
Abstract
Aims:
The heme oxygenase-1 (HO-1)/carbon monoxide (CO) pathway induced in astrocytes after ischemic brain injury promotes vascular endothelial growth factor (VEGF) expression to maintain and repair neurovascular function. Although HO-1-derived CO has been shown to induce hypoxia-inducible factor-1α (HIF-1α)-dependent VEGF expression, the underlying mechanism independent of HIF-1α remains to be elucidated.
Results:
HO-1 and VEGF were coexpressed in astrocytes of ischemic mouse brain tissues. Experiments with specific siRNAs and pharmacological activators/inhibitors of various target genes demonstrated that astrocytes pre-exposed to the CO-releasing compound, CORM-2, or transfected with HO-1 increased HIF-1α-independent VEGF expression via sequential activation of the following signal cascades; Ca2+/calmodulin-dependent protein kinase kinase β-mediated AMP-activated protein kinase (AMPK)α activation, AMPKα-induced increases in nicotinamide phosphoribosyltransferase (NAMPT) expression and cellular NAD+ level, sirtuin 1 (SIRT1)-dependent peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) stabilization and activation, and PGC-1α/estrogen-related receptor (ERR)α-mediated VEGF expression. All of these sequential events were blocked by an L-type voltage-gated Ca2+ channel inhibitor and Ca2+ chelators, but not by other Ca2+ channel inhibitors.
Innovation:
HO-1-derived CO elicits Ca2+ influx by activating L-type Ca2+ channels, which is a key player in HIF-1α-independent VEGF expression by activating the AMPKα-NAMPT-SIRT1-PGC-1α/ERRα pathway.
Conclusion:
Our results provide new mechanistic insight into the possible role for L-type Ca2+ channels in HO-1/CO-induced angiogenesis. Antioxid. Redox Signal. 27, 21–36.
Introduction
C
Heme oxygenase-1 (HO-1) and HO-2 are essential enzymes in heme catabolism that cleave heme to carbon monoxide (CO), biliverdin (which is rapidly converted to bilirubin), and Fe2+. HO-2 is constitutively expressed in neurons where it functions as an intrinsic protector (15), whereas HO-1 is strongly induced in astrocytes in response to hypoxia and stress and promotes neuroprotection and angiogenesis in the ischemic milieu (25). In addition, HO-derived CO fulfills important roles in neurotransmission, neurogenesis, and blood circulation in the brain (6, 45, 48). Thus, the brain HO/CO system mediates multiple cerebrovascular and neuronal functions, including cerebral circulation, the blood–brain barrier, neuroregeneration, and neuroprotection, in neurovascular units.
The astrocytic heme oxygenase-1 (HO-1)/carbon monoxide (CO) pathway maintains and repairs neurovascular function following cerebral ischemia, although the underlying mechanism is only partially understood. The present study demonstrates that HO-1-derived CO stimulates neurogenic and angiogenic vascular endothelial growth factor (VEGF) expression in astrocytes by enhancing L-type Ca2+ channel-mediated Ca2+ influx and subsequent hypoxia-inducible factor-1α-independent VEGF promoter activity. These data provide new mechanistic insight into a possible role of L-type Ca2+ channels in HO-1/CO-mediated induction of VEGF, an important player not only in angiogenesis but also in maintenance of neurovascular function in the brain.
Increasing evidence demonstrates that HO-derived CO promotes angiogenesis and neovascularization by regulating the proangiogenic VEGF expression (13, 16). One study demonstrated that CO induces VEGF expression in astrocytes by increasing hypoxia-inducible factor-1α (HIF-1α) protein levels (10). However, another study showed that HO-1-derived CO elevates VEGF expression in myocytes by activating p38MAPK-dependent Sp1 (40), suggesting that CO promotes HIF-1α-independent VEGF expression. Moreover, metabolic stress stimulates VEGF expression and angiogenesis in skeletal muscle cells by activating HIF-1α-independent and energy-sensing mechanisms linked to the peroxisome proliferator-activated receptor-γ (PPARγ) coactivator-1α (PGC-1α)/estrogen-related receptor α (ERRα) pathway (3). Activation of AMP-activated protein kinase (AMPK), a crucial cellular energy sensor, also increases VEGF-mediated angiogenesis by activating PGC-1α in a mouse model of hindlimb ischemia (28, 38). The metabolic sensors, PGC-1α and AMPK, are effectively activated by endogenous and exogenous CO (46, 56). These results suggest that the HO/CO pathway stimulates VEGF expression and angiogenesis through HIF-1α-dependent and independent mechanisms.
We hypothesized that HO-1-mediated CO promotes HIF-1α-independent VEGF expression in astrocytes after ischemic brain injury, possibly contributing to endogenous repair processes, such as angiogenesis, neuronal protection, and neurogenesis (30, 57). In the present study, we show that the HO-1/CO system stimulates L-type calcium channel-mediated Ca2+ influx, which activated the Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ)-dependent AMPKα axis in astrocytes. This, in turn, increased cellular levels of NAD+, a cosubstrate of the deacetylase sirtuin 1 (SIRT1), through nicotinamide phosphoribosyltransferase (NAMPT), resulting in deacetylation-dependent stabilization of PGC-1α and HIF-1α-independent VEGF expression in astrocytes. Thus, the HO-1/CO axis contributes to neovascularization and subsequent improvement of neurovascular function during the recovery phase after ischemic injury.
Results
HO-1, VEGF, and energy-sensing genes are expressed in the peri-infarct region of the mouse ischemic brain and in in vitro CO-pretreated astrocytes
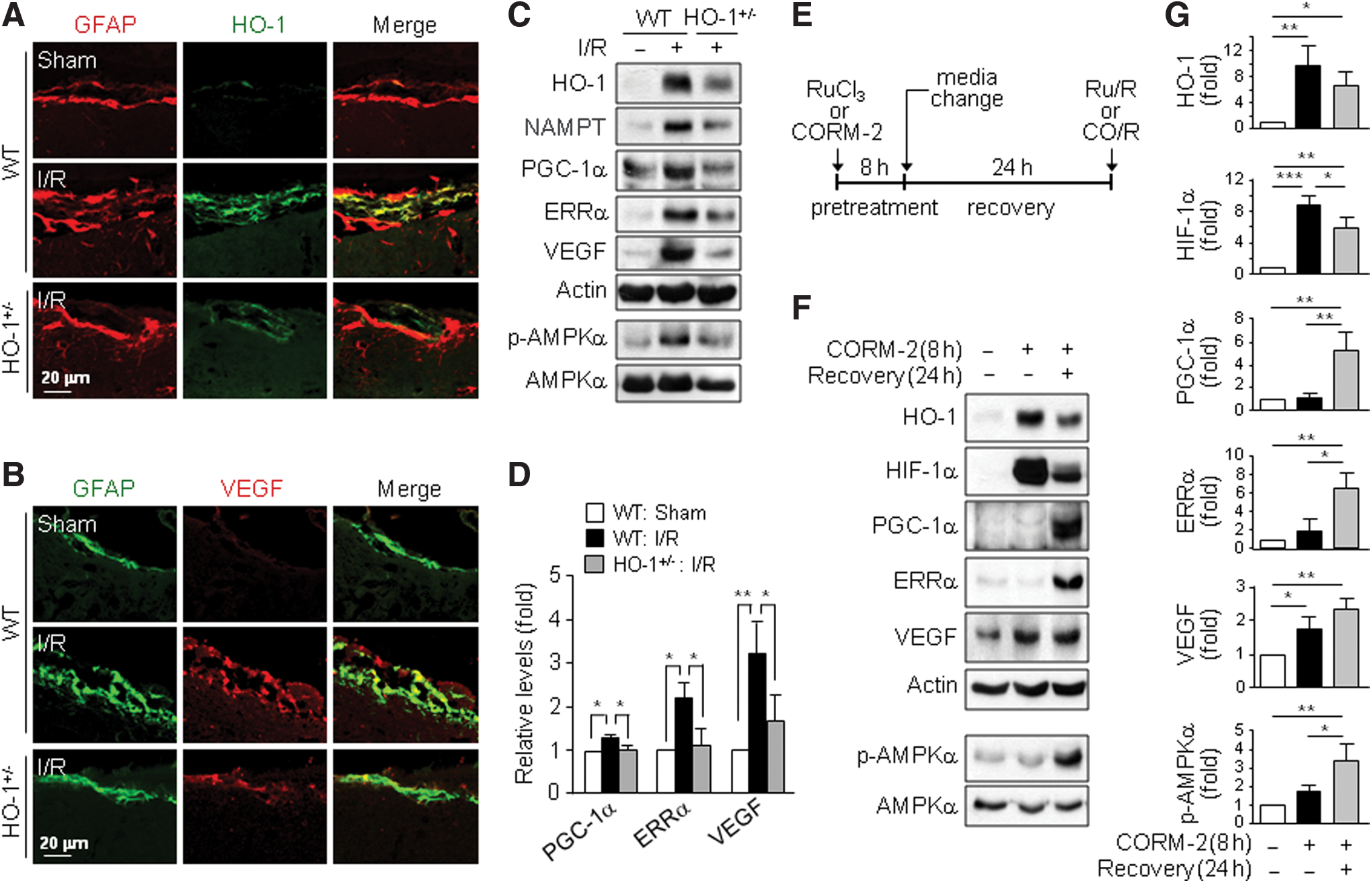
The HO-1/CO pathway protects against brain injury from ischemia/reperfusion (I/R) (47), probably via VEGF-mediated angiogenesis and neurogenesis (10, 30). We first investigated whether the HO-1/CO axis induces VEGF expression in mice subjected to I/R. HO-1 and VEGF expression increased significantly only in the peri-infarct brain region of wild-type (WT) mice compared with that in HO-1+/− mice (Supplementary Fig. S1A–C; Supplementary Data are available online at
CO/R induces HIF-1α-independent VEGF expression and angiogenesis
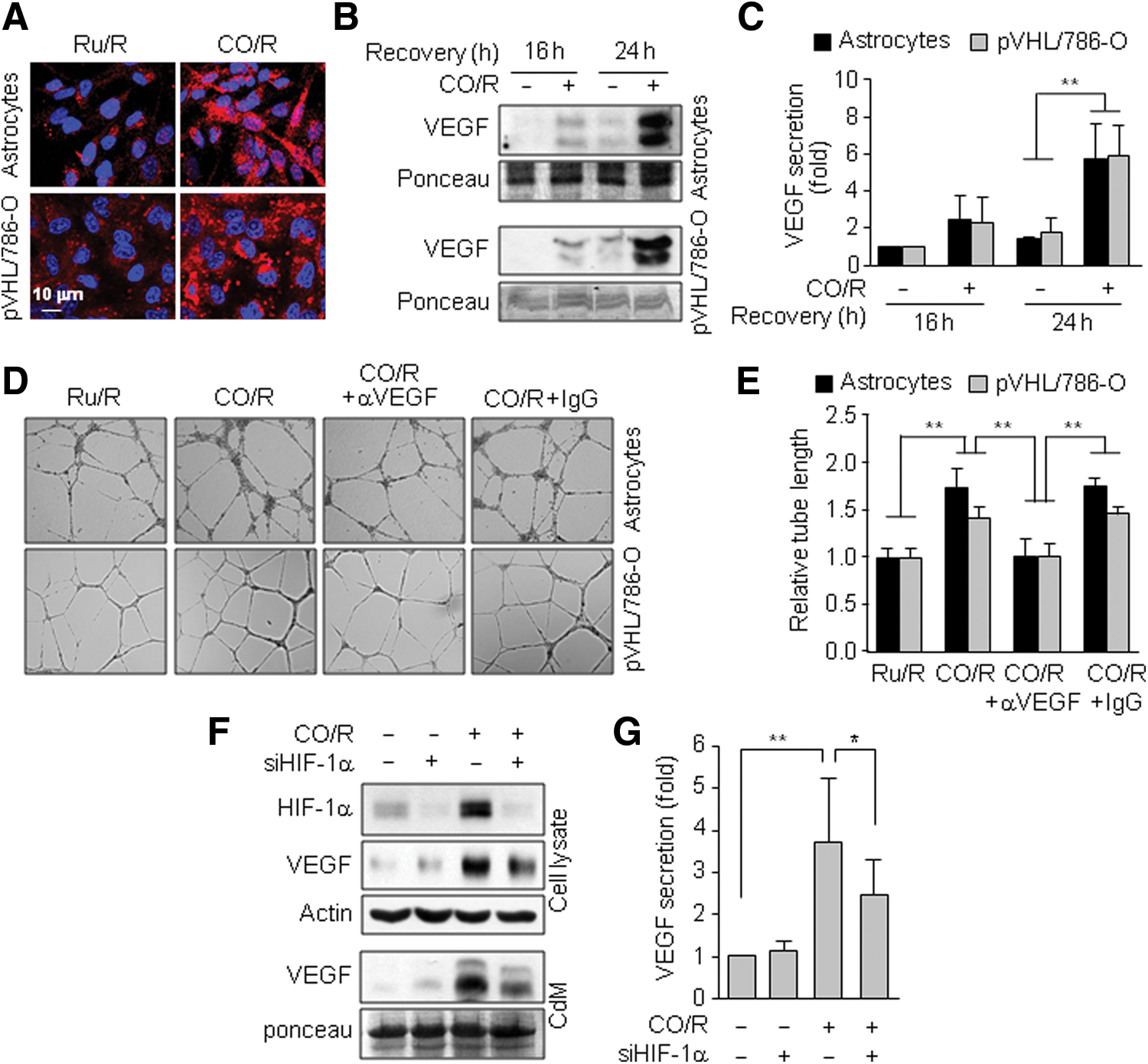
To further confirm a role of HIF-1α in VEGF expression under the CO/R condition, we examined VEGF levels in normal and HIF-1α-deficient cells. CO/R increased VEGF expression and secretion in both normal astrocytes and HIF-1α-deficient pVHL/786-O cells, as determined by immunocytochemistry and Western blotting (Fig. 2A–C). Moreover, conditioned media (CdM) from both cell types exposed to CO/R showed similar tube-forming activity, a typical phenotype of VEGF-induced in vitro angiogenesis (19), in primary human brain microvascular endothelial cells (HBMECs) and this increase was abrogated by preincubation with a neutralizing VEGF antibody (Fig. 2D, E), suggesting that CO/R induces VEGF expression and angiogenesis in an HIF-1α-independent manner. Knockdown of HIF-1α only partially decreased CO/R-induced VEGF expression and secretion by astrocytes (Fig. 2F, G), supporting the hypothesis that CO/R can stimulate VEGF expression via an HIF-1α-independent pathway.
CO/R induces VEGF expression via the AMPKα-mediated PGC-1α pathway
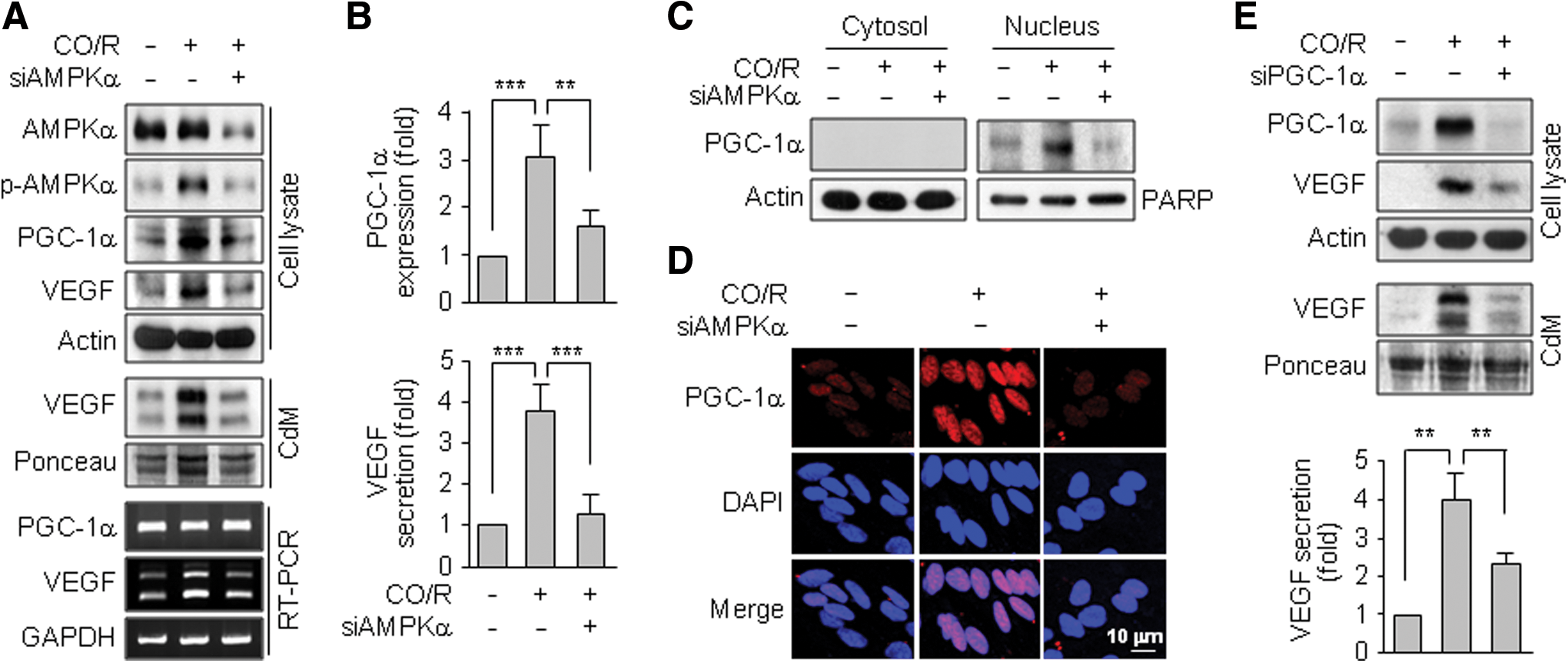
We examined a role of AMPKα in CO/R-induced VEGF expression using siRNA technology. Specific knockdown of AMPKα blocked the CO/R-induced increases in VEGF mRNA and protein, as well as p-AMPKα and PGC-1α protein, levels without altering the PGC-1α mRNA level (Fig. 3A, B), suggesting that CO/R increases AMPKα-dependent PGC-1α protein stability, which is an important axis of HIF-1α-independent VEGF expression (3, 28). As expected, CO/R promoted nuclear accumulation of PGC-1α, which was effectively inhibited by AMPKα knockdown (Fig. 3C, D). Moreover, PGC-1α knockdown decreased VEGF expression and secretion (Fig. 3E). These results suggest that CO/R activates AMPKα, leading to increased PGC-1α-mediated VEGF expression.
Positive feedback loop between HO-1/CO and AMPKα amplifies PGC-1α-mediated VEGF expression
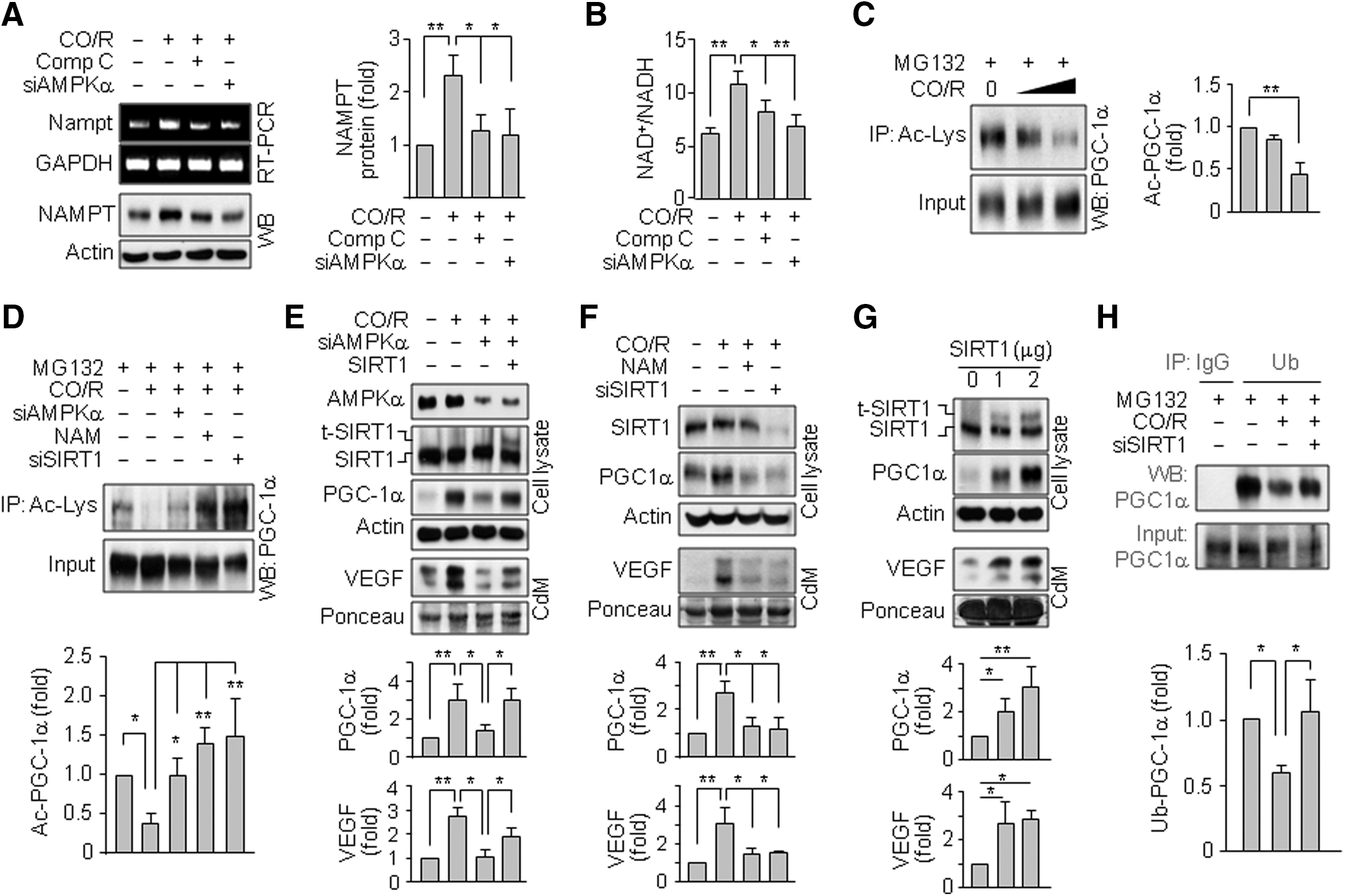
Because CO/R induced HO-1 (Supplementary Fig. S2), we examined a functional role of HO-1 in CO/R-induced increases in PGC-1α protein and AMPKα phosphorylation. CO/R elevated PGC-1α, p-AMPKα, and secreted VEGF levels and these increases were blocked by the HO inhibitor tin-protoporphyrin (SnPP) (Fig. 4A) or HO-1 knockdown (Fig. 4B). Moreover, HO-1 overexpression increased the levels of PGC-1α and p-AMPKα, as well as elevated VEGF secretion (Fig. 4C). Interestingly, the CO/R-induced increases in PGC-1α, HO-1, and secreted VEGF levels were effectively blocked by the AMPK inhibitor Compound C (Fig. 4D) or AMPKα knockdown (Fig. 4E). Conversely, activation of AMPK by 5-aminoimidazole-4-carboxamide-1-b-D-ribofuranoside (AICAR) elevated HO-1, PGC-1α, and secreted VEGF levels in a dose-dependent manner (Fig. 4F, G). These results suggest that a positive feedback loop exists between the CO/AMPKα and HO-1/CO axes, leading to amplification of PGC-1α-mediated VEGF secretion.

CO/R stimulates the CaMKKβ/AMPKα axis by activating L-type Ca2+ channels
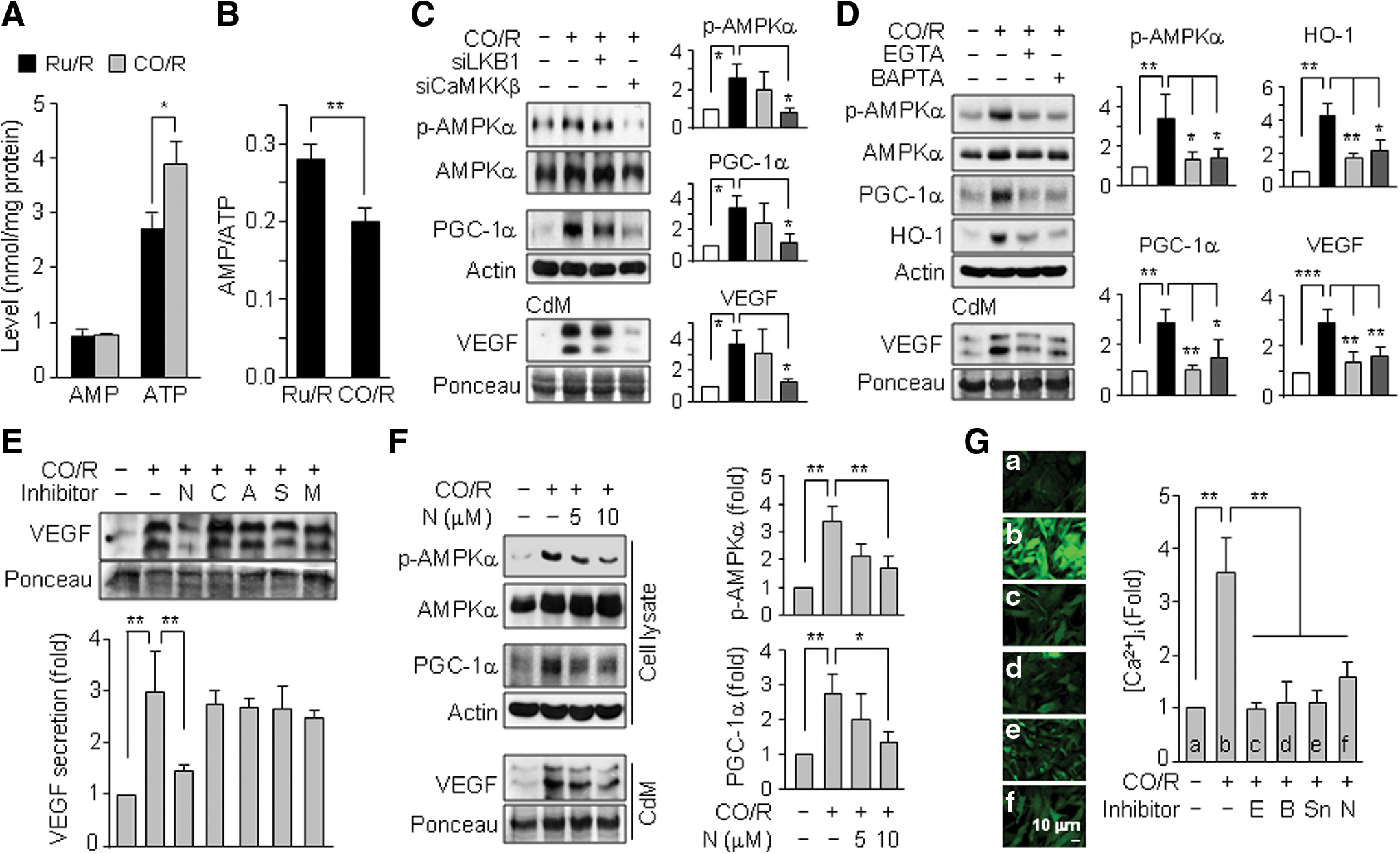
Since AMPKα was phosphorylated and activated by an increased AMP/ATP ratio, CaMKKβ, or LKB1 (18), we examined which mediator is responsible for CO/R-mediated AMPKα activation. CO/R significantly increased ATP levels, without changing AMP levels, in astrocytes, compared with those in control cells exposed to Ru/R (Fig. 5A), resulting in a decrease in the cellular AMP/ATP ratio (Fig. 5B). However, knockdown of CaMKKβ, but not LKB1, remarkably reduced the CO/R-induced increases in p-AMPKα, PGC-1α, and VEGF protein levels (Fig. 5C). These results suggest that CaMKKβ plays an important role in CO/R-mediated AMPKα activation. As CaMKKβ is activated by a rise in intracellular Ca2+ concentration [Ca2+]i (43), we next examined a possible role of [Ca2+]i in AMPKα-mediated VEGF expression signaling. Treatment with the extracellular and intracellular Ca2+ chelators, EGTA and BAPTA-AM, almost completely abolished the CO/R-induced increases in p-AMPKα, PGC-1α, HO-1, and VEGF protein levels (Fig. 5D). As extracellular Ca2+ influx is mainly regulated by various subtypes of voltage-gated Ca2+ channels (23), we examined which types of these channels are involved in CO/R-mediated VEGF expression. Among the different voltage-dependent Ca2+ channel blockers, the L-type Ca2+ channel blocker, nifedipine, but not inhibitors of N-type (ω-conotoxin GVIA), P/Q-type (ω-agatoxin TK), R-type (SNX 482), or T-type channels (mibefradil), suppressed the CO/R-evoked increase in VEGF secretion (Fig. 5E). Nifedipine also significantly decreased p-AMPKα, PGC-1α, and VEGF levels in a dose-dependent manner (Fig. 5F). Consistent with these data, the CO/R-mediated elevation of [Ca2+]i influx was abrogated by EGTA, BAPTA-AM, SnPP, and nifedipine (Fig. 5G). These findings suggest that CO/R increases Ca2+ influx via L-type Ca2+ channels, which in turn contributes to CaMKKβ-dependent activation of the AMPKα/PGC-1α pathway, leading to VEGF expression.
CO/R regulates PGC-1α deacetylation and ubiquitination via the AMPKα/SIRT1 axis
AMPK activates SIRT1 by increasing intracellular NAD+ levels and subsequently promotes post-translational activity of PGC-1α via deacetylation (9, 20, 62). Thus, we examined if CO/R regulates expression of nicotinamide (NAM) phosphoribosyltransferase (NAMPT, a rate-limiting enzyme in the NAD+ salvage pathway) and cellular NAD+ level. CO/R increased NAMPT expression and cellular NAD+/NADH ratio, which were blocked by Compound C or AMPKα knockdown (Fig. 6A, B), indicating that AMPKα mediates NAMPT expression and NAD+ synthesis under the CO/R condition. Interestingly, CO/R also evoked PGC-1α deacetylation (Fig. 6C), which was suppressed by AMPKα and SIRT1 knockdown or the SIRT1 inhibitor, NAM (Fig. 6D). The suppressive effects of AMPKα knockdown on PGC-1α level and VEGF secretion under the CO/R condition were rescued by SIRT1 overexpression (Fig. 6E). Treatment of astrocytes with NAM or SIRT1 siRNA blocked the CO/R-induced increases in PGC-1α and VEGF levels (Fig. 6F). On the other hand, ectopic expression of SIRT1 directly elevated PGC-1α and VEGF levels in a dose-dependent manner (Fig. 6G). To investigate how SIRT1 regulates PGC-1α protein levels, we examined the effects of CO/R and SIRT1 on ubiquitination of PGC-1α. CO/R decreased PGC-1α ubiquitination, and the effect was reversed by SIRT1 knockdown (Fig. 6H). These results suggest that CO/R-mediated AMPKα/SIRT1 activation increases PGC-1α protein stability and its biological activity by suppressing ubiquitination and promoting deacetylation, consequently resulting in VEGF expression.
CO/R activates the PGC-1α/ERRα/VEGF pathway
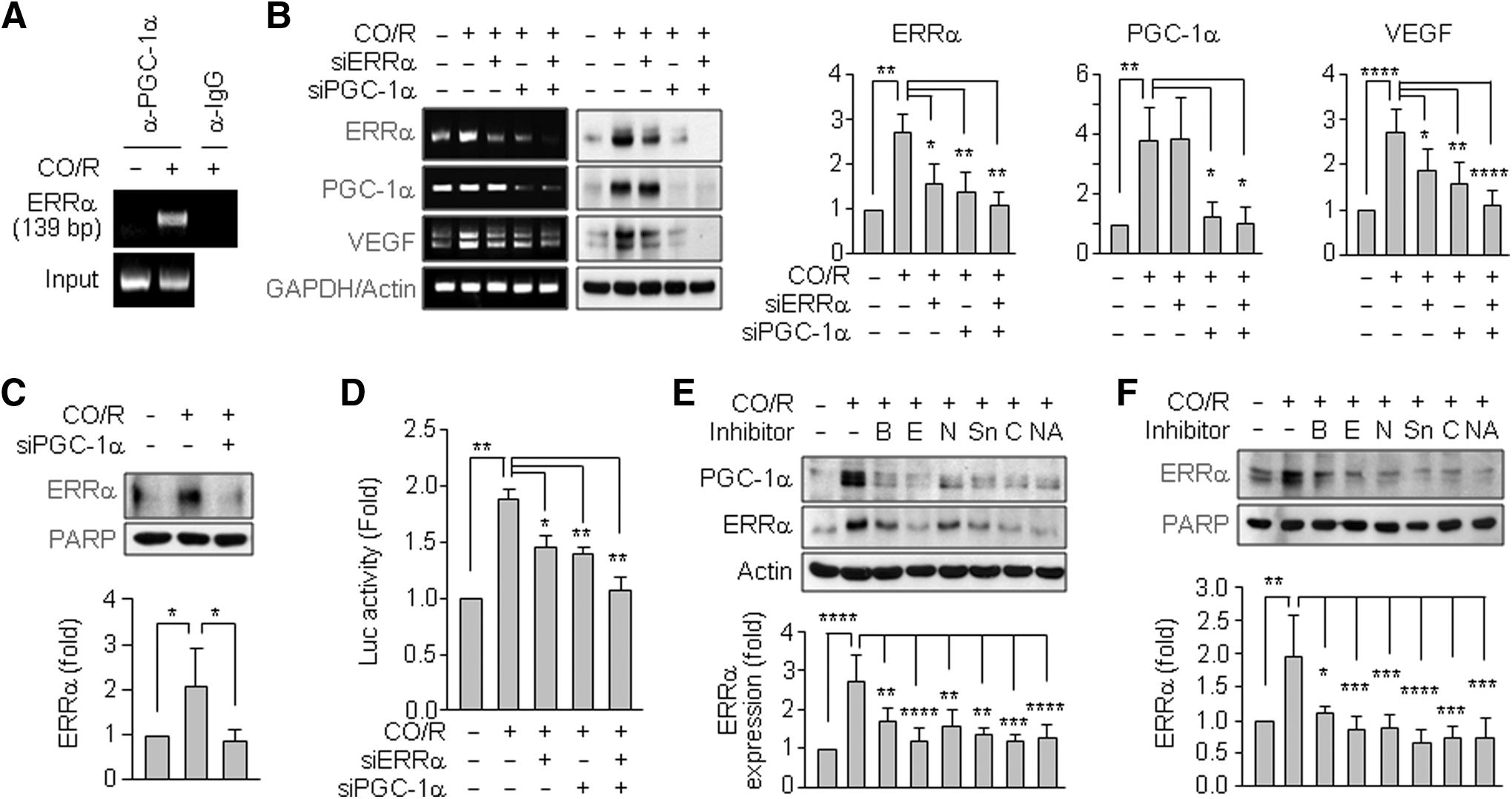
PGC-1α, a transcriptional coactivator, enhances VEGF expression by binding to the transcription factor, ERRα (3). We examined the effects of CO/R on PGC-1α/ERRα axis activity. A chromatin immunoprecipitation (ChIP) analysis showed that CO/R promotes binding activity of PGC-1α to the ERRα promoter through interaction with PPARγ in human astrocytes (Fig. 7A). In addition, CO/R increased ERRα mRNA and protein levels and nuclear accumulation of ERRα, which were suppressed by PGC-1α knockdown (Fig. 7B, C); however, PGC-1α levels did not decrease in response to ERRα knockdown (Fig. 7B). On the other hand, CO/R-mediated increases in VEGF gene expression and its promoter activity were suppressed by knockdown of PGC-1α or ERRα and synergistically abrogated by co-knockdown of PGC-1α and ERRα (Fig. 7B, D). We also examined the effects of [Ca2+]i on PGC-1α and ERRα expression in astrocytes. The increased PGC-1α and ERRα levels under the CO/R condition were abrogated by BAPTA-AM, EGTA, or nifedipine (Fig. 7E). Similar results were observed in the treatment with the HO-1/AMPKa/SIRT1 pathway blockers, SnPP, Compound C, or NAM (Fig. 7E). Nuclear accumulation of ERRα by CO/R was blocked by these inhibitors (Fig. 7F). These results suggest that CO/R regulates VEGF expression via the PGC-1α/ERRα axis by increasing Ca2+ influx via L-type Ca2+ channels.
The combination of exogenous CO and bilirubin mimics the functions of CO/R
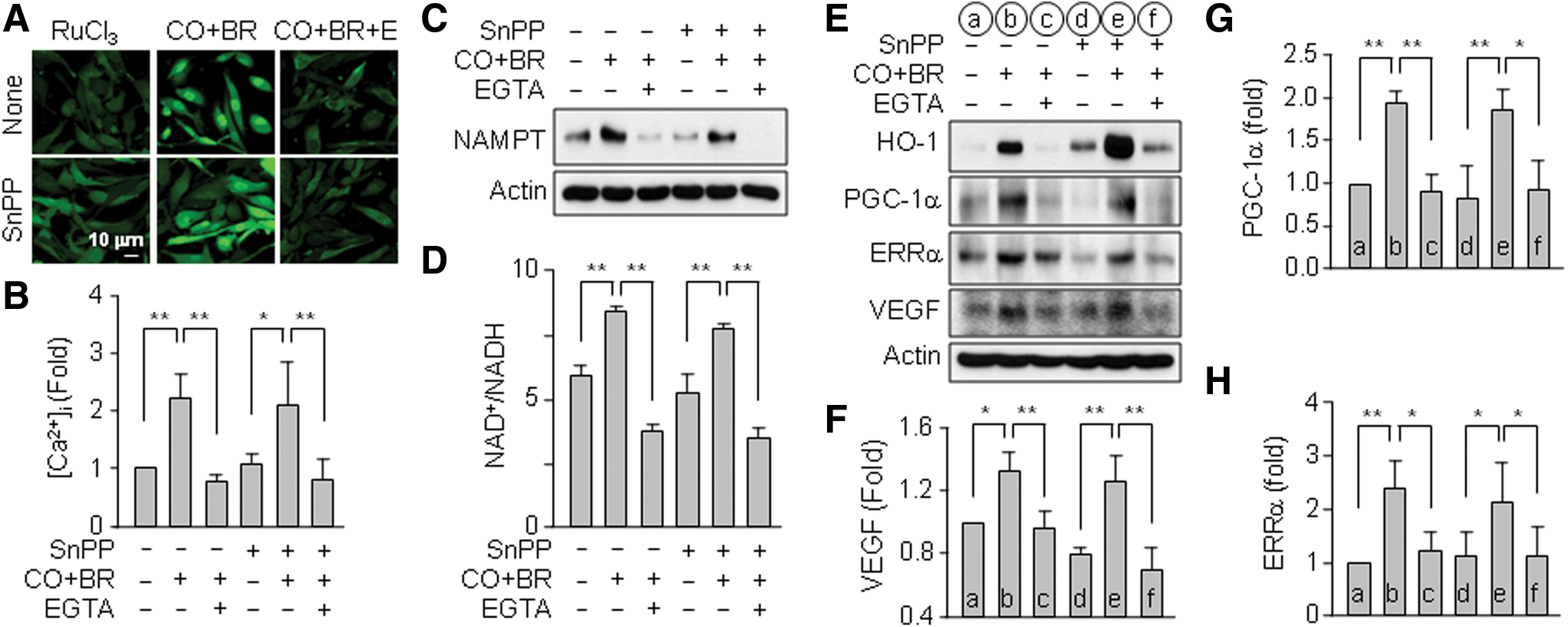
Because CO/R-mediated VEGF expression was dependent on HO-1 activity (Fig. 4A–C), we hypothesized that heme degradation products may be involved in the VEGF expression pathway. To assess this hypothesis, we examined the effects of CORM-2, bilirubin, and Fe2+ on Ca2+ influx and VEGF expression in astrocytes in the presence of SnPP to inhibit endogenous HO-1 activity. Significant induction of VEGF was observed when astrocytes were cotreated with CORM-2 and bilirubin compared with other combinations (Supplementary Fig. S3A, B). Similarly, the combination of CORM-2 and bilirubin was the most effective on the elevation of PGC-1α and ERRα protein levels (Supplementary Fig. S3A and B). Treatment with Fe2+ alone elevated, in part, the protein levels PGC-1α, ERRα, and VEGF (not statistically significant), but not HIF-1α (Supplementary Fig. S3A and B). However, the metal ion did not generate any notable synergistic effect in combination with other by-products (Supplementary Fig. S3A and B). Furthermore, the combination of CORM-2 and bilirubin increased Ca2+ influx, NAMPT expression, the NAD+/NADH ratio, and target protein (VEGF, PGC-1α, and ERRα) levels in astrocytes and these increases were effectively blocked by EGTA (Fig. 8A–H). These results indicate that the combination of CO and bilirubin may mimic CO/R-mediated Ca2+ influx, which is critically involved in HIF-1α-independent VEGF expression by stimulating the AMPKα-SIRT1-PGC-1α-ERRα pathway.
Discussion
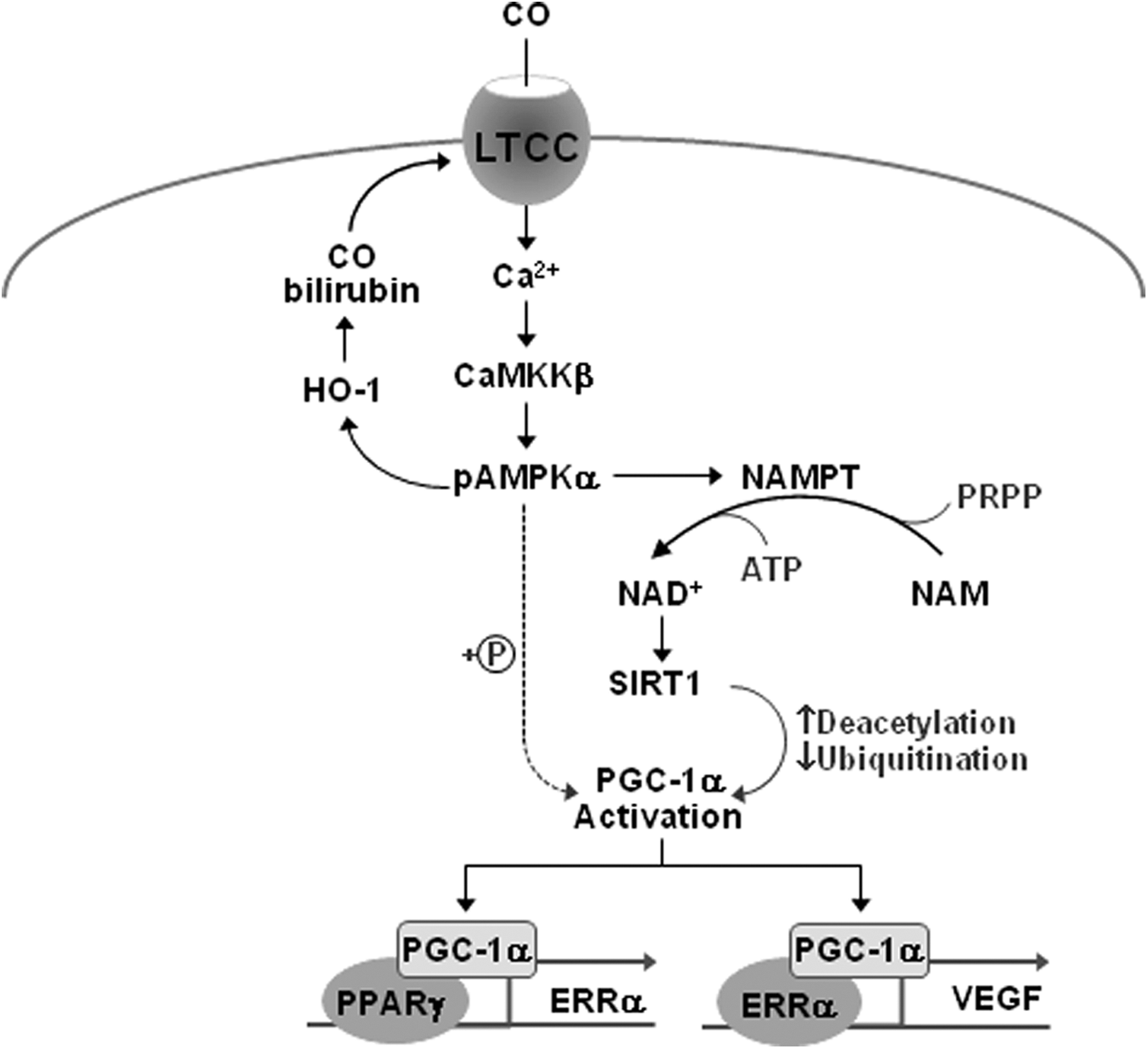
The HO/CO pathway serves as an intracellular messenger in the brain and vasculature (10, 25, 55). VEGF is a key angiogenic factor that promotes vascular leakage during the acute phase and neurogenesis during the delayed phase after ischemic injury (12, 63). In this study, we found that CO/R increased VEGF production at the transcriptional level in both primary astrocytes and HIF-1α-deficient pVHL/786-O cells, suggesting that CO/R upregulates VEGF expression in an HIF-1α-independent manner. Based on the present data, we propose that CO/R induces VEGF expression in an HIF-1α-independent manner in astrocytes through a sequential signal pathway as follows (Fig. 9). CO/R initially increased [Ca2+]i by activating L-type voltage-gated Ca2+ channels and subsequently activated the CaMKKβ/AMPKα axis. This axis was responsible for NAMPT expression and NAD+ synthesis, leading to SIRT1-dependent PGC-1α activation through decreased acetylation and ubiquitination. Activated PGC-1α elevated ERRα expression by increasing its promoter activity. Both PGC-1α and ERRα were involved in transcriptional upregulation of VEGF expression. These results indicate that the HO/CO axis induces VEGF expression by first activating L-type calcium channels and subsequently stimulating a Ca2+-dependent sequential signal cascade pathway, leading to coordinated neurovascular coupling.
Increasing evidence shows that CO promotes angiogenesis via expression of VEGF and other factors (16, 31). Although VEGF expression can be regulated by several factors, HIF-1α is a strong transcription factor, which is primarily regulated at the post-translational level, with increased protein stability dependent upon hypoxia (5, 26, 35, 51). Under normoxic conditions, HIF-1α protein is hydroxylated, binds the E3 ligase VHL protein, and is rapidly degraded by proteasomal proteolysis. However, HIF-1α is stabilized under hypoxic conditions and translocates into the nucleus, where it dimerizes with HIF-1β and binds to the hypoxia response element in the VEGF promoter, leading to induction of VEGF expression. Our previous study showed that HO-1-derived CO stimulates VEGF production by increasing HIF-1α protein level via two distinct mechanisms, such as translational stimulation and stabilization of the HIF-1α protein (10, 32). However, VEGF can be upregulated by the HIF-1α-independent pathway, which is associated with PGC-1α and ERRα, major regulators of mitochondrial function in response to exercise and other stimuli (3, 8, 38). Similarly, in this study, we found that CO/R stimulated VEGF expression by the Ca2+-dependent PGC-1α/ERRα axis that is evidently independent of HIF-1α. Thus, the HO-1/CO axis stimulates VEGF expression via HIF-1α-dependent and -independent mechanisms and particularly the latter is regulated by cytosolic Ca2+ signaling.
Two different effects of CO on [Ca2+]i-mediated cell function have been reported. HO-1-derived CO exerts cardioprotective properties by suppressing Ca2+ influx through inhibition of L-type Ca2+ channels (53). In fact, CO alters cardiac L-type Ca2+ channel function through redox modification of three cysteine residues in its cytoplasmic C-tail region by increasing formation of reactive oxygen species from mitochondrial respiratory complex III. On the other hand, CO promotes Ca2+ influx via N-type and L-type Ca2+ channels in the carotid body (17, 50). Moreover, CO activates intestinal L-type Ca2+ channels, probably by activating endothelial nitric oxide synthase (NOS), but not inducible or neuronal NOS (39). The discrepant effect of CO on L-type Ca2+ channel activation may be dependent upon cell type. Our present data show that L-type Ca2+ channels play a key role in CO/R-induced Ca2+ influx responsible for CaMKKβ-dependent AMPKα activation and VEGF expression in astrocytes. However, further investigation is needed to determine which L-type Ca2+ channel subtype, consisting of four main variants, is involved in CO/R-mediated VEGF expression.
Ca2+ is a main contributing factor to intracellular signal transmission through changes of [Ca2+]i. The catalytic activity of CaMK family is elegantly regulated in response to fluctuations in [Ca2+]i. Of this family, CaMKKα/β is highly expressed in the brain and initiates the signaling cascade by phosphorylation and activation of other CaMKs (2, 58). CaMKKβ is a physiological upstream activator of AMPK (60). In fact, extracellular Ca2+ influx induced by adiponectin is necessary for subsequent activation of CaMKKβ and the energy-sensing kinase AMPK in myocytes (27), suggesting that [Ca2+]i is an important regulator of metabolic homeostasis via CaMKKβ-dependent AMPK activation. However, AMPK can also be activated by LKB1 and an elevated AMP/ATP ratio (18, 61). In the present study, we found that CO/R activated AMPK in a CaMKKβ-dependent manner, with no association with intracellular AMP level or LKB1. Thus, it is possible that astrocytic Ca2+ influx induced by CO/R is an apical event in the regulation of energy-sensing metabolic homeostasis and neurovascular function via the CaMKKβ/AMPK axis in the ischemic brain microenvironment.
AMPK is an upstream activator of SIRT1, which is also a metabolic sensor and a gatekeeper of mitochondrial activity. AMPK activates SIRT1 by increasing the [NAD+/NADH] ratio via transcriptional induction of NAMPT, a key enzyme in mammalian NAD+ biosynthesis (11, 20). The NAMPT promoter region possesses putative binding sites for FoxO transcription factors, which are phosphorylated and activated by AMPK (22). Similarly, we found that CO/R increased AMPKα-dependent NAMPT expression and NAD+ synthesis, resulting in SIRT1-dependent deacetylation of PGC-1α. These results suggest that astrocytic Ca2+ influx by CO/R is an important contributor to activation of PGC-1α via the CaMKKβ/AMPKα/NAMPT/SIRT1 axis.
PGC-1α activity is regulated by balancing its acetylation state via the deacetylase SIRT1 and the acetyltransferase, GCN5 (29). Deacetylation of PGC-1α by SIRT1 enhances its biological activity to coactivate transcription factors, which in turn induce transcription of their target genes, including ERRα and VEGF. Astrocytes exposed to CO/R evoked PGC-1α deacetylation and VEGF production and these effects were reversed by AMPK or SIRT1 knockdown. This result suggests that CO/R-mediated PGC-1α activation is elicited through an AMPKα/SIRT1-dependent deacetylation mechanism. PGC-1α is rapidly degraded with a half-life of 0.3 h in the nucleus via the ubiquitin proteasome system (59). Our data show that CO/R increases the PGC-1α protein level, without changing the mRNA level, and also decreases PGC-1α ubiquitination in an AMPK/SIRT1-dependent manner, suggesting that CO/R inhibits ubiquitination-dependent proteasomal degradation of PGC-1α. Given our findings, it is possible that SIRT1 is a coordinative mediator of the CO/R-mediated increase in deacetylation and decrease in ubiquitination of PGC-1α, consequently leading to increased PGC-1α protein stability and activity. PGC-1α can also be activated by AMPK via phosphorylation at threonine-177 and serine-538 (28). Although PGC-1α phosphorylation was not directly determined under our experimental conditions, we cannot exclude the possibility that CO/R activates PGC-1α via an AMPKα-dependent phosphorylation mechanism. Importantly, AMPK-dependent PGC-1α activation stimulates HIF-α-independent VEGF expression and angiogenesis in coordination with ERRα (3, 49). Our findings, in conjunction with those of previous studies, suggest that astrocytic Ca2+ influx by CO/R stimulates HIF-1α-independent VEGF expression and angiogenesis via the CaMKKβ/AMPK/PGC-1α axis, resulting in delivery of metabolic substrates to facilitate the functions of neurovascular units in the ischemic brain region.
PGC-1α, a transcriptional coactivator, physically interacts with the transcription factors, PPARγ and ERRα (3, 14, 52). Both transcription factors are key regulators of mitochondrial energy metabolism and stimulate angiogenesis, suggesting that energy metabolism is coupled with angiogenesis. We demonstrated that CO/R elicits increased ERRα expression in a PGC-1α-dependent manner. This effect was likely to bind the PPARγ/PGC-1α complex to the putative PPARγ binding site (+583 to +539, TCAAAGG) in the ERRα promoter. Moreover, the PGC-1α/ERRα pathway elicits robust induction of VEGF transcription through binding to the conserved ERRα binding sites in the first intron of the VEGF gene (3). In this study, we found that CO/R-induced increases in PGC-1α, ERRα, and VEGF protein levels were simultaneously inhibited by extracellular and intracellular Ca2+ chelators and an L-type Ca2+ channel blocker. These findings suggest that CO/R powerfully induces PGC-1α/ERRα-dependent VEGF expression via a CaMKKβ/AMPKα/SIRT1 axis by activating L-type voltage-gated Ca2+ channels.
CO/R-mediated HO-1 degrades heme into its by-products, such as CO, bilirubin, and iron. Among them, CO and bilirubin are major bioactive by-products. CO promotes Ca2+ influx via N-type and L-type Ca2+ channels in the rat carotid body (17, 50), and bilirubin activates voltage-gated Ca2+ channels in rat neuronal cells, although the Ca2+ channel type has not been identified (54). In this study, we found that the combination of both bioactive products mimicked the CO/R-mediated increases in Ca2+ influx, NAD+ synthesis, and PGC-1α/ERRα-mediated VEGF expression. However, a further study should be performed to determine how CO and bilirubin interplay to open L-type voltage-gated Ca2+ channels.
Increasing evidence demonstrated that HO-1, an antioxidant enzyme for redox homeostasis, plays an important role in cytoprotection, antiapoptosis, and anti-inflammation, which contribute to establishing a protective or preventive state in a wide variety of human diseases (7). Thus, HO-1 has been suggested as one of the vitagenes, including heat shock proteins, antiapoptotic proteins, and antioxidant enzymes, which exert crucial functions for cell survival, redox homeostasis, and repair processes (7). HO-1 induction occurs together with the induction of HSP70 during various physiopathological conditions, including nitric oxide or nitrosative stress (7, 33, 34). The present study showed that CO/R increased VEGF expression by activating the AMPKα-NAMPT-SIRT1-PGC-1α/ERRα pathway via HO-1 induction. Although we did not directly evaluate it, CO can activate the vitagene network to protect cells from death in the acute phase of ischemic stroke. Therefore, we will further investigate the relationship between the HO/CO pathway and vitagene network.
It has been shown that basal levels of CO are about 5–8 × 10−8 M in cortical cerebral fluid of newborn pigs and may rise to 1 μM after induction of seizure (37). In infants with jaundice of unknown etiology, blood levels of bilirubin, as an index of CO production, were increased more than 12 mg/dl, that is, ∼200 μmoles/l (4), suggesting that more than 100 μM of CO can be circulated in blood, although mostly bound to hemoglobin, after production in certain local tissues (spleen or liver). The CO-releasing compound, CORM-2, is unstable in aqueous solution with a half-life of 1 min and liberates ∼0.7 mole of CO/mole (44), assuming that CO concentration in our cell culture system using 100 μM CORM-2 corresponds to 70 μM as an initial concentration. Therefore, our CO/R system using 100 μM CORM-2 can be related to the pathological or therapeutic conditions.
In conclusion, our present data demonstrate that the HO-1/CO axis, induced by I/R, increases angiogenesis via PGC-1α/ERRα-mediated, but HIF-1α-independent, VEGF expression in astrocytes. This pathway appears to be stimulated by a sequential cascade, such as extracellular Ca2+ influx via L-type voltage-gated Ca2+ channels, CaMKKβ-dependent AMPKα activation, NAMPT-mediated elevation of the [NAD+/NADH] ratio, and SIRT1 activation (Fig. 9). Thus, the HO-1/CO system may promote repair after ischemic brain injury by enhancing Ca2+-mediated, but HIF-1α-independent, VEGF expression and angiogenesis, which provides the injured area with sufficient nutrients and oxygen, leading to a coupled circuit between energy metabolism and angiogenesis.
Materials and Methods
Materials
Cell culture media and supplements were purchased from Invitrogen Life Technologies. Fetal bovine serum (FBS) was obtained from HyClone Laboratories. MG132, AICAR, Compound C, BAPTA-AM, and proteinase inhibitor cocktail were purchased from Enzo Life Sciences. CORM-2 (a CO-releasing compound, [Ru(CO)3Cl2]2), RuCl3 (a noncarbonyl inert control compound), nifedipine, EGTA, ATP, AMP, trichlorofluoromethane solution, mibefradil, iron(II) chloride, and bilirubin were purchased from Sigma. Sn(IV)-protoporphyrin IX dichloride (SnPP) was purchased from Frontier Scientific. ω-Conotoxin GVIA, ω-Agatoxin TK, and SNX 482 were purchased from Tocris Bioscience (Bristol, BS110QL). Fluo-4 (Thermo Fisher Scientific) and tetrabutylammonium hydrogen sulfate (TBAS; EMD Millipore) were used. siRNAs targeting human HIF-1α and SIRT1 were purchased from Dharmacon. siRNAs targeting human HO-1, AMPKα, ERRα, and PGC-1α were purchased from Santa Cruz Biotechnology.
Cell culture
Primary human brain astrocytes and HBMECs were purchased from the Applied Cell Biology Research Institute. Primary astrocytes were cultured in Dulbecco's Modified Eagle Medium supplemented with 10% FBS and used in passages 5–9. HBMECs were grown in M199 supplemented with 20% FBS, 3 ng/ml basic fibroblast growth factor (EMD Millipore), and 10 U/ml heparin (Sigma). HIF-1α-deficient pVHL/786-O cells (42) were prepared by stable transfection of 786-O renal carcinoma cells with a plasmid encoding myc-pVHL, which was selected using G418 (provided by Dr. Kyu-Won Kim, Seoul National University).
Cell treatment and CdM preparation
When astrocytes were grown up to 80% density with 10% FBS DMEM, media were changed into 0% FBS DMEM. Astrocytes were treated with RuCl3 (200 μM) or CORM-2 (100 μM) in the presence or absence of various chemicals for 8 h in serum-free DMEM, washed with fresh medium, and further cultured for 24 h in serum-free DMEM and collected medium for CdM. Collected CdM was concentrated ( × 3) through a centrifugal filter device (3 kDa cutoff; EMD Millipore). In addition, cells were transiently transfected with pcDNA3.1/HO-1 vector (provided by Dr. Jozef Dulak, Jagiellonian University), pcDNA3.1/SIRT1 (provided by Dr. Sungwoo Ryoo, Kangwon National University), or various siRNAs (50 nM) using Lipofectamine and Plus reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. After recovery for 12 h, cells were treated with RuCl3 (200 μM) or CORM-2 (100 μM) as described above.
Experimental animals using an MCAO model
WT and HO-1+/− mice (BALB/c mice, Jackson's Laboratory) were kept in standard conditions with water and food available ad libitum. All experimental procedures were carried out under a protocol approved by Kangwon National University's Animal Care and Use Committee and were in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. Eight-week-old male mice were anesthetized with a mixture of 2% isoflurane (Baxtor) in 70% N2O and 30% O2 (v/v) mixture via face mask. Anesthesia was maintained with 1.6% isoflurane. Bilateral common carotid arteries were isolated and occluded using nontraumatic aneurysm clips. A rectal temperature probe was introduced, and a heating pad maintained the body temperature at 37°C during the surgery. Focal cerebral ischemia was achieved by MCAO on the right side as previously described (21). Briefly, the right carotid artery was exposed through a midline cervical incision. The right external carotid artery was dissected free and isolated distally by coagulating its branches and placing a distal ligation before transection. A piece of 5-0 monofilament nylon suture (Ethicon; Johnson-Johnson), with its tip rounded by gentle heating and coated by 0.1% (w/v) poly-L-lysine, was inserted into the lumen of right external carotid artery stump and gently advanced 9 mm into the internal carotid artery from the bifurcation to occlude the ostium of MCAO. After 2 h of ischemia, the suture was pulled back and the animals were allowed to recover. Mice were kept for 24 h and sacrificed. Sham-operated animals were subjected to the same surgical procedures, except that the common carotid arteries were not occluded.
Tissue preparation and immunohistochemistry
For the histological analysis, mice were anesthetized with sodium pentobarbital (30 mg/kg, i.p.) and perfused transcardially with phosphate-buffered saline (PBS, pH 7.4), followed by 4% paraformaldehyde in PBS. The brains were removed and postfixed in the same fixative for 6 h at 4°C. Each specimen was then dehydrated through a series of graded ethanol solutions and embedded in paraffin using standard techniques. Paraffin-embedded brains were sectioned into 10-μm sections, and sections were mounted on slides coated with 2% Elmer's glue. The sections were treated with 0.3% H2O2 in PBS to block endogenous peroxidase activity for 30 min, and then incubated in 10% normal horse serum-supplemented PBS for 30 min. The sections were next incubated with a rabbit anti-VEGF antibody (1:100; Santa Cruz Biotechnology), a mouse anti-GFAP antibody (1:200; EMD Millipore), a rabbit anti-GFAP antibody (1:200; EMD Millipore), or a mouse anti-HO-1 antibody (1:150; Enzo Life Sciences) in PBS at room temperature. After washing three times for 10 min with PBS, the sections were incubated in a mixture of both Cy3-conjugated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch) and FITC-conjugated goat anti-mouse IgG (1:200; Jackson ImmunoResearch) for 2 h at room temperature. The immunoreactions were observed under the confocal microscope (Olympus FV1000).
2,3,5-Triphenyltetrazolium chloride staining
Mice were euthanized and perfused transcardially with 0.1 M PBS. Brains were removed, and 1-mm coronal sections were dissected from the frontal pole using a mouse brain slicer (brain Matrix; ASI Instruments). Six slices were selected according to the mouse brain atlas, including the main portion of the infarct. The slices were incubated for 30 min in 2% 2,3,5-triphenyltetrazolium chloride (TTC) solution (Sigma) at 37°C and fixed by immersion in 4% paraformaldehyde solution in PBS for 6 h. The brain tissues were cryoprotected by infiltration with 30% sucrose overnight. Images of TTC-stained brain sections were obtained using a digital camera (Sony).
Promoter vector construction and luciferase assay
Human VEGF promoter (−746 to +438) was produced by genomic polymerase chain reaction (PCR) from human endothelial cells. Primers used were 5′-CGG
Immunofluorescence staining
Astrocytes were fixed in 3.7% formaldehyde for 30 min at room temperature, washed gently, blocked, and incubated with the PGC-1α or ERRα primary antibody (SantaCruz Biotechnology) overnight at 4°C, followed by incubation with an Alexa Fluor antibody (Thermo Fisher Scientific). Nuclei were stained using DAPI (Thermo Fisher Scientific). Images were obtained with a confocal microscope.
Western blot analysis
Proteins from whole cell lysates and CdM were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and were analyzed by Western blot analysis as described previously (10). Antibodies used in this study were as follows: HIF-1α (BD Biosciences); Poly ADP ribose polymerase (PARP) (EMD Millipore); HO-1 (Enzo Life Biosciences); ERRα, PGC-1α, and SIRT1 (Santa Cruz Biotechnology); AMPKα, p-AMPKα (Thr172), and acetyl-lysine (Cell Signaling Technology); NAMPT (AdipoGen); VEGF (Thermo Fisher Scientific or Santa Cruz Biotechnology); ubiquitin (Thermo Fisher Scientific); and β-actin (Sigma). Each Western blot is a representative image of data obtained from at least three individual experiments.
Immunoprecipitation
Cell lysates (200 μg of protein in 1 ml) were incubated with an antibody for PGC-1α (1 μg/ml) or acetyl-lysine (1 μg/ml; Cell Signaling Technology) in TEG buffer (20 mM Tris-Cl pH 7.4, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol, containing 150 mM NaCl, and 0.1% Triton X-100) with constant rotation overnight at 4°C. Immune complexes were collected by centrifugation following incubation with protein G sepharose (50 μl; Thermo Fisher Scientific) and washed three times with TEG buffer. Immunoprecipitates were analyzed by SDS-PAGE, followed by Western blotting using the indicated antibodies.
Reverse transcriptase-PCR
Total RNAs were isolated from the indicated cells using TRIzol reagent (Thermo Fisher Scientific). Reverse transcriptase (RT)-PCR analysis was performed as described previously (10). The following sets of primers were used: human HO-1: 5′-CAGGCAGAGAATGCTGAG-3′ (forward) and 5′-GCTTCACATAGCGCTGCA-3′ (reverse), human ERRα: 5′-TGAGAAGCTCTATGCCATGCCTGA-3′ (forward) and 5′-ATAGAAATGGGCCAGCACTTTGCC-3′ (reverse), human PGC-1α: 5′-TCCCGATCACCATATTCC-3′ (forward) and 5′-TTCAAGAGCAGCAAAAGC-3′ (reverse), human NAMPT: 5′-GGATCCATGAATCCTGCGGCAGAAGC-3′ (forward) and 5′-CTCGAGATGATGTGCTGCTTCCAGTTC-3′ (reverse), human VEGF: 5′-GAGAATTCGGCCTCCGAAACCATGAACTTTCTGT-3′ (forward) and 5′-GAGCATGCCCTCCTGCCCGGCTCACCGC-3′ (reverse), human GAPDH: 5′-CAGGGCTGCTTTTAACTCTG-3′ (forward) and 5′-TAGAGGCAGGGATGATGTTC-3′ (reverse), mouse HO-1: 5′-GGCCCTGGAAGAGGAGATA-3′ (forward) and 5′-GCTGGATGTGCTTTTGGTG-3′ (reverse), and mouse actin: 5′-TCCTTCGTTGCCGGTCCACA-3′ (forward) and 5′-CGTCTCCGGAGTCCATCACA-3′ (reverse). PCR products were analyzed on 1.2% agarose gels and the gels were digitally imaged (BioImaging System).
AMP/ATP ratio measurement
Cell concentrations of ATP, ADP, and AMP were determined with the use of high-performance liquid chromatography. After centrifugation, 3 × 106 astrocytes were suspended in 300 μl of media with 20 μl of 1 M HClO4. HClO4 was removed by mixed-phase extraction employing 11.75:13.25 (v/v) of tri-n-octylamine and Freon 11. A 250 × 4.6-mm Zorbax Rx C8 column was sequentially equilibrated at a flow rate of 1.0 ml/min in methanol, H2O, buffer B (50 mM KH2PO4, 8 mM TBAS, and 40% [v/v] acetonitrile at pH 5.8), and buffer A (50 mM KH2PO4, 8 mM TBAS at pH 5.8). Adenine nucleotides were detected spectrophotometrically (254 nm). AMP and ATP (10, 50, 250 ppb) were used for standard.
Intracellular NAD+/NADH measurement
Intracellular NAD+ and NADH levels were measured using an NAD+/NADH quantification kit (BioVision) as manufacturer's instructions. Cells (0.5 × 106) were extracted with 800 μl of NAD+/NADH extraction buffer by two freeze/thaw cycles. To detect NADH, 400 μl the extracted samples was heated to 60°C for 30 min. Each 50 μl extracted sample with or without heating was added into a 96-well plate, then 50 μl NAD+ cycling mix was added. Five microliters NADH developer was added into the mix. This mix was incubated for 4 h and read at 450 nm.
Measurement of intracellular Ca2+
[Ca2+]i levels were measured by quantitative fluorescence imaging using the Ca2+-sensitive dye, Fluo-4. Cells plated on coverslips were incubated with 1 μM of Fluo-4 for the last 30 min of CO exposure. Subsequently, coverslips were viewed under a confocal microscope. Acquisition of live image frequency was every 5 s for 5 min.
ChIP assay and nuclear fraction assay
The ChIP assay was performed according to the protocol supplied by Millipore. Astrocytes were treated with Ru/R or CO/R and cross-linked with 1% formaldehyde for 10 min at room temperature. An antibody against PGC-1α (Santa Cruz Biotechnology) was used in the immunoprecipitation, and PCR was performed with following primers that target the binding site of the PPARγ/PGC-1α complex on the ERR promoter: forward primer: 5′-GGAGGGCTCTATGTCTGGGA-3′ and reverse primer: 5′-GTAAGTGGGGAGAGCCAAGG-3′. The PCR product was analyzed on a 1% agarose gel. Nuclear proteins were extracted as previously (10).
Tube formation assay
HBMECs (2 × 105 cells) were cultured on Matrigel-coated 24-well culture plates and incubated with CdM from astrocytes and pVHL/786-O cells exposed to reagents in M199 containing 1% FBS. Tube formation was observed using an inverted phase-contrast microscope, and images were captured with a video graphic system. The length of tube formation was measured using LabWorks 4.6 (UVP Products).
Data analysis and statistics
Quantification of protein band intensity obtained by Western blot was analyzed using ImageJ (
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korea Government (MSIP) (NRF-2011-0028790 and 2013 M3A9B6046563).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.