
Abstract
Aims:
Paraquat (PQT), a redox-active herbicide, is a free radical-producing molecule, causing damage particularly to the nervous system; thus, it is employed as an animal model for Parkinson's disease. However, its impact on peripheral nerve demyelination is still unknown. Our aim is to decipher the influence of PQT-induced reactive oxygen species (ROS) production on peripheral myelin.
Results:
We report that PQT provokes severe locomotor and sensory defects in mice. PQT elicited an oxidative stress in the nerve, resulting in an increase of lipid peroxidation and protein carbonylation, despite the induction of nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent antioxidant defenses. We observed a dramatic disorganization of myelin sheaths in the sciatic nerves, dysregulation of myelin gene expression, and aggregation of myelin proteins, a hallmark of demyelination. PQT altered myelin gene expression via liver X receptor (LXR) signaling, a negative regulator of peripheral myelin gene expression through its dialog with the Wnt/β-catenin pathway. PQT prevented β-catenin binding on myelin gene promoters, resulting in the inhibition of Wnt/β-catenin-dependent myelin gene expression. Wnt pathway activation by LiCl dampened the deleterious effects of PQT. LiCl blocked PQT-induced oxidative stress and reduced Schwann cell death. LiCl+PQT-treated mice had normal sensorimotor behaviors and a usual nerve structure.
Innovation:
We reveal that PQT damages the sciatic nerve by generating an oxidative stress, dysregulating LXR and Wnt/β-catenin pathways. The activation of Wnt signaling by LiCl reduced the deleterious effects of PQT on the nerve.
Conclusion:
We demonstrate that PQT instigates peripheral nerve demyelinating neuropathies by enhancing ROS production and deregulating LXR and Wnt pathways. Stimulating Wnt pathway could be a therapeutic strategy for neuropathy treatment. Antioxid. Redox Signal. 27, 168–183.
Introduction
P

Paraquat (PQT) is a herbicide that induces oxidative stress, causing fatal damage. We report that PQT exposure provokes a burst of reactive oxygen species production, leading to peripheral nerve demyelination and consequent severe locomotor defects. At the molecular level, we demonstrate, for the first time, that PQT exerts its effect on myelin gene expression by activating liver X receptor (LXR) and inhibiting Wnt/β-catenin signaling, two essential pathways in myelination. The activation of Wnt pathway by the mood stabilizer lithium chloride reduced the deleterious effects of PQT on the nerves to restore its functionality. Thus, Wnt modulation could be considered a promising strategy to prevent PQT-induced neuropathy.
The excessive production of ROS or the decrease of antioxidant defenses in cells is implicated in the pathogenesis of diseases such as diabetes, atherosclerosis, and neurodegeneration (11, 12, 18). Indeed, PQT administration to rodents provoked cognitive impairment (7) and various features of Parkinson's disease, including motor deficits, dopaminergic neuronal loss, and α-synuclein aggregation (31). Moreover, PQT ingestion has been reported to cause cerebral edema and brain damage. An examination of the brain by electron microscopy showed a destruction of myelin, with abundant myelin breakdown products, and astrocytic gliosis (21). So, the purpose of the present work is to study the implication of oxidative stress induced by PQT in peripheral myelin breakdown. Indeed, in diabetic peripheral neuropathy, considerable evidence suggests that oxidative stress plays a critical role in reduction of sciatic nerve conduction, as well as alteration of sciatic nerve and myelin morphology and function (19, 36, 48).
The deleterious effects of oxidative stress are counteracted by the activation of adaptative responses that enhance the expression of genes encoding antioxidant enzymes. Nuclear factor erythroid 2-related factor 2 (Nrf2) is the key transcriptional regulator of antioxidant enzymes (28, 33): Glucose-6-phosphate dehydrogenase (G6PDH), isocitrate dehydrogenase 1 (IDH1), 6-phosphogluconate dehydrogenase (6-PGDH), glutathione-S-transferase (GSTM5, MGST1 and 2), and vitagenes heme oxygenase (HMOX), superoxide dismutase (SOD2), and NADP(H) dehydrogenase quinone 1 (NQO1). G6PDH, 6PGDH, and IDH1 protect cells from oxidative stress by reducing nicotinamide adenine dinucleotide phosphate (NAPDH) that is implicated in the renewal of reduced glutathione, an essential reducer against oxidative stress in cells.
Schwann cells produce a myelin sheath around axons in the peripheral nervous system to allow accurate nerve conduction. Myelin is composed of cholesterol, lipids, and proteins (myelin protein zero [MPZ] and peripheral myelin protein 22 [PMP22]) (17). Chronic elevation of oxidative stress alters the structure of myelin proteins and lipids and has negative consequences on myelin structure and nerve function in peripheral neuropathies (20). Some epidemiological data pinpoint that PQT intoxication distorted peripheral nerve functions. A study conducted on patients admitted after PQT self-poisoning showed that they had significantly lower nerve conduction velocity. The parameters returned to normal after 6 weeks (27, 45). However, it remains unknown whether PQT-induced oxidative stress could lead to peripheral demyelinating neuropathy.
Here, we revealed that the exposure of mice to PQT elicited severe locomotor defects accompanied by a disorganization of myelin sheaths and a deregulation of myelin genes. PQT intoxication triggered an elevation of oxidative stress in the nerve, provoking lipid peroxidation and myelin protein aggregation leading to demyelination. Within Schwann cells, PQT intoxication disturbed the steady-state level of myelin gene expression by altering positive and negative signals for myelination. PQT increases liver X receptor (LXR)-mediated inhibition by preventing Wnt/β-catenin to stimulate myelin gene expression. Our results demonstrate that PQT poisoning targets sciatic nerve and myelin proteins, which have a damaging impact on myelination. This observation may explain the peripheral nerve defects observed in humans exposed to PQT. Moreover, we showed that pre-treatment of mice with a modulator of Wnt/beta-catenin pathway, lithium, also known to be a mood stabilizer, counteracts the deleterious effect of PQT. Indeed, lithium restores locomotion, myelin sheath organization, and the myelin protein expression through the activation of the Wnt/beta-catenin pathway. Thus, the Wnt/beta-catenin pathway could be considered a promising therapeutic target against the deleterious effects of PQT on the peripheral nervous system.
Results
PQT provokes locomotor defects
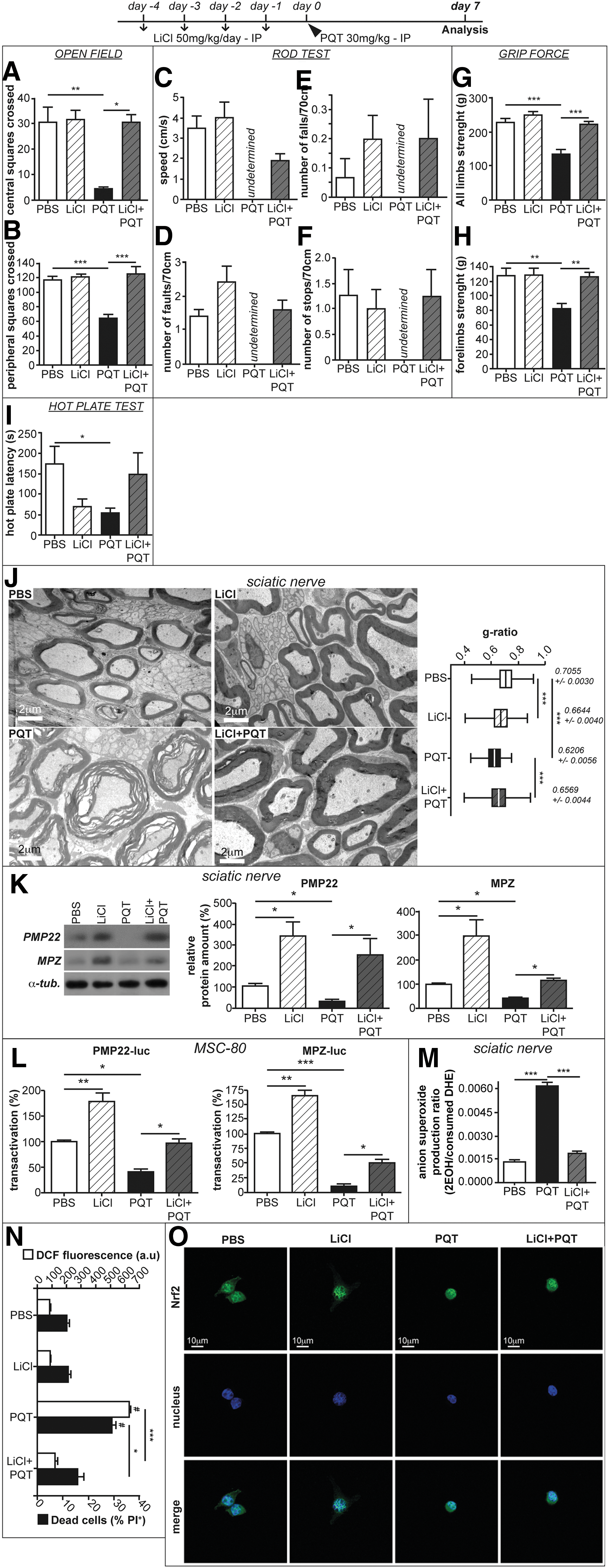
We first examined the behavioral defects induced by PQT intoxication. Mice received a single intraperitoneal injection of a low (10 mg/kg) or high (30 mg/kg) dose of PQT. Forty-eight hours and 1 week later, we recorded mouse ambulatory behavior in an open field. A significant decrease in the number of central (Fig. 2A) and peripheral (Fig. 2B) square crossings was found only in mice treated by a high dose of PQT. We then performed a Rod test to evaluate the locomotor skills of PQT-intoxicated mice. After 48 h, the mice that received a high dose of PQT were 2.5-fold slower than controls (Fig. 2C); the number of foot slips was increased by 3-fold (Fig. 2D). Moreover, we found that these mice fell three times during the rod crossing, whereas no falls were recorded for controls (Fig. 2E). PQT-mice also stopped twice more than the controls (Fig. 2F). Interestingly, 1 week after high-dose PQT administration, mice were no longer able to perform the rod test (Fig. 2C–F). Next, we performed a grip test to evaluate the muscular strength of the limbs. Mice that were intoxicated with a high dose of PQT displayed a significant decrease in the muscular strength of all the four limbs (Fig. 2G), including the forelimbs (Fig. 2H). Finally, we performed a hot plate test to assess the sensory behavior of the different groups of mice (Fig. 2I). Mice that received a high dose of PQT displayed after 1 week a significant decrease in the latency of leg withdrawal reflex compared with controls (50 s vs. 170 s). Taken together, all these data provided several lines of evidence that high-dose PQT intoxication induced severe motor and sensitive defects in mice.

PQT alters myelin structure
The alteration of locomotor and sensory activities prompted us to study the effect of PQT on myelin structure in the sciatic nerve of mice. Electron microscopy revealed that the myelin sheath structure of the sciatic nerves of mice treated with a low dose of PQT was unaffected (Fig. 2J). Interestingly, a high dose of PQT severely altered the myelin structure. We detected thicker myelin sheaths around the axons due to dramatic myelin sheath misfolding. In fact, we calculated the g-ratios (axon perimeter/nerve fiber perimeter) by using electron microscopy images. In phosphate-buffered saline (PBS)-treated mice, the average g-ratio was 0.7055 ± 0.0030; whereas in PQT-treated mice (30 mg/kg), it was significantly lower (g-ratio = 0.6206 ± 0.0056). By plotting the g-ratios of sciatic nerve as a function of their respective axonal diameters, we revealed an increase in myelin thickness regardless of the axonal size. These data show that the treatment with PQT (30 mg/kg) severely alters peripheral myelin structure in vivo.
PQT provokes ROS production, leading to depletion of GSH, lipid peroxidation, and protein carbonylation
Because PQT is known to dramatically induce ROS production, we analyzed the oxidative status in PQT-treated mice. Sciatic nerve superoxide production was assessed by quantifying oxidation of dihydroethidium (DHE) in 2-hydroxyethidium (2EOH) using high-performance liquid chromatography (HPLC) in either PBS-treated or PQT-intoxicated mice (Fig. 3A). PQT treatment (30 mg/kg) enhanced by threefold 2EOH production in the sciatic nerve when compared with the control group. Furthermore, PQT decreased the ratio of GSH/GSSG, confirming that PQT provokes oxidative stress in the sciatic nerves (Fig. 3B). It is likely that the elevation of oxidative stress leads to the accumulation of oxidative damage such as modification of lipids and proteins. Lipid peroxidation was revealed by the formation of a lipid peroxidation byproduct malonaldehyde (MDA). The levels of MDA in the sciatic nerves of PQT-treated mice were increased by twofold (Fig. 3C). In addition, we investigated protein carbonylation in sciatic nerves of PQT-intoxicated mice. We found a significant 2.5-fold increase in the overall level of protein carbonyls in sciatic nerves of PQT-treated mice compared with the control (Fig. 3D).
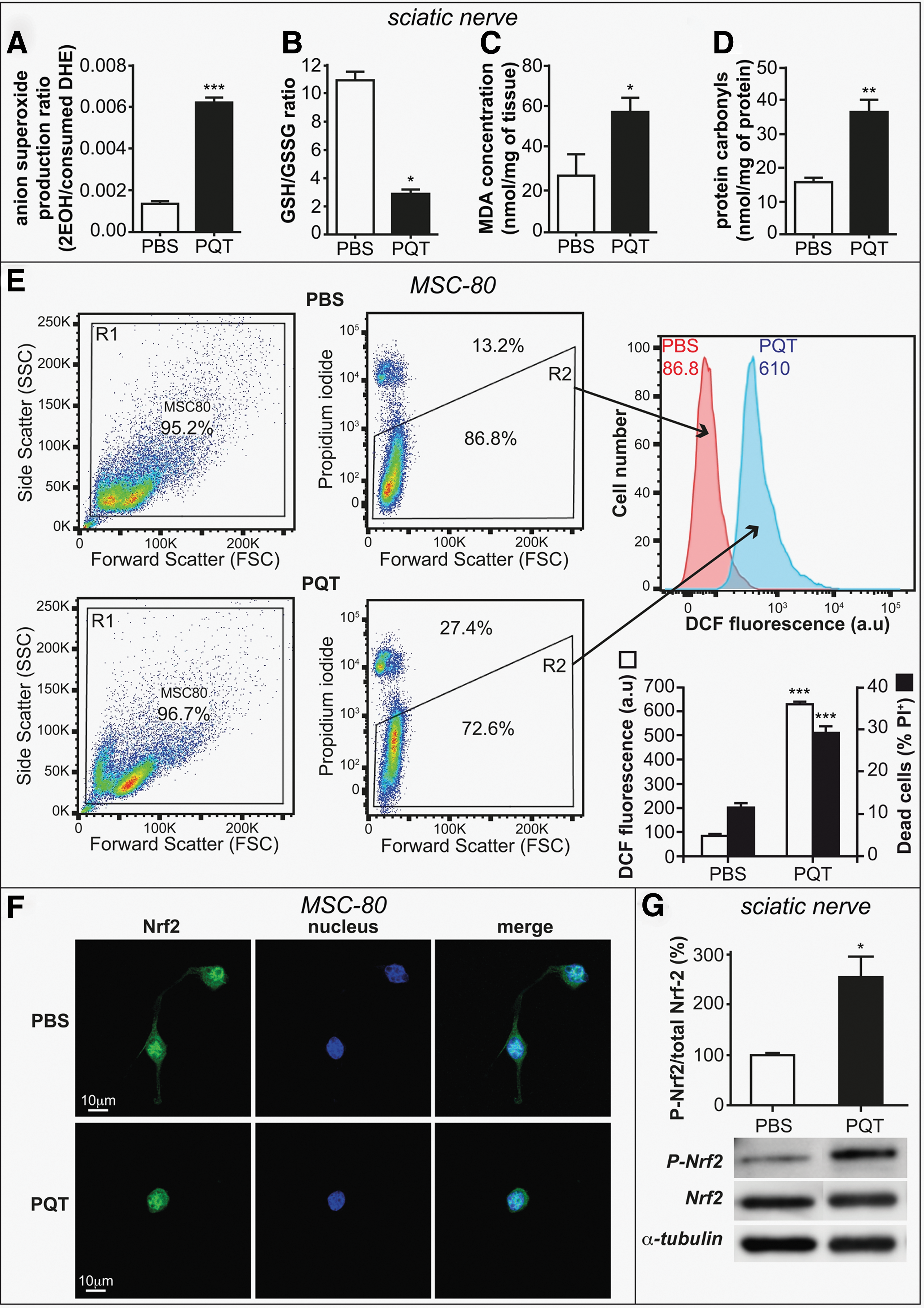
We also confirmed that PQT induced oxidative stress in the isolated Schwann cell line MSC80. We quantified the oxidative stress by means of a fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Fig. 3E). PQT (100 μM) enhanced ROS production after 24 h of treatment by sixfold, leading to an increase of MSC80 cell death by 3-fold. In addition, we studied the expression of Nrf2, a redox-sensitive transcription factor controlling antioxidant defenses and vitagenes. PQT (100 μM, 24 h) provoked the translocation of Nrf2 into the nucleus of mouse Schwann cell line (Fig. 3F) and increased by 2.5-fold the ratio of phospho-Nrf2/Nrf2 after 2 h of PQT administration in the sciatic nerves of mice (Fig. 3G).
PQT treatment enhances the adaptative response to oxidative stress
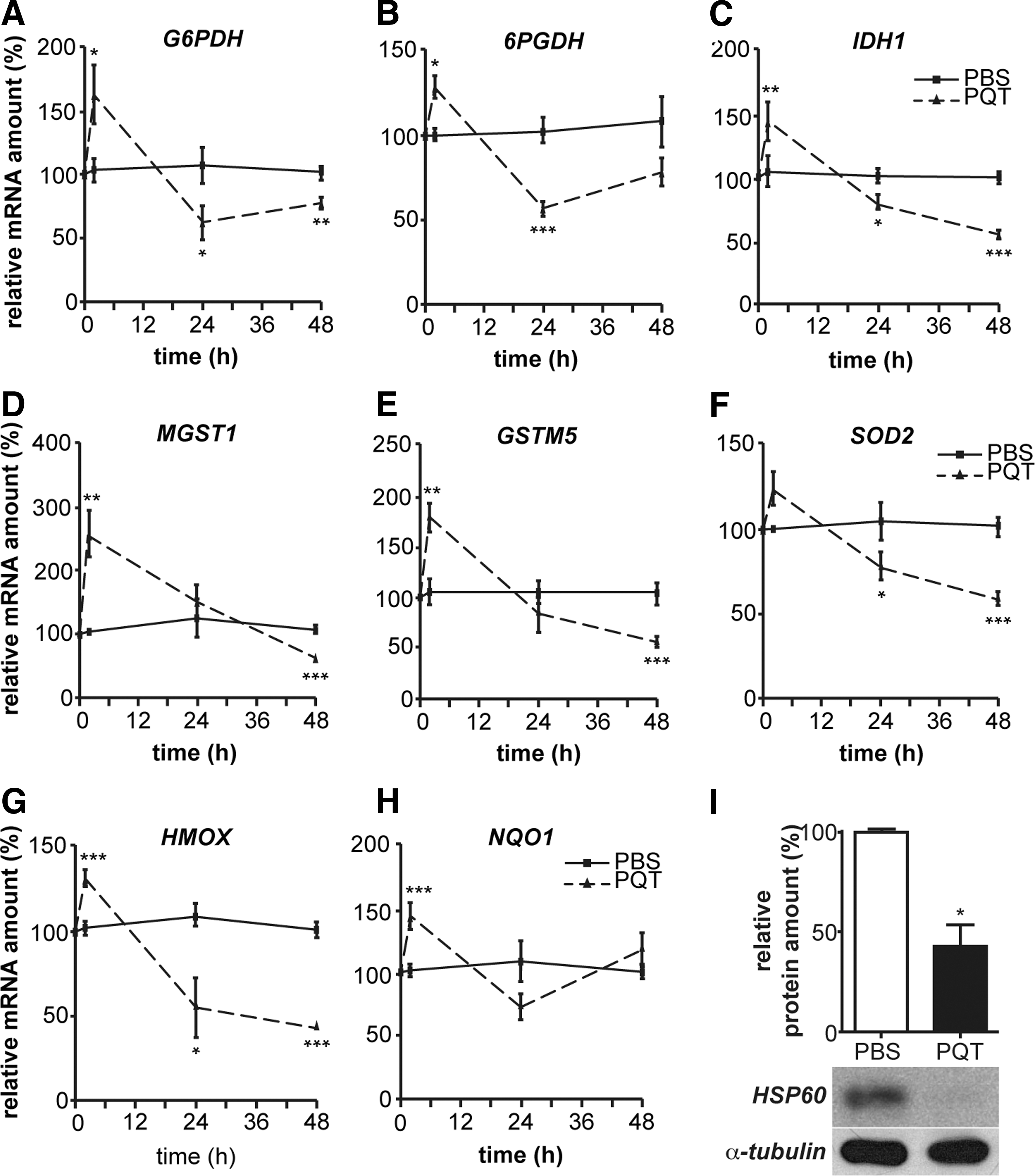
We investigated the adaptative molecular response to oxidative stress within the sciatic nerves of PQT-intoxicated mice. We analyzed by reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR) the expression of the enzymes related to glutathione metabolism. Two hours after PQT poisoning, we observed an increase in the mRNA amount of G6PDH (+58%), 6PGDH (+28%), IDH1 (+50%), MGST1 (+154%), GSTM5 (+65%), SOD2 (+24%), HMOX (+30%), and NQO1 (+43%) (Fig. 4A–H). Then, after 24 h, PQT intoxication decreased the amount of antioxidant transcripts: G6PDH (−46%), 6PGDH (−46%), IDH1 (−20%), SOD2 (−22%), HMOX (−27%), and NQO1 (−30%). As expected, after 48 h, all of antioxidant mRNA expression was decreased by PQT: G6PDH (−25%), 6PGDH (−25%), IDH1 (−50%), MGST1 (−15%), GSTM5 (−40%), SOD2 (−40%), and HMOX (−60%) (Fig. 4A–H). We also showed that the heat shock protein 60 (HSP60) protein amount, belonging to the vitagene network, is significantly decreased (−50%) after 48 h of PQT treatment (Fig. 4I). These findings revealed that the antioxidative response was not able to prevent PQT-induced oxidative stress.
PQT deregulates myelin expression in the sciatic nerve
We next examined the consequences of such an altered redox status on PMP22 and MPZ in the sciatic nerve of PQT-intoxicated mice. The expression of PMP22 transcript, assessed by RT-qPCR, was increased by 1.5- and 2-fold after 2 h and 24 h of PQT poisoning, respectively (Fig. 5A). It was abolished after 48 h. PMP22 protein level was diminished by 60% after 48 h and decreased until 1 week (−40%) (Fig. 5B). PQT intoxication did not significantly affect MPZ mRNA expression after 2 h and 24 h, but it decreased its expression after 48 h by 40% (Fig. 5C). At the protein level, PQT administration led to a 50% decrease of MPZ protein level after 48 h, which lasted until 1 week (−30%) (Fig. 5D).

Oxidative stress disrupts numerous cellular processes, among which is protein folding, leading to protein aggregation. So, we inquired whether PQT intoxication alters PMP22 aggregation. PMP22 contains hydrophobic domains that are detected by Kyte-Doolittle-Hydropathy software (not shown). We investigated PMP22 aggregation 1 week after PQT administration in the detergent-insoluble fraction of sciatic nerve protein extracts and observed a band migrating at 22 kDa related to PMP22 (Fig. 5E). Furthermore, slow-migrating anti-PMP22 reactive aggregates were detected at 37 kDa on the upper part of the gel in the sciatic nerve protein extracts of PQT-treated mice. This result showed that PQT, by generating oxidative stress, provokes oxidative post-translational modifications of PMP22, leading to myelin protein aggregation.
PQT inhibits myelin gene expression by deregulating LXR and Wnt/β-catenin pathways
To determine the action of PQT administration on myelin genes at the molecular level, we transiently transfected MSC80 cells with either PMP22-luciferase (Fig. 6A) or MPZ-luciferase (Fig. 6B) promoter constructs and incubated them with PQT or vehicle for 24 h. We showed that PQT disturbed myelin gene expression at the transcriptional level, as it drastically reduced the promoter activities of PMP22 and MPZ by 60% and 75%, respectively.

On the one hand, PQT, as a strong oxidative agent, is known to increase oxysterol production (22). On the other hand, we previously showed that LXRs and oxysterols inhibit peripheral myelin gene expression by inhibiting Wnt signaling (30). So, we hypothesized that PQT intoxication could alter LXR/oxysterol signaling to repress myelin gene expression. Thus, we first analyzed the effect of PQT intoxication on the LXR pathway in the Schwann cell line. A 24 h-treatment of MSC80 cells with PQT increased the mRNA amount of ATP binding cassette A1 (ABCA1) (+500%), a cholesterol transporter target of LXR (Fig. 6C). Furthermore, PQT also stimulated the expression of cytochrome 46A1 (CYP46A1) (+75%), the enzyme that converts cholesterol into 24(S) hydroxycholesterol, a ligand of LXR (Fig. 6D). Moreover, PQT increased the mRNA level of LXRβ, which is implicated in myelin gene repression (30) (Fig. 6E).
The stimulation of LXR pathway by PQT treatment inhibited Wnt/β-catenin signaling. Indeed, Axin 2 (a Wnt target gene) transcript was decreased by 60% (Fig. 6F) after PQT treatment. Dishevelled 2 (Dvl2) expression was reduced by 25% (Fig. 6G), and the expression of T cell factor 1 (TCF1) transcription factor was inhibited by 40% (Fig. 6H). At the protein level, PQT treatment also decreased active β-catenin protein amount (Fig. 6I), reducing the ratio of active β-catenin over total β-catenin.
To pinpoint the effects of PQT treatment on MPZ and PMP22 promoters, we investigated the presence of β-catenin at the level of two TCF/LEF response elements (TCFE) present in the MPZ promoter, and one TCFE located on the PMP22 promoter. ChIP analyses demonstrated that after PQT treatment, β-catenin recruitment on the MPZ promoter decreased by 80% at the level of the TCFE located at −8487/−8471 bp (Fig. 6J), and by 90% at the level of the second TCFE (−5487/−5461) (Fig. 6K). PQT treatment also decreased the recruitment of β-catenin on the PMP22 promoter (Fig. 6L). PQT did not affect the binding of IgG.
These observations were confirmed in vivo on the sciatic nerves of mice that were intoxicated with PQT. PQT increased the mRNA amount of ABCA1 by around 400% (Fig. 6M). We also detected an increase of the transcript level of CYP46A1 by ∼2.5-fold (Fig. 6N). Moreover, the mRNA amount of LXRβ was also increased on PQT intoxication in a time-dependent manner to reach almost 400% (Fig. 6O).
In the peripheral nerves, LXR crosstalks with Wnt signaling to fine-tune the myelination process (30). We detected an inhibition of Axin 2 transcript after 2 h (−50%) and 48 h (−90%) of PQT intoxication (Fig. 6P). Dvl2 expression was transiently enhanced by 90% after 2 h of PQT treatment, and it then decreased progressively in a time-dependent manner to reach −90% at 48 h (Fig. 6Q). The transcription factors TCF1 and TCF3 were also drastically down-regulated by PQT (Fig. 6R, S). We revealed a dramatic inhibition of active β-catenin protein expression and a decrease of the ratio of active β-catenin over total β-catenin (Fig. 6T). Our data show that PQT regulates signaling pathways governing myelin gene expression; it inhibits Wnt/β-catenin, whereas it activates LXR signaling that represses myelin genes (LXR).
Lithium, a Wnt/β-catenin modulator, prevents the deleterious effects of PQT on peripheral nerves
As the Wnt/β-catenin pathway is crucial for myelin preservation, we wondered whether its activation could overpass the deleterious effect of PQT. We mimicked Wnt activation by means of LiCl that inhibits GSK3β and increases the amount of active β-catenin (30, 32). Mice received a daily injection of LiCl (50 mg/kg) during 4 days before PQT poisoning (30 mg/kg). Interestingly, 1 week after PQT administration, LiCl restored the ambulatory behavior in the open field test. The mice treated with LiCl+PQT crossed the same number of squares as the PBS-treated ones (Fig. 7A, B). Moreover, PQT-intoxicated mice were not able to perform the rod test (Fig. 7C–F), whereas LiCl+PQT-treated mice succeeded. They had the same speed (Fig. 7C) and committed a similar number of faults (Fig. 7D) and stops (Fig. 7F) than PBS mice. The number of falls made by LiCl+PQT mice were equivalent to those made by PBS mice (Fig. 7E). Finally, the pre-treatment with LiCl improved the muscular strength of all limbs (Fig. 7G, H), as well as the sensory behavior (Fig. 7I) after PQT poisoning. It is noteworthy that we observed amelioration in the behavior of the LiCl+PQT mice as soon as 48 h after intoxication with PQT (Supplementary Fig. S1; Supplementary Data are available online at
Discussion
PQT is a commonly used herbicide despite its well established lung, renal, and neuronal toxicity (8, 27, 35). Patients ingesting PQT suffer from a transient alteration in the nerve conduction velocity. Our aim is to study the consequences of the administration of PQT on the peripheral nerve. One intraperitoneal injection of PQT (30 mg/kg, 48 h) severely alters the locomotor behavior of mice. The phenotype was more dramatic 1 week after PQT intoxication, accounting for the continuous ROS production elicited by a single PQT injection (4). In line with this, previous works highlighted the deleterious effect of high single doses of PQT (2, 8, 29, 37). In our 30 mg/kg PQT-treated mice, the alteration of locomotion suggests myelin abnormalities, leading to peripheral nerve defects. In fact, we observed that the myelin sheaths were misfolded, myelin gene expression was decreased, and PMP22 was aggregated.
We also confirmed that PQT generates massive ROS production observed in the sciatic nerves of PQT-treated mice and in the Schwann cell line incubated with PQT, despite the transient stimulation of Nrf2-related antioxidant genes and vitagenes (3, 43). In the sciatic nerve of PQT-treated mice, we detected a transient increase of antioxidant transcripts of enzymes involved in glutathione metabolism and cell detoxification. Their expression was decreased after 24 h, emphasizing the toxicity induced by PQT poisoning. The down-regulation of these vitagenes that are responsible for the increased adaptive ability to stresses (HMOX, HSP60, IDH1, NQO1, SOD, etc.) could account for the aggravation of the oxidative stress provoking demyelination associated with severe locomotor deficits. This phenomenon could lead to the death of Schwann cells.
We showed that this deleterious oxidative environment conducted lipid peroxidation, protein carbonylation, and aggregation of myelin proteins, among them being PMP22. In line with this, it is known that an increase in PMP22 insolubility in J trembler mice is causal for demyelination. Furthermore, PMP22 gene mutations and subsequent aggregation have been implicated in multiple mouse models of human peripheral neuropathies (14, 15, 20, 23, 41). The oxidative modification of proteins and their misfolding provoke their aggregation, which may have a critical role in neurodegenerative diseases and diabetic neuropathies (20). Finally, PQT induces axon degeneration of dorsal root ganglion neurons underlying peripheral neuropathies (13). These evidences could account for myelin disorders that were reported after PQT poisoning: myelin decompaction, repression of myelin genes, and subsequent behavioral defects.
As previously discussed, PQT-induced oxidative stress deregulates myelin gene expression. After a transient enhancement of MPZ and PMP22 mRNA expression, we observed a reduction of these genes 48 h after acute PQT exposure. MPZ and PMP22 are architectural proteins allowing the compaction of myelin sheaths. A slight modification in their expression initiates demyelinating neuropathies as observed in Charcot Marie Tooth diseases (38). By inhibiting myelin gene expression, PQT triggers the decompaction of myelin sheaths that become thicker. We showed that PQT acts directly on Schwann cells by decreasing the transactivation of MPZ and PMP22 promoters, suggesting a transcriptional effect of PQT on myelin genes. We have identified the mechanism by which PQT represses myelin gene expression. Indeed, it was already known that PQT enhances oxysterols synthesis as indices of oxidative stress (22). Interestingly, we showed that acute PQT administration enhanced the expression of the enzyme that converts cholesterol into oxysterol, the natural ligand of LXR. It, consequently, stimulated the LXR target gene ABCA1, confirming the activation of the LXR pathway that could participate in the inhibition of myelin gene expression in the sciatic nerves. Indeed, we previously showed that LXR signaling is able to repress peripheral myelin gene expression through its influence on the Wnt/β-catenin pathway, a positive regulator of the myelination process (30, 44). Besides its effect on myelin gene expression, LXR activation after PQT poisoning could also participate in the demyelinated phenotype by disrupting cholesterol homeostasis. Indeed, elevated cholesterol levels are essential for efficient targeting of MPZ from the endoplasmic reticulum to the surface of Schwann cells into the growing myelin sheath (42). As ABCA1 enhances cholesterol efflux from Schwann cells, the intracellular cholesterol levels are decreased and, as a consequence, MPZ trafficking is slowed. So, it appeared that PQT intoxication, by stimulating the LXR pathway, represses myelin gene expression, and concomitantly could impede the efficient targeting of myelin proteins to the membrane.
In Schwann cells, we also revealed that PQT treatment decreased the mRNA amount of several constituents of the Wnt/β-catenin pathway such as the transcription factors TCF1 and TCF3, Dvl2, Axin 2, and β-catenin. Furthermore, we highlighted that PQT down-regulates the active β-catenin amount that is able to bind to TCF/LEF transcription factors that are present in MPZ and PMP22 promoters. This provoked the decrease of β-catenin recruitment on TCF/LEF at the level of myelin gene promoters. In line with this, Wei et al. recently showed a decrease of the β-catenin amount that correlated with an exacerbation of oxidative stress in Parkinson disease (51).
As the inhibition of the Wnt/β-catenin pathway is detrimental for myelin sheath preservation (44), we wondered whether the stimulation of this pathway could counteract the deleterious effects of PQT. We have previously shown that the mood stabilizer, LiCl that mimics Wnt/β-catenin activation, has a pro-myelinating potential and neuroprotective potential (1, 30) Interestingly, LiCl pre-treatment reversed the toxic effects of PQT in mice and Schwann cells in vitro. LiCl restored the adequate structure of myelin sheaths, brought back to normal levels the expression of myelin genes, attenuated the oxidative stress elicited by PQT, decreased Schwann cell death, and stimulated the nuclear localization of Nrf2. In line with this, Alural et al. showed that lithium chloride promotes the nuclear translocation of Nrf2 by decreasing the PQT-induced expression of miR-34a that targets Nrf2 mRNA (1). The mice were able to adequately perform all the behavioral tests when they were pre-injected with LiCl. These data are in favor of a therapeutic potential of Wnt modulation after PQT poisoning.
In conclusion, we demonstrate that PQT is toxic not only for the central nervous system but also for the peripheral nerve. PQT provokes an intense oxidative stress that is not counteracted by the vitagene network. Schwann cell viability is then threatened. PQT dysregulates myelin genes through Wnt/β-catenin and LXR pathways, induces myelin protein PMP22 aggregation, and, consequently, disrupts the structure of myelin sheaths (Fig. 8). Interestingly the activation of the Wnt pathway by LiCl is able to dampen the demyelinating effects of PQT, thus opening an efficacious therapeutic avenue against PQT poisoning.

Materials and Methods
Animals and in vivo experiments
Eight-week-old male mice (C57BL/6) were purchased from Janvier. Independent groups of mice received (or not) daily pretreatment of LiCl injection for 4 days before receiving one intraperitoneal injection with PQT (10 or 30 mg/kg), LiCl (50 mg/kg), or a PBS from 2 h to 1 week before sacrifice. Sciatic nerves were collected and frozen in liquid nitrogen or fixed for histological analyses.
The behavioral tests (open field, rod test, grip force, and hot plate) were detailed in the Supplementary Materials and Methods section.
In vitro culture
The mouse Schwann cell line (MSC80) was maintained in Dulbecco's modified Eagle's medium (DMEM) that was supplemented with 10% decomplemented fetal calf serum (Hyclone-Perbio), 1% penicillin, 1% streptomycin, 1% glutamine, and 0.2% Fungizone (Gibco). All cultures were grown at 37°C in a humidified atmosphere of 5% CO2. MSC80 cells were incubated with PQT at the concentration of 100 μM in DMEM (34).
Plasmids and chemicals
MPZ-luc was provided by Dr. G Lemke (The Salk Institute, La Jolla, CA), and PMP22-luc was provided by Dr. P Patel (Baylor College of Medicine, Houston, TX). PQT was purchased from Sigma-Aldrich (ref. 36541).
Transient transfections
The mouse Schwann cell line (MSC80) was transiently transfected by using Effecten reagent (Effecten Transfection Reagent; Qiagen, Cat. No. 301425). Sixteen hours after transfection, the medium was replaced by DMEM containing PQT (100 μM) and/or lithium chloride or vehicle (PBS). Luciferase activity was determined by using the enzymatic method. The β-galactosidase activity was used to normalize the transfection efficiency (see Supplementary Materials and Methods section).
RT-qPCR experiments
Total RNA was obtained by using RNA NOW (Ozyme). One microgram was reverse transcribed with random primers from Promega and MMLV reverse transcriptase from Sigma-Aldrich (ref. M1302). qPCR was performed with standard protocols by using SYBRGreen ROX Mix (ABgene) as a fluorescent detection dye in ABI PRISM 7000 (see Supplementary Materials and Methods section).
Western blot
Protein content was determined by using the “RC DC” protein assay kit (Bio-Rad) with bovine serum albumin (BSA) as standard. Aliquots of 20 μg of total MSC80 extracts pre-treated with PBS, lithium (10 mM), or/and PQT (100 μM) were used for each sample. Homogenate proteins were separated on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto polyvinylidene difluoride membranes. Nonspecific binding sites in the transblots were blocked at 4°C overnight with 5% BSA in Tris buffer saline 1 × (TBS) with 0.1% Tween 20 (Invitrogen). Membranes were then incubated at 4°C overnight with the following primary antibodies diluted in a mixture of TBS-0.1% Tween: active β-catenin antibody (dephosphorylated) (1/250; Millipore), total β-catenin antibody (1/1000; BD Biosciences), MPZ antibody (1/1000; Abcam), PMP22 antibody (1/1000; Abcam), HSP60 (1/1000; Santa Cruz), and α-tubulin antibody (1/10,000 for normalization). They were then incubated at room temperature for 1 h with the appropriate secondary antibody diluted in 5% BSA/TBS-0.1% Tween (anti-rabbit at 1:20,000; anti-mouse at 1:20,000), followed by ECL Plus. Protein bands were visualized with an enhanced chemiluminescence detection system. Western blots were quantified by means of NIH Image J Software.
Protein aggregation
Frozen sciatic nerves from adult mice were crushed under liquid nitrogen; lysed in immunoprecipitation buffer (10 mM Tris–HCl [pH 7.5], 5 mM EDTA, 1% Nonidet P-40, 0.5% deoxycholate, and 150 mM NaCl) that was supplemented with protease inhibitors (14). For each lysate, at least six nerves from three mice per genotype were used. The lysates were microcentrifuged; the supernatant was removed; and the insoluble material was incubated with 10 mM Tris–HCl, 3% SDS for 10 min at room temperature. After a brief sonication, total protein concentrations were measured by DC assay (BioRad). Equal amounts of samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. Blots were blocked with 5% BSA/TBS-0.1% Tween and incubated with the indicated PMP22 primary antibody. They were then incubated at room temperature for 1 h with the appropriate secondary antibody diluted in 5% BSA/TBS-0.1% Tween (anti-rabbit at 1:20,000; anti-mouse at 1:20,000), followed by ECL Plus. Protein bands were visualized with an enhanced chemiluminescence detection system. Western blots were quantified by means of NIH Image J Software.
Chromatin immunoprecipitation
MSC80 cells (10 millions) were fixed with 1% formaldehyde for 10 min. The cross-linking was quenched with 0.125 M glycine for 10 min. Fixed chromatin was then isolated, which was later physically sheared by sonication to obtain DNA shearing of an average length of 300–600 base pairs. Immunoprecipitation was done by using Protein A/G magnetic nanobeads (Dynabeads Life Technologies). The antibodies used in ChIP assays are as follows: 5 μg of β-Catenin (BD transduction No. 610154) and 5 μg Rabbit IgG (Sigma I5006) (5) In addition, we have also validated our ChIP assays by performing Western blots each time by using one third volume of the precipitated samples (data not shown). Crosslinks were reversed by incubating samples at 65°C during 4 h. DNA was isolated by phenol/chloroform extraction and ethanol precipitation followed by purification using GENECLEAN BPbio columns. Genomic DNA (input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heated for de-crosslinking, followed by ethanol precipitation and column purification. RT-qPCRs were done as mentioned earlier with the primer pairs listed in the primers' table. Then, the percentage input was calculated, as mentioned in (16).
Lipid peroxidation
Lipid peroxidation assay kit (abcam) was used to detect MDA present in samples as described in the Supplementary Materials and Methods section.
Protein carbonylation
Sciatic nerves were homogenized in 5–10 ml of 100 mM Tris, pH 7.4 and then, tissues were centrifuged at 10,000 g for 15 min at 4°C. We removed the supernatant and stored it on ice. Then, the protein concentration of each sample was determined as previously described. The protein concentrations have to be adjusted to between 5 and 10 mg/ml. Briefly, 200 μl of each sample was mixed with 200 μl of 10 mM 2,4-dinitrophenylhydrazine in 2 N HCl or with 200 μl of 2 N HCl alone for the blank and incubated at room temperature for 1 h in the dark with vortexing every 15 min. Proteins were precipitated with 20% trichloroacetic acid (w/v), vortexed, and centrifuged (13,000 g for 3 min). The pellet was washed three times with 1 ml ethanol-ethyl acetate (1:1 v/v) before re-dissolving in 1 ml of 6 M guanidine HCl in 20 mM potassium phosphate adjusted to pH 2.3 with trifluoroacetic acid. The absorbance was measured in the supernatant at 360 nm and carbonyl content was calculated, using the molar absorption coefficient of 22,000 M −1 cm−1 relative to protein concentration (40).
Transmission electron microscopy
Mice were deeply anesthetized by an intraperitoneal injection of 40 mg/kg ketamine and 30 mg/kg xylazine and then intracardially perfused with 4% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 m phosphate buffer, pH 7.4. Tissues were dissected and immersed in the same fixative solution at 4°C overnight, washed in phosphate buffer, postfixed in 2% osmium tetroxide, dehydrated in graded ethanol series, and embedded in epoxy resin. For electron microscopy, ultrathin sections (50–90 nm) were cut on an ultramicrotome (8800 Ultrotome III; LKB Bromma) and collected on 300-mesh nickel grids. Staining was performed on drops of 4% aqueous uranyl acetate, followed by Reynolds's lead citrate (39). Ultrastructural analyses were performed in a JEOL jem-1011 electron microscope and digitalized with Digital Micrograph software. Image acquisition was performed at the Cochin Imaging Facility. Electron microscopy images were used for calculating the g-ratio and axon perimeter by using NIH Image J software. At least 100 randomly selected axons were analyzed per animal. At least three animals were used per group. Healthy axons were defined on the basis of the presence of intact membranes and the normal complement of organelles.
Assessment of cytosolic ROS by DCFH-DA
Cellular oxidative stress was measured by flow cytometry by using the DCFH-DA method. DCFH-DA diffuses through the cell membrane and is hydrolyzed enzymatically by intracellular esterase to non-fluorescent DCFH, which is rapidly oxidized to highly fluorescent 2′,7′-dichlorofluorescein. For measurement of ROS, MSC80 cells (250,000 cells/well in six-well plates) were incubated with paraquat dichloride in a time-course manner. After washing with PBS 1 × without Ca2+ and Na+, adherent cells were detached with trypsine-EDTA. Then, cells were suspended in medium and centrifuged at 400 g for 3 min. Subsequently, cell pellets were resuspended with the medium containing cells in suspension previously recovered. DCFH-DA stock (Molecular Probes by Life Technologies, D-399) was prepared in dimethyl sulfoxide, held under N2, and stored at 80°C to reduce premature oxidation. For DCFH-DA staining, 1 ml of MSC80 cells diluted in PBS was exposed to 0.5 μM DCFH-DA at 37°C for 30 min.
Flow cytometry analysis
We used an FACSCanto flow cytometer (Canto Leia II, BD FACSCanto™ Flow cytometer; BD Biosciences) that was equipped with a 488-nm argon laser to analyze all fluorescence signals of the labeled MSC80 cells. A total of 10 events were recorded at a light scatter to exclude debris and aggregate with a flow rate of less than 200 cells/second for each assay. Green fluorescence (DCF) and red fluorescence (PI) were detected in the FL-1 channel with a 530/30 nm band-pass filter, respectively. The compensations and the settings were adjusted according to the assay. Data were analyzed by using Flow Jo, single-cell analysis, Software.
In situ detection of superoxide by HPLC
Briefly and as previously described (10, 11, 25, 26), freshly isolated nerves were stroked with mortar and pestle and suspended in 40 μl of acetonitrile. The solution was then centrifuged at 12,000 g for 10 min at 4°C. The homogenate was dried under vacuum and analyzed by HPLC with fluorescence detectors (Jasco HPLC system, LC-2000 plus series). Quantification of DHE, 2 hydroxyethidium (2EOH), and ethidium concentrations was performed by a comparison of integrated peak areas between the obtained and standard curves of each product under identical chromatographic conditions as described. EOH and ethidium were monitored by fluorescence detection with excitation at 510 nm and emission at 595 nm, whereas DHE was monitored by ultraviolet absorption at 370 nm.
Statistical analysis
Unless otherwise specified, means of treatment groups were compared with one-way ANOVA. When the ANOVA showed that there were significant differences between the groups, Newman–Keuls test was used to identify the sources of these differences. p ≤ 0.05 was considered statistically significant. Two-group comparisons were performed by Student's t-test.
Footnotes
Acknowledgments
This work was funded by INSERM, CNRS, Paris Descartes University, ANR. M.H. and N.K. received PhD fellowships from the French Ministry of Research (MNRT). The authors greatly acknowledge Claire Mader from the Animal Facility of the Saints Pères, Jean-Maurice Petit from the Saints-Pères Imaging Facility, A. Schmitt from the Cochin Imaging Facility, and S. Dupuy from the Saints-Pères Flow Cytometer Facility.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.