
Abstract
Significance:
Therapeutic hypothermia is commonly applied to limit ischemic injury in organ transplantation, during cardiac and brain surgery and after cardiopulmonary resuscitation. In these procedures, the kidneys are particularly at risk for ischemia/reperfusion injury (IRI), likely due to their high rate of metabolism. Although hypothermia mitigates ischemic kidney injury, it is not a panacea. Residual mitochondrial failure is believed to be a key event triggering loss of cellular homeostasis, and potentially cell death. Subsequent rewarming generates large amounts of reactive oxygen species that aggravate organ injury.
Recent Advances:
Hibernators are able to withstand periods of profoundly reduced metabolism and body temperature (“torpor”), interspersed by brief periods of rewarming (“arousal”) without signs of organ injury. Specific adaptations allow maintenance of mitochondrial homeostasis, limit oxidative stress, and protect against cell death. These adaptations consist of active suppression of mitochondrial function and upregulation of anti-oxidant enzymes and anti-apoptotic pathways.
Critical Issues:
Unraveling the precise molecular mechanisms that allow hibernators to cycle through torpor and arousal without precipitating organ injury may translate into novel pharmacological approaches to limit IRI in patients.
Future Directions:
Although the precise signaling routes involved in natural hibernation are not yet fully understood, torpor-like hypothermic states with increased resistance to ischemia/reperfusion can be induced pharmacologically by 5′-adenosine monophosphate (5′-AMP), adenosine, and hydrogen sulfide (H2S) in non-hibernators. In this review, we compare the molecular effects of hypothermia in non-hibernators with natural and pharmacologically induced torpor, to delineate how safe and reversible metabolic suppression may provide resistance to renal IRI. Antioxid. Redox Signal. 27, 599–617.
Introduction
I
Mitochondria are the main endogenous source of ROS, formed by a reduction of oxygen in the electron transport chain (ETC), that lead to the generation of superoxide (O2 −). In addition, mitochondria generate nitric oxide (NO), which can react with O2 − to form the highly reactive radical peroxynitrite (ONOO−) (147). Mitochondrial dysfunction may therefore lead to loss of cellular homeostasis and cause subsequent organ injury. Although IRI is a detrimental event that underlies acute kidney injury (AKI), there are very few therapeutic options to successfully preclude IRI.
Suppression of metabolism might be particularly effective in this regard, as it halts the overwhelming production of noxious metabolites such as ROS and limits mitochondrial dysfunction during IRI. Therefore, therapeutic hypothermia (i.e., lowering the body temperature) is commonly used to limit renal IRI during cardiac surgery and kidney transplantation (72, 130). Although animal studies demonstrate that lowering body temperature reduces metabolism, decreases oxidative stress, and limits inflammation after an ischemic insult (25, 118, 142, 157), the clinical benefit of targeted temperature management is firmly debated, as results on the effect of therapeutic hypothermia on preventing organ injury and reducing mortality are conflicting (72, 130). A large meta-analysis comprising 2218 patients indicates that the use of therapeutic hypothermia does not prevent AKI or dialysis, although it is associated with a lower mortality rate (130). Likely, protective effects of therapeutic hypothermia are offset by an increased production of free radicals and induction of apoptosis secondary to mitochondrial dysfunction induced by rewarming (36).
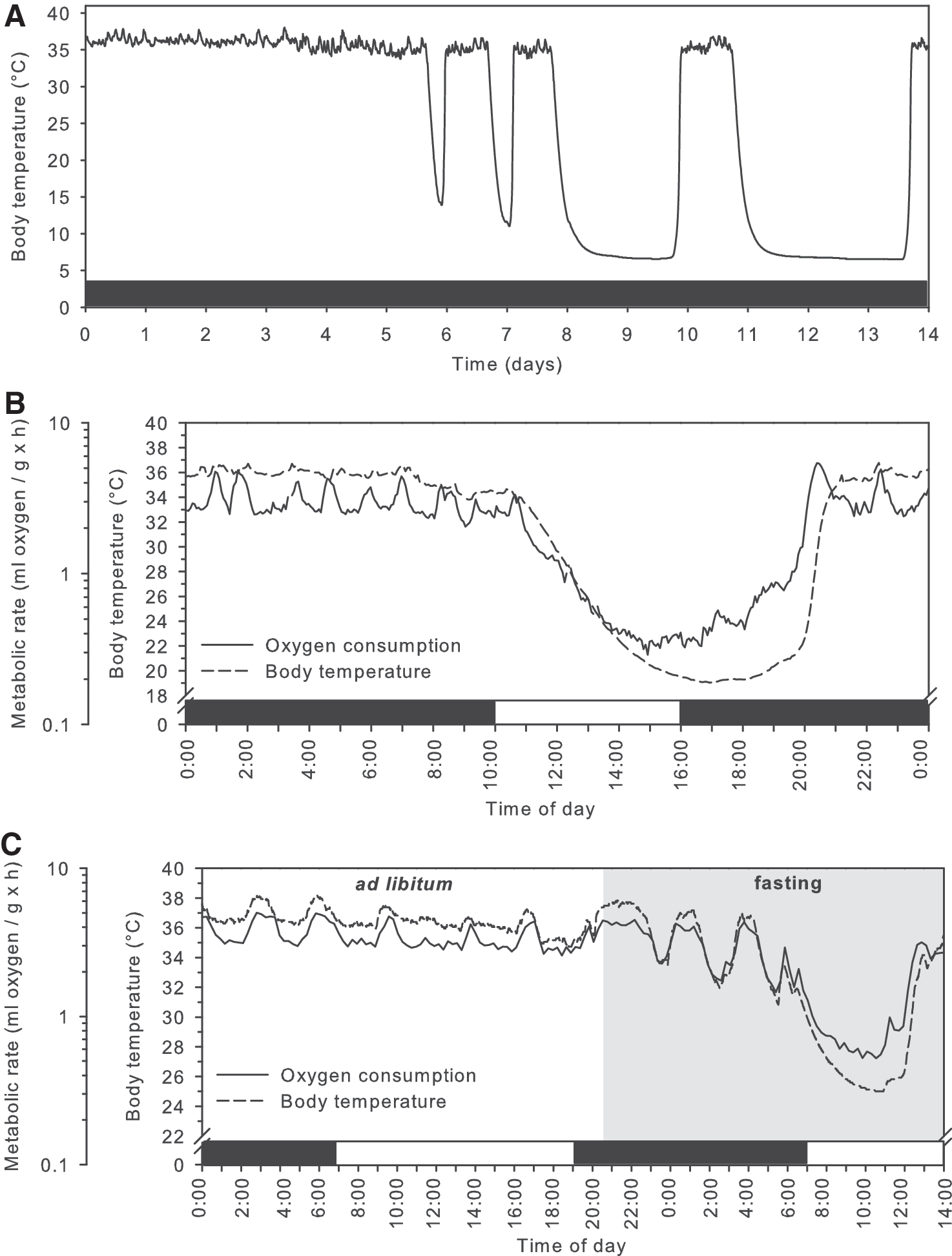
Nature has, however, provided us with excellent examples of animals that are able to cope with physiologically extreme conditions, including a profoundly lowered body temperature: mammalian hibernators. Hibernating animals endure repeated bouts of torpor, a physiological state characterized by profound reduction in metabolism, and, as a consequence, reduced body temperature. Torpor bouts are interspersed by interbout arousals during which metabolism and temperature rapidly return to euthermic levels. Based on differences in length and magnitude of reductions in metabolism and body temperature, three types of torpor use are recognized: (a) seasonal hibernation, (b) daily torpor, and (c) hibernation at relatively high body temperatures, as seen in bears. In seasonal hibernation, torpor bouts are characterized by strong reductions in metabolism of about 95–99%, which result in drops of body temperature to as low as −2°C to 4°C (1, 5, 58, 127). Torpor bouts last from several days to a month depending on the ambient temperature and the species (1, 58, 127); in addition to low metabolism and body temperature, ventilation rate, cardiac output, and renal function are also strongly depressed. In other generally smaller species, torpor may occur on a daily basis (<24-h bouts), during which metabolic rate is reduced by 70–95% and the body temperature typically falls to 15–30°C for several hours, after which the animal returns to normal physiology (see examples in Fig. 2) (46).

Hibernating bears deploy a different strategy; they lower metabolism by about 75%, and heart rate is reduced from 55 to as few as 9 beats per minute, during a torpor bout that seems to last the entire winter season, without arousals, although the body temperature cycles at multi-day intervals between 30°C and 36°C (a typical example is shown in Fig. 3) (143, 144). Irrespective of the strategy employed, animals that use torpor have a markedly increased resistance to I/R, as suggested by their ability to withstand repetitive torpor/arousal cycles without organ injury (Fig. 1) (2, 43, 54, 116, 136, 158).
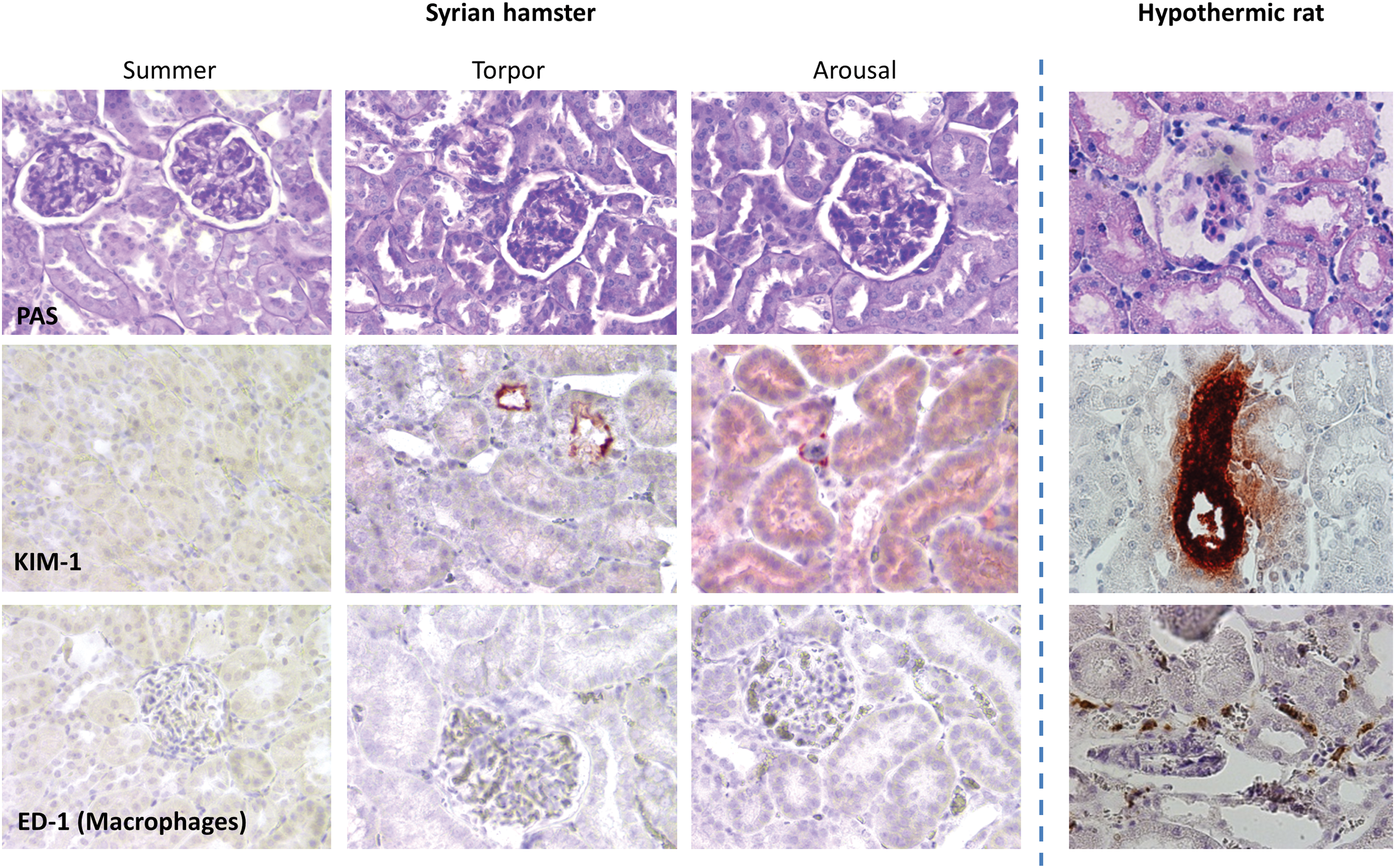
Renal tubular cells are well preserved throughout hibernation, including cell polarity, brush border structure, and the number and morphology of mitochondria, in contrast to loss of microvilli at the brush border after I/R in non-hibernators (110, 158). Moreover, hibernators even show increased resistance to experimental (warm) I/R when not in torpor (i.e., during arousal) (75, 102, 108) or during the summer period (34, 81). Thus, mammalian hibernators are able to withstand extreme physiological conditions, without loss of cellular integrity.
The molecular mechanisms of hibernation and daily torpor that cause reduced metabolism and preservation of cellular homeostasis are not well understood. Unraveling the underlying signaling pathways holds promise as potential drug targets to prevent IRI in humans. Several pharmacological compounds have been identified that can induce transient reductions in body temperature, which resemble the thermal effects of torpor, but these lack other features of natural torpor, including regulated reduction in metabolic rate. Nevertheless, these compounds, which include hydrogen sulfide (H2S) (9, 13, 16), adenosine (109, 148), and 5′-adenosine monophosphate (5′-AMP) (16, 131, 161), also appear to confer protection from some pathological conditions such as IRI. In this review, we compare the role of mitochondria in the induction of kidney injury during hypothermic I/R in non-hibernators with the maintenance of renal mitochondrial homeostasis during hibernation, and we describe how these effects may be pharmacologically exploited by mimicking torpor in non-hibernators.
Ischemic organ injury is initiated by mitochondrial dysfunction
I/R stimulates mitochondrial fragmentation and the formation and opening of mitochondrial permeability transition pores
Under physiological conditions, mitochondria appear as elongated tubular structures, which are maintained by a delicate balance between fusion and fission events (155). Disruption of this balance occurs in the early stages of (warm) I/R, as demonstrated by mitochondrial fragmentation in cultured human and mouse renal tubular cells, as well as in vivo in proximal tubular cells in mice (19). Suppression of Dynamin-related protein 1 (Drp1), a core protein in the mitochondrial fission machinery, attenuates mitochondrial fragmentation, cytochrome c release, caspase activation, and tubular cell apoptosis during AKI induced by I/R (19). Hence, rapid morphological changes such as mitochondrial fragmentation via Drp1-mediated fission events are likely to be important in the onset of IRI. It remains, however, unclear whether the disruption of mitochondrial morphology under such pathological conditions is a cause or consequence of disease (17, 121, 151). Although it is likely that mitochondrial fragmentation facilitates irreversible damage of the organelle, it does not represent a per se point-of-no-return (19).
A critical event in the onset of IRI, marking irreversible mitochondrial damage, involves the formation and opening of mitochondrial permeability transition pores (MPTPs). When formed, MPTPs allow diffusion of small molecules (up to 1.5 kDa) across the inner mitochondrial membrane, leading to leakage of anti-oxidants, protons, calcium (Ca2+), and other pro-apoptotic factors from the mitochondrial matrix, and equilibration of other ions and water between the matrix and the mitochondrial intermembrane space (62, 77, 115). MPTPs are formed in the mitochondrial membrane by fusion of the pro-apoptotic proteins Bax/Bak in the outer mitochondrial membrane, the adenine nucleotide translocase (ANT, or adenosine diphosphate [ADP]/ adenosine triphosphate [ATP] translocator), F1FO-ATP-synthase, and phosphate carrier (PiC) in the inner mitochondrial membrane, and cyclophilin D in the mitochondrial matrix (77). Formation and opening of MPTPs are, among other factors, induced by increased Ca2+ in the mitochondrial matrix, phosphate, free radicals, mitochondrial membrane depolarization, and alkalosis. Drp1 seems to facilitate Ca2+-induced opening of MPTP, as Drp1 overexpression increases the susceptibility to Ca2+-induced MPTP opening in COS epithelial cells (53). Conversely, acidosis and increased levels of ADP, magnesium (Mg2+), and cyclosporine A inhibit MPTP opening (62, 77).
Formation and opening of MPTPs leads to loss of mitochondrial membrane potential, respiratory chain uncoupling and reduces ATP synthesis. In a futile attempt to compensate for the reduced mitochondrial membrane potential, mitochondria actively pump protons from the matrix through the mitochondrial F1FO-ATPase, thereby turning from ATP producers into ATP consumers (62). Ultimately, the opening of MPTPs can progress into swelling and rupture of mitochondria, eventually leading to cell death (77). Hence, MPTP formation is at the center of a detrimental cycle of events that play a key role in the initiation of mitochondrial dysfunction induced by IRI.
Mitochondrial dysfunction induces renal inflammation
Inflammation plays an important role in the pathophysiology of renal IRI, which is mainly mediated by endothelial and epithelial cells, tissue resident macrophages, and dendritic cells (44, 80). Mitochondrial dysfunction contributes to the onset and progression of renal inflammation through several downstream mechanisms. Free radicals, which are mainly produced by dysfunctional mitochondria, can induce inflammation through upregulation of Toll-like receptor 4 (TLR4), transcriptional activation of nuclear factor kappa B (NF-κB), and activation of the Nod-like receptor protein 3 (NLRP3) inflammasome. In turn, this leads to caspase-1-mediated cleavage of pro-interleukin-1β (pro-IL-1β) into IL-1β (4, 74, 105, 133).
In addition to pro-inflammatory effects of free radicals, circulating (mitochondrial) damage-associated molecular patterns (DAMPs) activate the innate immune system through recognition by pattern recognition receptors (PRRs), such as TLR4 (74). Many DAMPs are proteins and nucleic acids that are released from injured and dying cells (154); examples of important DAMPs are high-mobility group box-1 (HMGB1), actin, mitochondrial formyl peptides, (mitochondrial) DNA, and ATP (74, 162). On activation of PRRs by DAMPs, downstream activation of NF-κB induces upregulation of cell adhesion molecules and stimulates the production of (chemotactic) cytokines (4, 133). The increased production of pro-inflammatory chemotactic cytokines, together with increased expression of adhesion molecules on the cell surface, attracts neutrophils and monocytes that, in turn, release additional free radicals and further potentiate the damage (55).
Therapeutic hypothermia
Hypothermia diminishes cellular ATP depletion during ischemia
Intracellular ATP levels are the result of the balance between ATP production, mainly by mitochondria through oxidative phosphorylation, and ATP consumption. Warm ischemia of the kidney (37°C, 40 min) leads to a 20-fold decrease in ATP and a 3-fold drop in ADP, whereas levels of downstream metabolites [i.e., AMP, adenosine, inosine, and (hypo)xanthine] rise, as demonstrated in rats (157). However, the deleterious effects of ischemia on ATP production by mitochondria are dampened by hypothermia (i.e., 32–33°C) and levels of adenylate high-energy phosphate (2 × ATP+ADP) are 33% higher as compared with warm ischemia (157).
Hypothermia may, in addition to its effect on ATP production, also decrease ATP consumption. The function of mitochondrial ETC complexes I and II is reduced by ∼70% already after 40 min of warm ischemia in rat kidneys (in situ), as measured after brief reperfusion at 25°C, whereas 24 h of cold ischemia of kidneys (ex vivo, at 4°C) leads to only ∼15% suppression of mitochondrial ETC complex I (6). In addition, the activity of complex II is only reduced after 48 h of cold ischemia, as measured after reperfusion and rewarming the kidney samples to 25°C (6). Although hypothermia seems to dampen the deleterious effects of I/R on mitochondrial function, it should be noted that a direct comparison between the effects of warm and cold ischemia may be influenced by methodological differences (i.e., in situ vs. ex vivo).
The reduced ATP production after (cold) I/R may be explained by a reduced expression of mitochondrial ETC complexes. Cold I/R of the kidney in vivo in rats (4°C, 40 min) followed by warm reperfusion (37°C, 18 h) leads to a reduced ATP production and a lowered protein level of ETC complexes I, II, III, and IV, as compared with warm non-ischemic kidneys (113). Unfortunately, it cannot be concluded whether the lowered protein levels are due to ischemia, hypothermia, or a combination of both (113). Taken together, the reduced production of ATP during warm ischemia is not matched by a similarly lowered consumption of ATP. Hypothermia seems to diminish mitochondrial injury and suppress ATP consumption to dampen the deleterious effects of I/R on cellular levels of ATP (156, 157), thereby improving the cellular energy balance.
Hypothermia inhibits mitochondrial fragmentation, but disrupts ion homeostasis, leading to the formation and opening of MPTPs during I/R
Although warm I/R leads to mitochondrial fragmentation that is mediated by Drp1 and may progress into irreversible mitochondrial damage (19), hypothermia inhibits the activation of Drp1 in response to I/R and lowers the subsequent cytochrome c release from mitochondria, as demonstrated in cerebral I/R in mouse (138). However, despite its positive effects on ATP levels during I/R and mitochondrial fragmentation, cooling negatively affects ion transport, thereby contributing to mitochondrial dysfunction by different mechanisms. Maintenance of ion homeostasis by ion pumps is one of the largest energy-consuming mechanisms in the cell, which not only depends on the amount of ATP available but is also sensitive to changes in temperature. Direct suppression of mitochondrial respiration by hypothermia, free radical damage to the ETC complexes, and inhibition of the citric acid cycle by free radicals impede mitochondrial ATP production during cold I/R, which, together with the direct inhibitory effects of hypothermia, may impair (active) ion transport.
The sodium-potassium pump (Na+/K+-ATPase) uses 5–40% of the total ATP turnover (depending on the cell type) and is, therefore, highly sensitive to changes in the amount of ATP available (52, 112). Dysfunction of the Na+/K+-ATPase results in accumulation of intracellular Na+ and leakage of K+ out of the cell, initiating cell membrane depolarization and triggering influx of Ca2+ into the cell through voltage-gated channels (60). Subsequently, high cytosolic Ca2+ levels lead to rapid uptake of Ca2+ into mitochondria by the mitochondrial Ca2+ uniporter (mCU), thereby resulting in increased mitochondrial Ca2+ levels (117). Influx of Ca2+ into the mitochondrial matrix increases respiratory rate, H+ extrusion, and ATP production, as well as free radicals. Hypothermia, thus, indirectly affects mitochondrial function via the impairment of ATP-dependent ion transporters and subsequent disruption of ion homeostasis.
Prolonged increased levels of Ca2+ in the mitochondrial matrix, moreover, induce the formation and opening of MPTPs in mitochondria, which triggers release of cytochrome c and subsequent activation of apoptotic pathways (Fig. 4) (62). Cytochrome c subsequently diffuses to the endoplasmic reticulum, where it binds to inositol triphosphate (InsP3) receptors, thereby further enhancing cellular Ca2+ levels by stimulating release from the endoplasmic reticulum (92). MPTPs, in addition, increase proton leakage, which leads to a loss of the mitochondrial membrane potential and a major loss of depolarization capacity (i.e., change in membrane potential in response to ADP addition) that, in turn, lowers ATP production, whereas free radical production is increased (39, 115).
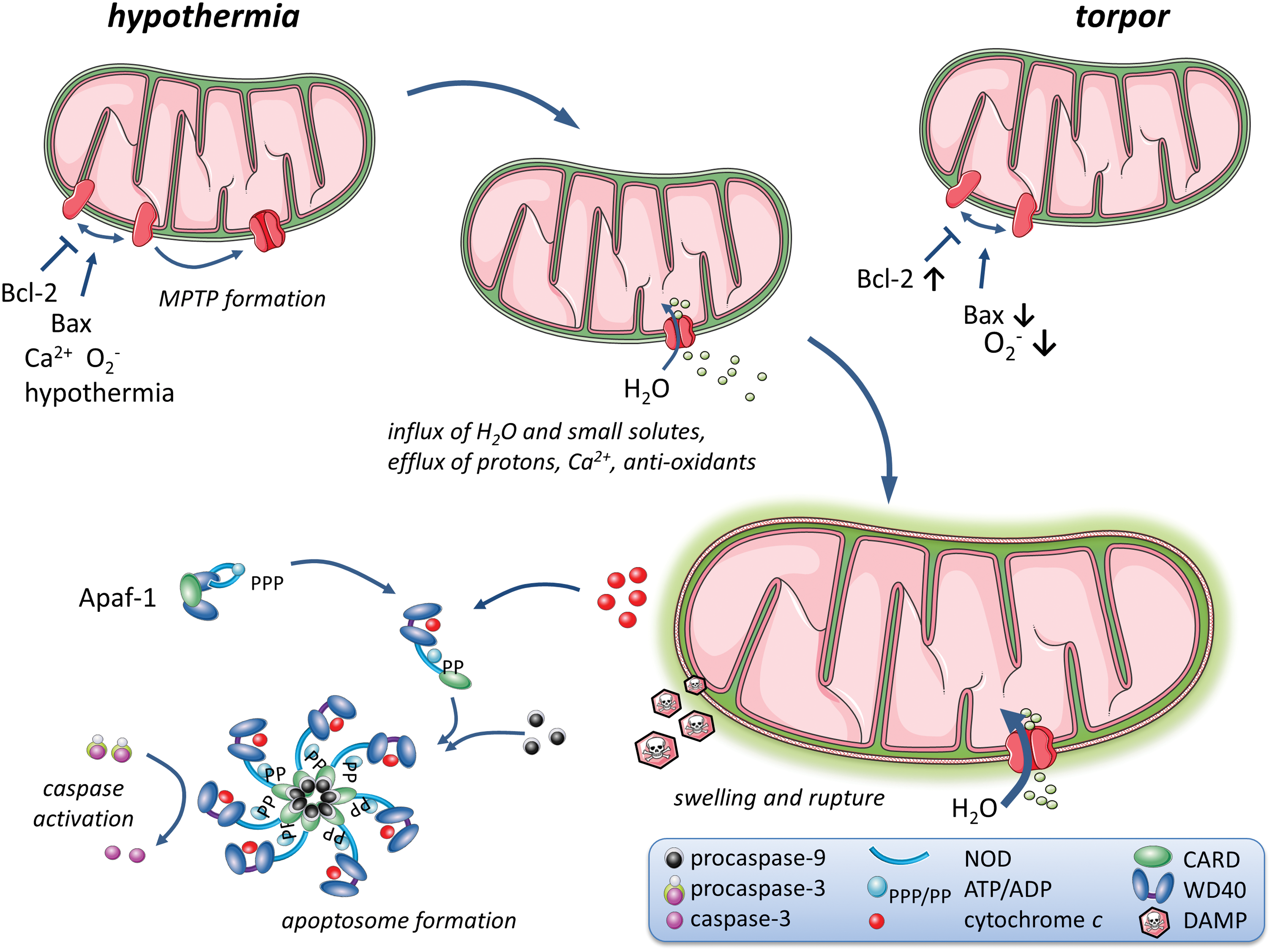
Excessive osmotic water entry mediated by MPTPs into the mitochondrial intermembrane space leads to swelling of mitochondria, which may ultimately lead to rupture of the outer mitochondrial membrane (62) and release of pro-apoptotic proteins, including cytochrome c, apoptosis-inducing factor (AIF), Smac/DIABLO, and apoptotic protease activating factor 1 (Apaf-1; consisting of caspase recruitment domain [CARD] and WD40 repeat) (77, 114, 115). Cytochrome c and Apaf-1 bind with procaspase-9 to form the apoptosome, which, in turn, recruits the caspases 1–4 and activates the caspase cascade during rewarming after cold ischemia (Fig. 4) (114, 115). Hypothermia-induced disruption of cellular ion homeostasis not only affects the membrane potential but also disrupts cellular morphology by stimulating influx of water in response to the cellular buildup of Na+ (107). As a consequence, swelling of vascular smooth muscle and endothelial cells, combined with disruption of the cytoskeleton secondary to ATP depletion, may impair renal perfusion due to an altered shape of capillaries, as revealed in rat renal cells (76).
At a later stage, uncontrolled swelling can lead to rupture and necrosis of cells, as demonstrated in rat glial cells (107). It is likely that a similar mechanism is also operable in the kidney, although this remains to be studied. Together, although hypothermia partly precludes mitochondrial injury induced by I/R, these beneficial effects are limited by the disruption of ion homeostasis and the formation of MPTPs. Collectively, these events lead to a combination of necrosis and necroptosis (regulated necrosis) during ischemia, followed by apoptosis during reperfusion and rewarming, due to the activation of the apoptotic pathways in sublethally injured cells (64).
Cold I/R leads to increased levels of free radicals
Opening of MPTPs, in the context of cold I/R, also contributes to the formation of free radicals due to loss of the ETC-component cytochrome c and anti-oxidants, such as reduced glutathione (GSH), from mitochondria (85). Cytochrome c functions as an electron transporter between ETC complexes III and IV. Hence, loss of cytochrome c leads to over-reduction of complex III, increasing electron leakage to O2 and forming O2 −, instead of complex IV reducing O2 to water (H2O) (65).
Cold I/R of rat kidney cells in vitro leads to increased amounts of mitochondrial O2 −, which diminishes toward baseline levels after 6 h of rewarming (93). Although most mitochondrial O2 − molecules are dismutated rapidly, the remainder may react with NO and form the highly reactive, oxidizing, and nitrating molecule peroxynitrite (ONOO−). Consumption of NO in the formation of peroxynitrite with O2 − may explain the lowered levels of NO after cold I/R in rat proximal tubular cells (NRK) (93). Moreover, either inhibition of NO production through the NO inhibitor L-NG-monomethyl arginine (L-NMMA) or inhibition of the generation of O2 − by overexpressing superoxide dismutase 2 (SOD2) precludes the formation of ONOO− from NO and O2 − and protects against cold storage-induced oxidative injury, maintains ETC complex I and II activity, and limits cell death (93). Thus, loss of cytochrome c and damage to ETC complexes I and II, due to the opening MPTPs, augment the generation of O2 −, which combines with NO to form highly reactive ONOO− during cold I/R.
Free radical production during cold I/R leads to lipid and protein damage
The increased generation of free radicals during cold I/R leads to damage to lipids, proteins, and nucleic acids, and it negatively affects ATP synthesis by inhibiting the citric acid cycle and damaging the ETC, as demonstrated in isolated perfused rat hearts (146). Although nitration of proteins leads to the formation of nitrotyrosine, oxidation of lipids and proteins leads to the formation of reactive aldehydes such as malondialdehyde through lipid oxidation and protein carbonyls by protein carbonylation.
Cold storage of rat kidney cells in vitro and cold I/R of the kidney in vivo lead to a strong increase in the levels of nitrotyrosine (93, 113). Similarly, ex vivo cold preservation of kidney grafts derived from living and brain death pigs leads to increased levels of nitrotyrosine, as compared with levels directly after harvesting the organ (126). Furthermore, ex vivo cold ischemia of rat kidney leads to a time-dependent increase in lipid peroxide levels and protein carbonyl content (29), and similar effects on protein oxidation are found after ex vivo cold preservation of the murine heart followed by in vivo rewarming and reperfusion (33). Unfortunately, due to the design of many studies, it is not possible to specifically attribute these effects to changes in temperature, ischemia, or reperfusion.
In addition, most studies do not take time of the day into account, although mitochondria exhibit a substantial circadian rhythm to adapt to changes in nutrient availability, energy supply, and cellular remodeling throughout the day. Melatonin reduces fission of mitochondria by suppressing the translocation of Drp1 and the mitochondrial fission 1 protein (Fis1) and upregulating the expression of the mitochondrial fusion proteins (mitofusins 1 and 2 [Mfn1/2]) and optic atrophy 1 (Opa1) (137). Moreover, under normal conditions, release of hydrogen peroxide (H2O2) from mitochondria occurs in an oscillatory, circadian rhythm, controlled by the enzymes peroxiredoxin III (PrxIII) and sulfiredoxin (69, 111). To our knowledge, however, no study thus far has described an effect of mitochondrial circadian rhythm on (renal) IRI. Together, although it remains to be determined what the relative contribution of changes in temperature, ischemia, reperfusion, and circadian rhythm to renal IRI are, cold I/R of the kidney leads to lipid and protein damage by nitration and oxidation induced by free radicals.
Cold I/R is marked by a reduction in enzymatic anti-oxidant activity
In addition to effects that lead to increased generation of free radicals, cold I/R affects the expression and activity of the anti-oxidant enzymes SOD and glutathione peroxidase (GPx) as well. SOD catalyzes the dismutation of O2 − into O2 or H2O2. H2O2, subsequently, can react with GSH to be converted into H2O and GSH disulfide, which is catalyzed by GPx (29, 97). SODs consists of a group of three anti-oxidant enzymes: SOD1 (or Copper-Zinc SOD [CuZn-SOD]) is located in cytoplasm, SOD2 (or Manganese SOD [MnSOD]) is located in mitochondria, and SOD3 (or extracellular SOD [EC-SOD]) is located extracellularly (160). GPx consists of a family of eight enzymes, of which GPx1 is the most abundant and is located in the cytoplasm (97).
Ex vivo cold preservation of the rat kidney induces an early upregulation in the levels of SOD at 6 h, which then falls to baseline levels after 12 h (29). Levels of SOD2 are not affected by cold I/R of the kidney in vivo in rats, although it does lead to a strong decrease in SOD2 activity (113). Similar results are obtained after warm I/R in mice, which suggests that ischemia, rather than hypothermia, causes the reduction in SOD2 activity (104). The effect of (cold) I/R on levels or activity of SOD3 has not yet been explored. The total activity of GPx, estimated by measuring the reduction of H2O2 using GSH as a reducing agent, is reduced after 24 h of ex vivo cold preservation of the rat kidney as compared with baseline (29). Thus, (cold) I/R leads to MPTP-mediated production of O2 − and leakage of anti-oxidants, as well as a reduction in enzymatic anti-oxidant activity.
Renal mitochondrial response in other (normothermic) pathophysiological states
In addition to (therapeutic) hypothermia, other diverse pathological states are associated with renal mitochondrial dysfunction under normothermic conditions. For example, diabetic nephropathy and hypertensive kidney injury are associated with an imbalance in redox regulation within the mitochondrial matrix of renal tubular cells.
In diabetic nephropathy, hyperglycemia leads to increased formation of free radicals, through different mechanisms. First, hyperglycemia exacerbates the formation of free radicals that are formed as byproducts of oxidative phosphorylation, as an increase in glucose reabsorption in the proximal tubules results in increased glycolysis and oxidative phosphorylation (59, 90). Second, hyperglycemia leads to upregulation of NOX4, an isoform of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase; a major source of O2 −) within the mitochondria in the renal cortex. Upregulation of NOX4, in turn, leads to glucose-mediated oxidative injury within glomerular mesangial cells (11, 48). Third, methylglyoxal, a byproduct of glycolysis, induces the formation of advanced glycation end-products (AGEs) that augments the formation of free radicals, thereby impairing the function of renal tubular cells (31, 90).
In non-diabetic, hypertensive renal injury, upregulation of NADPH oxidase in renal medullary cells leads to increased formation of free radicals, as demonstrated in a rat model of salt-sensitive hypertension (140). In turn, the increased free radical formation is associated with higher levels of renal injury as compared with control rats (140). Thus, renal mitochondrial dysfunction is not only provoked by hypothermia but also a prominent feature of normothermic disease states, including diabetic and hypertensive nephropathy.
Mammalian hibernation
Mitochondrial respiration is actively suppressed during torpor in hibernators
In contrast to non-hibernators, mitochondrial homeostasis is well preserved at low body temperatures during natural torpor. A key difference between hypothermia and hibernation is that on entrance into torpor, hibernators initially and profoundly reduce their metabolism, which only, subsequently, leads to a lowered body temperature (and not vice versa as in hypothermia) (125). On entrance into torpor, 13-lined ground squirrels actively suppress mitochondrial respiration by 70% (when measured ex vivo at 37°C), corresponding to a whole-animal oxygen consumption of less than 10% compared with summer active animals (95). In addition, levels of lactate in the liver are reduced during torpor in the 13-lined ground squirrel as compared with summer animals, which suggests that anaerobic metabolism is suppressed, or at least not activated when mitochondrial respiration is suppressed (120). Although the initial metabolic suppression during torpor is actively regulated, it is subsequently combined with a passive reduction of the metabolic rate due to the lowered body temperature during torpor (20, 58, 125) (summarized in Table 1).
AKI, acute kidney injury; ATP, adenosine triphosphate; Drp1, Dynamin-related protein 1; ETC, electron transport chain; GPx, glutathione peroxidase; GSH, glutathione; I/R, ischemia/reperfusion; MPTP, mitochondrial permeability transition pore; SOD, superoxide dismutase.
The exact mechanisms that lead to the profound reduction in mitochondrial respiration during torpor remain to be unraveled. However, recent studies provide some insight into changes that occur in the ETC during torpor, which may help to reveal the molecular mechanisms that lead to metabolic suppression during hibernation. Compared with arousal, torpor leads to a 40% lower activity of ETC complex I, a 60% lower activity of ETC complex II, but does not affect the activity of ETC complexes III, IV, or V, as measured in isolated mitochondria derived from 13-lined ground squirrel liver (20, 91, 95). Importantly, the main protein content of ETC complexes I and II is not different between torpor and arousal, thus suggesting that the lower mitochondrial function is due to regulatory changes (91).
The lower activity of complex II might be explained by a reduced substrate availability and direct inhibition during torpor. Levels of succinate are reduced in the liver during torpor in the 13-lined ground squirrel as compared with summer animals (120). Uptake of succinate into mitochondria is mediated by the dicarboxylate transporter. Experimental inhibition of the dicarboxylate transporter equally affects respiration in mitochondria during torpor and arousal, which suggests that a reduced function of the dicarboxylate transporter does not play a key role in the suppression of mitochondrial respiration in the liver during torpor in the 13-lined ground squirrel (30). However, levels of oxaloacetate, a direct inhibitor of complex II, are higher in the liver during torpor and arousal as compared with summer active 13-lined ground squirrels. Pre-incubation of mitochondria from torpid animals with isocitrate alleviates the inhibiting effects of oxaloacetate and increases mitochondrial respiration (3). Hence, the reduced mitochondrial respiration during torpor can, at least in part, be explained by direct inhibition of ETC complex II by increased levels of oxaloacetate.
In addition to substrate oxidation, phosphorylation of ADP occurs at a lower rate during torpor, whereas the reduced body temperature passively lowers proton leak during torpor (20). ADP phosphorylation by F1FOATPase is the main factor that reduces the mitochondrial membrane potential under phosphorylating conditions. Although non-hibernators may even reverse the function of ATP synthase in an attempt to maintain the mitochondrial membrane potential in response to opening of MPTPs, the latest data suggest that the activity of F1FOATPase is not different between torpor and arousal, as studied in liver mitochondria from the 13-lined ground squirrel (91). Presumably, a parallel reduction in substrate oxidation and proton leak allow for maintenance of the mitochondrial membrane potential at low temperatures (20 –22). Although the precise mechanisms that govern metabolic suppression during torpor remain to be identified, it may be due to a combination of suppression of mitochondrial respiration that comprises inhibition of ETC complexes by allosterism or post-translational modifications, as well as passive effects of low temperature on metabolism.
Consumption of ATP is suppressed during torpor
Specific adaptations to limit ATP consumption during torpor may enable preservation of cellular homeostasis during periods of reduced mitochondrial respiration with a lowered ATP production. Passive thermal effects and active suppression of ATP-consuming processes may allow net preservation of ATP levels. Data on the levels of ATP during torpor are, however, inconsistent among species and between tissues. Concentrations of ATP are well preserved in the brain of the Syrian hamster (86) and even increase in the liver in the 13-lined ground squirrel (120) during torpor, whereas levels of ATP in erythrocytes in the 13-lined ground squirrel (37) and skeletal muscle are markedly reduced, in both the golden-mantled squirrel and white-tailed prairie dog (41, 89).
Results from these different studies should be interpreted with caution, as ATP is rapidly metabolized and its tissue levels may be influenced by the body temperature and time between euthanasia and freezing of the sample (120). After maintenance of ion homeostasis, the main consumer of cellular ATP is the synthesis of protein and nucleic acids for cellular proliferation and maintenance of homeostasis (23). Cellular proliferation is markedly reduced during torpor. Intestinal epithelial cells, for example, have a fast turnover rate and enterocytes migrate in only 3–5 days from the crypts to the top of the villus in summer active animals, whereas this process can take more than 3 weeks during hibernation in the 13-lined ground squirrel (27).
In addition, protein and nucleic acid synthesis is globally suppressed during torpor (127). The rate of DNA synthesis is 24-fold lower in torpid arctic ground squirrels as compared with aroused animals and cells accumulate in the G2 phase of the cell cycle (73). Tissue-specific adaptations may further limit energy expenditure. For example, cofilin-2 can modulate ATP consumption in cardiac tissue by binding actin and is an inducer of apoptosis in mitochondria (8). Dephosphorylation of cofilin-2 during torpor in the heart tissue of ground squirrel stimulates binding of cofilin-2 to actin (50). Therefore, sequestering cofilin-2 into actin-cofilin rods likely prevents cofilin-2 from participating in ATP hydrolysis on actin turnover and precludes its localization in mitochondria where it can otherwise induce apoptosis (50). Furthermore, the activity of Na+/K+-ATPase in the liver, kidney, and skeletal muscle derived from torpid golden-mantled ground squirrels is reversibly reduced by phosphorylation of the enzyme and may enable preservation of ATP by reducing ATP consumption (89).
During hibernation in the 13-lined ground squirrel, glycogen accumulates in cardiac tissue and may serve as a substrate for anaerobic metabolism to maintain ATP production during periods of lowered oxygen supply (56). Increased glycogen storage may contribute to the resistance to ischemia-induced cardiac contracture of hearts from hibernating ground squirrels as compared with hearts from summer squirrels (56). Although the kidney also stores small amounts of glycogen, mainly in the renal medulla (47), it is unknown whether hibernation affects renal glycogen storage. (47) Thus, it appears that specific adaptations induced during torpor actively suppress both ATP production and consumption, in addition to passive effects of the lowered body temperature.
Anti-oxidant adaptations of hibernation
The reduced mitochondrial respiration during torpor may be expected to lead to a lowered generation of free radicals, although a reduced oxidative phosphorylation without suppression of proximal ETC complexes may well lead to electron leakage, thereby augmenting the generation of free radicals. However, the precise effects of torpor on the different ETC complexes and the generation of free radicals remain to be unraveled. Upregulation of specific anti-oxidant enzymes may limit oxidative injury during torpor-arousal cycles. However, it should be noted that in contrast to cold I/R followed by rewarming, arousal from torpor may not be associated with a gross fluctuation in tissue oxygenation; in the brain of arctic ground squirrels, there is a high arterial oxygen pressure and a low expression of hypoxia-inducible factor-1α (HIF-1α) during torpor (88).
In contrast to these observations, levels of conjugated dienes (i.e., hydrocarbon chains that are products of lipid peroxidation) and activation of the redox-sensitive transcription factor NF-κB are increased in the intestine of 13-lined ground squirrels during hibernation as compared with summer animals (26). Levels of conjugated dienes are particularly high during entrance into torpor and early in torpor bouts, which suggests that oxidative injury (lipid oxidation) mainly occurs during the early stages of torpor after a euthermic period, which is in sharp contrast to reperfusion after ischemia in non-hibernators (26). On arousal, the free radical production by complex I is actively suppressed, whereas passive thermal effects reduce free radical production from complex III as the squirrel is arousing from torpor (20). Because activity of complex I is the main source of free radical production on reperfusion in non-hibernators, the active suppression of complex I on arousal from torpor may allow mitochondrial respiration to increase while dampening the formation of free radicals.
In addition to the active suppression of complex I to limit the production of free radicals, hibernators appear to alter levels of some antioxidant enzymes during torpor-arousal cycles. Ascorbic acid, GSH peroxidase, and GSH reductase all catalyze the removal of H2O2. The level of ascorbic acid in the liver and plasma increases during torpor as compared with summer active European and arctic ground squirrels, respectively (24, 38). The activity of catalase, which catalyzes an alternative H2O2 detoxification pathway, is higher in the brain and heart, but lower in the liver during torpor, as compared with summer active 13-lined ground squirrels (103). On the other hand, the activity of catalase in serum increases on (early) arousal in Syrian hamsters, as compared with torpor and winter euthermic animals (100).
Serum levels of GSH, an anti-oxidant and co-factor for the production of GSH peroxidase, are augmented in arousal, as compared with torpor and winter euthermic Syrian hamsters (100). In line with this finding, the plasma level of GSH peroxidase increases on arousal as compared with torpor (100). The activity of GSH peroxidase in the liver is higher in torpor, whereas no change is observed in the heart or brain (103). The activity of GSH reductase, however, is reduced during torpor in the brain and intestinal mucosa, as compared with summer active 13-lined ground squirrels, whereas levels in the heart and liver are not affected by hibernation state (28, 100, 103).
Together, hibernation is associated with profound changes in different anti-oxidant systems. The use of different study designs and species, whereby different states of hibernation or summer euthermia may be used as a comparator, hampers the interpretation of the specific anti-oxidant enzymes that may be upregulated during torpor and arousal. Interestingly, although renal tubular cells from non-hibernators (i.e., rat) may be protected against oxidative injury by upregulation of MnSOD in mitochondria (93), no differences in SOD protein expression or activity were found between active and hibernating 13-lined ground squirrels in the liver, brain, and heart (103).
Specific adaptations, including upregulation of anti-oxidant mechanisms, combined with reduced levels of the pro-apoptotic p53 protein and caspase-3-activity and increased levels of the anti-apoptotic Bcl-XL, a member of the Bcl-2 family of proteins, as compared with the pro-apoptotic Bax throughout hibernation (i.e., torpor and arousal) may limit the formation of MPTP in mitochondria and cell death in hibernators (43) (Fig. 4). Taken together, these observations suggest that low body temperature during torpor may minimize organ injury in hibernators, whereas upregulation of non-enzymatic anti-oxidants, and perhaps also anti-oxidant enzymes, may reduce reperfusion injury by preventing generation of large amounts of free radicals, thus preserving mitochondrial homeostasis.
Ion homeostasis is maintained throughout hibernation
In non-hibernators, hypothermia and loss of ATP can lead to disruption of major ion transporters, which, in turn, can lead to mitochondrial dysfunction, activation of cell death pathways, and uncontrolled swelling of the cell. In hibernators, however, ion homeostasis is preserved, possibly through maintenance of ATP levels throughout torpor-arousal cycles combined with specific adaptations of ion pumps and channels (7, 42, 71, 89).
Low temperature per se decreases the activity of Na+/K+-ATPase, as illustrated in isolated erythrocytes in vitro (71). However, the inhibitory effect of hypothermia occurs more pronounced in guinea pigs (non-hibernators), as compared with 13-lined ground squirrels (71). In contrast to active transport, passive leakage is far more reduced at lower temperature in squirrels than in in guinea pigs (71).
Not only temperature but also torpor itself seems to influence ion transport; the Na+/K+-ATPase activity is reduced during torpor in the liver, kidney, and skeletal muscle, but not in the heart, when measured ex vivo at 25°C as compared with euthermic golden-mantled ground squirrels (89). The reduced Na+/K+-ATPase activity can be (partly) restored by incubating the extracts with protein kinase A and alkaline phosphatase, which suggests that post-translational modifications negatively affect the ion transporter (89). Furthermore, in addition to low temperature and torpor, seasonal changes may also affect ion transporters in hibernators. In the jerboa, a hibernating desert rodent, the activity and amount of Na+/K+-ATPase units in the medullary thick ascending limb of the nephron are reduced after cold exposure in winter euthermic and hibernating jerboas, as compared with summer euthermic animals (7). Moreover, season seems to differentially affect ion transporters in the kidney, as the amount and activity increase in cortical and outer medullary collecting ducts after cold exposure (7).
The reduced activity of Na+/K+-ATPase may potentially allow maintenance of ATP levels by lowering the consumption of ATP. Together, low temperature, torpor, and season all affect Na+/K+-ATPase in hibernating animals, presumably due to direct inhibitory effects, post-translational modifications, and changes in the amount of transporters. Data on other types of ion transporters at low temperature and during hibernation are scarce. Current data suggest a reduced activity of Ca2+ channels during torpor in cardiomyocytes and enhanced sequestration of Ca2+ by the sarcoplasmic and the endoplasmic reticulum, which may prevent excessive cytosolic Ca2+ levels (152). Whether such a mechanism is operable in other cell types remains to be studied.
Pharmacological induction of a torpor-like state
H2S induces a torpor-like state and protects against oxidative injury
Mimicking a state of torpor in non-hibernators by pharmacological agents may be clinically relevant for major (cardiac) surgery and renal transplantation, given the resistance to injury induced by repetitive cooling/rewarming and I/R in hibernators. A torpor-like state can be induced by H2S in small mammals, but not in large animals (9, 119). After induction of this torpor-like state in mice, metabolic rate is reduced to about 10% of the normal range, leading to a drop in body temperature to around 15°C. On arousal, no behavioral or functional differences are observed (9, 10).
An important difference from targeted temperature management is that H2S primarily reduces the metabolic rate, which is followed by a drop in body temperature and not vice versa. Interestingly, inhalation of H2S can induce protection against renal and myocardial IRI in non-hibernators, as demonstrated in rats (13, 124).
The mechanisms by which H2S protects against IRI include mitochondrial effects that lead to preservation of ATP levels, as well as activation of anti-oxidant and anti-inflammatory pathways such as phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), mammalian target of rapamycin (mTOR), and nuclear factor erythroid 2-related factor 2 (Nrf-2) (122). The mitochondrial effects of H2S comprise dose-dependent electron donation at coenzyme Q (a quinone) and inhibition of complex IV (Fig. 5).

Low concentrations of H2S (i.e., at low micromolar concentrations) can stimulate cellular respiration and hence, ATP production, by serving as an electron donor at the level of coenzyme Q in the ETC between complexes I and III (45, 49, 94). At high concentrations (i.e., at high micromolar concentrations), H2S reversibly and competitively binds to cytochrome c oxidase (complex IV), thereby inhibiting the effect of oxygen on the enzyme (68, 94). In addition, at these concentrations, electron donation at coenzyme Q by H2S leads to diversion of electrons toward ETC complex II, thereby reducing fumarate into succinate and, thus, partly reversing the function of the ETC (49). Together, these effects inhibit mitochondrial respiration and may potentially explain the reduced metabolism after inhalation of H2S.
Lowering the body temperature does not seem to be critical in H2S-induced protection against IRI, since H2S governs protection against warm renal IRI as well (83). However, no study has so far directly compared the effect of body temperature on the protective effects of H2S or 5′-AMP on IRI. Hence, although metabolic suppression leads to a lowered body temperature that does not seem to play an essential role in the protection against IRI, it remains to be determined whether lowering the body temperature augments or reduces the protective effects of H2S.
Endogenous H2S is produced by cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE), 3-mecaptopyruvate sulfurtransferase (3-MST), and D-amino acid oxidase, which are expressed at relatively high levels in the kidney, as compared with other organs (63). Endogenous production of H2S plays an important role in maintenance of mitochondrial homeostasis in I/R, as inhibition of either CSE or CBS leads to loss of mitochondrial ATP production and cell death under hypoxic conditions (45, 132, 141). In addition, H2S functions as an antioxidant by directly scavenging free radicals and blocking the Ca2+-mediated opening of MPTP channels, thereby preventing release of pro-apoptotic factors from mitochondria (70, 129).
The levels of CBS and H2S in lung tissue are increased during torpor, as compared with summer or interbout arousal in the Syrian hamster, and may potentially preclude cellular injury during torpor (134). Induction of endogenous H2S production (i.e., by dopamine) in non-hibernators does not lead to a torpor-like state, but leads to an increased plasma H2S level, a reduced ROS production and protects against hypothermia-induced renal injury (Fig. 5) (40). In summary, increasing H2S levels induces a torpor-like state and conveys anti-oxidant and anti-apoptotic effects, indicating that the induction of endogenous H2S production or administration of exogenous H2S constitutes a promising novel pharmacological strategy to limit (hypothermic) IRI.
5′-AMP can induce a reversible state of hypothermia with increased resistance to I/R
Plasma levels of endogenous 5′-AMP, a metabolite of ATP hydrolysis, are increased during fasting-induced daily torpor in mice. Moreover, both peripheral intravenous administration of 5′-AMP (Fig. 6) and its dephosphorylated metabolite adenosine can induce a reversible hypothermic state in mice (161). Although such a reversible hypothermic state that is accompanied with a reduced consciousness may mimic torpor, the fall in body temperature and heart rate during entrance into this state is much faster as compared with fasting-induced natural torpor (Fig. 7) (131). These observations suggest that 5′-AMP-induced hypothermia is not related to fasting-induced torpor.


The precise molecular mechanisms by which 5′-AMP induces hypothermia remain controversial and include effects that are mediated by adenosine receptors, activation of AMP-activated protein kinase (AMPK), and depletion of ATP by adenylate kinase. Further, both central and peripheral effects are suggested to be involved in the induction of hypothermia by 5′-AMP.
Activation of adenosine type-1 receptors (A1R) on temperature-sensitive hypothalamic neurons after dephosphorylation of 5′-AMP to adenosine may suppress the thermoregulatory responses that usually maintain body temperature (99). In line with this, entrance into natural torpor in the Arctic ground squirrel during its hibernation season can be blocked by an A1R antagonist, whereas an A1R agonist can induce a torpor-like state in winter-adapted arctic ground squirrels when injected intracerebrally, but not in summer squirrels (67). Interestingly, this effect of adenosine is not limited to natural hibernators, since an intracerebral injection of adenosine can induce a hypothermic torpor-like state in rats through activation of the A1R as well (148).
In line with 5′-AMP acting through adenosine receptors, administration of the non-selective adenosine receptor antagonist, aminophylline, abolishes the 5′-AMP-induced hypothermic state in mice (131). Further, peripheral intravenous administration of adenosine, ADP, or ATP induces a hypothermic response in mice (131).
Alternatively, regulation by 5′-AMPK may be involved in the effects of 5′-AMP on body temperature. Phosphorylation of AMPK by an increased [AMP]:[ATP] ratio induces activation of downstream signaling pathways that stimulate nutrient uptake, ATP generation, and a reduction in ATP consumption by suppressing metabolism (66, 82). In accordance with a role for AMPK, 5′-AMP is also able to induce a hypothermic state in ecto-5′-nucleotidase and adenosine receptor (A1, A2A, A2B or A3)-deficient mice (32). A third hypothesis is that excess (5-)AMP is converted together with ATP by adenylate kinase to ADP, thereby resulting in a net decline of ATP, leading to a reduced glycolysis and ultimately, disruption of ATP production (78). Thus, although the precise molecular mechanisms remain to be revealed, an injection of 5′-AMP leads to the induction of a reversible hypothermic state in non-hibernators.
Despite the uncertainty about its precise metabolic effects, endogenous (5-)AMP protects against IRI, which seems to mediate its effects on a cellular level, rather than in the central nervous system. Administration of exogenous 5′-AMP affords myocardial and cerebral protection against IRI in non-hibernators independent of lowered body temperature (79, 98, 139). Current data suggest that (5-)AMP induces protection against IRI not only via activation of AMPK but also by its metabolite adenosine. Activation of AMPK induces an energy-saving state to prevent lactate accumulation, activates protective pathways that result in the production of endothelial nitric oxide synthase (eNOS) and NO, and limits cellular injury during ischemia (14, 106). In line with this, an injection of the synthetic AMPK agonist, AICAR, protects against renal injury, macrophage infiltration, and kidney dysfunction after IRI in rats (79).
Mice deficient for CD73, an ectonucleotidase that metabolizes ATP into downstream metabolites (ADP, AMP, and, subsequently, adenosine), have an increased resistance to renal IRI as compared with wild-type animals. However, mice deficient for CD39, an ectonucleotidase that converts AMP into adenosine, suffer increased levels of renal injury after I/R, which could be reduced by administration of a soluble form of CD39 (apyrase) (84). Hence, it seems that not AMP, but its downstream metabolite adenosine is an important molecular mediator to protect against renal IRI.
Extracellular ATP acts as a DAMP and can induce an inflammatory response, although it is rapidly metabolized to its anti-inflammatory metabolite adenosine by the CD73/CD39 pathway mentioned earlier (12, 84). Subsequently, adenosine can be metabolized into inosine by adenosine deaminase (ADA), which is expressed in both an intracellular and an extracellular manner, or can be transported into the cell by endonucleotide transporters (ENT1, ENT2) (51).
Under hypoxic conditions in mice, extracellular concentrations of adenosine rise due to increased release of ATP, whereas the function of ENT1 and ENT2 is suppressed (51). Experimental inhibition of ENT, either pharmacologically or by using knock-out techniques, limits leukocyte influx and protects against AKI after I/R, which is likely due to augmented extracellular concentrations of adenosine (51). Adenosine exerts its immunosuppressive roles by activation of the adenosine type 2a receptor (A2AR) on dendritic cells, neutrophils, and T-cells, inducing an increase of intracellular cAMP and a reduced leukocyte function (35, 101, 150). In addition, the adenosine type 2b receptor on vascular endothelial cells, but not in tubular epithelial cells, protects against renal IRI by facilitating post-ischemic blood flow in the kidney (51). Thus, although the precise molecular pathways involved are not yet fully disclosed, (5-)AMP induces a reversible hypothermic state that is accompanied by increased resistance against renal I/R.
H2S interaction with 5′-AMP
Both H2S and 5′-AMP can induce a hypothermic, torpor-like state in rodents. However, there are currently no available data on whether these two pharmacological agents interact with each other. Recently, we demonstrated that although 5′-AMP leads to endogenous H2S production in the Syrian hamster, the induction of the torpor-like state does not depend on the production of endogenous H2S (40). Pharmacological inhibition of endogenous H2S by aminooxyacetic acid (AOAA), however, results in substantial glomerular and tubular injury during the torpor-like state induced by 5′-AMP (40). In contrast, inhibition of AMPK abolishes the protective effect of H2S on myocardial IRI in the rat (153). Thus, although both H2S and 5′-AMP can induce a torpor-like state and govern protection against IRI in non-hibernators, current evidence suggests that protection of cells from damage during induced torpor, by either H2S or AMP, depends on the interplay between both.
Conclusion
Therapeutic hypothermia, or targeted temperature management, is currently used to limit renal IRI in the setting of major cardiac surgery and renal transplantation. Although hypothermia may delay ATP depletion, it can lead to mitochondrial dysfunction and, as a consequence, increased free radical production. Continued loss of ATP during hypothermia may disrupt ion homeostasis and, ultimately, cause cell death, which leads to release of DAMPs that stimulate inflammation of the kidney. In contrast to non-hibernators, hibernating animals are able to cycle through torpor-arousal states with repetitive cooling/rewarming and withstand experimental I/R without signs of kidney injury.
Hibernators actively reduce their metabolic rate, which is followed by a decline in body temperature and not vice versa as in the case in therapeutic hypothermia. Suppression of metabolism seems key to preventing organ injury during natural torpor. Maintenance of mitochondrial homeostasis and reduced ATP consumption minimize loss of cellular levels of ATP during torpor and arousal. In addition, loss of ion homeostasis during torpor is well tolerated by hibernators, and there is an increase in anti-apoptotic and anti-oxidant defense pathways. Importantly, the reversible hypothermia that resembles natural torpor can be mimicked by H2S, adenosine, and 5′-AMP in non-hibernators, all of which increase resistance to renal IRI as well.
Unraveling the molecular mechanisms that allow hibernators to cycle through torpor and arousal without signs of renal injury and pharmacological strategies to mimic such a state in non-hibernators may provide a safe alternative to therapeutic hypothermia in clinical practice. Signal transduction pathways affected by natural torpor and pharmacologically induced torpor-like states may emerge as promising therapeutic targets to safely lower metabolism and protect against (renal) IRI in humans.