derives from the salvage pathways that involve the metabolism of the NAD+ precursors nicotinic acid, NAM, and the NAM riboside (NR) (56, 89).
Boosting NAD+ through the supplementation of NAM or NR enhances mitochondrial biogenesis and function in tissues in which NAD+ content and sirtuin activity are increased. The activation of NAD+–Sirt1/3 pathways in liver and skeletal muscles is associated with the protective effect of NAM and NR against HFD-induced metabolic dysfunction (16, 144). The activation of Sirt3 by NR also reduces neurite degeneration and hearing loss by noise exposure (15). NAM riboside is now commercially available with the brand name NIAGEN (Chromadex Incorporated, Irvine, CA), and, combined with pterostilbene, has been launched on the online market as a nutraceutical antiaging pill called Basis from Elysium Health.
All these agents are effective strategies to boost sirtuins and are the subjects of intensive preclinical and clinical research, but they are not selective for Sirt3.
More recently, honokiol—a natural biphenolic compound derived from the bark of magnolia trees, which protects from cardiac hypertrophy—has been identified as the first selective Sirt3 activator (102). This compound can enter the mitochondria, where it physically interacts with Sirt3, enhancing its affinity for NAD+, thus fostering deacetylase activity. Activated Sirt3 can, in turn, activate its own gene promoter via PGC-1α (102). This natural Sirt3-activating compound could be the key chemical template for developing synthetic Sirt3 agonists. The recent case of the Nobel Prize awarded for the discovery of artemisinin and its semi-synthetic derivatives for the treatment of malaria would suggest that this could be the right direction to go also with Sirt3.
Conclusions
Control of mitochondrial dynamics appears to be the most promising way to preserve mitochondria structural and functional integrity, and could prolong lifespan by protecting against organ damage. Although the existing findings are encouraging, our understanding is still quite basic with regard to various aspects of mitochondrial biology. Important knowledge gaps must be filled to properly advance our understanding of the role of upstream and downstream mechanisms in the regulation of mitochondrial functional and structural integrity.
Growing understanding of the role of Sirt3 in mitochondrial function has contributed to defining a new molecular target amenable to therapeutic intervention, which has the potential to prevent organ damage. Given the protective role of Sirt3 in aging-associated metabolic decline and the development of fibrotic processes in multiple organs, including the kidney, it is tempting to suggest that Sirt3 might have an important impact on healthy aging. Moreover, Sirt3 is a crucial regulator of mitochondrial functional integrity and a nonredundant player in renal tubular injury and repair in AKI. Agents that enhance Sirt3 may be attractive therapeutic strategies for preserving kidney function after an acute toxic insult through mitochondrial protection.
In contrast, experimental studies have documented the pathogenic role of mitochondrial dysfunction in CKD, whereas the involvement of Sirt3 in renal disease progression is still a matter of current investigation. The available data showing that Sirt3 plays a role in insulin resistance, because of its property of orchestrating the efficient use of available nutrients, could have clinical relevance for patients with type 2 diabetes with renal insufficiency before the onset of overt nephropathy, who are at high risk of progressing to ESRD (103).
Collectively, emerging data on the crucial role of mitochondria in the pathogenesis of several diseases, along with the profound impact of Sirt3 on the organelle's metabolic adaptive stress response, offer the rationale for gaining a better understanding of the intracellular mechanisms regulating metabolic adaptations in multiple organs. All these efforts pave the way for the search for selective Sirt3 activating compounds, which the current pharmacopeia is remarkably lacking.
Footnotes
Acknowledgments
The authors are deeply indebted to Professor Giuseppe Remuzzi for his extremely valuable critical insights and thoughtful suggestions that enhanced the quality of this article. The authors also thank Dr. Antonella Piccinelli for advice on preparing the figures and Manuela Passera for help with preparing the article.