
Abstract
Introduction
M
The classification of macrophage activation states dates back to the 1960s and is, to a large extent, based on the ex vivo response of macrophages to Th1 and Th2 cytokines [reviewed in Martinez and Gordon (137)]. In 2004, Mantovani et al. proposed the classification of activated or polarized macrophages into classical (M1: IFN-γ + TNF-α or LPS) polarization and three different types of alternative (M2) polarization based on the activating agent: M2a referred to IL-4 and IL-13 activation, M2b referred to immune complex activation, and M2c referred to IL-10 activation (136). Since then, several other activating agents have been described in macrophages, including CXCL4 being identified as an M4-polarizing agent (75). This macrophage classification system has been broadly utilized to characterize the heterogeneity of macrophages in atherosclerotic plaques in vivo (74, 198). While the M1 and M2 classification system is inadequate to capture the complexity and diversity of macrophage activation states (151), we will use these terms here for the sake of simplicity and clarity, referring to macrophages with predominantly proinflammatory proatherogenic properties as M1 and macrophage phenotypes with predominantly anti-inflammatory and inflammation-resolving properties as M2.
In addition to the assorted cellular functions, macrophages originate from at least two different sources during different developmental stages as well as homeostatic versus inflammatory conditions. During homeostasis, the majority of tissue-resident macrophage populations, including splenic red pulp macrophages, microglia, Kupffer cells, osteoclasts, Langerhans cells, and large peritoneal macrophages, originate from embryonic yolk sac-derived progenitor cells (54, 55, 79, 177, 231). However, in response to tissue injury, there is a vast infiltration of monocyte-derived macrophages that originate from bone marrow hematopoietic stem cells (125, 198, 237). This recruitment of blood monocytes to discrete anatomical locations, followed by their differentiation into mature macrophages, is a rate-limiting step in multiple physiological processes, including wound repair, pathogen clearance, and replacement of tissue-resident macrophages (43, 80), as well as pathophysiological processes such as obesity and atherosclerosis (147, 219). Interestingly, blood monocytes themselves are categorized into subsets that can be identified based on the expression of certain surface markers, with CD14hi:CD16lo monocytes in humans considered classical (inflammatory) monocytes that contribute to the monocyte-derived macrophage tissue population and CD14lo:CD16hi nonclassical (patrolling) monocytes that survey endothelial integrity (73). The two functionally distinct monocyte subsets have also been identified in the mouse based on the expression of Ly6C, CX3CR1, and CCR2, with Ly6Chi, CCR2+, and CX3CR1lo monocytes identified as the classical subset and Ly6Clo, CCR2−, and CX3CR1hi monocytes as nonclassical monocytes (70). Similar monocyte subsets have now been described in various species, including rats, pigs, cows, horses, and nonhuman primates (236).
The increased production of reactive oxygen species (ROS) in monocytes and macrophages is associated with changes in cell function, activation state, and survival (63). These events are usually associated with a shift in the balance between ROS production and cellular antioxidant systems such as the glutathione-dependent antioxidant system (85, 175). Under conditions that lead to elevated rates of hydrogen peroxide (H2O2) and organic peroxide (ROOH) production, cysteine residues become highly susceptible to thiol oxidation, S-nitrosylation, S-cysteinylation, and S-glutathionylation (77, 82). These thiol modifications, in turn, lead to changes in protein activities and/or localization and thus can act as signaling mechanisms to regulate cell function. Recently, our group has characterized a number of proteins that are S-glutathionylated and contribute to (dys)function of monocytes by enhancing infiltration of monocyte-derived macrophages (100, 101, 163, 207, 208). In this review, we will discuss the current state of research of protein-S-glutathionylation and thiol redox signaling in monocytes and macrophages. We will also discuss how thiol redox-dependent changes in cell functions relate to the progression of inflammatory diseases linked to monocyte and macrophage dysfunction, with a specific emphasis on atherosclerosis.
Protein thiol modifications in monocytes or macrophages
The reactivity of a protein thiol is largely determined by its pKa, whereby lower pKa and more acidic thiols are more easily oxidized (221). Under normal physiological conditions, most protein thiols are in the reduced state due to the highly reductive state of the cytosol (76). However, under conditions of oxidative stress, increased production of H2O2 and ROOH leads to thiol oxidation, producing proteins with sulfenic, sulfinic, and sulfonic acid residues (82, 144) (Fig. 1). In turn, oxidized thiol groups, particularly sulfenic acids, can react with glutathione (GSH) to form mixed disulfides or S-glutathionylated proteins (82) (Fig. 1, #1). Alternatively, deprotonation of protein thiols allows the molecule to exchange GSH with glutathione disulfide (GSSG) to produce an S-glutathionylated protein (82) (Fig. 1, #4). However, only a small proportion of cysteine thiols are deprotonated under physiological pH (82). Furthermore, the intracellular GSH:GSSG surpasses 100 in the absence of overt oxidative stress (72), suggesting that the transfer of the GSH moiety from GSSG to a protein thiol is highly unlikely to be a major intracellular mechanism of S-glutathionylation. A thiyl radical-mediated mechanism for protein-S-glutathionylation has also been proposed, which involves a protein thiyl radical (protein-S•) reacting with GSH and being stabilized by radical transfer to molecular oxygen to form superoxide (O2 −•) (186, 218) (Fig. 1, #2). Alternatively, GSH itself can form a sulfenic acid (GS-OH), which can react with protein sulfhydryl residues to generate S-glutathionylated proteins (82, 144) or GSH to form GSSG (Fig. 1, #3). It should be noted that under nitrosative stress, certain protein thiols and GSH can also form S-nitrosothiols, for example, S-nitrosoglutathione (GSNO), which can react with protein thiols to form mixed disulfides, resulting in S-glutathionylation (64). However, a discussion of S-nitrosylated proteins is beyond the scope of this review.
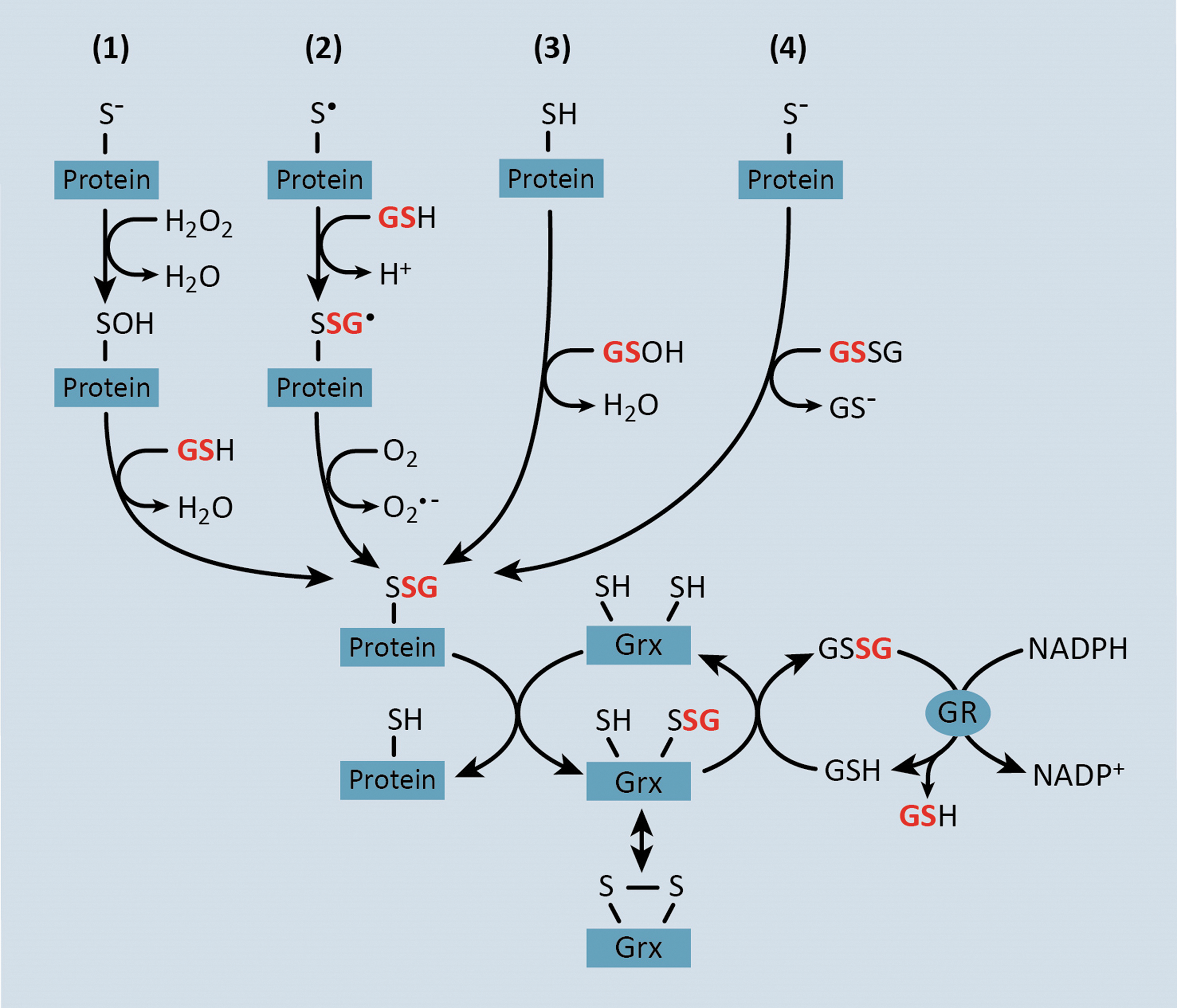
The concept that S-glutathionylated proteins could serve as thiol redox signaling proteins was first postulated by Mieyal et al. in 1995 (145). The criteria for S-glutathionylation as a regulatory mechanism are (i) S-glutathionylation occurs at a specific cysteine residue, (ii) S-glutathionylation changes the function of the modified protein, (iii) S-glutathionylation occurs on endogenous proteins in response to a physiological stimulus, (iv) the extent of thiol modification corresponds with the level of the physiological stimulus, and (v) S-glutathionylation is reversible (143). In fact, protein-S-glutathionylation is emerging as a major reversible post-translational cysteine modification that under conditions of prolonged oxidative stress, can lead to the dysregulation of proteins involved in transcription, DNA synthesis, protein turnover, apoptosis, signal transduction, and mitochondrial function (5, 45, 66, 132, 182). The reversal of protein-S-glutathionylation is catalyzed by glutaredoxin (Grx) enzymes whereby Grx catalyzes the transfer of the GSH adduct from substrate proteins and onto its own cysteine residue (Fig. 1). Grx can then return to the reduced state upon donation of the GSH group to another GSH molecule, thereby producing GSSG (Fig. 1). GSSG, in turn, is reduced back to two molecules of GSH by glutathione reductase (GR), which uses electrons from NADPH (Fig. 1).
Several proteomic approaches have been developed to characterize protein-S-glutathionylation in various cell types, including bacteria, protozoa, plants, and mammalian cells and tissues (29, 52, 65, 98, 127, 153). More recently, our group and others have used proteomic approaches to characterize protein-S-glutathionylation in monocytes and macrophages (148, 187, 209). These proteomic approaches rely on trapping protein thiols in their native redox state by the addition of a thiol-alkylating agent such as N-ethyl maleimide (NEM) (34). After trapping free protein thiols, oxidized thiol residues are labeled directly with chemoselective probes to identify proteins with modified thiol residues. For example, dimedone (5,5-dimethyl-1,3-cyclohexadione) has been used to label and map protein sulfenic acid residues in cells (34), and a biotinylated dimedone analog has been used to affinity purify and identify sulfenated proteins in a proteomic screen using rat cardiac myocytes and heart tissue (30). In addition, GSH can be enzymatically removed from proteins in vitro. The resulting free thiols can then be labeled with specific thiol-reactive probes and analyzed by mass spectrometry to identify proteins with S-glutathionylated thiol residues (3, 88). In monocytes and macrophages, there have been no studies using biotinylated dimedone to identify sulfenated proteins; however, Su et al. recently identified potentially S-glutathionylated proteins in the RAW264.7 macrophage-like cell line by selectively deglutathionylating NEM-treated cell lysates and enriching proteins with a thiol-affinity resin (187). Enriched proteins were then labeled with an isobaric tag (iTRAQ) and examined by mass spectrometry (187). This study identified 265 proteins that are sensitive to S-glutathionylation in response to H2O2 treatment (187). Since the levels of H2O2 in this study were supraphysiological, it remains to be determined whether the proteins identified in this study are bona fide S-glutathionylated proteins under physiological conditions or will meet any of the other criteria set forth by Mieyal et al. for thiol redox signaling (143). In contrast, McGarry et al. have used a tandem mass tag labeling protocol along with MS/MS analysis to identify 724 S-glutathionylated proteins in mouse liver lysates under physiological conditions (139). More recently and more relevant to monocytes and macrophages, our group has used a mass spectrometry-based proteomics approach to identify S-glutathionylated proteins in both the THP-1 monocyte cell line and purified mouse peritoneal macrophages under baseline conditions as well as under conditions of metabolic stress induced by hyperglycemia and high low-density lipoprotein levels (HG+LDL) (209). In this study, we selectively deglutathionylated NEM-treated monocyte or macrophage lysates with recombinant Grx and labeled the reduced thiol residues with iodoacetamide conjugated to desthiobiotin. These labeled proteins were then affinity purified and examined by LC-MS/MS (209). We identified over 100 proteins that were S-glutathionylated under physiological conditions in monocytes or macrophages, many of which showed changes in their S-glutathionylation status in response to metabolic stress (209).
In addition to proteomic screens, there are now a growing number of reports for individual proteins being S-glutathionylated in either monocytes or macrophages under physiological conditions. For example, our group has shown that metabolic stress leads to an increase in global protein-S-glutathionylation levels (207), and we identified β-actin, mitogen-activated protein kinase (MAPK) phosphatase 1 (MKP-1), and 14-3-3ζ as S-glutathionylated proteins in THP-1 monocytes (100, 101, 207). MKP-1 activity is modulated via modification of Cys258, while Cys25 of 14-3-3ζ is crucial for its effects on monocyte chemotaxis (100, 101). In addition to these three proteins, we have recently identified several other S-glutathionylated proteins in metabolically stressed THP-1 monocytes, including 14-3-3γ, S100α (S100 calcium-binding protein A1), histone H3, β-tubulin, PKM (pyruvate kinase muscle isozyme), HSP90β (heat shock protein 90B), HSP60, glutathione peroxidase (GPX), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), cofilin, and ATP5A1 (ATP synthase, α subunit, isoform 1) (209). Rinna et al. showed that (similar to MKP-1) protein tyrosine phosphatase 1B (PTP1B) is S-glutathionylated in a rat alveolar macrophage cell line, NR8383 (168), and PTEN (phosphatase and tensin homolog deleted from chromosome 10) protein has also been identified as an S-glutathionylated protein in these cells (40). NHE1 (Na+-H+ exchanger isoform 1) is S-glutathionylated in primary human monocytes (110), while caspase-1 S-glutathionylation occurs in isolated peritoneal macrophages (141), although it should be noted that S-glutathionylated caspase-1 was only detected in macrophages from mice deficient in the superoxide dismutase 1 (SOD1) enzyme (see section on Prevention or Reversal of Protein Thiol Modifications) (141).
Regulation of Protein Function by S-Glutathionylation
Post-translational modifications (PTMs) of specific protein residues are a widely conserved mechanism for altering protein activity, stability, and/or localization. In signal transduction, phosphorylation of serine, threonine, or tyrosine residues is the primary PTM utilized for orchestrating multidirectional cascades that operate in a highly specific and temporal manner (47). This is made possible by the use of enzymes, namely kinases and phosphatases, which provide the specificity and reversibility of this PTM. Reversible changes in the redox state of specific cysteine thiols by protein-S-glutathionylation also serve as a PTM that affects protein activity or function, and recent studies have aimed at predicting protein-S-glutathionylation motifs in silico (32, 191). Similar to kinase-mediated phosphorylation, transthiolation and thiol oxidation in response to intracellular ROS drive S-glutathionylation of protein thiols at specific residues (Figs. 1 and 2). Alternatively, S-glutathionylation of thiol residues can be reversed by the activity of Grx enzymes, which resembles removal of phosphate by protein phosphatases (Fig. 2).

A number of proteins that are involved in signal transduction (I-κB, PKCα, MKP-1, and PTP1B), cell adhesion (actin), and Ca2+ signaling (SERCA) have been shown to be regulated by both phosphorylation and S-glutathionylation (Fig. 2) (1, 20, 26, 31, 41, 67, 83, 101, 171, 179, 199, 213, 217). In certain cases, phosphorylation or S-glutathionylation of independent residues has similar effects on protein function. For example, phosphorylation or S-glutathionylation of SERCA increases SERCA activity (1, 83). In addition, β-actin S-glutathionylation at Cys374 or β-actin phosphorylation at Tyr53 decreases actin polymerization (171, 199, 213). However, in many cases, phosphorylation or S-glutathionylation of proteins has opposing effects on signal transduction. For instance, S-glutathionylation of inhibitor of nuclear factor-κB (NF-κB), IκB, stabilizes IκB and diminishes NF-κB activity (179), whereas phosphorylation of IκB leads to its proteasomal destruction and enhanced NF-κB transcriptional activation (31). It is therefore conceivable that under certain physiological conditions, these two types of PTMs could mutually regulate the activity or localization of certain proteins given that these modifications are both reversible and exclusive to distinct amino acid residues.
Mechanisms Promoting Protein-S-Glutathionylation
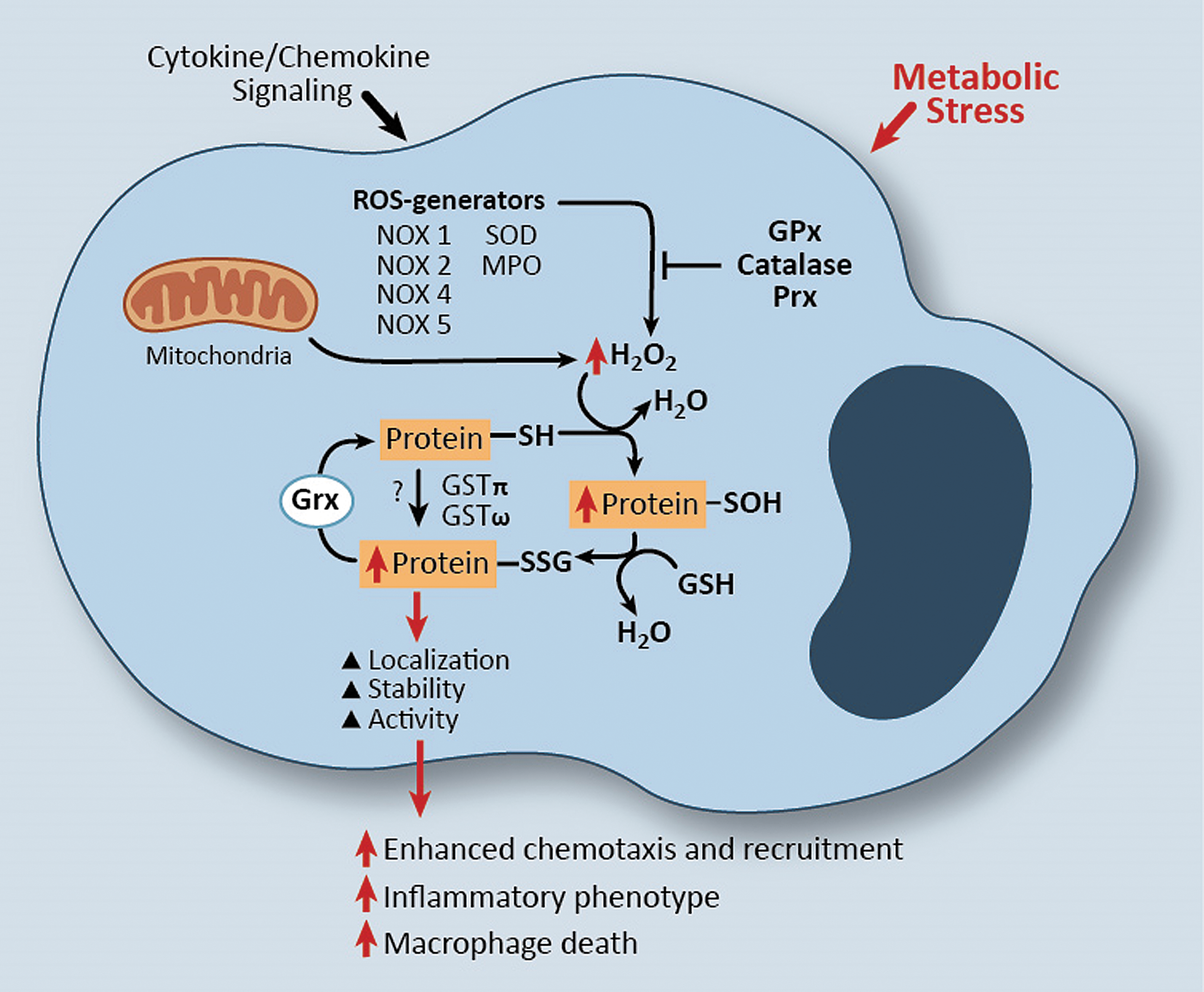
In addition to protein-S-glutathionylation resulting from H2O2-mediated oxidation of reactive thiol residue(s) (Fig. 1), recent studies suggest that certain glutathione S-transferase (GST) enzymes, including GSTπ, can also catalyze protein-S-glutathionylation (see the GSTs linked to protein-S-glutathionylation section). Thus, GST-catalyzed protein-S-glutathionylation requires GST, GSH, and a protein containing a reactive cysteine thiol residue. Like most other cell types, monocytes and macrophages possess the enzymatic repertoire for protein-S-glutathionylation to occur by either of the aforementioned mechanisms, that is, mediated by mitochondrial-generated H2O2, NADPH oxidase (NOX) enzymes or other ROS-generating enzymes, as well as catalyzed by either GSTπ or GSTω (159, 164) (Fig. 3).
Mitochondrial ROS associated with protein-S-glutathionylation
The majority of mitochondrial ROS (mROS) are derived from superoxide (O2 −•), which is generated through a one-electron reduction of oxygen (O2) (150). These superoxide anions are efficiently dismutated into H2O2 by mitochondrial SOD, which is expressed at high levels in the mitochondria (150). Thus, H2O2 produced in the mitochondria would have the potential to interact with cysteine thiols of mitochondrial proteins. In addition, H2O2 is able to pass through aquaporin transmembrane proteins (59), which could allow mitochondrial H2O2 to access protein thiols in other subcellular compartments. Aquaporin-2 (AQP2), AQP6, AQP8, and AQP9 are localized to intracellular vesicles (7, 106), with AQP8 and AQP9 shown by electron microscopy to localize to the inner mitochondrial membrane (IMM) (7, 27). AQP9 is expressed in both human monocytes and human monocyte-derived macrophages (44); however, studies to identify an aquaporin protein responsible for H2O2 escape from the mitochondria through the outer mitochondrial membrane (OMM) are lacking thus far. Alternatively, ROS and RNS are thought to exit the mitochondria through the mitochondrial permeability transition pore (mPTP), where there is cross talk between mROS and other pro-oxidant enzymes outside the mitochondria (see “NOXs associated with protein-S-glutathionylation”). The mPTP comprises the voltage-dependent anion channel at the OMM and the adenine nucleotide translocase (ANT) and cyclophilin D (CypD) proteins at the IMM (42). Interestingly, Sloan et al. showed that heart tissue from diabetic rats have more thiol oxidation when compared with nondiabetic animals (183), and thiol cross-linking agents such as diamide increase thiol oxidation of ANT at Cys56 and cause mPTP opening (39).
In addition to elevated production of ROS, including H2O2, mitochondria also contain GSH at a similar concentration to that in the cytosol (167). GSH, which is synthesized in the cytosol, diffuses across the OMM where it is carried across the IMM by different proteins dependent upon the cell type. 2-Oxoglutarate carrier and dicarboxylate carrier were identified as GSH carrier proteins in the liver and kidney, whereas tricarboxylate carrier was identified in brain mitochondria and astrocytes (132, 167). It is still unclear what contribution these two mechanisms play in GSH transport into monocyte or macrophage mitochondria.
Due to the high H2O2 levels and GSH presence, mitochondria have a microenvironment that favors protein-S-glutathionylation reactions (131). In fact, several mitochondrial proteins are known to be targets for S-glutathionylation, including mitochondrial complex 1 subunits, uncoupling protein 2 (UCP2), and UCP3 (131). In addition, the α subunit of ATP synthase is S-glutathionylated in heart tissue from dogs that experience heart failure, and the enzyme is S-glutathionylated in vitro at both Cys244 and Cys294, although under conditions of artificially high oxidative stress (214). Our group has recently shown that the α subunit of ATP synthase undergoes S-glutathionylation in THP-1 monocytes cultured under physiological conditions (5 mM glucose) as well as THP-1 cells cultured under metabolic stress (25 mM glucose +100 μg/ml LDL) (209).
NOXs associated with protein-S-glutathionylation
NOX enzymes are transmembrane protein complexes that transport electrons from NADPH across membranes to reduce oxygen to generate superoxide (O2 −) and H2O2. The superoxide (O2 −) generated by NOX enzymes can also be further converted to H2O2 either by spontaneous dismutation or catalyzed by superoxide dismutase (SOD). H2O2 can be converted to hydroxyl radicals (.OH) and hydroxyl anions (OH−) by iron-catalyzed reactions or to hypochlorous acid (HOCl) by myeloperoxidase (MPO) (63). To date, seven Nox family members (Nox 1–5 and Duox 1–2) have been characterized and found to differ in their tissue distribution (21), with four of these family members (NOX1, NOX2, NOX3, and NOX4) reported to be expressed in monocytes or macrophages (21, 62, 115, 174). NOX enzyme function is regulated both by subcellular localization and interaction with regulatory proteins that help localize ROS production and activation of specific redox signaling pathways that mediate various cellular functions (211).
NOX2, also known as gp91 phagocytic oxidase (gp91phox), is the most well-characterized catalytic subunit of NOX enzymes in macrophages and is localized to both intracellular and plasma membranes where it directly produces superoxide into the extracellular space or phagolysosome for pathogen killing. The oxidative burst mounted by activation of NOX2 is the major ROS-generating process in macrophages and was originally described by Rossi and Zatti in the early 1960s (170, 232). Activation of NOX2 occurs through a complex series of protein–protein interactions with regulatory proteins that include p22phox, p47phox, p67phox, and the GTPase Rac, which are required for stabilization and translocation (21, 190). NOX1 was the first homolog of NOX2 to be characterized, and the NOX1 and NOX2 proteins display 56% identity (18, 189). The NOX1 protein is localized to endosomes and, similar to NOX2, produces ROS in the form of superoxide (211). Additionally, NOX1 and NOX2 share some regulatory proteins for their functionality (21). NOX1 or NOX3 expression can be induced in RAW264.7 macrophage-like cells in response to TLR agonists, that is, LPS and nonmethylated CpG DNA, as well as RANKL (receptor activator of NF-κB ligand) stimulation (103, 104, 118, 174). In addition, induction of NOX1 expression occurs in response to ethanol exposure in alveolar macrophages (229).
NOX4 expression has been characterized in multiple cell types, including kidney, vascular endothelial cells, and cardiac tissue and cardiomyocytes, with localization to the endoplasmic reticulum (6), mitochondria (23), and nucleus (113). Several reports suggest that NOX4 is also expressed in both human and murine myeloid cells (78, 115, 116). However, there are conflicting reports about which myeloid cells express NOX4 and under what conditions (178). For example, our laboratory previously characterized the inducible nature of NOX4 in human monocyte-derived macrophages stimulated with oxidized LDL (115) and in THP-1 cells exposed to metabolic stress and monocyte chemoattractant protein-1 (MCP-1) (207). NOX4 is also detectable by RT-PCR in mouse bone marrow-derived macrophages and in murine thioglycollate-elicited peritoneal macrophages (data unpublished). Unlike NOX1 and NOX2, NOX4 activation can occur upon interaction with the p22phox regulatory protein only whereby the NOX4-p22phox complex generates primarily H2O2 (21).
Although there have been no definitive studies to address which NOX protein is responsible for changes in global protein-S-glutathionylation, several independent studies have demonstrated that one or more of the NOX proteins are essential for the S-glutathionylation of certain target proteins. For example, a peptide inhibitor of NOX2, Nox2ds-tat, blocks S-glutathionylation of endothelial nitric oxide synthase (eNOS) in human lung microvascular endothelial cells (222), and eNOS S-glutathionylation is significantly diminished in aorta and heart tissue of angiotensin-II-treated NOX2−/− and p47phox−/− mice (112). In isolated triad vesicles (T-tubules attached to sarcoplasmic reticulum vesicles) from skeletal muscle, the NOX inhibitor, apocynin, blocks S-glutathionylation of ryanodine receptor type 1 (RyR1) (87). However, it is uncertain which NOX protein contributes to RyR1 S-glutathionylation as the authors noted that both NOX2 and NOX4 are present in cardiac muscle tissue. In THP-1 monocytes, our group has shown that siRNA directed against NOX4 mitigated the enhanced β-actin-S-glutathionylation induced by metabolic stress (207).
Importantly, studies have shown that cross talk occurs between NOX enzymes and mROS and that this cross talk affects protein-S-glutathionylation. Khan et al. showed that intermittent hypoxia of PC-12 cells leads to S-glutathionylation of both the 75 kD mitochondrial Complex 1 protein (NDUFS1) and the 50 kD mitochondrial Complex 1 protein (NDUFV1), which diminishes Complex 1 function—this increased S-glutathionylation of both proteins is blocked by apocynin or siRNA-mediated depletion of NOX2 (99). Alternatively, mROS, generated by phorbol esters or myxothiazole, activates NOX2 in isolated human neutrophils by enhancing membrane localization of phox regulatory subunits (112). This translocation and NOX activation are blocked by sanglifehrin A (SfA), an inhibitor of the mPTP, and mROS-induced activation of NOX activity is impaired in whole blood from CypD−/− mice (112). To our knowledge, there have been no studies to assess cross talk between mROS and NOX enzymes in monocytes or macrophages.
GSTs linked to protein-S-glutathionylation
GSTs are a class of isoenzymes that catalyze the conjugation of reduced GSH to reactive sulfur groups on various molecules such as xenobiotics (84). GSTs are catalytically active as homo- or heterodimers and are divided into seven classes based on sequence similarity (84, 200). There are increasing amounts of data, suggesting that two GST enzymes, GSTπ and GSTω1, play a role in protein-S-glutathionylation. Both enzymes are expressed in mouse monocytes and macrophages, as well as human monocytes (14).
Although GST has been reported to bind GSH and produces a thiolate anion (GS−) at physiological pH (81), the mechanism for GST-mediated protein-S-glutathionylation is still unclear. GSTπ itself can be S-glutathionylated at Cys47 and Cys101, and mutation of these residues reduces GSTπ activity in vitro (203). Alternatively, GSTπ interacts with both peroxiredoxin 1 (Prx1) and 1-cys peroxiredoxin (1-cysPrx or Prx VI) (105, 135), and GSTπ can S-glutathionylate 1-cys peroxiredoxin (1-cysPrx or Prx VI) in vitro (135). However, the role of this GSTπ S-glutathionylation or the interaction of GSTπ with Prx proteins in facilitating S-glutathionylation of target proteins remains unknown. Nonetheless, mouse embryonic fibroblasts (MEFs) from GSTπ−/− mice that are treated with PABA/NO, a prodrug that releases NO, or a mimetic of oxidized GSH, NOV-002, exhibit lower global protein-S-glutathionylation, including lower levels of β-actin S-glutathionylation, when compared with MEFs from wild-type (WT) mice (203). However, it should be noted that in the absence of nitrosative or oxidative stress, there was very little detectable protein-S-glutathionylation in either WT or GSTπ−/− MEFs (203), suggesting that H2O2-induced protein sulfenic acid residues are still necessary for GST-mediated S-glutathionylation. This notion is supported by data from McGarry et al. showing that very few proteins from liver tissue are differentially S-glutathionylated when comparing GSTπ−/− mice with WT mice (139).
Using a yeast two-hybrid assay, GSTπ was reported to interact with AMP-activated protein kinase (AMPK), an interaction that occurs in vitro and in liver tissue lysates (108). In vitro, GSTπ facilitates the S-glutathionylation of AMPK in the presence of low concentrations of GSH, and this S-glutathionylation enhances AMPK kinase activity independent of AMPK phosphorylation (108). In addition, inhibition of GSTπ in C10 murine alveolar epithelial cells with TLK-199 or siRNA blocks S-glutathionylation of the Fas ligand (FasL) in response to FasL stimulation (10), suggesting that GSTπ plays a role in the FasL autoregulatory loop involving protein-S-glutathionylation. In contrast to GSTπ, GSTω1 catalyzes the removal of GSH from a peptide substrate in vitro, and overexpression of GSTω1 leads to a reduction in protein-S-glutathionylation in T47-D breast cancer cells under normal culture conditions (142). However, overexpression of GSTω1 leads to accelerated protein-S-glutathionylation in response to treatment with GSNO, a result that the authors attributed to increased GSSG levels in the presence GSTω1 (142). In the Raw 264.7 macrophage cell line, aflatoxin B1 treatment increases GSTω1 expression as well as S-glutathionylation of several proteins, a result that is mitigated by siRNA-mediated depletion of GSTω1 in these cells (159). The role that GST proteins play in facilitating protein-S-glutathionylation and how GST-mediated effects on S-glutathionylation are related to ROS-mediated thiol oxidative stress remain open questions.
Additional enzymes linked to protein-S-glutathionylation
In addition to mROS and NOX proteins, MPO is a pro-oxidant enzyme that has been linked to protein-S-glutathionylation. MPO is an enzyme stored in azurophilic granules of phagocytic cells, which is released into the extracellular fluid in the setting of inflammation. The major product of MPO is HOCl, which forms upon its interaction with H2O2 and chloride (Cl−). Although MPO itself has not been directly linked to any S-glutathionylated proteins, Stacey et al. used a redox proteomic assay to show that treatment of HUVECs with HOCl or chloramines results in a number of proteins with oxidized thiol residues (185). Additionally, cyclophilin A is S-glutathionylated at Cys161 in response to chloramines (185). Last, the flavoprotein sulfhydryl oxidase enzyme (QSOX) has been listed as an enzyme with the potential to S-glutathionylate target proteins (144, 161); however, there have been no studies to address this possibility in monocytes and macrophages or other cell types to this point.
Prevention or Reversal of Protein Thiol Modifications
For thiol redox-sensitive signaling pathways to function properly, changes in protein thiols must be controlled and reversible. To maintain thiol redox homeostasis, monocytes and macrophages contain a large array of antioxidant enzymes to prevent or reverse protein-S-glutathionylation, as well as small-molecule ROS scavengers such as ascorbic acid, vitamin E, and GSH. These antioxidant enzymes can be divided into either enzymatic ROS scavengers or deglutathionylating enzymes, that is, enzymes that remove GSH from S-glutathionylated proteins. Intracellular ROS scavengers include SOD, catalase, and substrate-specific peroxidases such as GPX enzymes, while Grx enzymes catalyze deglutathionylation reactions.
Enzymatic ROS scavengers
Superoxide dismutase
Three SODs have been characterized in mammalian cells: cytosolic CuZn-SOD (SOD1), mitochondrial Mn-SOD (SOD2), and extracellular EC-SOD (SOD3). Each of these enzymes catalyzes the dismutation of superoxide O2 •− into H2O2 and O2. Although SOD enzymes can generate H2O2 as a by-product, at least one study has shown that increased SOD levels prevent forskolin-induced S-glutathionylation of the Na+-K+ pump β1 protein in isolated rabbit ventricular myocytes (220). Similarly, SOD1 or SOD2 overexpression blocks NO-induced S-glutathionylation of the SERCA protein in smooth muscle cells (1). Interestingly, a recent study reported that SOD2 itself undergoes S-glutathionylation in NRK-52E (normal rat kidney proximal tubular cells) when treated with GSNO, thereby decreasing SOD2 activity (158).
In the monocytic cell lines, U937 and THP-1, changes in expression of SOD proteins were detected during 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced monocyte-to-macrophage differentiation, whereby TPA treatment reduces SOD1 and SOD2 expression while increasing SOD3 expression in a PKC-dependent manner (97). SOD1 activity is also reduced in rat peritoneal macrophages in response to LPS in a time- and dose-dependent manner and this reduction is blocked by inhibitors of NO synthase (94). In contrast, others have reported that SOD2, but not SOD1, mRNA expression and activity increased in mouse peritoneal macrophages in response to LPS exposure (205). The role of SOD enzymes in protein-S-glutathionylation in monocytes and macrophages or the regulation of SOD by S-glutathionylation in monocytes and macrophages has not been examined in any detail thus far.
Catalase
The catalase enzyme, identified over 100 years ago, is responsible for the metabolism of H2O2 into H2O and O2 in peroxisomes (107, 129). In vitro, SOD and catalase enzymes block RyR2-S-glutathionylation in isolated cardiac SR vesicles (172), and reducing catalase activity in lung extracts enhances oxidation of cysteine residues in the proteasomal regulatory subunits, Rpn1 and Rpn2 (238). Similarly, increased oxidation of AMPKα cysteine residues occurs in lung tissue from mice treated with a pharmacologic inhibitor of catalase, aminotriazole (ATZ), or in lungs from acatalasemic mice (239). In contrast, overexpression of catalase prevents (i) UV light-induced mitochondrial pyruvate dehydrogenase-S-glutathionylation in HeLa cells (91), (ii) p65 NF-κB-S-glutathionylation in an endothelial cell line exposed to two different carbon monoxide (CO) donors, tricarbonyl dichlororuthenium (II) dimer and methylene chloride, and (iii) STAT3-S-glutathionylation in bovine aorta endothelial cells exposed CO donors (226).
Catalase expression and activity in monocytes and macrophages have been explored using human blood monocytes and alveolar macrophages, as well as the THP-1 cell line. Alveolar macrophages, which generate higher ROS levels than blood monocytes, have higher catalase levels than blood monocytes and an inability to activate p38 MAP kinase, which can be overcome by ATZ (28). However, this increased catalase expression may be cell-type dependent since blood monocyte-derived macrophages stimulated with GM-CSF have high catalase levels, similar to alveolar macrophages, whereas monocyte-derived macrophages stimulated with M-CSF do not (109). Similar to alveolar macrophages and GM-CSF-stimulated macrophages, TPA-induced monocyte-to-macrophage differentiation of U937 cells results in decreased catalase expression (225). However, catalase itself enhances monocyte-to-macrophage differentiation of THP-1 cells induced by all-trans retinoic acid (50).
GSH peroxidases
GPxs are a family of enzymes that utilize GSH as an electron donor for the metabolism of H2O2 and lipid peroxides, converting them into water and corresponding alcohols, respectively (25). There are eight GPx enzymes in mammalian cells. These enzymes possess a reactive selenocysteine residue (GPx1–4) or a reactive cysteine residue (GPx6–8) in their catalytic domain that facilitates reactivity—human GPx6 possesses a selenocysteine residue, whereas rodent GPx6 possesses a cysteine residue. Thus, GPx enzymes themselves are S-glutathionylated during the course of their reaction with H2O2 (25). GPx activity is significantly higher in alveolar macrophages when compared with blood monocytes (28); however, the only data linking mammalian GPxs to changes in protein thiol modifications are that increased eNOS-S-glutathionylation within heart tissue occurs in GPx1−/− mice in an age-dependent manner (155).
Protein deglutathionylation
Reversal of protein-S-glutathionylation (i.e., deglutathionylation) is almost exclusively determined by the activity of Grx enzymes, which are members of the thioredoxin superfamily of thiol-disulfide oxidoreductases (35). There are six Grx isoforms expressed in human cells, which are generated from four independent genes. Grx1 and Grx2 both possess a dithiol catalytic domain, although Grx2 isoforms (comprising Grx2a, Grx2b, and Grx2c splice variants) have a slightly modified dithiol domain. Grx3 and Grx5 possess monothiol catalytic domains (124). Grx1 and Grx3 are primarily localized to the cytosol, although Grx1 also localizes to the IMM and the nucleus under certain conditions (124). Grx2a and Grx5 both have mitochondrial signaling peptides, causing mitochondrial localization, whereas Grx2b and Grx2c lack a mitochondrial signal and are localized to the cytosol and the nucleus (124). Expression of each Grx isoform is detected at the mRNA level by oligonucleotide microarray chips in Mac-1+ cells from mouse bone marrow (111) as well as in mouse monocytes and macrophages (14). In addition, all Grx mRNA isoforms are present in human blood monocytes and monocyte-derived macrophages (128, 138, 184). Takashima et al. reported that PMA-induced monocyte-to-macrophage differentiation promotes an increase in Grx1 expression (197); however, the relevance of this increase to physiological conditions is still unclear as human blood monocytes and monocyte-derived macrophages expressed similar Grx1 levels as measured on Affymetrix chips (128, 138, 184). While both Grx1 and Grx2 have been shown to play a role in protein-S-glutathionylation, Grx5 protein plays a role in iron–sulfur cluster transfer in cells (123) and its role, if any, in protein thiol signaling is still unclear. Similarly, the effects of Grx3 on protein-S-glutathionylation remain unknown.
Grx1 is the most well-characterized Grx enzyme, and several proteins undergo Grx1-mediated changes in S-glutathionylation. In vitro, purified Grx1 removes S-glutathione from hepatocyte proteins and reverses the S-glutathionylation-mediated inactivation of PTP1B (20, 96). Alternatively, Grx1 facilitates the in vitro S-glutathionylation of GAPDH, β-actin, and PTP1B in the presence of GSH thiyl radicals (GS•) (186). Under physiological conditions, the Grx1 enzyme shows a strong preference for catalyzing deglutathionylation in the presence of NADPH and GR (124, 144). For example, Grx1−/− mice display increased global protein-S-glutathionylation levels in lung tissue in response to cigarette smoke, to antigen-stimulated allergic inflammation with ovalbumin, or to oropharyngeal exposure to LPS (2, 36, 90). Additionally, MEFs isolated from Grx1−/− mice have elevated protein-S-glutathionylation in response to H2O2 (89), and siRNA-mediated depletion of Grx1 blocks fibroblast growth factor-induced deglutathionylation of β-actin in NIH3T3 cells (213). In lung tissue and mouse tracheal epithelial cells, loss of Grx1, that is, Grx1−/− cells, increases S-glutathionylation and augmentation of FasL in response to Pseudomonas aeruginosa infection, and Grx1 overexpression in lung tissue had the opposite effect on FasL-S-glutathionylation and activity (9). Grx1 similarly regulated FasL-S-glutathionylation and activity in C10 cells in response to FasL itself (8). Interestingly, alveolar macrophages isolated from Grx1−/− mice have similar levels of global protein-S-glutathionylation when compared with isolated alveolar macrophages from WT mice, even when mice are exposed to oropharyngeal LPS (2).
Similar to Grx1, Grx2a selectively deglutathionylates synthetically S-glutathionylated RNase or BSA proteins and deglutathionylates mitochondrial complex 1 proteins in vitro in the presence of high GSH/GSSG ratios (22, 68, 95). However, Grx2a promotes S-glutathionylation of mitochondrial complex 1 proteins and GAPDH in vitro in the presence of low GSH/GSSG ratios (22, 68). Thus, the effect of Grx enzymes on protein-S-glutathionylation depends on the ratio of GSH/GSSG in cells. In vivo, the role of Grx2 is also complex. Grx2−/− mice have elevated levels of global protein-S-glutathionylation as well as increased levels of S-glutathionylated β-actin and two different mitochondrial complex proteins in lens tissue (223), suggesting that Grx2 promotes protein deglutathionylation in this tissue. However, overexpression of Grx2a in mouse heart tissue promotes S-glutathionylation of mitochondrial proteins, especially when animals are treated with the cardiotoxic anticancer drug, doxorubicin (51). Thus, the effect of Grx2 on protein-S-glutathionylation may be cell, tissue, and possibly context dependent, as well as dependent on the level of reduced GSH in the cellular or tissue milieu.
Cross Talk Between Thiol Modifications and Signal Transduction Pathways in Monocytes/Macrophages
MKPs, which inactivate members of the MAPK signaling pathway, such as ERK and p38, are dual specificity phosphatases (DUSPs) that (like other protein tyrosine phosphatases) contain a conserved C(X)5R motif in their catalytic domain (93). Importantly, the cysteine residue of the C(X)5R motif can be S-glutathionylated in both PTP1B and MKP-1 (20, 101). In macrophages, increased MKP-1 levels are thought to control proliferation and activation in response to M-CSF and LPS, respectively, both of which require ERK 1/2 signaling (38). Our group has shown that in THP-1 monocytes and primary human blood monocytes, oxidative and metabolic stress promotes S-glutathionylation of MKP-1, thereby inhibiting its phosphatase activity and targeting it for proteasomal degradation. Loss of MKP-1 results in hyperactivation of ERK and p38 via increased phosphorylation in response to MCP-1 and increases monocyte adhesion and chemotaxis. Overexpression of Grx1, however, protects MKP-1 from S-glutathionylation-dependent inactivation/degradation and normalizes monocyte adhesion and chemotaxis in response to MCP-1 under conditions of metabolic stress (Fig. 4) (101).
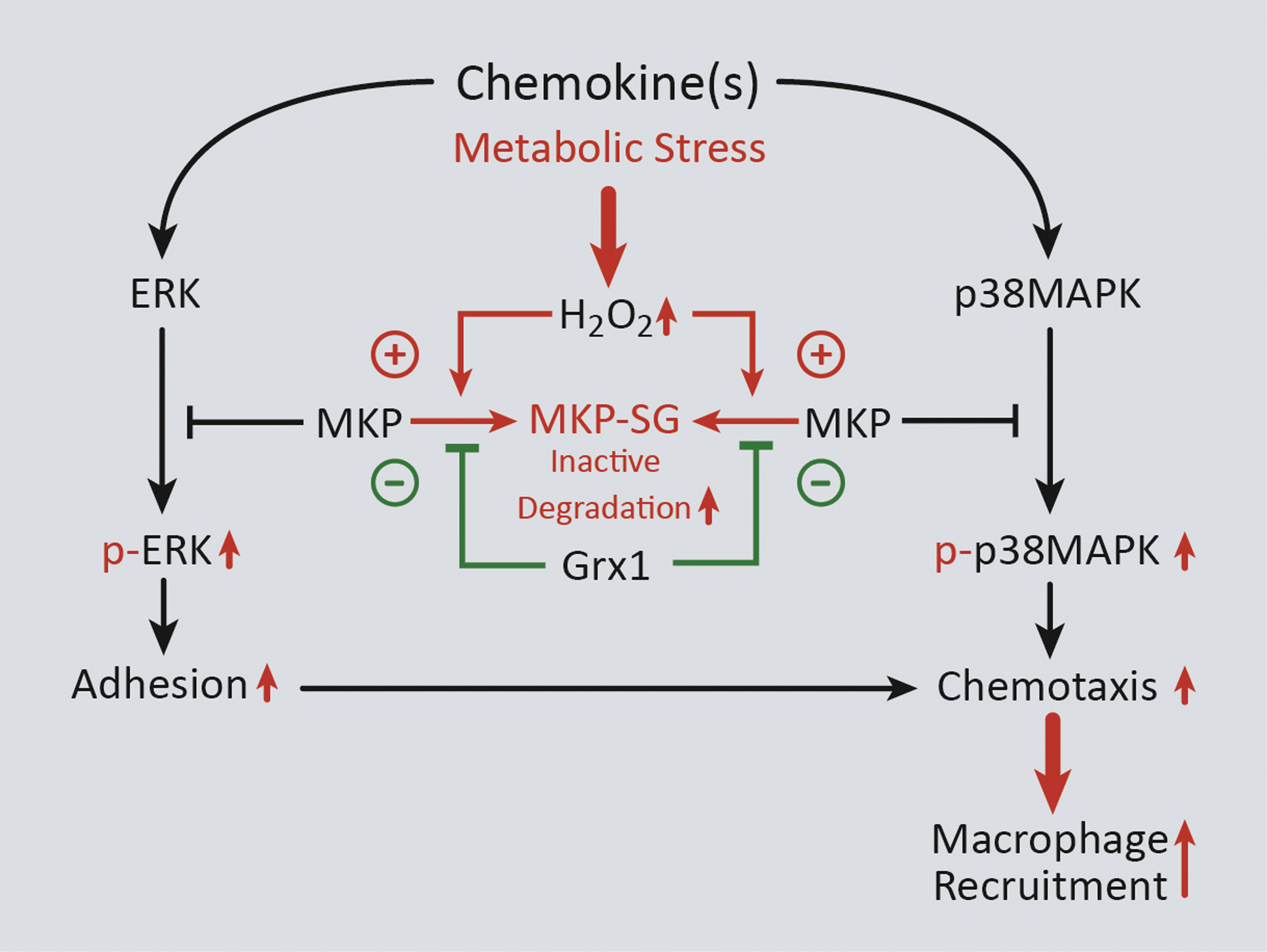
Recently, we have shown that another DUSP family member, Slingshot Homolog 1L (SSH1L), also plays a role in accelerated monocyte chemotaxis caused by metabolic stress. The mechanism by which this occurs is through increased S-glutathionylation and degradation of 14-3-3ζ, a binding partner of SSH1L that maintains SSH1L in an inactive state (102). S-glutathionylation and degradation of 14-3-3ζ result in the release of SSH1L and subsequent dephosphorylation of the Cofilin protein (100). Cofilin is an actin-binding protein that (in its dephosphorylated state) promotes actin depolymerization and actin turnover in response to various extracellular stimuli (146). Thus, dephosphorylation of Cofilin in response to metabolic stress in monocytes results in accelerated chemotaxis (100). Importantly, we showed that overexpression of Grx1 reverses the metabolic stress-induced changes in cofilin phosphorylation (100). The overlap and contribution of S-glutathionylation of these two DUSP proteins, as well as other DUSP proteins, to enhanced monocyte migration induced by metabolic stress are still under investigation.
In addition to thiol modifications regulating DUSP proteins, recent studies have demonstrated a complex feedback loop between Grx1-regulated protein-S-glutathionylation and the NF-κB signaling pathway in macrophages and other cell types. NF-κB is a transcriptional activator comprising heterodimers of the Rel family of proteins (i.e., p50, p52, cRel, RelA, and RelB), which is activated by the proteasomal destruction of IκB proteins in response to phosphorylation by the IκB kinase (IKK) complex (130). The IKK complex comprises three proteins: IKKα, IKKβ, and NF-κB essential modulator (130). Stimulation of RAW264.7 macrophage-like cells with activators of the NF-κB signaling pathway, including IL-1β, TNFα, and LPS, induces RelA binding to the Grx1 promoter and causes upregulation of Grx1 (4). In addition, oropharyngeal administration of LPS causes Grx1 upregulation in alveolar macrophages (2). In turn, Grx1 has been reported to regulate S-glutathionylation of multiple proteins in the IKK/IκB/NF-κB pathway. Lung tissue and lung homogenates from Grx1−/− mice have elevated global protein-S-glutathionylation and IKKα-S-glutathionylation levels, respectively, in response to cigarette smoke, as well as enhanced NF-κB activity, suggesting that Grx1 inhibits CS-induced IKKα-S-glutathionylation and NF-κB signaling (36). Similarly, siRNA-mediated depletion of Grx1 exacerbates IL-17-induced S-glutathionylation of both IKKα and RelA, thereby enhancing NF-κB signaling in C10 cells (154). In contrast, IKKβ-S-glutathionylation caused by H2O2 decreases IKKβ activity and blocks NF-κB signaling in response to TNFα (166). Furthermore, overexpression of Grx1 blocks these effects, whereas Grx1 siRNA exacerbates the H2O2-mediated inhibition of TNFα (166). These data are similar to data obtained in alveolar macrophages, whereby genetic ablation of Grx1 prevents LPS-induced NF-κB nuclear translocation (2).
Protein Thiol Redox Signaling and Atherosclerosis
Redox signaling and oxidative stress play key roles in monocyte and macrophage biology in atherogenesis from the formation of the earliest stage lesions, that is, fatty streaks, to development of advanced plaques (121, 122). Subendothelial deposition of lipoproteins in atheroprone regions of the arteries is one of the earliest pathogenic events in atherosclerosis (195). Activation of endothelial cells by modified lipoproteins elicits the influx of monocytes via expression of adhesion molecules into the subendothelial space, which subsequently differentiate to macrophages that can be activated and/or internalize lipoproteins to form lipid-loaded foam cells, a key histological feature of atherosclerotic lesion (195). The death of these macrophages is a major contributor to the development of necrotic core, a well-recognized histological feature of instability in advanced atherosclerotic plaques (Fig. 5).
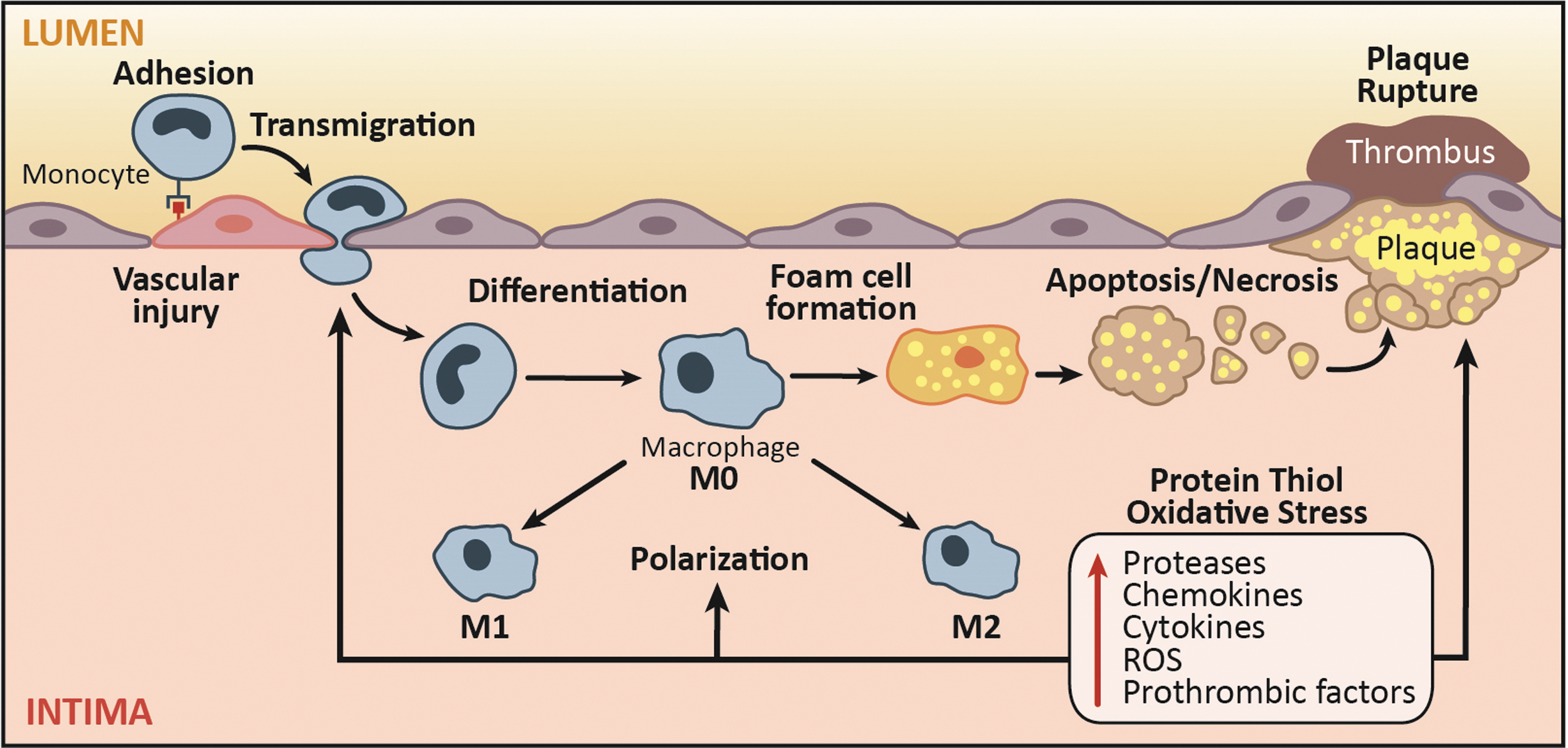
Role of ROS and protein-S-glutathionylation in monocyte recruitment and differentiation (plaque initiation)
Monocytosis has long been recognized as an independent predictor of ischemic coronary artery disease in humans (156), and studies in hyperlipidemic mouse models have shown that both classical Ly6Chi and nonclassical Ly6Clo monocyte subsets are recruited into the atherosclerotic lesions and contribute to plaque progression (196). Furthermore, high-fat feeding of apoE-deficient mice increases the number of circulating Ly6Chi monocytes and their infiltration into atherosclerotic lesions (192). However, monocyte subsets appear to have different efficiency in entering into atherosclerotic lesions in specific regions of the arteries (162), and the contribution of the major human monocyte subsets, that is, classical CD14hi/CD16lo and nonclassical CD14lo/CD16hi, to the pathogenesis of atherosclerosis and their relative efficiency in recruitment into the atherosclerotic plaques have yet to be determined.
Oxidative stress contributes to various proatherogenic aspects of monocyte recruitment and differentiation. Inhibition of NOX-mediated oxidative stress by apocynin or administration of the antioxidant catalase ameliorates the hyperhomocysteinemia-induced Ly6Chi monocytosis as well as the enhanced recruitment of the inflammatory monocytes into atherosclerotic lesions in apoE-deficient mice (233). Our in vivo studies have confirmed that thiol oxidative stress induced by metabolic stress enhances recruitment of monocyte-derived macrophages into MCP-1-loaded Matrigel plugs and is strongly correlated with atherosclerotic lesion size and macrophage content in aortic root of high-fat diet-fed LDL receptor null (LDL-R−/−) mice (163). We have shown that metabolic stress by hypercholesterolemia and hyperglycemia primes monocytes into a hypermigratory and proatherogenic phenotype, which is dependent on the induction of NOX4-mediated H2O2 production (207). This priming effect is associated with S-glutathionylation of several proteins, including actin (115, 116), MKP-1 (101), and 14-3-3ζ (100), which appear to accelerate monocyte chemotaxis via increasing actin remodeling (207). Prevention of protein-S-glutathionylation by transgenic overexpression of cytoplasmic Grx is sufficient to eliminate the metabolic stress-induced priming effect in monocytes (207). Last, mROS in plaque macrophages has also been shown to accelerate atherogenesis by promoting NF-κB-mediated induction of MCP1, which triggers enhanced influx of monocytes into the aortic root lesions in LDL-R-null mice fed a high-fat diet. Ectopic expression of catalase in the mitochondria of macrophages suppressed monocyte recruitment as well as atherosclerotic progression (216).
Role of ROS and protein-S-glutathionylation in plaque expansion and macrophage activation
Sustained recruitment of monocyte-derived macrophages, as a result of the imbalance between the proinflammatory and inflammation-resolving mechanisms, is a critical pathogenic aspect of atherogenesis (195). Numerous microenvironmental factors, including cholesterol crystals (53), oxidized LDL (OxLDL) (13, 212), and hypoxia (60, 61), as well as cytokines and growth factors [e.g., GM-CSF (188)], can promote a proinflammatory state in macrophages and potentially contribute to the sustained nonresolving inflammation in atherosclerosis. Although the exact role of M1 and M2-polarized macrophages in atherosclerosis has yet to be defined, observational evidence suggests that the relative abundance of M1-polarized macrophages in lesions, in particular, in the unstable region of the plaque, that is, shoulder (19), is associated with plaque vulnerability [recently reviewed in Chinetti-Gbaguidi et al. (33)]. More recently, an additional activation state of macrophages, termed M4 polarization, has been described, which is induced by platelet chemokine CXCL4, and demonstrates a unique transcriptome distinct from M1 and M2 polarization (56, 74, 75). M4-polarized macrophages, defined based on the coexpression of MMP-7 and S100A8, which seems to be exclusive to this activation state, have been identified in human coronary atherosclerotic plaques (56). Additionally, the abundance of M4-polarized macrophages, but not the CXCL4 plasma level, has been associated with plaque vulnerability (57). However, the exact functional role of CXCL4-induced M4 polarization in atherogenesis as well as its signaling mechanisms remains to be defined (74).
The role of ROS and thiol redox signaling in regulation of macrophage polarization state in atherosclerosis is not yet well elucidated. M2-polarized macrophages have been shown to have reduced and delayed ROS production compared with M1-polarized macrophages (160). Downregulation of NOX-2-mediated ROS generation produces a reductive microenvironment within the phagosome, which is critical for the phagocytic activity associated with the tissue repair function of M2-polarized macrophages (17). Additionally, deficiency of p47phox, a scaffolding protein for the NOX complex, renders macrophages hyper-responsive to IL-4-induced M2 polarization (230). Likewise, SOD1 overexpression promotes M2 polarization of alveolar macrophages (86). Together, these ex vivo studies support the significance of attenuated ROS production in development of M2 polarization state, which favors resolution of inflammation.
Role of ROS and protein-S-glutathionylation in macrophage death
In early lesions, macrophage death is rarely detected and occurs mostly through apoptosis, which does not elicit an inflammatory response and has been shown to suppress atherogenesis in mice models by limiting the plaque cellularity (69, 202, 235). However, macrophage death is frequent in advanced lesions, particularly within or in the vicinity of the necrotic core (202, 235), and may be induced by a number of mechanisms, including FasL-mediated cell death, free cholesterol cytotoxicity (24, 58, 227, 228), OxLDL cytotoxicity (11, 12, 215), or endoplasmic reticulum stress (48, 49, 134, 152, 181, 201, 206, 234, 235). Interestingly, FasL-mediated cell death is a central apoptotic mechanism of macrophages (227) and smooth muscle cells in atherosclerosis (71). Binding by FasL as well as caspase-dependent degradation of Grx1 mediates the S-glutathionylation of FasL in lung epithelial cells and promotes the formation of death-inducing complexes and apoptosis (8, 82). However, the role of S-glutathionylation of Fas in the regulation of macrophage apoptosis has not been determined.
Increased oxidative stress in advanced plaques appears to play a key role in macrophage apoptosis both as a direct trigger and as a mediator of ER stress-induced cytotoxicity (193). A multihit model proposed by Tabas et al. suggests that mild ER stress, which is not sufficient to induce apoptosis by itself, reduces the threshold to other proapoptotic stressors and promotes macrophage apoptosis in atherosclerotic lesions (193). ER stress can induce intracellular ROS production through multiple mechanisms. Increased generation of disulfide bonds as a result of ER stress may deplete the reduced GSH to cleave the disulfide bonds (133). ER stress may also enhance mROS formation through induction of oxidative phosphorylation to meet the energy demands associated with ER stress-induced protein modifications (133). Additionally, ER stress may result in calcium leakage from ER, which inhibits complex III and IV and results in enhanced oxygen consumption rate and ROS generation by the electron transport chain (133). ER stress may also induce calcium and calmodulin-dependent protein kinase II-mediated induction of NOX-2, enhancing ROS generation (119). Alternatively, nitrosative stress induces S-glutathionylation of protein disulfide isomerase, thereby activating the unfolded protein response and creating ER stress in cancer cells (204). This ER stress, if left uncorrected, could lead to macrophage death through various mechanisms (173).
Oxidative stress is implicated in OxLDL cytotoxicity, which unlike the native LDL causes a robust increase in intracellular ROS generation as well as protein-S-glutathionylation (37, 149, 165, 215). Lysophosphatidylcholine is a component of oxLDL, which causes macrophage death through NOX-mediated ROS production (157). OxLDL results in depletion of intracellular GSH and caspase-independent macrophage cell death (15), and we reported that siRNA-mediated depletion of Grx1 sensitized macrophages to oxLDL-mediated cytotoxicity (215). We have also reported that OxLDL induces NOX4-mediated ROS generation in human macrophages via the MEK1/ERK pathway and this is required for OxLDL cytotoxicity (115).
Role of ROS and protein-S-glutathionylation in efferocytosis and necroptosis
In contrast to the early lesions, macrophage apoptosis in advanced lesions accentuates necrotic core expansion, recruitment of monocytes, and production of proinflammatory cytokines, all of which are features associated with plaque instability (69, 180, 202, 235). The unfavorable effect of apoptosis in advanced lesions appears to be secondary to the ineffective clearance of the apoptotic cells as a result of either impaired macrophage efferocytosis or the enhanced rate of apoptosis in advanced plaques, which leads to secondary (postapoptotic) necrosis of cells, thereby exaggerating the inflammatory response (69, 176, 193 –195). Efferocytosis induces a robust increase in NOX-mediated ROS production and promotes apoptosis of the RAW264.7 cell line (117). Oxidative stress induced by exogenous H2O2 also impairs efferocytosis in human and murine macrophages (140).
Programmed necrosis or necroptosis is a recently described and regulated form of cell death, which is mediated by formation of the necrosome complex, containing receptor-interacting protein 3 (RIP3), RIP1, and caspase-8. There is a delicate interplay between the two forms of regulated cell death, necroptosis and apoptosis, which is regulated by the balance between caspase-8 and RIP3 (126). RIP3-dependent cell death contributes to development of advanced atherosclerotic plaques in LDL receptor knockout mice without a significant effect on the development of early lesions (126). Induction of a proinflammatory response by necrotic cells, which are more difficult to clear by efferocytosis compared with apoptotic cells, has been suggested to underlie the proatherogenic role of necroptosis in advanced lesion (126). Proinflammatory stimuli, such as TNF-α and low-dose LPS, have been shown to induce macrophage necroptosis (16, 169), which is mediated by mROS formation (169). The role of ROS in regulation of macrophage necroptosis in atherosclerosis remains unexplored.
Conclusions
While we are still in the early stages of understanding how thiol redox signaling affects monocytes and macrophage function and how dysregulation of thiol redox signaling pathways contributes to disease onset and progression, recent studies suggest that a large number of proteins undergo changes in S-glutathionylation in response to pathophysiological conditions. Further understanding of the mechanisms that control this novel signaling paradigm in monocytes and macrophages, as well as identifying S-glutathionylated proteins that contribute to pathogenesis, is likely to provide novel molecular targets to combat diseases associated with monocyte or macrophage dysfunction, including atherosclerosis.
Funding
Reto Asmis is supported by grants from the National Institutes of Health (RO1 AT006885 and RO1 HL115858).