
Abstract
Aims:
Mitochondrial dysfunction has emerged as a major contributing factor to endothelial dysfunction and vascular disease, but the key mechanisms underlying mitochondrial dysfunction-induced endothelial dysfunction remain to be elucidated. In this study, we aim at determining whether mitochondrial dysfunction in endothelial cells plays a key role in vascular disease, by examining the phenotype of endothelial-specific CR6-interacting factor 1 (CRIF1) knockout mice. We also used siRNA-mediated downregulation of CRIF1 gene in the endothelial cells to study about the in vitro pathophysiological underlying mechanisms.
Results:
Downregulation of CRIF1 in endothelial cells caused disturbances of mitochondrial oxidative phosphorylation complexes and membrane potential, leading to enhanced mitochondrial reactive oxygen species production. Gene silencing of CRIF1 results in decreased SIRT1 expression along with increased endothelial nitric oxide synthase (eNOS) acetylation, leading to reduced nitric oxide production both in vitro and in vivo. Endothelium-dependent vasorelaxation of aortic rings from CRIF1 knockout (KO) mice was considerably less than in wild-type mice, and it was partially recovered by Sirt1 overexpression in CRIF1 KO mice.
Innovation:
Our results show for the first time a relationship between mitochondrial dysfunction and impaired vascular function induced in CRIF1 deficiency conditions and also the possible underlying pathway involved.
Conclusion:
These findings indicate that CRIF1 plays an important role in maintaining mitochondrial and endothelial function through its effects on the SIRT1-eNOS pathway. Antioxid. Redox Signal. 27, 234–249.
Introduction
E
Even though it is known that CR6-interacting factor 1 (CRIF1) deficiency is a crucial factor leading to mitochondrial dysfunction in endothelial cells, it is not clear how it leads to impaired vascular function in cardiovascular disease pathogenesis. We showed that CRIF1 deficiency induces oxidative stress and mitochondrial dysfunction in endothelial cells, leading to downregulated SIRT1 levels accompanied with increased endothelial nitric oxide synthase (eNOS) acetylation and decreased nitric oxide production that promote impaired vascular function. eNOS is identified as an SIRT1 substrate under oxidative stress, highlighting the beneficial effect of SIRT1 on endothelial vascular biology for prevention of oxidative stress-induced endothelial dysfunction.
Mitochondria occupy at least 5% of endothelial cell volume and contribute to ATP generation in a process called oxidative phosphorylation (OXPHOS). Even though mitochondria are not the major source of energy production for endothelial cells, under certain conditions, the reliance of endothelial cells on OXPHOS is increased, and the contribution of mitochondrial respiration cannot be ignored (11). In addition to ATP generation, among the five OXPHOS complexes embedded in the inner mitochondrial membrane, complexes I (NADH dehydrogenase) and III (ubiquinol- cytochrome oxidoreductase) produce significantly high levels of ROS, even under physiological conditions. Thus, damaged mitochondria causing excessive production of mitochondrial ROS, and aggravating mitochondrial dysfunction may lead to various cardiovascular diseases such as heart disease (11) and atherosclerosis (12, 48) by impairing their normal function. Therefore, maintenance of normal mitochondrial function is essential to ensure maintenance of ROS contents, efficient ROS scavenging and to prevent oxidative stress-related events. There is a clear relationship between mitochondrial dysfunction, mitochondrial ROS production, and endothelial dysfunction (8, 28).
Sirtuins are a highly conserved family of proteins that possess either mono-ADP-ribosyl transferase or deacetylase activity. Mammals have seven sirtuins (SIRT1-7) localized throughout the cell; all SIRT family proteins regulate cellular metabolism and mitochondrial function according to their dependence on nicotinamide adenine dinucleotide (NAD+). Silent information regulator factor 2-related enzyme 1 (sirtuin 1, SIRT1) is an (NAD)-dependent deacetylase that helps regulate many physiological functions, including gene transcription, cell senescence, energy metabolism, and oxidative stress (47). It is also well known for deacetylating histones, nonhistone proteins, and other transcription factors. SIRT1 also has a significant role in modulating endothelial cell functions. Of particular importance is its ability to physically interact with endothelial nitric oxide synthase (eNOS), resulting in eNOS deacetylation and its enhanced activation (34). Lysine acetylation-deacetylation has been recognized as a novel post-translational mechanism for the regulation of eNOS activity. The importance of this post-translational modification of eNOS lies in the fact that eNOS is activated only after its SIRT1-mediated deacetylation at the lysine residue. Thus, for the regulation of eNOS enzymatic activity, there should be a fine tuning between acetylation-deacetylation mechanisms (24). Further, eNOS activation results in generation of nitric oxide (NO), one of the most important factors regulating vascular homeostasis. Inhibition of SIRT1 in the arterial endothelium results in the suppression of endothelium-dependent vasodilation and a decrease in bioavailable NO (34). Notably, ROS-mediated depletion of cellular NO level is known to promote endothelial dysfunction (44). A previous study demonstrated that an increase in ROS levels can both directly and indirectly control SIRT1 enzymatic activity (42).
CR6-interacting factor 1 (CRIF1) is a mitochondrial protein that is essential for the production of OXPHOS polypeptides within the mitochondria and their subsequent insertion into the inner mitochondrial membrane (27), and CRIF1 deficiency is a major factor underlying mitochondrial dysfunction in endothelial cells (35). We hypothesized that downregulation of CRIF1 leads to oxidative stress-induced mitochondrial dysfunction and subsequent endothelial dysfunction via the SIRT1-eNOS pathway. CRIF1 deficiency induces excessive mitochondrial ROS production and subsequent mitochondrial dysfunction, leading to endothelial activation (35). Previous studies support the relationship between the SIRT1 and eNOS pathway. SIRT1 is shown to regulate endothelial function, and, hence, any alteration in endothelial SIRT1 will affect normal vascular physiology (1, 24). It has been shown that oxidative stress downregulates SIRT1, leading to acetylation of eNOS, which results in reduced NO-mediated signaling and endothelial dysfunction. Human umbilical vein endothelial cells (HUVECs) exposed to oxidative stress showed decreased SIRT1 levels, concomitant with increased eNOS acetylation (1). Our results demonstrate that knockdown of CRIF1 in HUVECs induces mitochondrial dysfunction and reduces SIRT1 expression along with decreased eNOS phosphorylation and increased eNOS acetylation, thereby promoting endothelial dysfunction. In this context, it is important to note that administration of a mitochondria-specific antioxidant abolished the detrimental effects of ROS, recovering the SIRT1-eNOS axis pathway and redox signaling. These findings provide proof of concept regarding the pivotal contribution of mitochondrial oxidative damage toward disease pathogenesis.
Results
Endothelial-specific CRIF1 knockout mice showed short life span and mitochondrial dysfunction
To investigate the role of CRIF1 and vascular function, we first attempted to establish primary OXPHOS deficiency in vascular endothelial cells in vivo by using endothelial-specific CRIF knockout (CRIF1 KO) mice. We crossed conditional CRIF1 loxp mice (CRIF1 flox/flox) with mice expressing a Cre recombinase gene under the control of the endothelial-specific receptor tyrosine kinase promoter (Tek-Cre). Knockout of the CRIF1 gene was confirmed by immunohistochemistry in aorta and tissue Western blot in various tissues (Fig. 1A, B and Supplementary Fig. S5; Supplementary Data are available online at
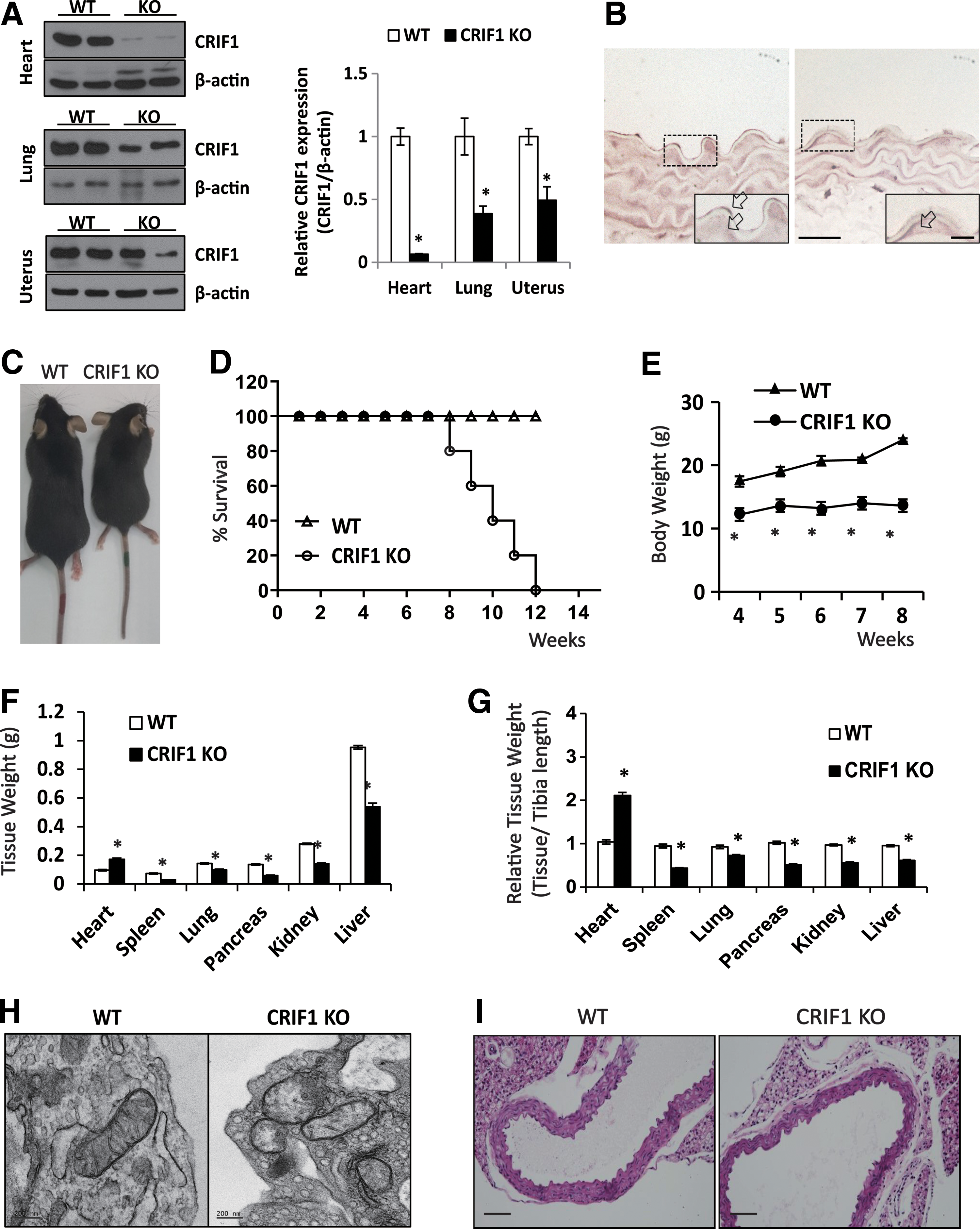
For identifying the heart function, we performed echocardiography in 8 week-aged mice. CRIF1 KO mice showed reduced EF (ejection fraction) and FS (fractional shortening) as compared with the WT mice, suggesting LV systolic dysfunction in CRIF1 KO mice (Supplementary Fig. S1A, B). We also confirmed pathology in brain, pancreas, spleen, lung, kidney, etc. (data not shown). Although some tissues showed abnormal morphology, there were no observed phenotypic changes that could lead to death. These results lead us to believe that dysfunctional heart may be the causative factor for premature death of EC CRIF1 KO mice. Collectively, these data suggest that homozygous loss of CRIF1 in endothelial cell leads to marked phenotypic changes in the heart, which leads to death. To test whether the mitochondrial dysfunction affects vascular physiologic and pathologic functions, we verified mitochondrial abnormalities in endothelial cells from aorta in 10-week-old WT and CRIF1 KO mice by using transmission electron microscopy (TEM). TEM images of mitochondria in the vascular endothelial cells showed noticeable morphological abnormalities such as distorted or fewer cristae in CRIF1 KO mice (Fig. 1H).
To identify the morphological or pathological changes of vascular structure between WT and CRIF1 KO mice, we performed hematoxyline and eosin staining (H&E staining) in 3-, 4-, and 5 week-aged mice (Fig. 1I). In the CRIF1 KO aorta, there was no difference in the tunica intima structure, but the length of tunica media and perimeter was shorter than in WT mice (Supplementary Fig. S2A). And the aorta size was not changed in CRIF1 KO mice even though grown, which was distinct from WT mice (Supplementary Fig. S2A). Distribution analysis of tunica media length showed a shift in peak frequency toward a higher value in WT mice than CRIF1 KO mice, according to the age of the mice (Supplementary Fig. S2B). But the relative length of tunica media and perimeter, normalized by tibia length or body weight, was moderately similar to other tissues (Fig. 1G and Supplementary Fig. S2A). This comprehensive analysis of the heart and aorta in CRIF1 KO mice indicated that loss of CRIF1 in endothelial cells results in a marked alteration of heart and aorta biology.
CRIF1 siRNA decreases OXPHOS subunits and impairs mitochondrial function in endothelial cells
We previously showed that OXPHOS complex synthesis was defective due to failed translation and insertion of newly synthesized OXPHOS complexes in CRIF1-deficient MS1 cells (27, 35). To identify the role of mitochondrial CRIF1 in endothelial cells, we examined the mitochondrial functions in HUVECs after CRIF1 knockdown. As shown in Figure 2A and Supplementary Figure S6, CRIF1 protein levels were markedly reduced 48 h after transfection with siRNA targeting CRIF1. Similar to CRIF1-deficient MS1 cells, the silencing of CRIF1 downregulated the protein expressions of OXPHOS complex subunits, especially that of Complex I (NDUFA9), Complex II (SDHA), Complex III (UQCRC2), and Complex IV (COX 4) (Fig. 2A and Supplementary Fig. S6). To assess mitochondrial abnormalities, we examined the mitochondria of CRIF1-deficient HUVECs by using TEM. The mitochondrial TEM image of CRIF1-deficient HUVECs showed an abnormal structure of cristae and encapsulated mitochondria by a double membrane, such as a mitophagy (Fig. 2B), suggesting that CRIF1 plays a critical role in both OXPHOS complex formation and mitochondrial structure in endothelial cells.

To directly estimate the mitochondrial function in CRIF1-deficient cells, the mitochondrial oxygen consumption rate (OCR) was measured by using the Seahorse XF24 extracellular flux analyzer. CRIF1-deficient cells showed lower OCR under basal conditions before exposure to oligomycin, which inhibits ATP synthase, and caused the expected decrease in OCR in both control and CRIF1-silenced endothelial cells (Fig. 2C). To measure maximal mitochondrial respiratory capacity, we treated carbonyl cyanide m-chlorophenyl hydrazone (CCCP), an uncoupler of mitochondrial respiration in cells. Cells usually increase OCR in response to CCCP to maintain proton gradients and mitochondrial function. CRIF1-silenced cells exhibited less of an increase in OCR after CCCP treatment compared with control cells, suggesting that these cells have less spare respiratory capacity than control cells. According to the previous report, CRIF1-deleted mouse embryonic fibroblast cells showed aberrant synthesis and defective assembly of OXPHOS complex I, III, and IV, leading to impaired mitochondrial respiration but enhanced glycolysis (27). Severe dysfunction of mitochondrial respiration leads to energy crisis in the cells and finally results in cell death. On the contrary, in mild dysfunction of mitochondrial respiration, glycolysis is substituted for deficiency of mitochondrial ATP supplement via metabolic switch and the cells can survive without undergoing cell death (40). To verify this theory, along with measuring the mitochondrial OCR, we measured the extracellular acidification rate (ECAR), which is a measure of glycolysis. As shown in Figure 2D, siCRIF1-transfected cells showed enhanced glycolysis. Moreover, staining with tetramethylrhodamine (TMRE) dye showed decreased mitochondrial membrane potential (MMP) in CRIF1-deficient cells. Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) was used as a positive control, which is known to decrease the MMP (Fig. 2E). Accompanied with impaired mitochondrial function, the dynamics of mitochondrial fusion-fission proteins were altered (Fig. 3A, B and Supplementary Fig. S7). Overall, mitotracker-red and DAPI staining showed that mitochondrial fragmentation was increased in CRIF1 knockdown HUVEC cells (Fig. 3B). These results suggest that CRIF1 deficiency induces mitochondrial dysfunction in endothelial cells.

CRIF1 siRNA decreases SIRT1 expression via mitochondrial ROS
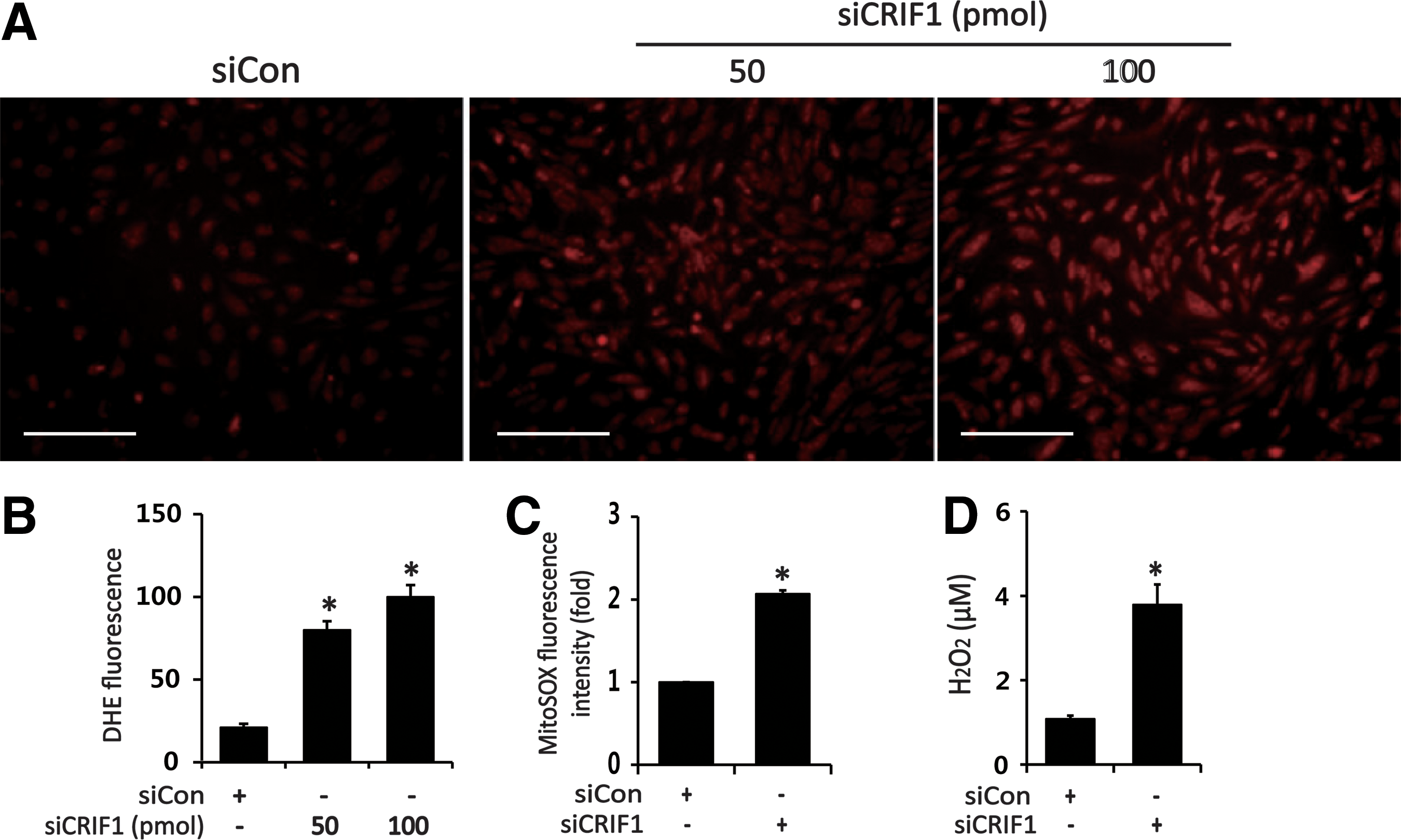
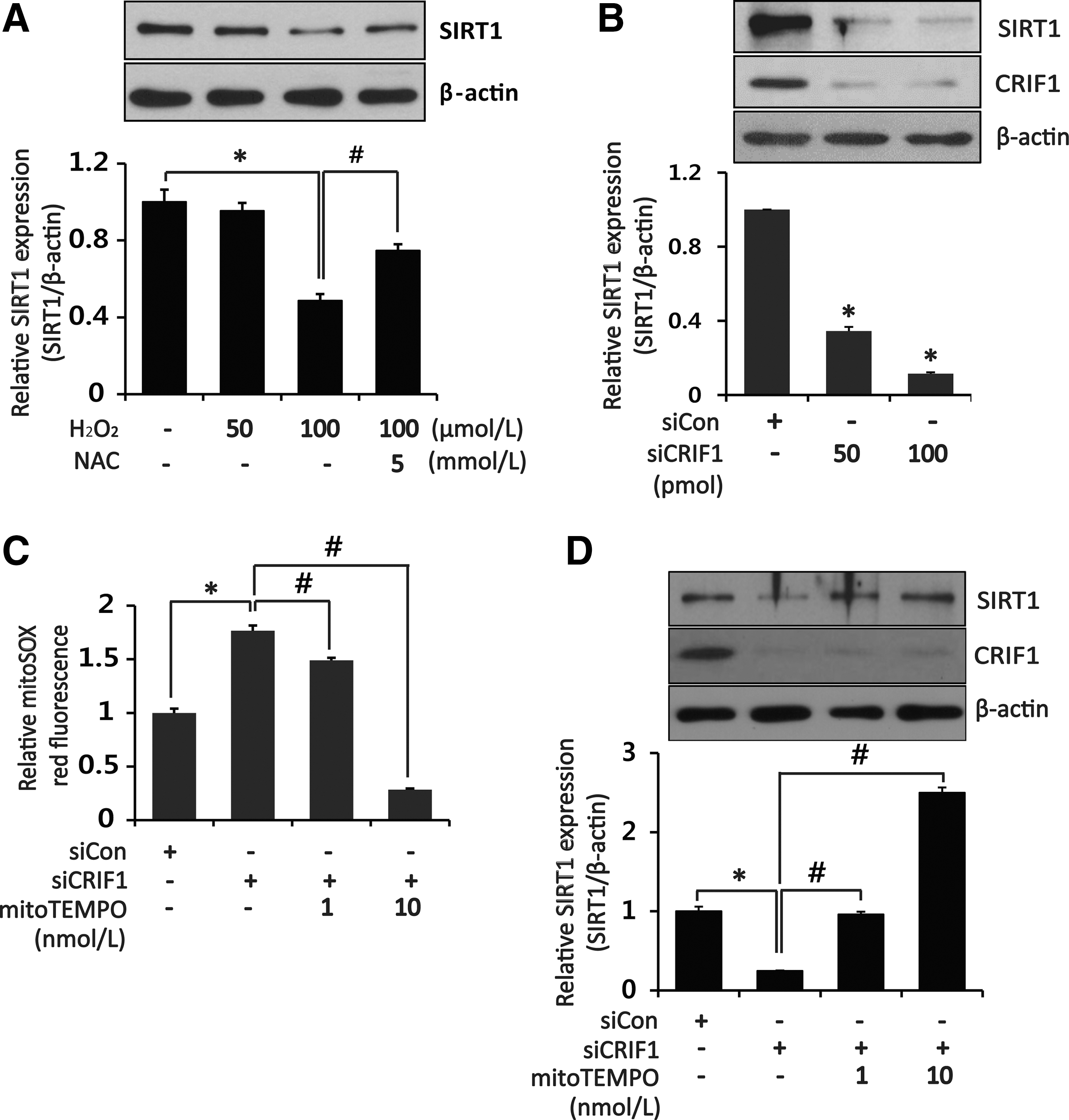
Considering the relationship between mitochondrial dysfunction and ROS generation, we next examined the effect of CRIF1 knockdown on mitochondrial ROS production in HUVECs. CRIF1 knockdown induced a marked increase in MitoSOX fluorescence intensity compared with control siRNA (Fig. 4C). Along with increased mitochondrial ROS levels, intracellular ROS levels were also augmented in CRIF1-deficient cells compared with control cells as measured by dihydroethidium (DHE) fluorescence intensity (Fig. 4A, B). Similarly, markedly higher levels of hydrogen peroxide (H2O2) were released from CRIF1-silenced cells compared with the control cells (Fig. 4D). Previous reports showed that oxidative stress decreases SIRT1 expression in the hippocampus and cerebral cortex, and direct modification of SIRT1 by ROS can mediate its proteasomal degradation (6). Therefore, we next examined whether CRIF1 deficiency-induced oxidative stress affects SIRT1 expression in HUVECs. We first established the direct effect of oxidative stress on SIRT1 by measuring SIRT1 expression in HUVECs 24 h after H2O2 treatment. As shown in Figure 5A and Supplementary Figure S8, H2O2 downregulated SIRT1 protein expression in a dose-dependent manner, which was significantly attenuated by pretreatment with the antioxidant N-acetyl-
CRIF1 siRNA decreases eNOS activity via decreased SIRT1 expression in endothelial cells
eNOS plays an important role in endothelial function and is inactivated by ROS (19). In addition, eNOS can physically interact with SIRT1 to stimulate eNOS upregulation through acetylation of lysine residues and by maintaining endothelial function (34). Moreover, de-acetylation of eNOS by SIRT1 occurs through its phosphorylation and finally enhances NO production (7). To identify the effect of mitochondrial dysfunction on eNOS activity, we first examined the eNOS phosphorylation in CRIF1-deleted HUVECs. eNOS phosphorylation was significantly decreased in a dose-dependent manner in siCRIF1-treated HUVECs (Fig. 6A and Supplementary Fig. S9). Next, we determined whether CRIF1 knockdown modifies SIRT1-mediated eNOS deacetylation, thereby rendering eNOS inactive. We found that eNOS acetylation was significantly increased in CRIF1-silenced cells (Fig. 6B, D and Supplementary Fig. S9). Furthermore, MitoTEMPO pretreatment (10 nM) for 1 h or adenovirus-mediated SIRT overexpression inhibited the increase in eNOS acetylation on lysine residues caused by CRIF1 knockdown (Fig. 6B, D and Supplementary Fig. S9). Finally, eNOS-derived NO production was similarly decreased in CRIF1-deficient HUVECs and recovered in CRIF1-deficient HUVECs that were treated with MitoTEMPO or adeno SIRT1 (Fig. 6C, E). Taken together, these results suggest that CRIF1 deficiency increased eNOS acetylation at lysine residues via decreased SIRT1 expression and reduced eNOS phosphorylation and NO bioavailability in endothelial cells.

CRIF1 deficiency decreases mitochondrial function and eNOS activity in vivo
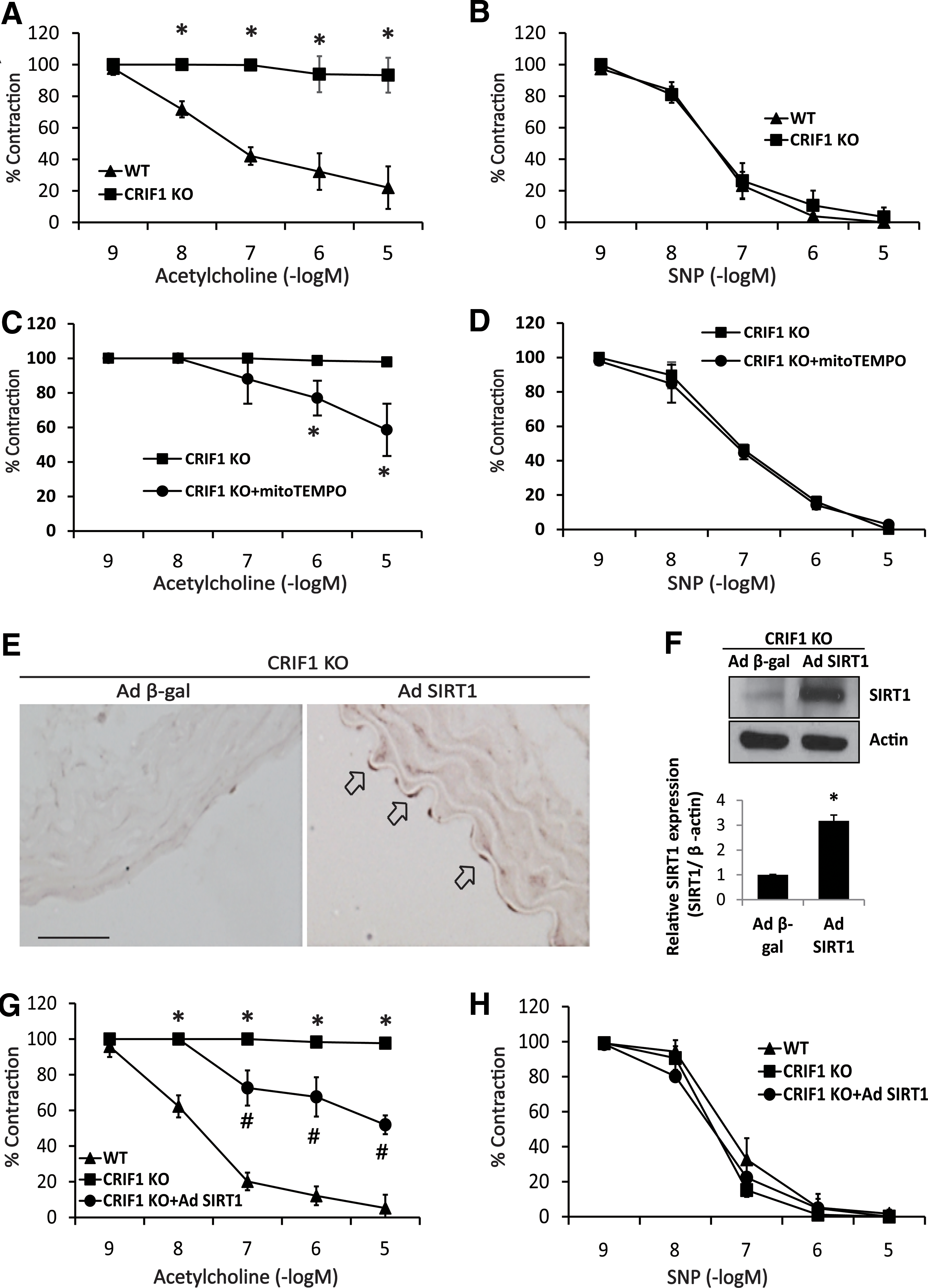
The function of CRIF1 was investigated in vivo by using conditional knockout mice that were generated by homologous recombination. Interestingly, there was a considerable decrease in the OXPHOS complexes II, III, IV, and V and CRIF1 proteins in the endothelial cells from CRIF1 KO mice as compared with the WT mice (Fig. 7A and Supplementary Fig. S10). In addition, mitochondrial ROS markedly was increased in CRIF1 KO mice (Fig. 7B).
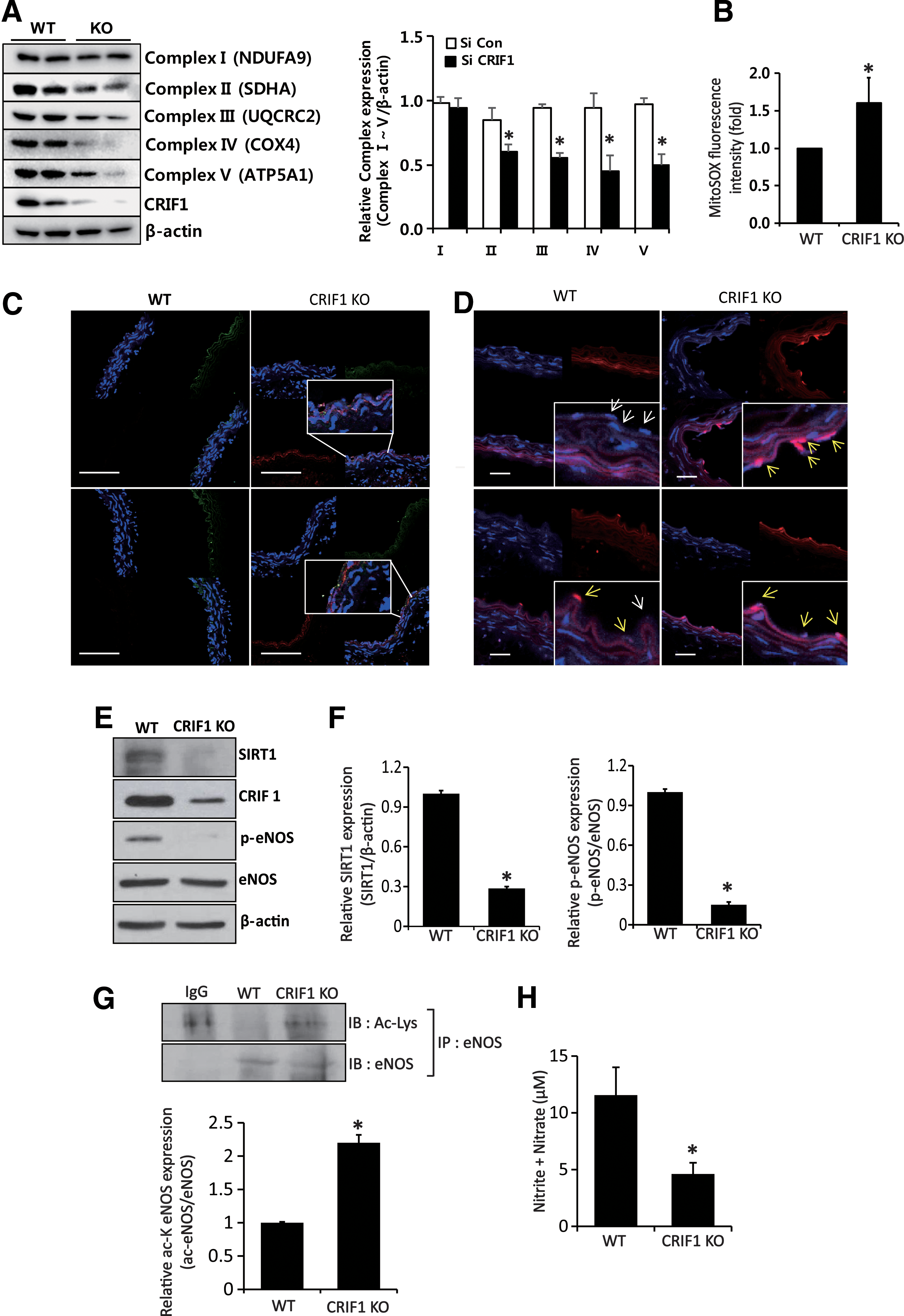
To confirm the vascular oxidative stress in vivo, we performed 4-hydroxynonenal (4-HNE) immunofluorescence staining or MitoSOX staining, which represents the lipid peroxidation levels or mitochondrial ROS contents, respectively, in the aorta of control and EC-specific CRIF1 KO mice. As shown in Figure 7C, 4-HNE and endothelial cell marker protein, CD31 double immunofluorescence staining with nucleic acid marker, DAPI was performed to identify both localization and quantification of lipid peroxidation in the aorta. The result showed no lipid peroxidation in endothelial cells from control mice, but considerable 4-HNE was detected in EC CRIF1 KO mice, which were restricted in endothelial cells (4-HNE merged with CD31). As 4-HNE staining means lipid peroxidation that is an indirect method for oxidative stress, we performed mitoSOX staining for showing the mitochondrial ROS contents directly. MitoSOX is more potent and specific to the mitochondria. As shown in Figure 7D, mitoSOX staining is mild in the aorta of control mice, but it is dramatically increased in EC CRIF1 KO mice. These results confirm that vascular oxidative stress was evident in CRIF1 KO animals, which was consistent with the in vitro data using the HUVEC cell line.
We also determined whether eNOS modification and SIRT1 expression levels were altered in CRIF1 KO mice. eNOS phosphorylation and SIRT1 expression levels were significantly decreased in the aorta (Fig. 7E, F), and eNOS lysine residue acetylation was also markedly increased in CRIF1 KO mice aorta compared with WT mice (Fig. 7G and Supplementary Fig. S10). Furthermore, NO production was decreased in plasma from CRIF1 KO mice compared with WT mice (Fig. 7H).
CRIF1 deficiency leads to vascular dysfunction in mice
To explore the physiological relevance of CRIF1 deficiency with vascular function, we first investigated the role of CRIF1 in mediating the physiological effects of SIRT1 on vascular reactivity. Endothelium-dependent vasorelaxation induced by acetylcholine was completely blocked in aortic rings from CRIF1 KO mice (Fig. 8A). In contrast, endothelium-independent vasodilation induced by the NO donor sodium nitroprusside (SNP) was similar in WT and CRIF1 KO mice (Fig. 8B). To know the functional role of ROS and SIRT1 in the impaired endothelium-dependent relaxation, MitoTEMPO was treated and adSIRT1 was infected in vascular aortic rings ex vivo of CRIF1 KO mice. As shown in Figure 8C and D, MitoTEMPO treatment partially recovered impaired endothelium-dependent vasorelaxation (EDR) in the aortic rings of CRIF1 KO mice. SIRT1 expression was reduced in the aorta of CRIF1 KO mice (Supplementary Fig. S4), and adenoviral gene transfer with adSIRT1 (3.0 × 108 pfu) for 24 h showed SIRT1 overexpression in the endothelial layer of the aorta from CRIF1 KO mice (Fig. 8E, F, and Supplementary Fig. S11). Similar to MitoTEMPO treatment, SIRT1 overexpression partially recovered impaired EDR in the aortic rings of CRIF1 KO mice (Fig. 8G, H). Endothelium-independent relaxation induced by SNP was similar in WT, CRIF1 KO mice and in MitoTEMPO-treated and SIRT1-overexpressed aorta (Fig. 8D, H). These findings indicate that impaired EDR in aortic rings from CRIF1 KO mice can either partially or to some extent be recovered by the SIRT1 overexpression, which suggests that SIRT1 is a key modulator in CRIF1 KO-induced endothelial dysfunction.
Discussion
Oxidative stress plays a crucial role in cardiovascular disease pathogenesis. All cardiovascular risk factors are linked to oxidative stress and endothelial dysfunction (5, 32). In the setting of oxidative stress, enzymatic production of free oxygen radicals, oxygen ions, and peroxide overwhelms the available antioxidant defense systems (3). Superoxide (O2 −) reacts with NO and leads to a loss of NO bioactivity. The resulting peroxynitrite (ONOO−) may cause eNOS dysfunction and, thus, reduce NO production. Reduced vascular NO bioavailability is the major mechanism of endothelial dysfunction observed in cardiovascular diseases (14, 15). Mitochondrial ROS production is an important cause of oxidative stress and mitochondrial disease. The molecular mechanism of vascular function in the setting of mitochondrial disease has only been partly identified, despite considerable pathophysiologic evidence for the relationship between vascular disease and mitochondrial dysfunction. Here, we demonstrated that CRIF1 deficiency-induced mitochondrial dysfunction is critical for endothelial dysfunction as evidenced by the profound loss of SIRT1 expression, eNOS activity, and endothelium-dependent vasomotor function, suggesting that CRIF1 deficiency-induced mitochondrial dysfunction is critical for the initiation and development of vascular conditions such as cardiovascular disease and atherosclerosis.
CRIF1 is a nuclear protein that interacts with growth arrest and DNA damage inducible (Gadd45), Nur77, and signal transducer and activator of transcription 3 (STAT3) (9, 30, 36). However, recent studies have shown that CRIF1 interacts with the 39S subunit of the mitoribosome, contributing to the synthesis and insertion of OXPHOS subunits in vivo and 39S subunit components in the mammalian mitochondrial ribosome (18, 27). Indeed, in this study, CRIF1 downregulation caused reduced OXPHOS expression along with an increased mitochondrial and intracellular ROS levels. These data are in agreement with recent reports showing that CRIF1 deficiency leads to impaired OXPHOS complex function and mitochondrial dysfunction, because CRIF1 regulates the translation and insertion of 13 polypeptide subunits that comprise mitochondrial OXPHOS complexes I, III, IV, and V (27). In addition, because there is a high density of OXPHOS complexes in the cristae of the inner membrane of normal mitochondria (46), CRIF1-deficient cells have abnormal structural changes, including distorted cristae (27). We used the HUVEC cell line and a tissue-specific KO mouse to show that ultrastructural abnormalities in the cristae were consistent with the findings that were suggestive of impaired OXPHOS complexes (e.g., low levels of basal and stimulated mitochondrial oxygen consumption and a depolarization of the inner mitochondrial membrane). Collectively, these results indicate that defective synthesized mitochondrial OXPHOS polypeptides may not fully form, leading to distorted and reduced cristae.
Sirtuins may play a critical role in vascular endothelial homeostasis controlling angiogenesis, vascular relaxation, and endothelial function (34, 38). SIRT1 is one of the seven sirtuins (SIRT1-7) found in different subcellular locations, including the nucleus (SIRT1, SIRT6, and SIRT7), cytosol (SIRT2), and mitochondria (SIRT3, SIRT4, and SIRT5) (13). The SIRT1/AMP-activated protein kinase (AMPK)/peroxisome proliferator-activated receptor-γ-coactivator1 (PGC1)/estrogen-related receptor-α (ERR) signaling pathway is involved in mitochondrial biogenesis, including the respiratory chain and mitochondrial antioxidant defenses (41). Indeed, mice with an SIRT1 deficiency, presumably due to a loss of mitochondrial function, develop postweaning cardiomyopathy that is characterized by cardiac hypertrophy (2, 29). In addition, morphological and functional mitochondrial abnormalities were seen in conjunction with decreased SIRT1 expression in SIRT1-deficient mice (37). Interestingly, cardiac hypertrophy and fibrosis, low body weight gain, and poor development were observed in endothelial-specific CRIF1 KO mice. Based on these findings, we suggest that these defects in CRIF1 KO mice were due to both decreased SIRT1 protein expression and mitochondrial dysfunction and destruction.
One of the most fundamental questions regarding SIRT1 downregulation is how the protein's expression is regulated by CRIF1 deficiency. Under oxidative stress, amino acid side chain oxidation results in reversible and irreversible modifications to proteins. The processes of disulfide bonds, S-nitrosylation, and S-glutathionylation are reversible and can be easily converted by thioredoxin and glutaredoxin enzymes. On the other hand, irreversible modifications, including carbonylation and tyrosine nitration, are believed to reduce protein function and degradation (16). It was recently shown that the reactive oxygen moiety H2O2 can post-translationally modify SIRT1 and affect its activity in endothelial cells (6, 23). Our results are consistent with these findings and show that SIRT1 protein expression is significantly decreased by H2O2. Thus, oxidative stress affects endothelial SIRT1, thereby inhibiting its activity. Here, we demonstrate an additional mechanism involving the administration of a mitochondria-targeting antioxidant. These results are consistent with previous evidence (21, 25, 33) in endothelial and vascular smooth muscle cells showing that reduced SIRT1 activity led to increased ROS production. We showed a reduction of mitochondrial ROS by a mitochondria-specific antioxidant and a consequent recovery of SIRT1 protein expression.
SIRT1 has been identified as a critical regulator of endothelial function (39). SIRT1 has also been shown to bind eNOS and to regulate its posttranscriptional activation. eNOS activation leads to NO release that exerts vasoprotective and cardioprotective effects in endothelial cells by regulating blood flow, inhibiting platelet aggregation and inflammatory cell adhesion (4). In the light of our data showing reduced SIRT1 levels in response to oxidative stress, we hypothesized that it would cause increased eNOS acetylation in endothelial cells (acetylation leads to eNOS inactivation). Indeed, increased eNOS acetylation in CRIF1-silenced HUVECs occurred in response to a downregulation of SIRT1 levels. This is supported by other observations that SIRT1 activation promotes endothelial-dependent vasodilatation by targeting eNOS for deacetylation to enhance NO production (34). Consistent with these findings, in our study, adenovirus-mediated SIRT1 overexpression partially recovered endothelial-dependent vasodilatation in CRIF1 KO mice. Therefore, the mechanism proposed for endothelial dysfunction after SIRT1 inhibition involves increased eNOS acetylation, leading to decreased enzyme activity.
In summary, our findings demonstrate that CRIF1 deficiency induces oxidative stress and mitochondrial dysfunction in endothelial cells. This leads to downregulated SIRT1 levels that are concomitant with increased eNOS acetylation and decreased NO production. CRIF1 deficiency promotes impaired vascular function. Identification of eNOS as an SIRT1 substrate under oxidative stress is important in understanding the beneficial effect of SIRT1 on endothelial vascular biology with regard to preventing oxidative stress-induced endothelial dysfunction, an early step in cardiovascular disease pathogenesis.
Materials and Methods
Cell culture and transfection
HUVECs were purchased from Clonetics (San Diego, CA) and cultured in Endothelial Growth Medium-2 from Lonza (Walkersville, MD) according to the manufacturer's instructions at 37°C with 5% CO2. Sub-confluent, proliferating HUVECs at passages 2–8 were used. HUVECs were transfected with short interfering RNA (siRNA) for CRIF1 (human siRNA sequence: sense-5′-UGGAGGCCGAAGAACGCGAAUGGUA-3′ and antisense-5′-UACCAUUCGCGUUCUUCGGCCUCCA-3′) and negative control siRNA by using Lipofectamine 2000 reagent from Invitrogen (Carlsbad, CA) as per the manufacturer's recommendations. The cells were incubated at 37°C in a 5% CO2 incubator for 48 h for gene knockdown.
Antibodies, immunoprecipitation, and immunoblotting
Anti-CRIF1, anti-SIRT1, anti-MFN-1, anti-TOM22, and anti-FIS-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-acetyl lysine and anti-COX-4 antibodies were from Cell Signaling Technology (Beverly, MA). Anti-4HNE and anti-CD31 were from Abcam. Anti p-eNOS, anti-OPA-1, and anti-DRP-1 antibodies were from BD Biosciences (San Jose, CA). Antibodies against OXPHOS complex subunits (NDUFA9, SDHA, UQCRC2, and ATP5A1) were purchased from Invitrogen. Lysine acetylation of eNOS was detected by immunoblotting immunoprecipitated eNOS with an acetyl lysine (Ac-K) antibody. Immunoprecipitations of eNOS were performed by incubating 2 μg antibody with 1 mg cell lysate or tissue homogenate overnight, followed by addition of 30-μl protein A-Sepharose slurry from Amersham Biosciences (Little Chalfont, UK) for 4 h. After washing, the immunoprecipitates were boiled in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, separated by electrophoresis, transferred to a nitrocellulose filter, and probed with eNOS and Ac-K antibodies and the appropriate peroxidase-conjugated secondary antibodies from Santa Cruz Biotechnology. Chemiluminescent signals were developed by using Super Signal West Pico or Femto Substrate from Thermo Fisher Scientific (Waltham, MA). Western blotting of 30 μg of whole-cell lysates or tissue homogenate from 8-week-aged WT and CRIF1 KO mice was similarly performed by using appropriate primary and secondary antibodies. Mitochondrial isolation from whole cells was performed by using a mitochondria isolation kit for cultured cells from Thermo Fisher Scientific. Blots were imaged, and band densities were quantified with a Gel Doc 2000 Chemi Doc system by using the Quantity One software (Bio-Rad, Hercules, CA). Values were normalized to a β-actin loading control.
H2O2 treatment in HUVECs
HUVECs were treated with or without 5 mM of NAC for 1 h in six-well plates, followed by treatment with 50 or 100 μM of H2O2 for 24 h, after which the cells were harvested for Western blot analysis.
ROS measurement
MitoSOX red and DHE dyes from Invitrogen were used to detect mitochondrial and intracellular ROS production, respectively. After 48 h of transfection with control or CRIF1 siRNA, cells were washed with phosphate-buffered saline (PBS) and trypsinized, and the resulting cell suspension was incubated in complete medium with 3 μM MitoSOX at 37°C for 15 min in the dark. During the incubation period, each sample was agitated every 5 min to ensure that the reagent reacted sufficiently with the ROS. To reduce the fluorescence background, each sample was washed twice with PBS before detecting the fluorescence intensity of MitoSOX red by using the fluorescence reader Fluoroskan Ascent (Thermo Fisher Scientific) at excitation and emission wavelengths of 530 and 590 nm, respectively. For intracellular ROS detection, 48 h after transfection with control or CRIF1 siRNA, cells were washed twice with PBS and incubated in complete medium with 1 μM DHE at 37°C for 30 min in the dark. After a subsequent wash with PBS, images of DHE-stained cells were obtained with an Olympus fluorescence microscope (Olympus, Tokyo, Japan).
Measurement of MMP
Changes in MMP after transfection with control or CRIF1 siRNA were measured by using TMRE dye. TMRE is a membrane potential-sensitive dye that translocates into mitochondria. Its fluorescence intensity is directly proportional to the MMP. After 48 h of transfection, cells were washed twice with PBS and trypsinized before the suspensions were collected. TMRE (100 nM) was added to each sample, and the cells were incubated in complete medium at 37°C for 15 min in the dark. FCCP (10 μM) was added to one sample as a positive control in each experiment. During the incubation period, each sample was agitated every 5 min. To reduce the fluorescence background, each sample was washed twice in PBS before measurement with a fluorescence reader from Fluoroskan Ascent Thermo Fisher Scientific at excitation and emission wavelengths of 530 and 590 nm, respectively.
OCR, ECAR
OCR was measured by using a Seahorse XF-24 analyzer from Seahorse Bioscience (North Billerica, MA). The day before OCR measurement, the sensor cartridge was calibrated with calibration buffer at 37°C. After 48 h of transfection with control or CRIF1 siRNA, cells were washed twice with XF assay media without sodium bicarbonate and phenol red and incubated in a 37°C incubator for 45 min up to 1 h before calibration. Three readings were taken after the addition of each mitochondrial inhibitor and before injection of the subsequent inhibitors. The mitochondrial inhibitors used were oligomycin (2 μg/ml), CCCP (10 μM), and rotenone (2 μM). OCR and ECAR were automatically calculated and recorded by the sensor cartridge and Seahorse XF-24 software. The plates were saved, and the protein concentrations were calculated to confirm that there were approximately equal numbers of cells in each well.
H2O2 measurement
H2O2 produced by cells was measured in conditioned medium as previously described (26). Amplex red hydrogen peroxide assay kits from Molecular Probes/Invitrogen were used according to the manufacturer's instructions to measure H2O2 released from the cells. Amplex Red was stored frozen at −20°C as a 10 mM DMSO stock. Horseradish peroxidase (HRP, 10 U/ml) was dissolved in 1 × reaction buffer and stored at −20°C. The reaction mixture contained 50 μM Amplex Red and 0.1 U/ml HRP in Krebs-Ringer phosphate (KRPG) buffer (145 mM NaCl, 5.7 mM sodium phosphate, 4.86 mM KCl, 0.54 mM CaCl2, 1.22 mM MgSO4, 5.5 mM glucose, pH 7.35). Each reaction had a volume of 100 μl. Then, 20 μl 1.5 × 104 cells suspended in KRPG buffer was added to the 100-μl reaction mixture. For the negative control, 20 μl KRPG buffer without cells was added to a separate 100 μl of reaction mixture. Amplex Red is specifically sensitive to H2O2. Oxidation of Amplex Red by H2O2 in the presence of HRP produces highly fluorescent resorufin. The level of resorufin produced was quantified in a fluorescence reader Fluoroskan Ascent from Thermo Fisher Scientific at excitation and emission wavelengths of 530 and 590 nm, respectively.
Nitrite and nitrate measurements
The NO metabolites nitrite and nitrate were measured in phenol red-free medium or mouse serum as previously described (24). Nitrite and nitrate, which are stable breakdown products of NO, were quantified by using a commercially available kit nitrate/nitrite fluorometric assay kit from Cayman Chemicals (Ann Arbor, MI) according to the manufacturer's instructions. The phenol red-free medium or mouse serum was deproteinized by using a 10-kDa cutoff filter. After subtraction of background fluorescence, values were normalized to obtain the total nitrite/nitrate amount.
Mouse studies
The Animal Care Committee of Chungnam National University approved the animal care and experimental procedures used in this study. Floxed CRIF1 (CRIF1 flox/flox) mice were generated as previously described (27). Tek-Cre mice were purchased from Jackson Laboratory (Bar Harbor, ME). To identify the genotype, PCR was performed with specific primers and extracted genomic DNA from tail snips. Body weight was measured once a week. Mice were maintained in a controlled environment (12-h light/12-h dark cycle; humidity 50–60%; ambient temperature 23°C) and fed ad libitum with food from Harlan (Indianapolis, IN). All mouse experiments were performed in the animal facility according to institutional guidelines, and the experimental protocols were approved by the institutional review board of Chungnam National University.
Histological and morphometric analysis
The aorta from 8-week-aged WT and CRIF1 KO mice were fixed in 10% neutralized formalin for 16 h, washed, and embedded in paraffin. Paraffin sections of the heart were deparaffinized, rehydrated, and stained with hematoxylin/eosin. For immunohistochemistry, deparaffinized mouse aortic ring sections were permeabilized and processed by using a Vectastain Universal Quick Kit (PK-8800; Vector Laboratories, Burlingame, CA). Primary antibodies against SIRT1 or CRIF1 were used at a 1:50 dilution, followed by incubation with biotinylated secondary antibody, streptavidin peroxidase solution, DAB peroxidase substrate, and hematoxylin counterstaining. Sections were digitally imaged with an Olympus fluorescence microscope (Olympus).
Transmission electron microscopy
TEM was performed as previously described (26). Briefly, tissues or cells were fixed in 1% glutaraldehyde at 4°C and then washed with 0.1 M cacodylate buffer at 4°C. After washing five times, the tissue was postfixed with 1% OsO4 in an 0.1 M cacodylate buffer (pH 7.2) containing 0.1% CaCl2 for 1 h at 4°C. Samples were dehydrated with serial ethanol and propylene oxide treatment and embedded in Embed-812 (EMS, Hatfield, PA). The resin was then polymerized at 60°C for 36 h. Tissue was sectioned by using an EM UC6 ultramicrotome (LEICA, Wetzlar, Germany) and stained with 4% uranyl acetate and citrate. The samples were observed with a Tecnai G2 Spirit Twin transmission electron microscope (FEI Company, Hillsboro, OR) and a JEM ARM 1300S high-voltage electron microscope (JEOL, Tokyo, Japan).
Echocardiography
Echocardiographic measurements were performed by using a Vivid E9 with an XD clear (GE) instrument with a 13-MHz linear microprobe (GE VIVID 7) in 8 week-aged WT and CRIF1 KO mice. Mice were anesthetized intraperitoneally with Ketamine (106.26 mg/kg body weight) and Xylazine (8.63 mg/kg body weight), and their chests were shaved. The hearts were examined by using the M-mode guided by a short-axis view of the two-dimensional mode. The FS and EF were automatically obtained as the formula, FS = ([LVEDd-LVESd]/LVEDd) × 100 (%) and EF = ([LVEDd2-LVESd2]/LVEDd2) × 100 (%).
Isolation of primary endothelial cell
To purify primary endothelial cells, we performed a procedure as described in a previous paper with modification (43). First, we prepared anti-CD31 antibody-conjugated magnetic beads (Dynabeads) and then isolated mouse pulmonary endothelial cells. Total lung was excised from 8 week-aged male mice and minced in collagenase buffer (DMEM with 1 mg/ml collagenase type I [Worthington]). The minced tissues were digested at 37°C for 40 min with gentle shaking and then filtered through a 100 μm filter. Digested cells were removed by centrifugation at 700 g for 5 min and incubated with RBC lysis buffer. The remaining cells were washed twice with PBS. The cell suspension was transferred to a 5 ml round-bottom polystyrene tube, and anti-CD31 antibody-conjugated Dynabeads was added. After incubation for 15 min at room temperature and washing with PBS, the sample was lysed with RIPA buffer and Western blot was performed.
MitoSOX staining in vivo
MitoSOX staining was performed as described in a previous paper (10) with modification. After obtaining the aorta, the sample was rinsed with 1 × PBS and incubated with 5 μg/ml of MitoSOX solution and DAPI for 10 min at RT with shaking. After subsequent washing of the aorta with 1 × PBS three times, a cryoblock of 20 μm thickness was cut. Immunofluorescent images were taken on an Olympus fluorescence microscope (Olympus).
Vascular reactivity
Endothelium-dependent vasorelaxation from 8-week-aged WT and CRIF1 KO mice was measured in aortic rings as previously described (45). Mice were sacrificed via a sodium pentobarbital overdose. A mid-sternal split was quickly performed, and the descending thoracic aorta was carefully excised and placed in ice-cold Krebs buffer (118.3 mM NaCl/4.7 mM KCl/2.5 mM CaCl2/1.2 mM KH2PO4/25 mM NaHCO3/1.2 mM MgSO4/11 mM glucose/0.0026 mM CaNa2 EDTA). The aorta was cleaned of excess fat, cut transversely into 5–10 rings (2.0–3.0 mm), and maintained at 37°C and pH 7.4. Endothelium-dependent vasodilation was determined by generating dose–response curves to acetylcholine. Vasorelaxation evoked by acetylcholine was expressed as a percent contraction that was determined by the percentage of inhibition to the preconstricted tension. Endothelium-independent vasodilation was measured by the vasorelaxation evoked by cumulative SNP in rings that were preconstricted with phenylephrine (10−6 M).
Ex vivo adenoviral infections and MitoTEMPO treatment
Endothelium-specific gene transfer or MitoTEMPO treatment was achieved ex vivo by incubating freshly isolated aortas from mice, suturing them at one end, adding 3.0 × 108 pfu of appropriate adenoviral stock or 10 nM MitoTEMPO, and incubating them at 37°C for 4 h as previously described (22). The solution was removed, and then the aorta was incubated for 24 h before sectioning into rings. The viruses used have been previously described (34).
Statistical analysis
Statistical analysis was performed by using SPSS (Version 17.0) statistical software (SPSS, Inc., Chicago, IL). Differences between two groups were evaluated by using t-tests. For multiple comparisons, a one-way analysis of variance was performed, and Tukey's tests were carried out for post hoc analyses. Data are presented as mean ± SEM. p ≤ 0.05 was considered statistically significant. All data are representative of at least three independent experiments.
Footnotes
Acknowledgments
The authors thank K.I. and Young Rae Kim for providing the adeno-SIRT1 and for critically reading the article. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2014R1A6A1029617, NRF-2015R1D1A1A01061516).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.