
Abstract
Significance:
Nitric oxide (NO)-dependent signaling is critical to many cellular functions and physiological processes. Soluble guanylyl cyclase (sGC) acts as an NO receptor and mediates the majority of NO functions. The signaling between NO and sGC is strongly altered by reactive oxygen and nitrogen species.
Recent Advances:
Besides NO scavenging, sGC is affected by oxidation/loss of sGC heme, oxidation, or nitrosation of cysteine residues and phosphorylation. Apo-sGC or sGC containing oxidized heme is targeted for degradation. sGC transcription and the stability of sGC mRNA are also affected by oxidative stress.
Critical Issues:
Studies cited in this review suggest the existence of compensatory processes that adapt cellular processes to diminished sGC function under conditions of short-term or moderate oxidative stress. Alternative splicing of sGC transcripts is discussed as a mechanism with the potential to both enhance and reduce sGC function. The expression of α1 isoform B, a functional and stable splice variant of human α1 sGC subunit, is proposed as one of such compensatory mechanisms. The expression of dysfunctional splice isoforms is discussed as a contributor to decreased sGC function in vascular disease.
Future Directions:
Targeting the process of sGC splicing may be an important approach to maintain the composition of sGC transcripts that are expressed in healthy tissues under normal conditions. Emerging new strategies that allow for targeted manipulations of RNA splicing offer opportunities to use this approach as a preventive measure and to control the composition of sGC splice isoforms. Rational management of expressed sGC splice forms may be a valuable complementary treatment strategy for existing sGC-directed therapies. Antioxid. Redox Signal. 26, 122–136.
Introduction
S
Most of the physiological effects of NO are mediated by the NO-sensitive soluble guanylyl cyclase (GTP pyrophospate-lyase, sGC). sGC is an enzyme-linked receptor for NO that is composed of one α and one β subunit. Two separate genes for each subunit (α1, α2, β1, and β2) are found in mammals, but only the ubiquitously expressed α1β1 and the less abundant α2β1 heterodimers have been detected in vivo (71). The domain organization of most abundant α1 and β1 subunits is depicted in Figure 1. The C-terminal half of both subunits is necessary and sufficient to form a functional catalytic site with low cyclic guanosine monophosphate (cGMP)-forming activity (24). The NO receptor portion of sGC is located in the N-terminal region of the enzyme (25). Although the function of the N-terminal domain of the α subunit is not entirely understood, it is clear that the N-terminal domain of the β subunit is essential in harboring the ferrous heme prosthetic group. It is to this heme moiety that NO binds with a high affinity.
Although the sGC–heme complex has many features that are similar to the heme–hemoglobin complex (i.e., a ferrous heme coordinated by a histidine residue), sGC exhibits an exceptional selectivity toward NO. Both functional (35) and direct binding (63, 126) studies demonstrate that sGC binds NO with a K D of ∼50 nM. Moreover, the binding is very fast and reaches a diffusion limited rate (139). On the other hand, sGC does not bind oxygen at physiologic pressure and concentrations (128), whereas the 250 μM K D for carbon monoxide (CO) (62) is well outside of the physiological range of CO.
The ferrous status of sGC heme is essential for NO sensing, since in the ferric form the affinity for NO is very low (128). The binding of NO to sGC heme triggers a number of conformational changes that eventually result in several hundred-fold enhancements of the cGMP-forming activity in the catalytic region of sGC (53). The exact nature of these conformational changes is currently the subject of intense investigation (14, 16, 59, 105, 130). This set of properties makes sGC uniquely sensitive to transient low nanomolar concentrations of NO that are typically produced under normal physiologic conditions (40). This ligand binding step is then amplified through the synthesis of the secondary messenger cGMP. However, these very same properties make the NO–sGC communication very susceptible to the effects of various reactive oxygen species (ROS).
Ever since the early reports suggested that sGC-dependent synthesis of cGMP may be activated by hydrogen peroxide (H2O2) (135) or superoxide dismutase (SOD) (75), the complexity of the relationship between NO-dependent activation of sGC and ROS was recognized. The term Reactive Oxygen Species is rather amorphous and is generally used in reference to the initial species that are generated by oxygen reduction (superoxide and H2O2) and their secondary reaction products. Some of these ROS have the capacity to directly react with NO with various rates and to form reactive nitrogen species (RNS). Among ROS, superoxide anion (O2 −) has the highest reaction rate with NO (84). In fact, the reaction between superoxide anion and NO is up to 100 times faster than the interaction between NO and sGC (84, 127). Therefore, under oxidative stress, when the level of generated superoxide exceeds the capacity of SODs to inactivate it, the bioavailabilty of NO (and therefore activation of sGC) is reduced, whereas the level of peroxynitrite increases.
Although it is generally understood that ROS is not a single entity, but a conglomerate of species with various redox potentials, different specificities and reaction rates, it is often impossible to discern the specific agent that is the major contributor to the investigated biological effect (30, 137). With such stipulation, this review attempts to catalog the effects of major ROS and RNS on sGC activity and expression.
As described next, cellular oxidative conditions and elevated ROS/RNS not only affect the bioavailability of NO but also, in many instances, affect the activity of sGC or change the redox status of sGC heme. Besides these direct effects on sGC function, ROS and RNS cause post-translational modifications, facilitate sGC degradation, affect the synthesis or stability of sGC mRNA, and alter the composition of expressed variants of sGC splice forms. This article provides a concise overview of some of the known effects of oxidative stress and ROS on sGC function and expression, with special emphasis on the regulation of sGC splicing.
ROS/RNS and sGC
Molecular oxygen has been recognized since the 1950s as a factor that greatly amplifies the toxicity from radiation-dependent cellular damage. The discovery of SOD class of enzymes (51, 91) that possess the ability to scavenge the superoxide provided the first evidence that oxygen-derived free radicals such as O2 −, H2O2, and hydroxyl radical (•OH) may be responsible for the toxicity of oxygen. Hydroxyl radical (•OH) is, by far, the most active among these three. However, the reactions that generate •OH from H2O2 via Fenton chemistry are slow, whereas deactivation through interaction with virtually any biological molecule is very fast. The current view is that this combination of factors makes the distance traveled by •OH very small, therefore diminishing its biological impact on the cell (84, 137). O2 − and H2O2 are more stable, and the diffusion rates for these agents are more suitable for the creation of a larger and more lasting oxidative environment (58, 81, 117).
Effects of O2 −
The effect of O2 − on sGC activity appears to be highly dependent on the mode that has been applied to generate or modulate the level of O2 −. For example, when rat aorta was exposed to O2 − generated by xantine/xantine oxidase (X/XO) reaction, the activity of sGC in the aortic lysate (133) was not affected. However, when endogenous generation of superoxide was induced in platelets (78) or rat aortic rings (52), NO-dependent activation of sGC decreased. However, such experiments are opened to interpretation due to possible interference from other oxidative species or through interaction with endogenous NO. Therefore, more conclusive results were sought when sGC-enriched fractions from the lysates of human platelets (10) or purified recombinant sGC (77) were exposed to O2 − generated by the X/XO system. In both reports, basal and NO-dependent sGC activities were attenuated. Moreover, O2 − inhibited the activation of sGC by the NO-independent stimulator YC-1 (77). Interestingly, Brune et al. observed that the heme-free sGC was also inhibited by O2 − (10) and suggested that the inhibitory effect of O2 − is not related to changes in the heme status of sGC.
Using an opposite approach, Friebe and colleagues demonstrated that addition of SOD to sGC preparation results in enhanced sGC activity, even without the addition of NO (32). They demonstrated that SOD-mediated stimulation of sGC is due to the elimination of NO scavenging via the reaction with superoxide. In contrast to studies by Brune et al. (10), the SOD-dependent stimulation was not observed with the heme-deficient enzyme.
Effects of ONOO−
As mentioned earlier, when O2 − levels overwhelm the scavenging capacity of SODs, the reaction between O2 − and NO generates peroxynitrite (ONOO−), a highly reactive oxidant. Under physiological redox conditions, ONOO− is efficiently inactivated by reactions with cellular thiols, such as glutathione (93). However, under oxidative stress, when the reducing capacity of cellular thiols is exhausted, ONOO− causes substantial oxidative damage and leads to lipid oxidation, DNA damage, protein nitration, and so forth (22, 84).
Interestingly, two rather opposite effects of peroxynitrite on the function of sGC are reported.
A substantial number of studies demonstrate that ONOO− negatively affects sGC activity and function. Exposure of rat aortic rings to SIN-1 was shown to impair endothelium- and NO-dependent vasorelaxation of isolated blood vessels (54, 116). Pre-treatment with ONOO− was also reported to inhibit NO-dependent activation of sGC. At the same time, the vasodilation of rat aorta in response to BAY58-2667 (116) or of rat iliac arteries in response to related BAY60-2770 (123) was enhanced after ONOO− treatment. Since both BAY58-2667 and BAY60-2770 are NO- and heme-independent activators of apo-sGC or sGC with oxidized heme, these studies clearly indicate that oxidation (or loss) of sGC heme takes place after exposure to ONOO−. Moreover, Stasch et al. demonstrated that exposure of endothelial cells to ONOO− not only decreased NO-induced cGMP synthesis but also diminished the level of sGC protein (116). The studies by Weber et al. (133) demonstrated that 4-h incubation of rat aorta with a flux of 0.13 μM/min of exogenous ONOO− had no effect on sGC-dependent cGMP synthesis. Diminished NO-dependent cGMP synthesis was observed only when aortas were incubated and when there was a much higher flux of 7.5 μM/min ONOO− (31, 133).
On the other hand, several studies indicated that ONOO− may stimulate sGC activity and function. Collaborative studies by Mayer et al. reported that in the presence of glutathione ONOO− acts as a potent sGC activator at a concentration above 10 μM (68, 69). The activation of sGC was not observed in the absence of glutathione (68, 69). A similar dependency on glutathione was observed when sGC activity was measured in endothelial cells exposed to SIN-1, a dual donor of NO and O2 − (69). The authors suggested that formation of nitrosothiol is an essential step in activation of sGC by peroxynitrite. This conclusion was independently corroborated by Wolin's group, which demonstrated that bovine coronary and pulmonary arteries relax in response to ONOO− (43, 138). They also detected the generation of NO and nitrosothiols from ONOO− when incubated with aortic tissues or glutathione-containing buffer. It should be noted that in these studies, the consequences of ONOO- treatment on NO-dependent activation of sGC was not evaluated.
The contrast between these two different sets of studies may reflect the biphasic effect of NOO− on sGC. When the pool of cellular thiols is high, an acute (several minutes) increase in ONOO− may be accompanied by its conversion into NO or nitrosothiols, resulting in a short-term activation of sGC (43, 68, 69, 138). However, sustained fluxes of high ONOO− are typically generated under inflammatory conditions, when the pool of intracellular thiols is usually depleted. It is reasonable to assume that under such conditions, longer (>0.5 h) exposure to exogenous ONOO− (54, 116, 133) or chronic generation of endogenous ONOO− under inflammatory conditions and/or vascular dysfunctions (116) causes oxidation of sGC heme and impaired sGC function.
Effect of sGC nitrosation
Elevated cellular oxidative stress is coupled with a higher content of RNS. Moreover, chronic administration of some nitrovasodilators, such as nitroglycerin, was shown to increase both oxidative and nitrosative stress (80, 89). An earlier study by Waldman et al. suggested that desensitization of sGC from thoracic aorta or human coronary artery to nitrovasodilators is due to a relatively stable molecular alteration of sGC that makes the enzyme specifically less sensitive to NO, but does not change the response to NO-independent activation by the heme-replacing protoporphyrin IX (131). Sayed et al. were the first to test whether nitrosation of sGC cysteines accounts for sGC desensitization. They demonstrated S-nitrosated sGC in primary aortic smooth muscle cells (SMCs) exposed to S-nitrosocysteine, in human umbilical vein endothelial cells treated with vascular endothelial growth factor, and in isolated aorta after sustained exposure to acetylcholine (97). These studies demonstrated that S-nitrosated sGC has impaired NO-stimulated activity and suggested the involvement of cysteine 243 in α1 and cysteine 122 in β1 sGC. In a later study, S-nitrosation of sGC was directly linked to the development of tolerance to nitroglycerin (98).
Accumulation of nitrosated β1 sGC was also reported in vivo in transgenic mice that overexpress eNOS in endothelial cells (83). In this article, nitrosation also resulted in lower sGC activation by NO in lungs. It should be pointed out that such a loss of sGC activity was observed only in the lungs, where the higher oxygenation is more permissive for generation of RNS. Mayer et al. also reported partial inactivation of sGC by stoichiometric S-nitrosation, when S-nitrosating agent dinitrosyl iron complexes (DNIC) were used (67). These authors also emphasized the critical role of cellular thiols in the outcome of sGC response to S-nitrosating agents. They demonstrated that in the presence of glutathione DNIC act as sGC activators, whereas in the absence of glutathione S-nitrosation desensitizes sGC. This study clearly indicates that the state of the cellular thiol pool, which is rapidly exhausted under oxidative conditions, predetermines the outcome of the nitrosative pressure on sGC function.
Effect of H2O2
Biologically generated H2O2 is predominantly the product of superoxide deactivation. SOD-catalyzed breakdown of O2 − is the primary mean of H2O2 synthesis, with oxidation of O2 − by reduced iron/sulfur clusters playing a secondary, but measurable, role (137). Although H2O2 is a two-electron oxidant, it usually reacts poorly with most biological molecules. The most common reactions with participation of H2O2 are the oxidation of cellular and protein thiols and the formation of hydroxyl radicals via Fenton chemistry. Interestingly, sGC contains a large number of cysteines that could be potential targets for H2O2. Moreover, sGC heme contains a coordinated ferrous iron that can potentially catalyze the Haber–Weiss conversion of H2O2 into hydroxyl ion and •OH (46).
Generation of •OH should result in oxidative damage to the immediate vicinity of the sGC heme pocket and oxidation of heme. Surprisingly, despite such apparently favorable conditions for sGC damage by H2O2, there is little evidence that such damage takes place. In fact, the opposite is true. Since White et al. reported that H2O2 elevates sGC activity in lung homogenates (135), a substantial number of reports described a rather complex relationship between sGC activity and expression and H2O2 exposure. Endothelium-independent sGC-dependent relaxation to H2O2 has been extensively documented by using bovine pulmonary or coronary arteries (12, 13, 43). This relaxation cannot be attributed exclusively to the disulfide linking, dimerization, and activation of PKGIα caused by H2O2 (11), since elevated cGMP production in isolated blood vessels after exposure to H2O2 has been reported (12, 13). The mechanistic details of H2O2-dependent activation of sGC are not entirely understood, but the participation of catalase has been proposed. The compound I species of catalase has been postulated as the crucial component for sGC activation (12, 13, 17). Oxidation of sGC heme does not appear to be essential for H2O2-dependent activation of sGC, since administration of ODQ, a specific sGC heme oxidant, did not substantially alter the relaxation of bovine arteries induced by H2O2 (43).
A number of reports indicate that the potential benefit of sGC-dependent vasorelaxation in response to H2O2 is limited due to the decrease of bioavailable NO caused by H2O2. Diminished NO-dependent vasorelaxation after the treatment with exogenous H2O2 was reported for rabbit aortic rings (124) and denuded rat iliac arteries (122). Diminished NO-dependent cGMP synthesis after exposure to H2O2 was also recorded in various models, including primary cultures of rat aortic SMC (36), rat pulmonary artery endothelial cells (EC) (124), or SMCs of fetal lamb pulmonary arteries (134). At the same time, some studies showed that H2O2 does not alter cGMP synthesis in response to BAY41-2272 (122, 124) or BAY60-2770 (122), supporting the notion that the redox state of sGC is not affected by H2O2.
Post-translational modifications of sGC may also be important in mediating the effect of H2O2. Treatment with H2O2 of PC12 cells, rat aortic SMCs, and rat aortic tissue caused phosphorylation of the Tyr192 of the β1 sGC subunit (72). Finally, prolonged exposure to H2O2 affects the levels of sGC protein and mRNA, as described further in this review.
Effect of complex oxidative stress on sGC function
NO/sGC/cGMP signaling is essential for various cardiovascular processes, many of which are directly affected by ROS. For example, ROS negatively affect the functions of smooth muscle, their growth, differentiation, and death (44, 118). ROS are also regarded as a risk factor for many cardiovascular disorders. Increased scavenging of NO due to inactivation by O2 − and generation of peroxynitrite is a strong contributing factor to vascular dysfunctions. However, the dysfunctions of NO/cGMP signaling in pathological conditions can only partially be attributed to the scavenging of NO. As discussed next, ROS directly affect the function and redox state of sGC and alter sGC expression.
Substantial progress in the understanding of sGC status under conditions of oxidative stress was made after the development of heme-independent sGC activators, such as BAY58-2667 (113) or HMR 1766 (101). These compounds strongly activate sGC, which lacks heme or has an oxidized ferric heme (102, 115). Therefore, the combination of NO donors and/or heme-independent activators provides a comprehensive probing of the status of sGC heme in cells and tissues from animals with various conditions that are associated with elevated oxidative stress. This type of analysis revealed that oxidation of sGC heme is a common response in many pathological conditions. Diminished NO response that coincides with elevated response to heme-independent activators was observed in aorta from hyperlipidemic watanabe heritable hyperlipidemic (WHHL) rabbits, mesocolon arteries from type II diabetic patients, and aortic rings from spontaneously hypertensive rats (SHR) (116). A similar response was observed ex vivo with primary rat vascular smooth muscle cells (VSMCs), in which oxidative stress was caused by menadione or rotenone (140), or in monkey coronary arteries after hypoxia or hypoxia/reoxygenation injury (121).
Heme oxidation is not the only change in sGC status that is reported by various groups. Multiple reports indicate that sGC thiol groups are also targeted during oxidative conditions. For example, chronic exposure to aldosterone of bovine VSMC or COS-7 cells expressing sGC affects NO-induced cGMP synthesis and decreases sGC activity, presumably due to the higher level of O2 − generated by NADPH oxidase. Proteomic analysis identified a specific peptide of β1 containing various degrees of oxidation of the β1-Cys122 residue (60). Nitrosation of sGC cysteine is another post-translational modification that is often encountered under oxidative conditions. Elevated sGC nitrosation was reported in rats with hypertension caused by chronic AngII administration or rat A7r5 SMC line exposed to AngII (21). sGC nitrosation was also reported in HUVECs treated by vascular endothelial growth factor (VEGF), or in rat aorta after sustained exposure to acetylcholine (97). With the exception of sustained treatment to acetylcholine, these experimental models are not associated with a substantial increase in NO production.
Effect of ROS/RNS on sGC expression
As describe earlier, a diminished response of sGC to NO or heme-dependent stimulators and an elevated response to heme-independent sGC activators are commonly reported effects of ROS or RNS on sGC function. Another often-reported effect observed under various pathological conditions or experimental models of oxidative and nitrosative stress is the decreased level of sGC protein in affected tissues or treated cells.
Diminished levels of sGC subunits were reported in cell culture after treatments with a number of pro-inflammatory agents (3, 85, 90, 103, 120). Lower levels of sGC protein were also recorded after chronic exposure of rat VSMCs (29, 86) or endothelial cells (116) to NO donors. The treatment of isolated rat aorta or cultured VSMC with a wide spectrum of ROS-generating systems (H2O2, O2 − via XO or menadione sodium bisulphate) reduced the levels of both sGC subunits (36). Diminished sGC protein is also consistently reported in various cardiovascular samples of lean (5, 92, 96) and obese (45) SHR. Multiple studies demonstrate that the hypertensive phenotype of SHR animals is associated with an increased level of ROS. It has been also proposed that an age-dependent increase of ROS, especially O2 −, is responsible for the lower level of sGC in corpus cavernosum of aging rats (111). The expression of β1 sGC was diminished in rat aorta at late stages of experimental heart failure (6), which was most likely caused by an elevated level of O2 − and could be partially reversed by the anti-oxidant action of vitamin E.
At least two different mechanisms have been implicated in the loss of sGC protein under oxidative/nitrosative conditions. Meuer and colleagues demonstrated that ubiquitin-dependent degradation is an important mechanism in controlling sGC level (73). They reported that oxidation of sGC by ODQ in rat aortic samples and cultured cells leads to robust ubiquitination of sGC followed by degradation via the proteasomal pathway. It is not entirely clear whether oxidized or heme-deficient sGC is preferentially ubiquitinylated. In vitro studies performed with purified sGC from Manduca sexta demonstrated that ferric sGC heme is not as tightly bound as ferrous heme and therefore may be lost soon after oxidation (33). Recent studies by Thoonen et al. provided additional support to the notion that cellular heme-deficient sGC is not stable (125). They reported that the Apo-sGC mouse strain expressing heme-free sGC is expressed at a lower level than the wild-type or heterozygote controls. Studies performed by Ghosh et al. (37, 38) demonstrated the importance of heat shock protein 90 (Hsp90) for the proper heme insertion during maturation of sGC. Interestingly, an earlier study reported proteasomal-dependent degradation of sGC in cells treated with Hsp90 inhibitors radicicol and geldanamycin (88). Accumulation of misfolded sGC lacking heme is most likely the reason for sGC degradation in case of Hsp90 deficiency and may be the mechanism of sGC degradation under oxidative stress.
Another likely mechanism of sGC protein reduction is associated with the decreased level of sGC transcripts under various oxidative/nitrosative conditions. It was reported that chronic exposure to NO donors diminishes the level of sGC transcript in rat pulmonary artery SMCs (29, 104) or rat medullary interstitial cells (129). Isolated rat aorta and cultured VSMC treated with ROS-generating agents reduced the steady-state levels of sGC mRNA (36). The lowering of sGC mRNA level is not restricted only to in vitro systems. For example, the level of sGC transcripts was significantly diminished in SHR animals as compared with control rats (96), especially at advanced age (47).
The exact mechanisms that contribute to decreased steady-state level of sGC transcript are only starting to emerge. The work by Gerassimou et al. suggests that the transcription of sGC genes may be directly affected by oxidative stress. The authors demonstrated that the extent of reduction of the α1 sGC transcript in response to H2O2 treatment matched very well the reduction of α1 promoter-driven transcription (36). Previous studies of sGC promoter function revealed a number of transcription factors that are essential for regulation of basal sGC expression (61, 109, 110). Although NFκB and CCAAT-BF are particularly important in regulating sGC expression under inflammatory conditions, it remains to be determined whether the same factors are responsible for lower transcription of α1 gene in conditions of oxidative stress.
A number of studies also demonstrated that processes that are responsible for stabilization of sGC mRNA are essential for regulation of the steady-state level of sGC mRNA and may be directly affected by oxidative stress. A series of studies by Kloss and colleagues revealed that human-antigen R (HuR) protein binds the adenine- and uracil-rich elements that are located in the 3′-untranslated regions (UTR) of α1 and β1 sGC transcripts and contributes to stabilization of sGC transcript. It has been demonstrated that decreased HuR levels increase the rate of α1 and β1 mRNAs degradation in rat aortic cells (48, 50). In SHRs, the reduction of HuR protein is associated with decreased α1 and β1 sGC mRNA and contributes to the development of age-dependent hypertension (49). Priviero et al. also correlated decreased sGC activity and protein in mesenteric arteries of SHR animals with a lower level of HuR (92). A decreased level of HuR in murine pulmonary VSMCs cultured after chronic hypoxia was accompanied by the reduction of α1 sGC mRNA (23).
Multiple studies demonstrated that the level of HuR protein changes under oxidative conditions and downregulates the steady-state level of different target mRNA (1, 26). The currently proposed mechanism suggests that thiol-dependent redox regulation of transcription factor activator protein 1 (AP-1), which occurs under oxidative stress, decreases the expression of HuR. This, in turn, destabilized mRNAs targeted by HuR protein (74).
Reduced sGC expression was described in response to O2 −; H2O2, chronic exposure to NO donors, and nitrosative agents, as well as in different animal models associated with increased oxidative stress. However, a number of reports document increased expression of sGC under certain conditions. For example, Kober et al. reported that short-term exposure to O2 − (X/XO system) resulted in increased expression of sGC protein in freshly isolated rat aortic rings (52). Increased expression of sGC protein was also observed in cholesterol-fed rabbits (57) and in rats and rabbits after a 3-day administration of nitroglycerine (79). Martin-Garrido et al. reported that exposure of rat aorta and of cultured rat VSMC to H2O2 increased the expression of β1, but not α1 protein via the stabilization of β1 transcript due to higher level of HuR (65).
These apparently contradictory observations are, at least in part, explained by the studies of sGC expression in aorta of rats with experimentally induced heart failure. Bauersachs et al. demonstrated that 8 weeks after heart failure was induced, Ach-dependent vasoresponse was diminished, whereas sGC expression was increased (4). However, such a compensatory increase in sGC expression was not observed after 12 weeks of chronic heart failure (6) and a lower level of β1 sGC was reported. These studies suggest that under moderate or short-term oxidative pressure, sGC dysfunction may be compensated by increased sGC expression. Longer or more damaging oxidative stress leads to lower expression of sGC gene and increased degradation of damaged sGC.
To summarize, downregulation of sGC expression and activity under oxidative stress or chronic vascular conditions include inhibition of transcription (3, 87, 90, 104, 120), destabilization of mRNA (49), and protein destabilization in an increased oxidative environment (7, 19, 28). Moderate or short-term oxidative stress may induce a compensatory mechanism, which remains to be fully understood.
sGC Splice Variants and Oxidative Stress
Alternative splicing of sGC transcripts and expression of alternative splice isoforms of sGC emerge as important mechanisms that contribute to the regulation of sGC expression in conditions of oxidative stress. It is estimated that more than 60% of human genes undergo alternative splicing, making it one of the most important post-transcriptional regulatory mechanisms (132). Due to alternative splicing, the same precursor RNA generates different transcripts via incorporation or exclusion of various exon sequences. Variations in UTRs may affect the stability and/or processing of individual transcripts via introduction or elimination of binding sites for regulatory proteins. On the other hand, changes in the coding sequence induced by mRNA splicing may generate polypeptide products with altered functional properties. A number of published reports demonstrated that all human sGC genes undergo alternative splicing (9, 15, 18, 106, 108). Analysis of sGC mRNA transcripts in the GenBank database reveals a remarkable diversity of human splice isoforms.
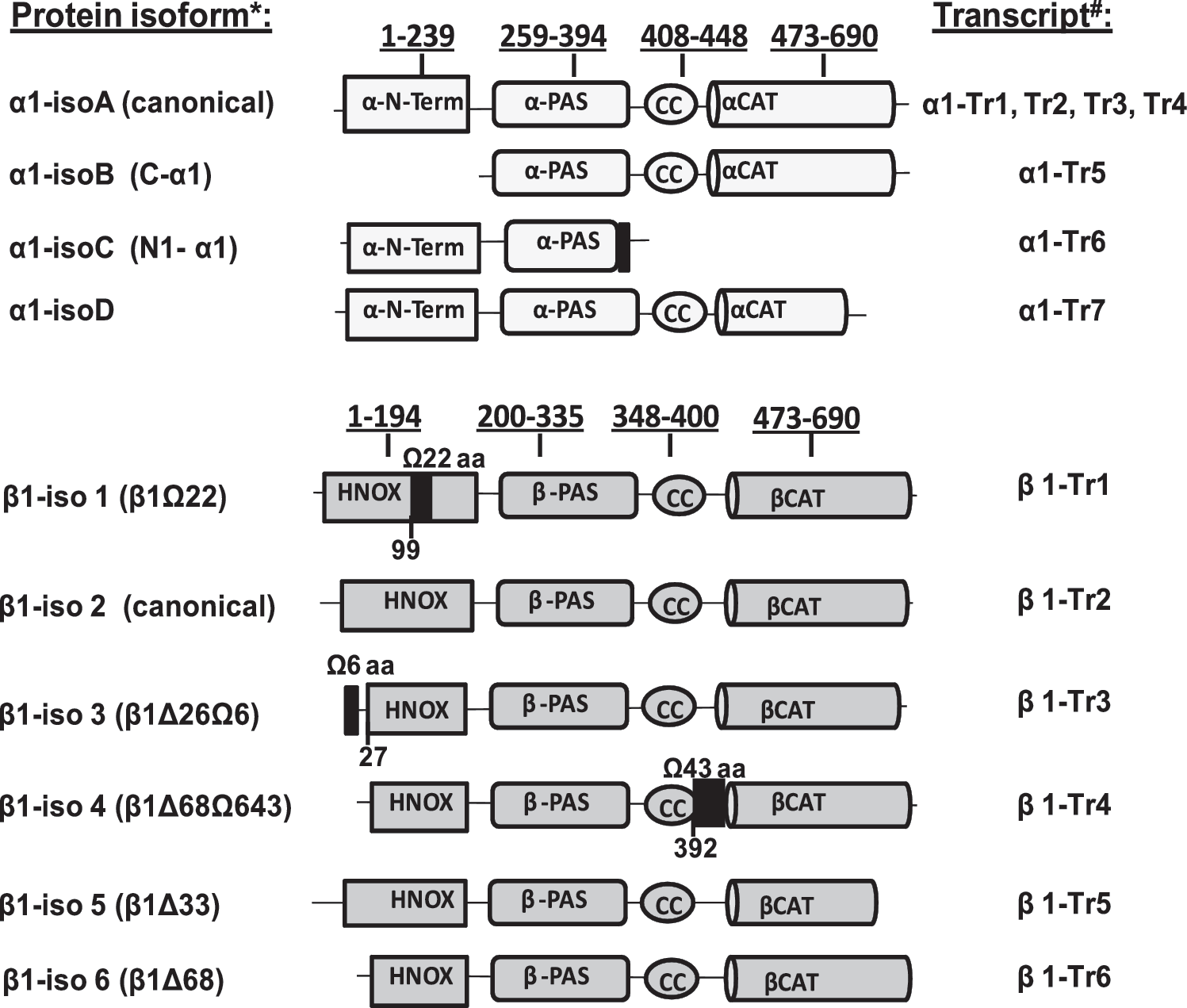
NCBI nucleotide database identifies 7 alternatively spliced transcripts for human GUCY1A3 gene of the α1 subunit (α1-Tr1 to α1-Tr7) (64, 107). All α1 transcripts can be sub-divided into two groups based on the encoded protein splice isoforms. The first group includes transcripts 1–4, which encode identical 690 amino acid protein isoform A (canonical α1 sGC) and differ from each other only in their 5′- and 3′-UTR sequences. The second group contains transcripts 5–7, each encoding a different polypeptide named, respectively, isoforms B, C, and D. The domain organization of sGC α1 splice protein variants is presented in Figure 1. These splice variants were characterized biochemically (56, 64, 108).
Based on GeneBank database, in addition to canonical β1 sGC transcript, five more alternative splice isoforms can be identified (64, 107). Domain organization of β1 splice protein variants (Fig. 1) indicates that, with the exception of the canonical β1 subunit (β1-iso2), all alternative splice isoforms of human β1 sGC contain deletions or insertions in the heme-binding domain. Although these β1 splice isoforms are not yet characterized functionally, the location of these insertions and deletions strongly suggest that they will generate sGC with impaired or absent NO-dependent stimulation of cGMP-forming activity.
It appears that the diversity of α1 and β1 sGC splice isoforms is more characteristic for humans. Among mammals, only mice and rats benefit from a sufficiently deep sequencing of the transcriptom to judge with any certainty the diversity of sGC splice forms. As far as the existing RNA-seq data suggest, rats do not have alternative splice transcripts, resulting in an alternatively spliced protein for either subunit (107). There are no predicted splice isoform for the β1 subunit in mice and only one α1 splice transcript that encodes the homolog of the human α1-isoC isoform.
Ritter et al. (94) reported the presence of three alternative α1 sGC splice transcripts in human heart, brain, artery, and B-cells. The translational start site was deleted in these splice transcripts, and no alternative sGC splice protein variants were reported at the time. Nevertheless, it was clear that elevated expression of one of the α1 splice transcripts correlated with decreased sGC activity in immortalized B-lymphocytes.
Later, our laboratories identified several splice variants encoding various polypeptides with deletions in C- and N-termini of sGC (108). We demonstrated that these transcripts are differentially expressed in normal human tissues. Functional characterization of the α1-isoB and α1-isoC splice isoforms revealed that co-expression of these isoforms together with canonical α1/β1 sGC alters the functional properties of sGC. The α1-isoC splice variant, which lost the catalytic domain due to splicing, displayed properties of a dominant negative mutant and inhibited the activity of the α1/β1 sGC heterodimer.
In contrast, the α-isoB, which contains a 240 residue deletion of the N-terminal regulatory domain and an intact catalytic region (Fig. 1), was fully functional and formed an active heterodimer with the β1 subunit, which was robustly activated by NO and allosteric stimulator BAY41-2272. The α1-isoB variant was much less sensitive to degradation induced by ODQ-dependent oxidation in human neuroblastoma cell culture. Under the oxidative pressure from 10 μM ODQ, canonical α1 and β1 splice variants (α1-isoA and β1-iso2) had an estimated half-life of ∼4.3 and 4.6 h, respectively. On the contrary, the α1-isoB splice isoform was stable and its expression increased after cell treatment with ODQ (Fig. 2) (108). Moreover, the expression of this oxidation/degradation-resistant α1-isoB splice isoform stabilized the endogenous β1 subunit and extended its half-life to ∼9.6 h.

The expression of endogenous α1-isoB splice protein was detected in human breast carcinoma MDA468 (20) and, to a lesser extent, in human neuroblastoma BE2 cells (20, 108). Interestingly, the α1-isoB splice isoform was also detected in differentiating human embryonic stem (hES) cells (106). Undifferentiated hES cells do not express sGC (76). However, when hES were allowed to differentiate, the expression of both canonical α1-isoA and α1-isoB splice isoforms increased. In fact, at early stages of differentiation, the 55 kDa α1-isoB constituted more than half of the expressed α1 sGC (106). The expression of this oxidation/degradation-resistant α1-isoB isoform is not restricted just to cell cultures. A 55 kDa α1-isoB immunoreactive band was previously reported in human brain samples (42). Without the benefit of the current bioinformatics data, this 55 kDa band was considered the product of a degradation process that occurs in vivo, but not in vitro. In a recent study, we reported that human aorta also exhibits a high level of 55 kDa α1-isoB protein in human aorta (64).
Besides the transcripts for canonical α1 and β1 subunits and functional α1-isoB variant, a number of transcripts coding for an additional splice variant of both α1 and β1 subunits were detected in normal human aorta (64). It was reported that healthy human aorta expresses mRNA for isoforms C and D of the α1 subunit. The alternative transcripts 3, 5, and 6 for the β1 subunit were also detected. The α1-iso D protein was expressed and characterized biochemically. It was demonstrated that α1-iso D has a significantly impaired capacity for activation by NO or allosteric regulators (64). Biochemical characterization of the α1-isoC splice protein demonstrated that it has a dominant-negative effect on sGC activity due to its lost catalytic region, but preserved capacity to heterodimerize with β1 sGC (108). The proteins coded by the alternative transcripts β1-Tr 3, 5, and 6 are also expected to be dysfunctional. They have missing regions in the catalytic domain or deletions/insertions in the heme-binding region (Fig. 1).
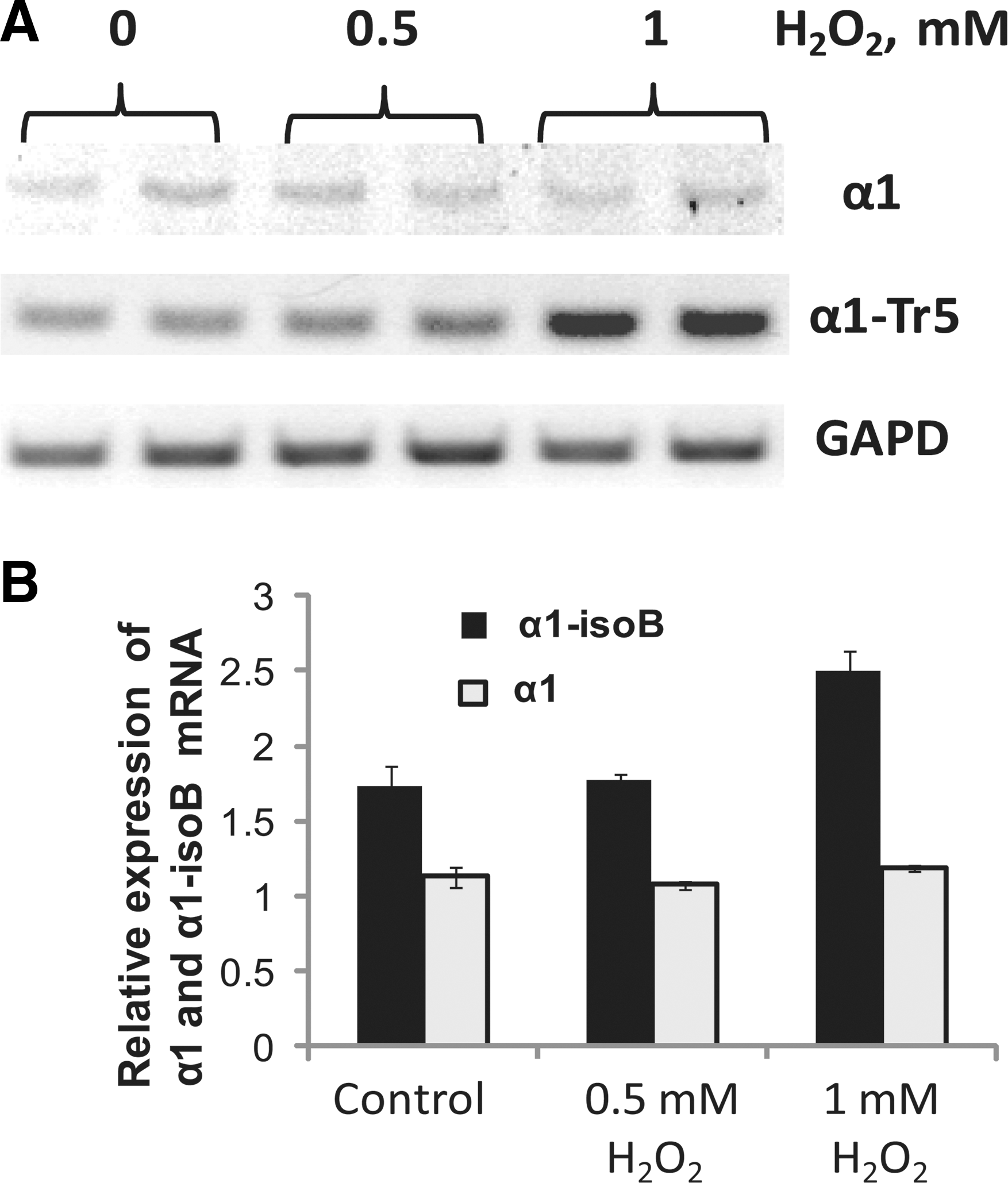
Previous studies revealed that the expression of human α1 sGC splice mRNA may be directly affected by cellular redox conditions. For example, the level of mRNA transcript encoding the canonical α1 sGC (α1-isoA) decreased in MDA468 and BE2 cells exposed to ODQ or H2O2. On the contrary, the expression of the mRNA and protein of oxidation/degradation-resistant α1-isoB splice isoform substantially increased in these cellular models (20). This feature is not restricted only to immortalized or transformed cells. As reported here, the treatment with H2O2 increased the expression of α1-isoB splice isoform mRNA in the early passage primary human aortic SMCs (Fig. 3).
Increasing evidence points to ROS and RNS as important causal factors in the pathogenesis of aortic aneurysm (70). Aneurysm is a chronic inflammatory condition of the aorta with characteristic structural deficiencies of the vascular wall. In one of our previous studies, we analyzed the composition of canonical and alternative splice variants (64). When commercial quantitative reverse transcription polymerase chain reaction (RT-PCR) probes and primers that recognize regions shared by all α1 or β1 splice variant were used, a 2-3-fold increase in α1 and β1 signals was observed in aortas with aneurysm (64).These data show the same trend as the increased expression of sGC in cholesterol-fed rabbits (57), rats and rabbits with chronic administration of nitroglycerin (79) or elevated expression of β1 sGC in rat aorta at early stages of heart failure (4).
The expression profile of the spliced α1 and β1 transcripts in healthy human aortas and aortas with aneurysm was previously reported (64). In that study, a semi-quantitative RT-PCR analysis based on primers specific for each individual splice form was performed. This approach provided a more accurate picture of the abundance and composition of the splice transcripts in normal aortas and aortas with aneurysm (64). It was determined that aortas with aneurysm have a much higher level of transcripts coding for dysfunctional α1-IsoD and α1-IsoC splice variant. A significant difference between the expression profile for the β1 splice transcripts in normal and diseased aortas was also observed (64). The analysis demonstrated that the level of transcripts that code for dysfunctional isoforms β1-iso1, β1-iso4, and β1-iso6 was also higher in aortas with aneurysm.
The expression of canonical full-length α1 and β1 sGC protein was lower in samples with aneurysm (64) (Fig. 4A). Higher sGC protein degradation in conditions of oxidative stress in aortas with aneurysm is probably the main reason why the level of these proteins did not change, despite the twofold increase in the level of canonical α1 transcript. The shift in the expression profile toward dysfunctional sGC splice forms is probably another factor that contributes to diminished α1 and β1 protein in samples with aneurysm. A recent study of mice expressing only heme-deficient Apo-sGC clearly demonstrated that heme deficiency results in a lower level of sGC protein (125). A similar mechanism probably decreases the expression of sGC in aorta with aneurysm.

The measurements of sGC activity in control samples and in samples with aneurysm more directly points to increased content of heme-deficient sGC in aortas with aneurysm. It was reported that in aortas with aneurysm, NO-dependent sGC activity constitutes 64% ± 4% of sGC activity of control aortas (64) (Fig. 4B). This correlates with the lower expression of α1 and β1 sGC and the higher content of dysfunctional sGC. However, the fraction of BAY58-2667-stimuated activity in aortas with aneurysm is higher and constitutes 86% ± 6% of the control (64) (Fig. 4B). The increase in the fraction of BAY58-2667-stimulated sGC supports the notion that samples with aneurysm have an elevated content of sGC with dysfunctional heme.
In the same study, the expression of the oxidation/degradation-resistant α1-IsoB isoform in human aorta was analyzed. The level of α1-IsoB mRNA was lower in samples with aneurysm than in control samples. Correspondingly, the α1-isoB protein was expressed in most normal aortas. In fact, in some samples, this was the major form of the α1 subunit. On the contrary, no signal or only traces of α1-isoB were detected in samples with aneurysm (64) (Fig. 4A). This comparison suggests that α1-isoB may have an important protective function. It is possible that the individuals with higher expression of α1-isoB are less prone to the development of vascular dysfunction, whereas individuals lacking this expression are more susceptible to them.
The exact mechanism behind the changes in sGC splicing under oxidative conditions is not well defined. Initial studies in this area identified a number of splice factors that may be directly responsible for the skipping of the human exon 3 of the α1 gene, which results in the transcript for the α1-isoB protein (20). In silico analysis suggests the existence of several cis-regulatory sequences around the alternative splice site that potentially bind serine/arginine-rich splice factors (SR) or heterogeneous nuclear ribonucleoproteins (hnRNPs) (Fig. 5A). The predicted hnRNP proteins are the polypyrimidine tract-binding protein PTBP1 and the hnRNP A2/B1 isoforms.

In a previous report, we demonstrated that the cells exposed overnight to H2O2 or cultured in glucose-containing media in the presence of exogenous glucose oxidase (GO) showed no changes in the level of the SR splice factor (20). The level of HuR protein, which stabilizes sGC mRNA, was also not affected. However, a dramatic reduction of PTBP1 and hnRNP A2/B1 proteins was observed (Fig. 5B) (20). A similar H2O2-induced reduction was previously reported for closely related hnRNP C1 and D proteins (41).
In another study, the hnRNP A1 homolog was hyperphosphorylated by the stress-induced MKK/p38-pathway, which led to the accumulation of hnRNP A1 in the cytoplasm and affected the splicing in the nucleus (39). A similar crosstalk between hnRNP A2/B1 proteins and p38 kinase is also possible under conditions of oxidative stress, which is a well-established activator of p38 kinase (112). Oxidation of cysteine residues of PTB and hnRNP A2/B1 splice factors is another likely contributor to the splice switching of sGC transcripts. Previous studies demonstrated that the effect of H2O2 or GO on PTB in a number of cell cultures was mimicked by bis(2-hydroxyethyl)disulfide (HEDS), a thiol-specific oxidant (20) (Fig. 5). On the other hand, hnRNP A2/B1 was identified as one of the proteins in Hepa 1–6 cells, which contain thiols that are prone to oxidation in response to cell treatments with GO (34).
These pilot studies suggested that PTBP1 and hnRNP A2/B1 may be directly involved in the cellular response, which results in switching to a different set of sGC transcript in response to an acute oxidative episode and/or a chronic low-level oxidative environment that is associated with various pathological conditions (Fig. 6).
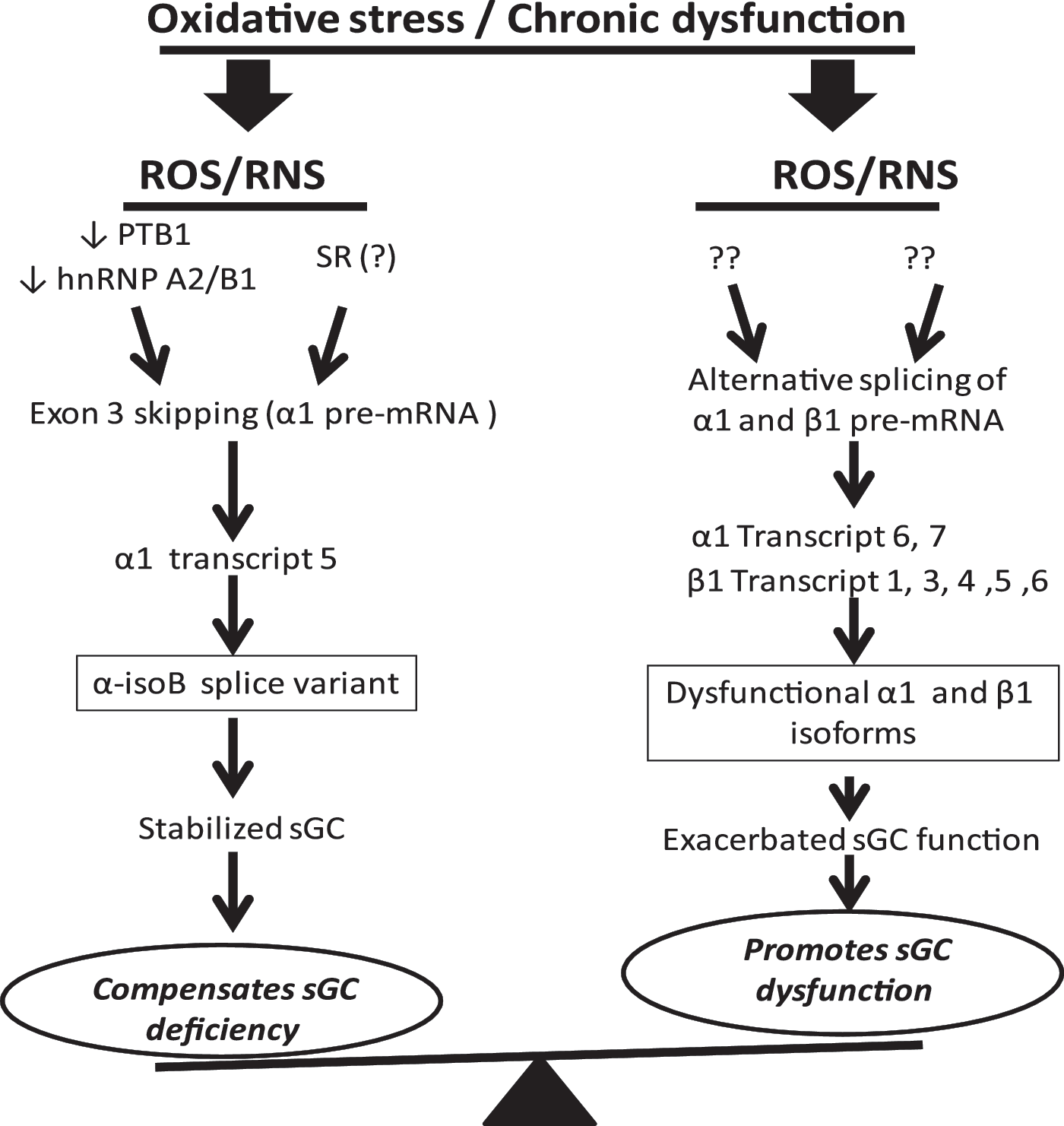
In summary, accumulated evidence allows us to propose that the splicing of sGC is tuned to changes in cellular oxidative condition and plays a dual role in regulation of sGC function (Fig. 6). On the one hand, alternative splicing promotes sGC function via the expression of a stable and functional α1-isoB isoform. On the other hand, the expression of dysfunctional isoforms α1-isoC, α1-isoD, β1-iso1, β1-iso3, β1-iso4, β1-iso5, and β1-iso6 may exacerbate ROS-dependent damage to sGC function and negatively affect sGC stability and activity. Changes in sGC splicing in aortas with aneurysm, where the composition of sGC splice transcripts is altered in favor of transcripts encoding for non-functional or dysfunctional sGC subunit isoforms, is most probably just an example of similar processes taking place in other tissues or under other chronic pathological conditions. These data suggest that sGC splicing is an important, previously unrecognized factor contributing to the development of vascular disease and other pathologies. Oxidative stress persists in diseased blood vessels and increases with age. Increased expression of α1-isoB may be a positive factor contributing to the maintenance of sGC function under oxidative stress conditions. On the contrary, diminished expression of α1-isoB compounded by increased expression of dysfunctional sGC splice forms may be exacerbating the dysfunction of NO/cGMP signaling in diseased tissues.
Future Directions
Targeting the process of sGC splicing in vasculature under oxidative stress may be an important preventive approach to maintain the composition of sGC transcripts expressed in healthy vessels and/or a complementary treatment to existing sGC-targeting therapies. Successful strategies that allow targeted manipulations of RNA splicing are now starting to emerge (136). Modified antisense oligos that are resistant to exonucleases are being developed to manipulate the ratio of expressed transcripts via targeting of alternative RNA splicing. In this approach, the splice-switching oligonucleotides act as competitors for the regulatory sites on the pre-mRNA transcript and sequester the dynamic components of the spliceosome complex responsible for each splicing event. Such targeted competition shifts the splicing process toward the preferred alternative splice site. This approach has been successfully applied to facilitate the expression of a preferred protein isoform and to alter the gene function in a desired manner (8, 100). This strategy was used to achieve exon exclusion, intron retention, exon shuffling, or usage of alternative 5′ and 3′ splice sites (66). Currently, a number of different synthetic oligonucleotides that are resistant to RNase H degradation and high hybridization efficiency and specificity to a specific target were developed (8). The use of modified oligos enabled application of this technique for splicing correction in clinically relevant cultured human cell lines and mouse models (2, 95, 99). Moreover, stable morpholino-oligonucleotides are currently being evaluated in clinical trials for the treatments of β-thalassemia and cystic fibrosis (100). Therefore, understanding the mechanisms that govern the splice switching of sGC transcripts in conditions of oxidative stress or pathological conditions should be beneficial for the development of therapies that modulate sGC function by restoring the composition of sGC splice transcripts featured in healthy tissues under normal conditions.
Footnotes
Acknowledgments
This work was supported by the University of Texas Bridge grant (I.G.S.) and the American Heart Association (grant 15GRNT25700101, E.S.M.). The authors would like to thank Filip Jelen, Elena Bogatenkova, Vladislav Sharin, Eva Golunski, Susan Laing, Anthony Estrera, Gilbert J. Cote, Wen Zhu, Anthony Thomas, Alexander Kots, Kalpana Mujoo, and Ferid Murad for their technical and intellectual contributions to the studies related to sGC splicing.