
Abstract
Significance:
Soluble guanylate cyclase (sGC) is an intracellular enzyme that plays a primary role in sensing nitric oxide (NO) and transducing its multiple signaling effects in mammals.
Recent Advances:
The chaperone heat shock protein 90 (hsp90) associates with signaling proteins in cells, including sGC, where it helps to drive heme insertion into the sGC-β1 subunit. This allows sGC-β1 to associate with a partner sGC-α1 subunit and mature into an NO-responsive active form.
Critical Issues:
In this article, we review evidence to date regarding the mechanisms that modulate sGC activity by a pathway where binding of hsp90 or sGC agonist to heme-free sGC dictates the assembly and fate of an active sGC heterodimer, both by NO and heme-dependent or heme-independent pathways.
Future Directions:
We discuss some therapeutic implications of the NO-sGC-hsp90 nexus and its potential as a marker of inflammatory disease. Antioxid. Redox Signal. 26, 182–190.
Introduction
T
Heat shock protein 90 (Hsp90) is a ubiquitously expressed ATP-dependent chaperone that helps to fold, stabilize, or modify the functions of select client proteins (42, 46). Hsp90 functions through its subdomain molecular motions and its inherent ATPase activity to help control client protein maturation, trafficking, and lifetime in cells (14, 62, 82). The molecular-level impacts of hsp90 on various client proteins are just beginning to be elucidated (38, 59, 64, 74). Hsp90 is known to associate with several heme proteins and the ascribed outcomes include assisting in protein maturation, stabilization, function, or activity and in shaping enzyme product distribution (Table 1) (50). Hsp90 was first reported to associate with sGC in 2003 (90), and subsequent cell and molecular studies identified possible regions of protein interaction (58) and the possible functional roles for the interaction (increase in the stabilization and activity of sGC) (2, 3, 24, 25, 67, 95), but did not explore how these outcomes may come about. In this review, we discuss recent work that reveals how hsp90, heme, NO, pharmacologic sGC agonists, and related cellular processes may govern sGC function in unexpected new ways.
Eukaryotic heme synthesis and hsp90-driven cytosolic heme insertions
Heme proteins are involved in a remarkable array of biological functions, including cell energetics, oxygen transport and storage, numerous enzymatic transformations, cell signaling, and host defense (60, 61). In all cases, the heme cofactor is essential for function, but because free heme is reactive, its production is tightly regulated (83). The specific steps of heme biosynthesis are well known and take place in both the cytosolic and mitochondrial compartments of a cell (13, 79, 92), with the final three steps occurring in the mitochondria (Fig. 1). Contrastingly, little is known about how heme is transported out of mitochondria or how heme insertion occurs in soluble proteins in the cytosol (17, 73). However, recent studies from our group (23, 25) as well as pioneering studies from Osawa's group (6) have shown specific involvement of chaperon hsp90 in cytosolic heme insertion into soluble proteins. Osawa and colleagues showed for the first time that hsp90 is required for cellular heme insertion into neuronal NO synthase (nNOS). Later, Ghosh et al. uncovered a role for hsp90 in inducible NOS (iNOS) heme insertion (23). In the latter case, hsp90 was shown to primarily associate with an apo-iNOS monomer in cells, and then found to drive heme insertion into the apoenzyme by an ATP-dependent process, after which hsp90 interaction with the heme-replete mature iNOS fell apart. Recently, it was found that heme insertion into the β subunit of sGC is also hsp90 dependent (25). Given that sGC structural homologs (H-NOX) and NOS have markedly different protein structures and heme environments (10, 48), these findings hint that hsp90 may play a broader role in heme protein maturation than was previously realized. In the sGC studies, hsp90 associated primarily with the heme-free form of sGC-β1 in cells, but the association fell apart once heme became inserted. Although the association did not require hsp90 to have an intact ATPase activity, as judged from results using hsp90 inhibitors, radicicol or novobiocin, or by using an ATPase-defective hsp90 mutant (D88N), an intact ATPase activity was essential to actually drive heme insertion into the apo-sGCβ1. Thus, the model suggested for hsp90 function in sGC maturation mimics the model proposed for driving heme insertion into apo-iNOS (Fig. 2). These similarities imply that hsp90 may operate through a common mechanism to target and stabilize heme-free forms of client heme proteins, and then enable their maturation by driving heme insertion in an ATP-dependent process.

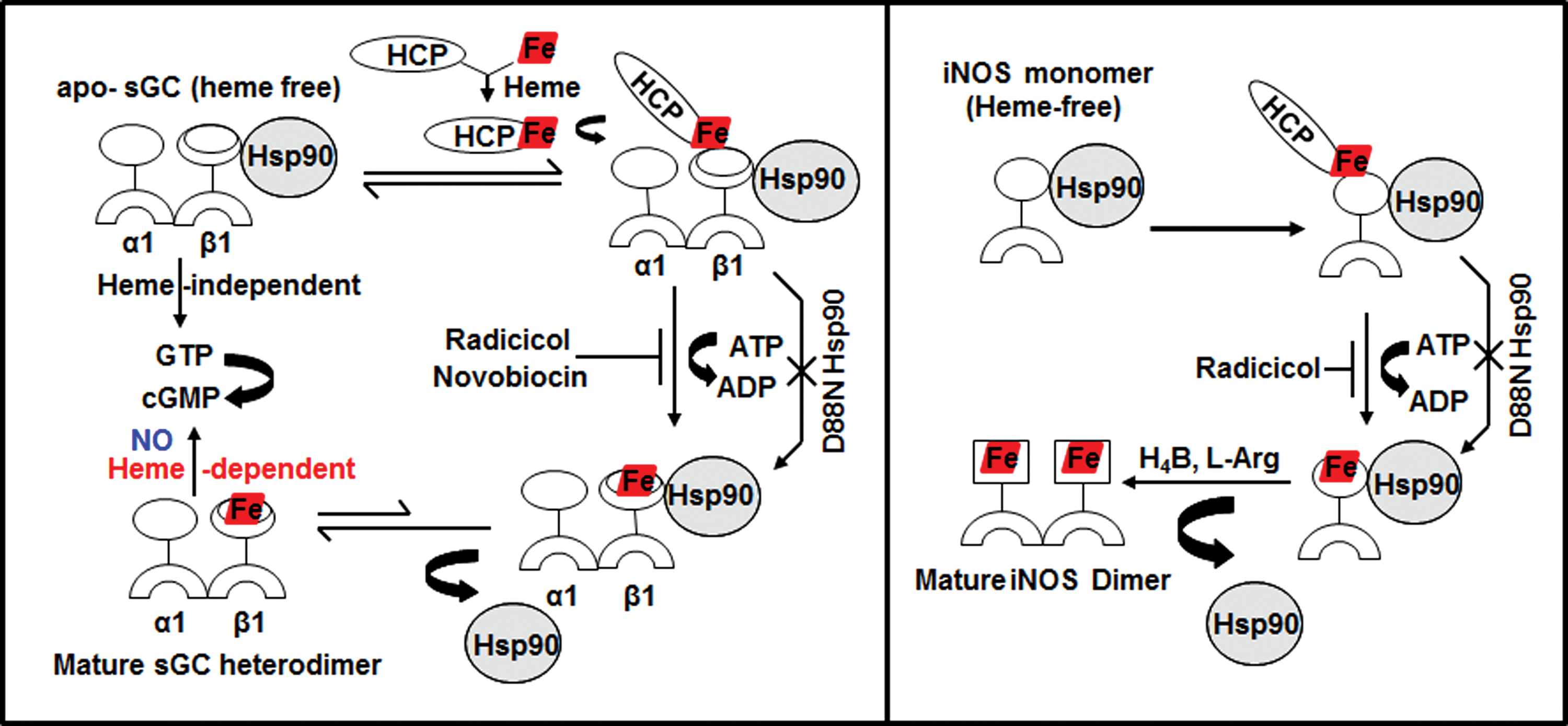
Relevance of apo-sGC/NOS-hsp90 interactions in cells
sGC is a key enzyme of the NO signaling pathway and is a therapeutic target in cardiopulmonary disease, with several sGC agonists currently in clinical development (29, 54, 69, 75 –77, 80). NO produced locally from NOS enzymes acts as a messenger that crosses cell membranes, binding to sGC heme, thereby promoting the synthesis of the second messenger, cGMP, which in turn produces vasorelaxation in smooth muscle and participates in a number of downstream signaling pathways (22, 45). Impaired NO and cGMP signaling has been implicated in the pathogenesis of a number of cardiovascular diseases ranging from pulmonary arterial hypertension (PAH) to cardiac hypertrophy and heart failure (9, 40, 47). Studies suggest that the interaction between apo-sGC or apo-NOS and hsp90 in cells is vital for heme insertion into both proteins (23, 25), which in turn is required for their maturation as biologically functional enzymes. Thus, any endogenous or pharmacologic disruption of their hsp90 interactions would be expected to antagonize their heme insertion and limit their maturation and biological function. In fact, disruption or dysregulation of their hsp90 binding would likely have a synergistic impact on any cellular processes that depend on NO signaling because it would diminish both production of the signal molecule (NO) by NOS enzymes and the capacity of the sGC to respond to NO (to make cyclic GMP). Thus, heme insertion in NOS and sGC is of dual significance in the NO signaling cascades. Currently, it is unknown if any of the hsp90 inhibitors being developed for cancer therapy (85) also impact maturation of sGC or iNOS by inhibiting their heme insertions, but this possibility should be considered. In addition, future work stands to explore if the interactions of hsp90 with apo-sGC or apo-iNOS might be manipulated in a positive way to ensure that sufficient or greater cGMP production occurs to restore proper functioning of biological NO signaling cascades in disease states.
NO-triggered heme insertion into sGC-β1, mutually exclusive hsp90 interaction versus sGC-α1β1 heterodimer formation, and a novel activation pathway for the sGC activator, BAY 60-2770
A recent study revealed that exposing cultured cells to a low dose of NO triggered hsp90-assisted heme insertion into the population of apo-sGC-β1 that existed in the cells (24) (Fig. 3). This occurred in cells that naturally express sGC and in cells transfected to transiently express sGC. The NO effect was relatively rapid, becoming complete within 2 min of exposure, and more slowly reversed with time if the NO exposure was continued. Remarkably, the rapid heme insertion into apo-sGC-β1 was linked to a rapid and reversible swapping of its protein binding partners, that is, from its initial hsp90 partner to its sGC-α1 subunit partner, thus forming a mature sGC-α1β1 heterodimer (Fig. 3). All these NO effects required that hsp90 had an active ATPase activity and that sufficient cellular heme was available and was insertable into the apo-sGC-β1 (i.e., an sGC-β1 heme binding mutant expressed in the cells did not alter its protein associations in response to NO). Overall, the study suggested that a dynamic and previously overlooked interplay may exist between NO, cellular heme, sGC, and hsp90, which can quickly increase the population of mature sGC heterodimers in cells when they encounter exposure to NO.

Within the same study, the pharmacologic sGC activator, BAY 60-2770, was found to mimic all the changes in sGC-β1 protein associations and consequent increase in sGC heterodimer formation that took place when the cells were given NO (Fig. 3). However, in contrast to the NO effect, the ability of BAY 60-2770 to do so did not require an intact hsp90 ATPase activity, nor that cellular heme be available or be insertable into the apo-sGC-β1 (24). These differences make sense if the BAY 60-2770 acts through its binding within the heme pocket of the apo-sGC-β1, which is where activators of this structural class are known to bind (77). Crystal structures of BAY 60-2770 bound to an sGC protein homolog (Nostoc enzyme) show that it binds in the protein's heme pocket and causes discreet changes in the protein structure that are thought to mimic the structural changes that take place when NO binds to heme-replete sGC-β1 when it is in complex as an sGC-α1β1 heterodimer (24, 39). These structural changes within sGC-β1 are thought in turn to cause a cascade of structural changes within the sGC heterodimer that lead to activation of enzyme catalysis (20, 89). In this context, it is remarkable to imagine that BAY 60-2770 binding within the heme pocket of the apo-sGC-β1, when it is in complex with hsp90, may cause similar, or perhaps distinct, structural changes within the β1 subunit that lead it to dissociate from hsp90 and to associate instead with sGC-α1. Although untested, this activation process by BAY 60-2770 would appear to differ from how it may activate an sGC heterodimer, where it must presumably bind to a heme-free sGC-β1 subunit that is already complexed with an sGC-α1 partner instead of hsp90. These different possibilities should be further explored and perhaps could also be investigated from a pharmacore development viewpoint, given that oxidatively damaged forms of sGC that respond to BAY 60-2770 appear to accumulate during several common inflammatory diseases (78).
Molecular aspects of the hsp90-sGC-β1 interaction
Early studies that employed antibody pulldown or affinity tag capture methods to study the hsp90-sGC interaction in mammalian cells reported that their association involved the M domain of hsp90 and two regions within sGC-β1, one in its per-arnt-sim (PAS) domain (amino acids 204–244) and another in its coiled-coil domain (amino acids 379–408) (58). Subsequent studies showed that both of these structural regions in sGC-β1 enable sGC-α1β1 heterodimerization (65, 97). Thus, in retrospect, the early studies also support the concept that sGC-β1 binding to hsp90 or to sGC-α1 may be mutually exclusive and have a reciprocal relationship.
A more recent study (67) used fluorescence polarization and hydrogen–deuterium exchange mass spectrometry to investigate the binding interactions between purified hsp90, sGC-β1, and some of their individual domains. The results suggest that the hsp90 M domain interacts primarily with two regions in apo-sGC-β1 that are located in the middle and C-terminal end of its PAS domain (Fig. 4). Based on a structural modeling and docking analysis (Fig. 4), the authors proposed that the interactions could create a sort of bidentate structural scaffold that (i) prevents apo-sGC-β1 from interacting with its partner sGC-α1 subunit and (ii) could facilitate structural change in the apo-sGC-β1 heme domain that may allow heme insertion. Because hsp90 can bind heme directly (41), it could also conceivably participate in furnishing heme to the apo-sGC-β1 during the maturation process. In these ways, hsp90 actions during sGC maturation would mimic how it influences protein associations and small ligand binding events in other hsp90 client proteins, particularly in clients that contain a PAS domain, including the aryl hydrocarbon receptor and HIF1α (44, 53, 87). Going forward, it will be important to test if the hsp90-apo-sGC-β1 interactions identified in the biophysical studies with purified proteins actually facilitate sGC maturation in an intact cell system. In addition, because cochaperone proteins are likely to aid the hsp90-driven heme insertion into sGC-β1, understanding their identities and participation would create a more comprehensive picture of how sGC maturation is regulated.

sGC-β1 heme binding is also sensitive to the redox state of the heme, with the affinity of sGC-β1 being greater toward ferrous heme than toward ferric (19). Given that sGC is thought to lose the oxidized ferric heme under high oxidant stress (19), it is vital to determine the mechanisms involved. For example, Might hsp90 play a role in aiding ferric heme loss from its sGC-β1 client protein? Might hsp90 hold the ferric heme transiently, only to transfer it back to apo-sGC-β1 as ferrous heme if the redox state of the cell returns to normal conditions? These questions may be addressed in future studies. At this point, the studies raise the possibility that the apo-sGCβ1-hsp90 interactions facilitate a dynamic heme transfer among the participating protein components, which in turn might be influenced by factors, including sGC protein modifications or changes in the intracellular and heme redox states, perhaps in a manner similar to what has been found to occur in other systems (63).
Impact of NO on heme protein maturation and its relevance to diseases
In cells, NO can affect heme availability and heme insertion in various ways. NO is known to cause a prolonged and significant loss of intracellular iron in cells once iNOS is expressed (36, 93). Moreover, NO is known to directly inhibit the mitochondrial enzyme, ferrochelatase, which is responsible for iron insertion into the newly synthesized porphyrin (21). Waheed et al. found that NO exposure blocks heme insertion in a number of apoprotein targets expressed in mammalian cells, including NOS enzymes, catalase, cytochrome P450, and hemoglobin (91), thus suggesting that NO can act as a global inhibitor of heme insertion. Manifestations could possibly occur in any disease characterized by chronically elevated NO levels, for example, in the acute malarial anemia prevalent among children of Sub-Saharan Africa (1, 33) or in pulmonary inflammatory diseases such as asthma (26, 35). As sGC is abundantly expressed in human airway smooth muscle cells, it is logical to predict that high airway NO levels may inhibit sGC heme insertion, resulting in increased amounts of heme-free sGC in a diseased state such as asthma. This may result in desensitization of sGC toward its natural activator NO, and further studies should aim to address these possibilities.
S-nitrosation of sGC and its relevance to NO desensitization or sGC deactivation
During prolonged NO exposure, sGC ultimately decreases its cGMP output by a process known as NO desensitization (55, 72). One potential mechanism of desensitization is through the S-nitrosation of Cys residues that are located in close proximity to the heme (4, 15, 43, 49, 68). S-nitrosation (SNO) is an NO-dependent post-translational modification that may directly alter protein structure and function (8, 28, 52). In sGC, it is thought to occur when NO, in addition to binding to the heme cofactor, ultimately modifies free thiols in the protein. The full-length sGC-β1 contains 34 cysteine residues (18), but only three of these (C78, C122, and C174) in sGC-β1 have been characterized within a minimum functional ligand-binding heme domain of sGC (amino acids 1-194) to study cysteine S-nitrosation (32). Using the biotin switch method, activity assays, and infrared spectroscopy, Sayed et. al (68), Frenhoff et. al (16), and Liu et. al (43) demonstrated that C78 and C122 on the β1 subunit are the key residues for sGC desensitization via S-nitrosation. One report (16) suggested that SNO modification of sGC cysteine residues (Cys 78 and Cys 122) actually involves the sGC heme moiety participating in the redox reaction, resulting in a deactivated ferric sGC that cannot respond toward NO.
A recent study (24) suggests an additional way that SNO modifications of sGC-β1 might be associated with NO desensitization: The buildup of SNO-sGC in cells exposed to NO was found to correlate with the kinetics of hsp90 association with sGC-β1 and a concomitant loss of the sGC-α1β1 heterodimer population. This suggests that the SNO modification(s) might alter the sGC-β1 in ways that weaken the heterodimer interaction, thus leading to NO desensitization. It also raises questions regarding the role of hsp90 association with the SNO-sGC-β1 postactivation, for example, Is it a natural process that helps to protect sGC-β1 from degradation (56)? Does it facilitate denitrosation? Is it accompanied by any hsp90-driven changes in sGC-β1 heme occupancy as described above (30, 51)? Besides the mechanisms of desensitization that possibly involve SNO modification of sGC-β1, additional mechanisms include (i) ferrous heme oxidation to ferric (27, 71, 96) and (ii) direct heme nitrosylation by NO (86).
Regarding the natural activation/deactivation cycle that sGC engages in during signal transduction, previous studies show that there are different time frames for deactivation, and it is still unclear if simple NO dissociation from the sGC heme directly leads to deactivation. In vitro deactivation of sGC enzyme occurs within minutes after NO dissociation (7, 34), whereas in vivo cell sGC activity can deflect within seconds after it is activated by NO (5), suggesting that additional factors are present in cells to speed sGC deactivation. Whether fast deactivation involves allosteric mechanisms, protein partners, or even SNO modifications are all intriguing possibilities that need to be addressed.
Heme-free or desensitized sGC in cells/tissues and potential therapeutic importance of sGC activators
Increased oxidant stress that occurs under inflammatory conditions (81) is thought to cause oxidation and loss of the sGC heme, thus creating a population of sGC that is insensitive to NO (19). This has increased interest toward developing drug candidates that can activate sGC independent of NO or its heme (37, 76, 78) and has piqued interest in the cellular mechanisms that may control the heme content, protein associations, and activity of sGC. Indeed, it is becoming evident that sGC can exist as a blend of heme-containing and heme-free populations in cells even under normal culture conditions (24, 25), and growing evidence suggests that this equilibrium shifts to favor the heme-free form under pathophysiological conditions characterized by increased oxidant stress (78, 84). The presence of heme-free sGC in a variety of transformed and primary cell types (24, 25) may reflect the original finding by Ignarro and colleagues who first purified sGC from bovine lung in a heme-deficient form (31, 57) and also support results of Roy et al. (66) that showed heme-free sGC normally exists in cells. Under these circumstances, activators of soluble guanylyl cyclase that can modulate sGC activity in a selective manner either by targeting the heme-containing (BAY 41-2272) or heme-free form (BAY 58-2770 or BAY 60-2770) of the enzyme possess considerable therapeutic potential (70, 76, 77). Such sGC agonists are of current interest as treatment for cardiovascular and cardiopulmonary diseases.
Molecular mechanisms that underlie pharmacological activation of the heme-free sGC appear to involve the triggering of sGC heterodimerization, which occurs, for example, in response to BAY 60-2770 insertion into the sGC-β1 subunit (Fig. 3). Thus, although the relative importance of this process would depend on the relative abundance of the apo-sGCβ1-hsp90 complex in cells, it may or may not need to involve hsp90 for an effective drug response (24, 25). Regarding the heme-dependent sGC activators, BAY 41-2272 is a structural analog to the commercial drug, Adempas or Riociguat (BAY 63-2521), which was recently approved by the FDA (54) to treat pulmonary hypertension in humans and has since been approved in Europe with expected worldwide approval. The success of this heme-dependent sGC activator in treating pulmonary hypertension (PAH) patients may be attributed to the low NO levels (94) that prevail in the diseased pulmonary arteries. This, coupled with the fact that Adempas works well in synergy with low NO levels (40, 94), makes it an effective drug in PAH. In contrast, diseases that are characterized by chronic high NO levels (26) may induce an increase in the heme-free sGC-β1. Under these conditions, heme-independent sGC agonists such as BAY 58-2667 or BAY 60-2770 can become more valuable because of their ability to selectively activate the oxidized/heme-free form of sGC (70, 77). However, since the natural state of sGC may be a blend of heme-containing and heme-free states, perhaps the most effective therapy might have both types of sGC drugs used in combination, with the relative ratio reflective of the individual and the extent of disease progression.
Heme-free sGC and sGC-hsp90 interactions as markers in cardiopulmonary diseases
Studies (24, 25) suggest that observing hsp90-sGCβ1 association in cells may itself indicate that heme-free sGC-β1 is present. The concept that the sGCβ1-hsp90 interaction is mutually exclusive with sGC-α1β1 heterodimerization (24) means it could be a marker of sGC status and activity. A predominant sGC heterodimer may be indicative of healthy condition, whereas a weak heterodimer with a strong sGCβ1-hsp90 component would indicate a diseased state. This paradigm may have potential applications in the pathogenesis of diseased states in organs where sGC is present (for example, lung, brain, or intestine) and its vasodilatory function holds biological relevance. The effects of high oxidant stress or of high NO can possibly be followed as markers or fingerprints in clinical diagnosis to detect the extent of a cardiopulmonary disease. For example, finding a lower amount of sGC-α1β1 heterodimer with a relatively increased amount of sGCβ1-hsp90 complex, perhaps coupled to SNO modification of the sGC-β1 subunit, would imply dysfunctional sGC and may indicate the severity of the disease and allow subsequent monitoring of treatment response. These concepts could help achieve better clinical diagnosis and outcomes across a wide spectrum of diseases.
Footnotes
Acknowledgments
Dr. D.J.S. is supported by NIH grants, HL081064 and GM097041, and Bayer grants for target award.