
Abstract
Significance:
Secreted proteins are important both as signaling molecules and potential biomarkers.
Recent Advances:
Protein can undergo different types of oxidation, both in physiological conditions or under oxidative stress. Several redox proteomics techniques have been successfully applied to the identification of glutathionylated proteins, an oxidative post-translational modification consisting in the formation of a mixed disulfide between a protein cysteine and glutathione. Redox proteomics has also been used to study other forms of protein oxidation.
Critical Issues:
Because of the highest proportion of free cysteines in the cytosol, redox proteomics of protein thiols has focused, so far, on intracellular proteins. However, plasma proteins, such as transthyretin and albumin, have been described as glutathionylated or cysteinylated. The present review discusses the redox state of protein cysteines in relation to their cellular distribution. We describe the various approaches used to detect secreted glutathionylated proteins, the only thiol modification studied so far in secreted proteins, and the specific problems presented in the study of the secretome.
Future Directions:
This review focusses on glutathionylated proteins secreted under inflammatory conditions and that may act as soluble mediators (cytokines). Future studies on the redox secretome (including other forms of oxidation) might identify new soluble mediators and biomarkers of oxidative stress. Antioxid. Redox Signal. 26, 299–312.
Redox State of Proteins in the Context of Their Localization
Redox proteomics of the thiol proteome
A
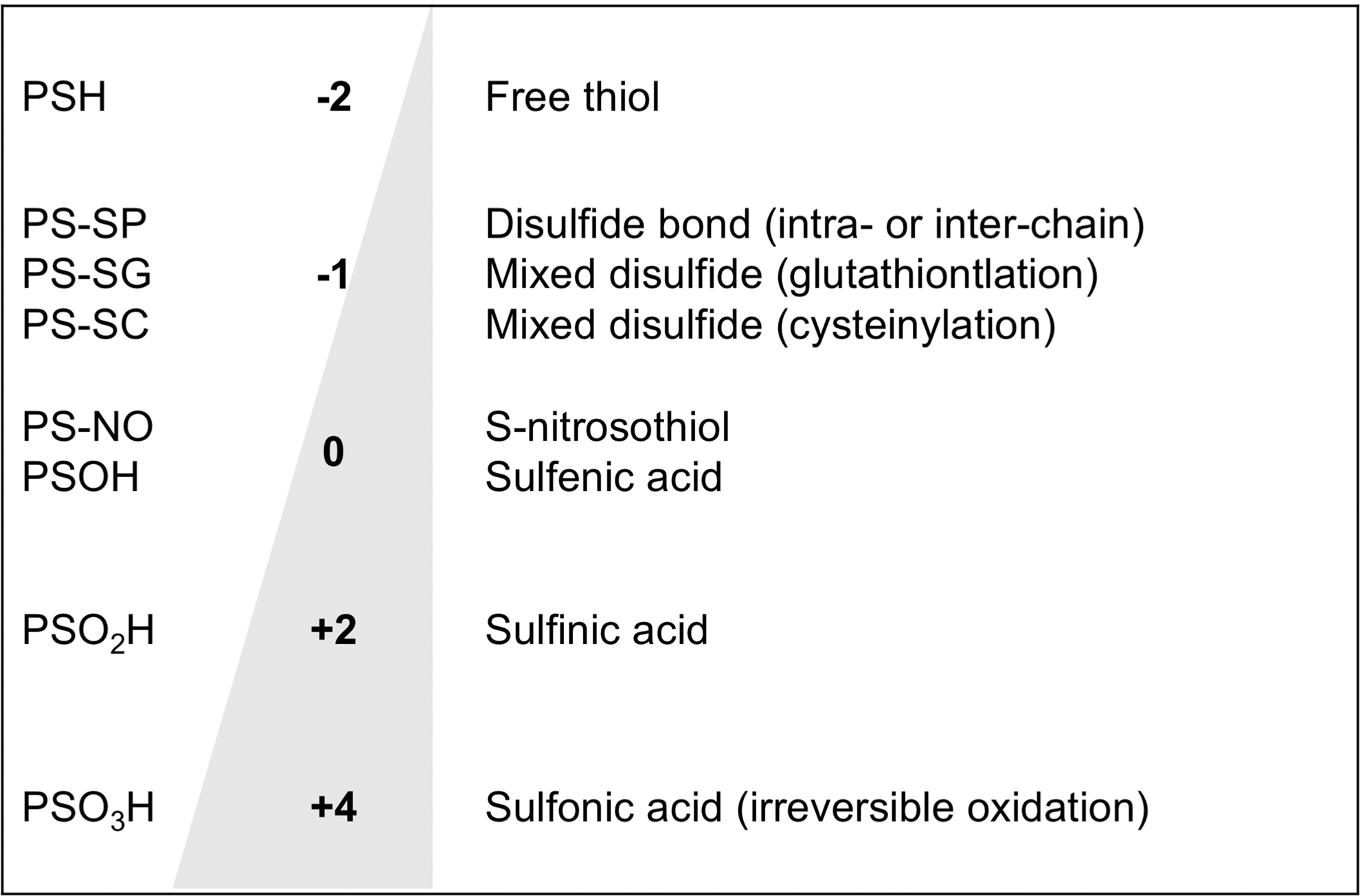
This review will deal with the redox states of protein thiols. Biochemistry textbooks and protein databases classify cysteines in proteins as either free or engaged in a disulfide bond. However, “free” cysteines can be reversibly or irreversibly oxidized to various forms. In particular, in addition to forming structural disulfides, cysteines can form transient reversible disulfides with another protein cysteine, free cysteine (cysteinylation), or the cysteine in the tripeptide glutathione (GSH; glutamyl-cysteinyl-glycine). Figure 1 lists the most important oxidation state of sulfur in cysteine. These forms of oxidation, due to their reversibility, are of great interest in the context of redox regulation as a means by which the redox state of the cell can regulate protein functions (43 –45).
Specific proteins undergoing glutathionylation were described even before the term “proteomics” became popular (in the late 1990s, according to Google Books Ngram Viewer—
Most redox proteomic studies published to date report the identification of cytosolic proteins because the analyzed samples were cell lysates. Studies by the group of Murphy, specifically focusing on the mitochondria, have identified several mitochondrial proteins susceptible to thiol oxidation (60). Protein abundance is also a key factor in the identification of oxidized proteins. For instance, when we tried to identify specific biochemical pathways targeted by protein glutathionylation, we could identify glycolytic enzymes, as shown in Figure 2, suggesting that glycolysis is potentially regulated by the redox state of protein thiols. In fact, Figure 2 also shows that many of the glycolytic enzymes undergoing glutathionylation were known in the literature for their enzymatic activity to be affected by thiol oxidation. This would predict that a shift in the GSH:oxidized GSH (GSSG) ratio may affect the overall anaerobic energy metabolism. However, the focus on glycolytic enzymes may be due to the fact that they are abundant proteins, which are preferentially identified by most proteomics techniques (75).

Subcellular localization of the thiol redox state
This section will discuss the redox state of protein cysteines in different cellular compartments. It will provide some reason for the high prevalence of cytosolic proteins among those undergoing transient posttranslational modifications (PTM), including glutathionylation, and possibly identify understudied areas worth exploring.
Cytosol
The intracellular environment is considered a highly reducing one, mainly due to the 1–10 mM concentrations of GSH, at a high GSH:GSSG ratio (up to 100:1). This is an unfavorable condition for the formation of disulfide bonds, and in fact, structural disulfide bonds are normally formed in the endoplasmic reticulum that is a more oxidizing environment (GSH:GSSG, 1:1) (50).
Plasma
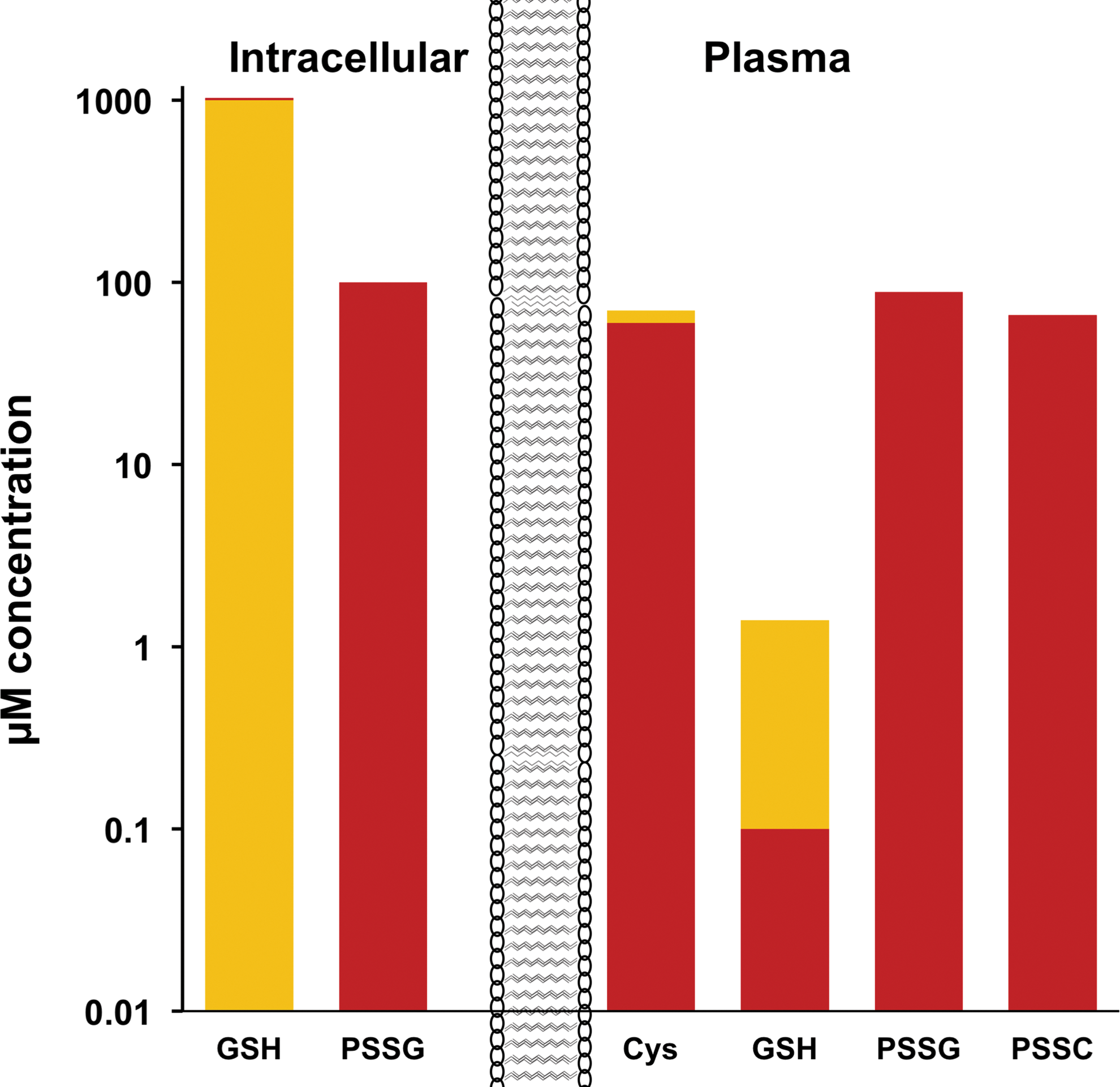
On the contrary, the extracellular environment is oxidizing. The main free thiol in serum/plasma is represented by cysteine but that is only in the range of 10 μM and with a reduced:oxidized (cysteine/cystine) ratio of 1:6 (9). GSH is also present in plasma but in the range of 1 μM (1000-fold less than in the cell) and at a GSH:GSSG ratio around 1 (9). Other studies also reported that plasma GSH+GSSG is present at at least 10-fold lower concentrations than Cys+cystine, but while cysteine is mainly oxidized (Cys:cystine ratio around 1:10), glutathione is more reduced, even extracellularly (GSH:GSSG ratio of 10:2) (57). The same study shows that significant amounts of cysteine and GSH are present in the oxidized form in plasma as mixed disulfides with proteins: 40% of total plasma cysteine is present in cysteinylated proteins, and 70% of plasma GSH is oxidized as mixed disulfides to form glutathionylated proteins (57); by comparison, Brigelius et al. (14, 15) reported that in the normal liver, 1% of intracellular GSH is present as glutathionylated proteins. Hence, even if total plasma glutathione is 1:10 of total plasma cysteine/cystine and is predominantly in a reduced state, the plasma concentration of glutathionylated proteins is slightly higher than that of cysteinylated proteins, raising the interesting question of whether protein glutathionylation occurs before secretion. Figure 3 summarizes the concentration and redox state of thiols in the cell and in plasma that, although coming from the studies cited above and obtained in different models, can provide an idea of the order of magnitude of the different redox states of cysteines.
Structural disulfides in cytoplasmic versus secretory proteins
Most studies on protein structure point out that cytosolic proteins have no disulfides, while extracellular proteins have no or little free cysteines (34, 36, 90). Butera et al. (17) have calculated that, based on UniProt annotations for 4000 proteins, about half of the disulfide bonds are in membrane proteins and the other half in proteins containing a secretion signal sequence, but about 5% disulfide bonds are found in nucleoplasmic or cytoplasmic proteins.
Studies using diagonal electrophoresis in primary cardiac myocytes or a neuronal cell line have shown that cytosolic proteins can form disulfide bonds upon exposure to oxidants, such as diamide or hydrogen peroxide (13, 30). These studies introduce the class of transient disulfides, a concept that Linke and Jakob summarized with the title “Not every disulfide lasts forever” (62). Disulfides also vary in their surface exposure, and while the most surface exposed are easily reduced, those buried in the three-dimensional structure of the protein require prior denaturation of the protein (16). Accessibility is also important for a cysteine to be susceptible to glutathionylation because of the size of GSH (307 kDa). This is even more important when glutathionylation occurs by thiol–disulfide exchange with GSSG (612 kDa) (45), and even more if this reaction is catalyzed by glutaredoxin (63). It is likely that transient disulfides are also surface exposed. These disulfides can be intraprotein or interprotein ones, and Brennan et al. noted that many of them imply associations with other proteins (13).
Protein thiols on the plasma membrane
The plasma membrane is at the interface of the intracellular environment and the extracellular one. We might expect, therefore, that protein thiols on the inner side are prevalently reduced and the ones on the exofacial side prevalently oxidized. We are not aware of studies attempting a quantification of the redox state on protein thiols on the two sides of the membrane.
However, a few articles reporting the existence of cell surface, exofacial, thiols have received some attention. Early studies on lymphocytes showed that their surface thiols increased upon mitogenic stimulation (58). Studies overexpressing or inhibiting protein disulfide isomerase showed that this enzyme is important for keeping exofacial thiols in the reduced state (53). Thus, an active mechanism could be involved in the presence of free thiols on the exofacial side of the plasma membrane despite the extracellular environment being oxidizing. Redox proteomics has been successfully applied to identify membrane protein whose exofacial thiols are redox sensitive (55, 56).
The Secretome
With the term “secretome,” we refer to proteins that are released in the cell culture supernatant or found in circulation. We are aware that a conservative use of the term “secretion” would restrict its use to proteins that are secreted through the classical pathway and have a signal sequence for targeting to secretion via the Golgi system. However, the term “nonclassical secretion” is often used to define nonclassical mechanisms; hence, our use of the term “secretion” instead of “release” in this review.
Oxidized proteins in circulation
To our knowledge, only a few plasma proteins have been studied in terms of their redox state.
Serum albumin is a major plasma protein, probably the most abundant secretory protein, and present in serum at concentrations of 30–50 mg/ml. Human serum albumin has 17 cysteines of which only one (Cys34) is not engaged in a structural disulfide. Due to the low concentrations of free thiols in plasma, albumin may well be the main thiol antioxidant in circulation (76, 94). This can undergo oxidation to form a mixed disulfide with cysteine (cysteinylation), homocysteine (32), or a sulfinic acid (66). Increased levels of cysteinylated albumin have been associated with intrauterine growth restriction (3).
Transthyretin is a transport protein (the name originates from “transports thyroxine and retinol”), also involved in amyloidosis (83). It can be found in circulation in the 0.5 mg/ml range as S-cysteinylated, S-cysteinglycinylated, and glutathionylated (42, 84).
High-mobility group protein box 1 (HMGB1) is a nuclear protein that can be released and act as a proinflammatory danger signal (97). HMGB1 has 3 redox-active cysteines (Cys23, Cys45, and Cys106). The different redox states of these cysteines also regulate the proinflammatory activity of HMGB1 (103). In circulation, it can be found with an intramolecular disulfide between Cys23 and Cys45 or sulfonylated on all three cysteines, and its oxidation is affected by acetaminophen poisoning (103).
Pentraxin 3 (PTX3) is an inflammatory protein related to C-reactive protein that is elevated in a number of infectious, inflammatory, and cardiovascular diseases (12). Formation of disulfide-linked oligomers of PTX3 has been reported, and one study has shown that measuring its oxidation state can provide a superior biomarker for sepsis than measurement of total PTX3 (29).
Studies Specifically Addressing the Proteomic Identification of the Redox Secretome
Despite redox proteomics has been applied to the thiol proteome looking at different forms of thiol oxidation (S-nitrosylation, transient disulfides, and mixed disulfides), so far high-throughput proteomics of the thiol secretome has only been applied to protein glutathionylation. However, these are very recent studies, and hopefully, they will be followed up by investigations on other types of thiol oxidation. In particular, given the high concentration of extracellular cysteine, it will be important to identify the proteins released as cysteinylated.
We have specifically asked if proteins can be released as glutathionylated by inflammatory macrophages. To answer the question, we have used a strategy using biotinylated GSH ethyl ester (BioGEE), originally used to identify glutathionylated proteins in HeLa cells (88). The principle is based on labeling the intracellular GSH pool with biotinylated GSH. However, because GSH is poorly cell permeable, its ethyl ester is used (98). This reagent is now commercially available and has been used in more than 150 articles on protein glutathionylation.
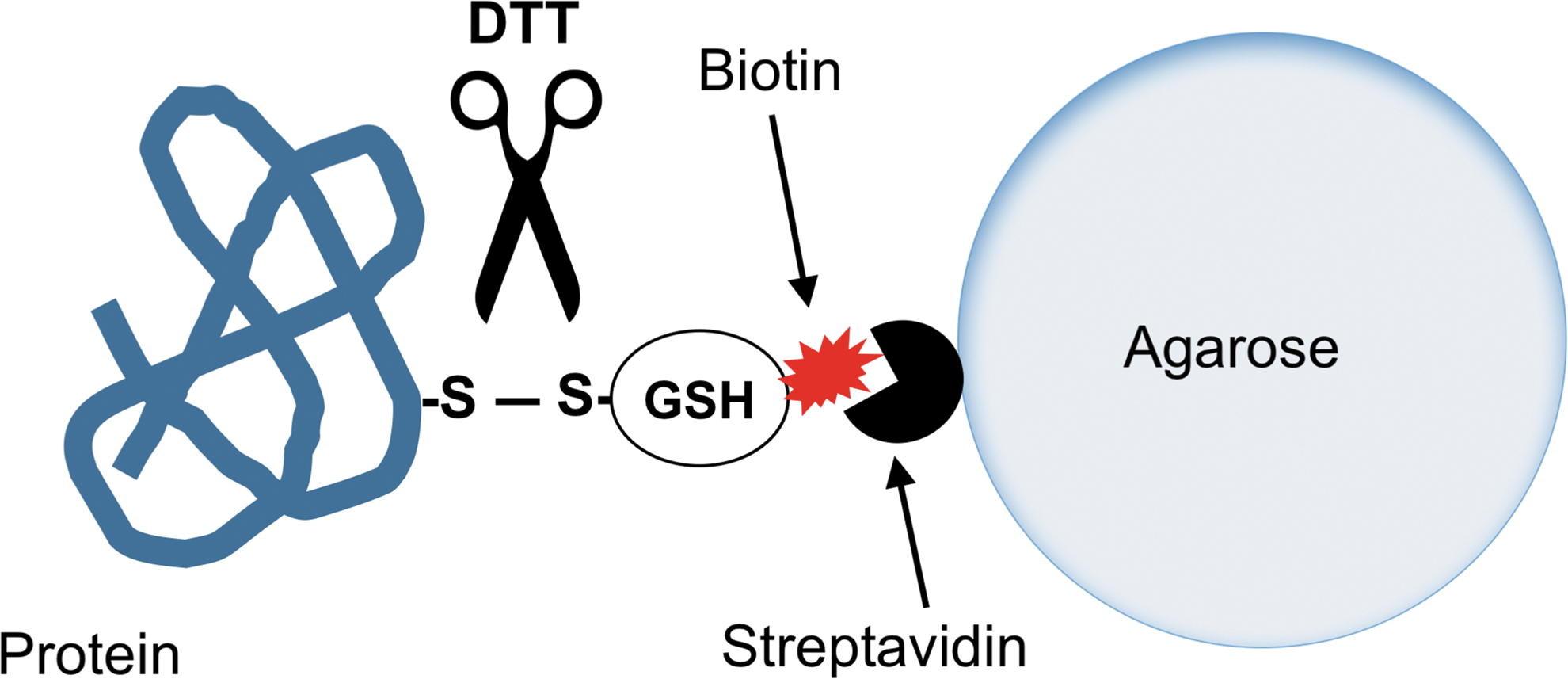
The experimental design is based on previous publications from other groups. Basically, cells are preincubated for 2 h with BioGEE, washed, and then cultured in reduced-serum medium (Opti-MEM; not completely serum free and not defined as it contains a proprietary mixture of growth factors) with or without an inflammatory stimulus (lipopolysaccharide, LPS). After harvesting the supernatant, free thiols are blocked with an alkylating agent (such as N-ethyl maleimide, NEM) to prevent thiol–disulfide exchanges. Then, proteins are bound on streptavidin–agarose and analyzed by mass spectrometry. The proteins on the beads can be directly digested with trypsin for analysis of tryptic peptides or eluted with dithiothreitol (DTT). The latter approach should reduce the chances of identifying proteins nonspecifically binding to streptavidin or agarose and enrich those that are attached to streptavidin through a disulfide bond (Fig. 4).
Once a list of proteins is generated by the mass spectrometry (MS) analysis, one has to bear in mind that this proteomics strategy is not quantitative but highly sensitive and has a number of bias; for instance, the proteins identified could still be bound nonspecifically to the beads or be a result of the nonspecific leakage from a small number of dead cells. Having identified a protein in an LPS-stimulated supernatant does not imply that its secretion is induced by LPS, also because there will always be a small amount of secretion by resting cells, or, again, due to a small amount of dead cells. Therefore, it is imperative that each protein is validated. In our studies, we normally provide the list of the identified proteins as supplementary data but report only those validated in the main body of the article (23). This makes it possible for others to eventually validate other proteins of interest.
In fact, to claim that a protein in the list of those identified by MS is released as glutathionylated and its release is induced by LPS, both arms of the claim need to be confirmed. Typically, a Western blot is required to measure the protein in the supernatant of LPS-stimulated versus unstimulated cells. This should be done with a proper number of replicates and, possibly, followed by densitometry and statistical analysis (23). Of course, if an enzyme-linked immunosorbent assay (ELISA) or any other assay (such as an enzymatic assay in case the protein is an enzyme) is available, that would be preferable as it may be more quantitative than a Western blot. Demonstrating that a protein is secreted as glutathionylated is more difficult as it requires immunoprecipitation with an antibody against the specific protein followed by nonreducing gel electrophoresis and Western blot with an anti-GSH antibody. Although we have used this approach, we feel it is not always satisfactory due to the presence of nonspecific bands and the nonoptimal migration in nonreducing conditions that does not allow an exact estimate of the molecular weight of the various bands. A control where the sample is run under reducing conditions, to remove GSH from the protein, will help. Finally, checking on a database, such as UniProt, that the “glutathionylated” proteins identified actually have a free cysteine available for glutathionylation will avoid embarrassing mistakes.
In some cases, of course, the glutathionylated protein may have already been detected by the MS analysis of the proteins or their peptides at the stage of the identification. Identification of a glutathionylated peptide will also provide the additional information of which cysteine is glutathionylated. However, many protocols for MS analysis include a reducing step, making thus impossible to identify glutathionylated peptides.
Proteins in the Redox Secretome
Glutathionylated proteins
Table 1 lists proteins secreted as glutathionylated by LPS-stimulated macrophages identified as described above. Below, we discuss some of them.
Redoxins: Three proteins in the list are involved in redox regulation and were the first ones we validated. In fact, we had previously identified by redox proteomics intracellular peroxiredoxin (PRDX) 1 (37) and thioredoxin (TRX) 1 (18) among the proteins undergoing glutathionylation in human lymphocytes.
Thioredoxin is a protein thiol–disulfide oxidoreductase catalyzing a thiol–disulfide exchange reaction. Although its most important function is to keep ribonucleotide reductase in the reduced state, and thus is essential for DNA synthesis (49), it has a broad substrate specificity and can reduce hundreds of proteins (73) with the overall reaction (i) below:
(i) TRX-(SH)2 + protein oxidized → TRX-S-S + protein reduced.
The oxidized TRX is then reduced back by the NADPH-dependent selenoprotein enzyme TRX reductase.
Under oxidative conditions, such as in the absence of TRX reductase to regenerate TRX in its reduced state, TRX can act as an oxidant (39) with the reverse of the reaction above. TRX has long been known to be secreted and was originally described by the group of Yodoi as a leukemia cell product that stimulates lymphocyte proliferation (89, 96).
Peroxiredoxins are TRX peroxidases and use reduced TRX as the electron donor in eliminating H2O2. During its catalytic action, PRDX is first oxidized by H2O2 and then reduced by TRX-(SH)2 with the resulting reaction (ii) below:
(ii) TRX-(SH)2 + H2O2 → TRX-S-S + H2O2.
PRDXs have a very high specificity for TRX and, in general, cannot use other proteins as substrates.
It is very interesting that both PRDXs and their substrate are coreleased under inflammatory conditions. We have hypothesized elsewhere that, because the extracellular environment is an oxidizing one and lacking glutathione and NADPH, these two proteins could act in concert to oxidize other proteins (81).
Secretion of PRDXs has also been reported previously when these proteins were originally termed “natural killer cell-enhancing factors” (40).
Profilin and vimentin were also identified among the intracellular glutathionylated proteins in our pioneering redox proteomics study (37). Profilin has already been reported by a previous study in the human immunodeficiency virus 1 (HIV-1)-infected, monocyte-derived macrophage secretome, although that study did not investigate its redox state (54). Like profilin, vimentin was also identified previously in the macrophage secretome, although its redox state was not investigated (54, 68).
Other forms of thiol oxidation
In addition to glutathionylation, disulfide-linked homodimerization has also been reported in secreted proteins. We found that PRDX1 and PRDX2 need to form disulfide-linked homodimers to be secreted. Mutation of the cysteine involved in homodimerization, but not of other cysteines, prevents their secretion (70).
A similar observation was made for fibroblast growth factor 1 (FGF1), where dimerization occurs during its secretion (67), and mutation of that cysteine involved in homodimerization blocks it (51).
In the case of PRDX1 and PRDX2, disulfide-linked dimerization targets them for secretion via the exosomal route, while FGF1 is then secreted as part of a larger multimeric complex (67).
Secretion of carbonylated proteins
In terms of irreversible oxidative damage, redox proteomics studies on oxidatively damaged proteins by the group of Butterfield have found increased carbonylation in some circulating protein in Alzheimer's disease, including haptoglobin β chain, serotransferrin, and α2-macroglobulin (28).
The problem of cell lysis contamination in secretome studies
Whenever studying nonclassical secretion of proteins, the problem arises as to whether their presence in the cell culture supernatant is due to secretion or if it is an artifact due to leakage from a small amount of dying cells in the culture. This problem is often addressed with a shortcut, where cell toxicity is measured by trypan blue exclusion or assays, where cell viability is assessed by a colorimetric or fluorometric metabolic assay, such as those using the oxidation of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT). The risk that release of the protein may be due to nonspecific leakage from dead cells is then ruled out with statements, such as “cell viability was over 95%” or “cell death was less than 5%.” However, the real question is whether that 5% (or even a 0.5%) of cell death can account for the observation that a protein is present in the secretome.
We have addressed this problem by assessing not only the cell viability, as described above, but also the percentage of cell death that would be required to account for the amount of protein released in the supernatant. For instance, if the cells are cultured in 24-well plates in 1 ml of culture medium that is then analyzed to measure the secreted proteins, parallel samples should be set up in the same way and the cells then lysed in 1 ml of buffer. Then, comparable volumes/dilutions of the cell lysate or the supernatant should be analyzed by Western blot. Using this approach to the study of PRDX1 secretion, for instance, we calculated that at least 40% cell death would be required to account for the observed release of PRDX1 (23).
An alternative approach is to measure the release of an irrelevant intracellular protein, such as actin. Although we have done so (23), this may not work in all experimental models as in some cases actin may be also secreted (54). In general, we strongly recommend a combination of all these methods to rule out nonspecific leakage.
Methods of Proteomics Analysis with Specific Consideration to Problems Arising from the Study of Secreted Proteins
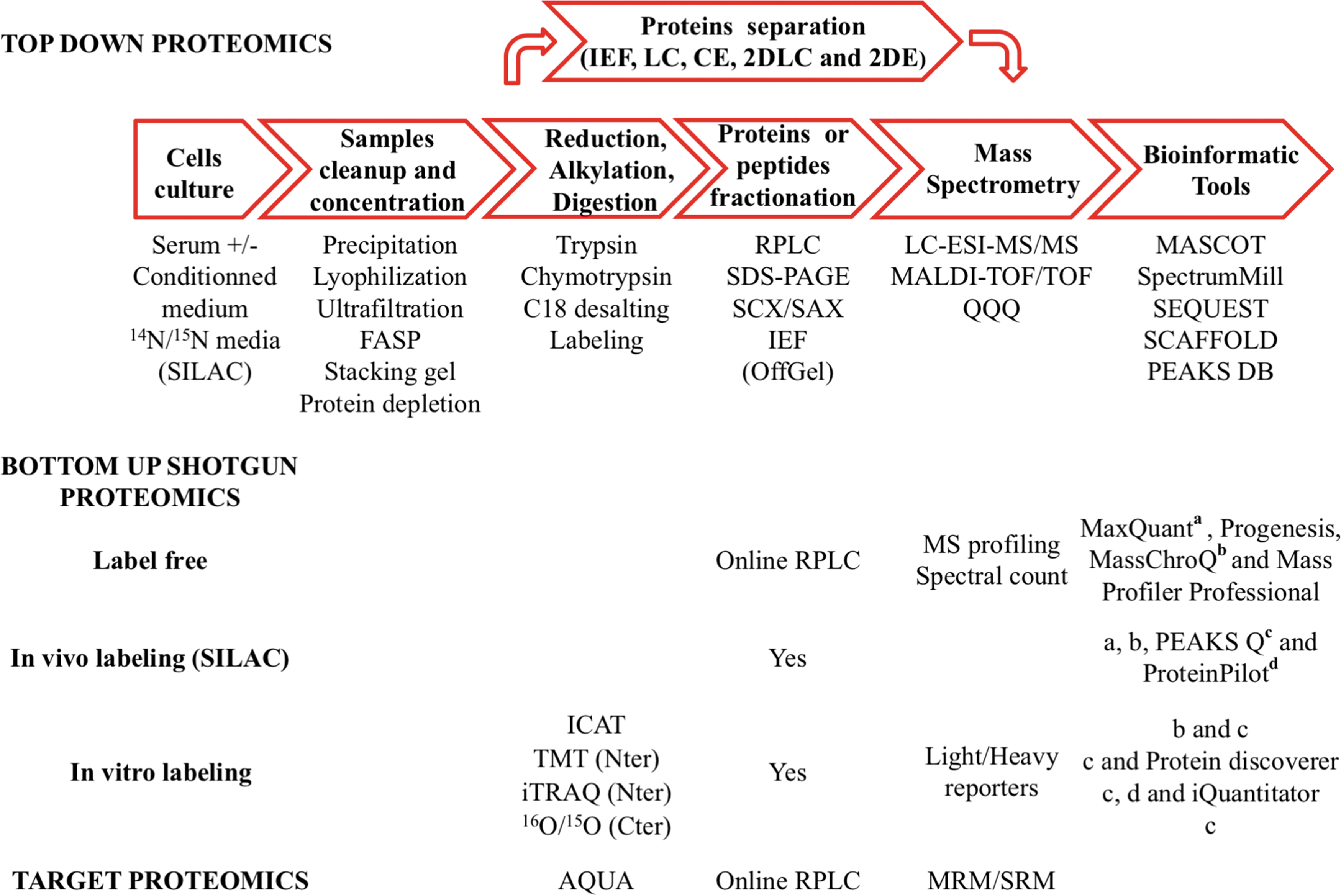
A wide range of methodologies are used to characterize cellular secretomes (69). Among them, the recent advances in liquid chromatography–tandem mass spectrometry (LC-MS/MS)-based shotgun proteomic techniques have provided powerful tools for targeted or nontargeted protein identification and quantification strategies. Conditioned media, with or without serum supplementation, have often been used in secretomics, and unfortunately in such complex biological samples, the proteins of interest are generally present in small amounts, are highly diluted, and may be masked by more abundant serum proteins. To overcome the problem of large dynamic range between low- and high-concentration proteins, the strategies of sample preparation and MS-based analysis should be adapted (Fig. 5 for a global view of the possible strategies). Prefractionation of proteins (top-down approach) or peptides (bottom-up proteomics) with high-resolution separation methods can be applied to decrease the complexity of samples. Use of the new generation of high-resolution mass spectrometry (MS) can allow quantitative, in-depth, large-scale protein expression and characterization in complex mixtures. All the different aspects (cell culture, sample preparation, fractionation methodologies, MS technologies, quantitative profiling of dynamic secretomes, and data analysis) are discussed below.
Culture media and sample preparation
Here, we discuss some general considerations of cell culture, sample concentration, and cleanup that should be done with the minimum number of steps to limit the loss of protein before prefractionation. To avoid contamination of low-abundance proteins secreted by cells, a serum-free culture medium should be used, whenever possible; phenol red-free media are also preferred as phenol red may interfere with MS analysis and very difficult to eliminate. Conditioned medium should be spin at high speed (20,000 g at 4°C) to eliminate cell debris. Concentration of large volumes of medium can be done by freeze-drying (10) or ultrafiltration, for instance, on Amicon Ultra-15 with 3-kDa cutoff membrane (Millipore); this filtration device also enables desalting as culture media have many chemical compounds, such as salts and amino acids, which interfere with MS through ion suppression; ultrafiltration also enables buffer exchange before fractionation or protease in-filter digestion, described as filter-aided sample preparation (FASP) (100). Protein precipitation using organic solvents (such acetone) or trichloroacetic acid is another option to achieve both protein concentration and elimination of undesirable molecules (46), although subsequent resolubilization of the precipitated proteins may be difficult.
Before proteomic analysis, some authors used multiple immunoaffinity depletion methods (e.g., ProteoPrep 20; Sigma) to remove some of the most abundant plasma proteins (91) or Protein Equalizer technology, such as combinatorial peptide ligand library (ProteoMiner; Bio-Rad) (77, 92); sometimes, the two technologies are used to allow enrichment of proteins in biological fluids for in-depth proteomic analysis (11). Independent of the strategy used, proteins need to be reduced, alkylated, digested by proteases (trypsin, chymotrypsin, and LysC), labeled, and desalted on C18 before MS analysis.
Quantitative proteomic profiling by mass spectrometry
Due to the complexity and broad dynamic range of proteins in cell secretomes, prefractionation techniques are necessary to reduce the complexity of conditioned medium and enhance proteome coverage enabling the identification of more proteins by MS. The two main proteomic approaches, top down and bottom up, are presented in this section. Particular attention will be given to shotgun proteomics with peptide labeling or label-free emerging strategies that take advantage of the most recent advances in separation technology, high scan speed, and high-resolution analyzers. We will also mention targeted proteomics by multiple/selected reaction monitoring (MRM/SRM).
Top-down proteomics
In the off-line strategy, proteins are first separated by gel-based methods, liquid chromatography, or mixed methods, digested by proteases, and then analyzed by MS (nanoLC-MS/MS, MALDI-TOF, and MALDI-TOF/TOF).
The classical proteomic approach involves 2-dimensional gel electrophoresis (2-DE) and difference gel electrophoresis, which enables protein separation by isoelectric focusing, according to their isoelectric points in the first dimension, and sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), according to their molecular weight in the second dimension (38, 106). Protein spots are then visualized by staining with Coomassie blue, silver nitrate, or fluorescent dyes (such as SYPRO Ruby). After gel imaging, gels are quantitatively analyzed using an image analysis software, and the spots of interest are excised from gel and treated as described above before identification by MS.
A combination of multidimensional separation methods, including C4 reverse-phase liquid chromatography (35), off-gel electrophoresis (65), and SDS-PAGE (74), can be used to separate proteins in the first dimension. Collected fractions or gel slices are then subjected to trypsin digestion. Peptides are subsequently desalted and analyzed online by nanoLC-MS/MS with an electrospray ionization interface (ESI).
Capillary electrophoresis (CE) can be directly interfaced with mass spectrometers via ESI and used for intact protein characterization, taking advantage of an excellent resolution size-sorted capacity and a good sensitivity (105). Top-down approaches using CE allow elucidation of complex PTM to generate information on protein isoforms (59). However, top-down proteomics allows limited proteome coverage identification compared with a bottom-up approach.
Bottom-up shotgun proteomics
Bottom-up shotgun is a modern strategy, now widely used in large-scale, detailed, relative quantitative protein profiling in complex mixtures. Recent advances in MS led to the development of several methods for relative protein quantification, including isotopic labeling: stable isotope labeling by amino acids in cell culture (SILAC), isobaric tags for absolute and relative quantification (iTRAQ), isobaric tandem mass tags (TMT), isotope-coded affinity tags (ICAT), and 16O/15O. Alternative methods include MRM/SMR or label-free methods based on peptide intensity measurements.
SILAC is based on in vivo incorporation of isotope-labeled amino acids (Lys and Arg) into de novo synthesized proteins during cell culture (72). After treatment, cell culture media from the two samples are combined and subjected to sample preparation as described above. Several orthogonal separation techniques can be used: Purified proteins are separated by SDS-PAGE (74), and in-gel trypsin-digested or desalted peptides are first fractionated by strong cation exchange (SCX) (87). Then, all subsequent fractions, either from SDS-PAGE or SCX, are subjected to reverse-phase nanoLC-MS/MS analysis.
Other in vitro label-based techniques have also been used: for example, addition of light/heavy chemicals to each protein cysteinyl residue with ICAT reagents (47), differential labeling at the N-terminal group of digested peptides with iTRAQ (80) or TMT reagents (87), and labeling of digested peptides at the C-terminal with 16O/15O (11). Note that in these approaches, all labeled peptides are chemically identical and coelute during the chromatographic separation step but have different masses that can be resolved by MS.
In the ICAT method, proteins from two biological samples (e.g., control cells and treated cells) are labeled independently and mixed before trypsin digestion, and ICAT-labeled peptides are isolated by affinity biotin tag; enriched peptides are separated online by nanoLC-MS/MS or off-line by MALDI-TOF/TOF. The relative quantification of protein amount is determined by the ratio of the peptide pairs and MS/MS fragmentation-based peptide/protein identification.
The iTRAQ and TMT methods are particularly attractive quantification techniques in shotgun proteomics, mainly due to the possibility of labeling up to 8 (iTRAQ-8plex; SCIEX) or 10 (10plex TMT; Thermo Scientific) different biological samples, which can then be simultaneously analyzed in a single experiment. In brief, the sample workflow is the following: Purified protein extracts are reduced, alkylated, and digested, and the peptides contained in each sample are labeled by reagents and mixed together before prefractionation by different techniques (off-gel electrophoresis) (93). Then, all the fractions obtained are analyzed by high-resolution nanoLC-MS/MS, which identifies peptides and the relative abundance of protein in the different samples using the ratio of reporter ions released during the fragmentation step. For either top-down or bottom-up proteomics, dedicated database search algorithms (MASCOT; Matrix Science; SPECTRUM MILL; Agilent Technologies; SEQUEST; Thermo Finnigan; and PEAKS DB; Bioinformatics Solutions, Inc.) are used for protein identification. Also for both in vitro and in vivo labeling approaches, dedicated bioinformatic tools are used for the relative protein quantification: iQuantitator (85) for iTRAQ and MaxQuant (97) and MassChroQ (98) for SILAC are cited as examples (see Fig. 5 for more details).
Besides labeling-based techniques, label-free quantification has become the new generation in shotgun proteomic strategies, taking advantage of recent advances in separation technology and MS. At present, identification of 5000 proteins is attainable in a standard 90-min gradient with nanoLC ultra-high-pressure high-pressure liquid chromatography system coupled online to modern high-resolution mass spectrometers (5, 82). Two MS label-free data analysis pipelines can be applied. The first uses the spectral count, which is based on the frequency number of identified total MS/MS spectra of the same protein in each of the data sets (27). The second approach is based on measurement of changes in chromatographic peptide ion intensity, detecting peptides by MS, matching the corresponding peptides across multiple LC/MS runs using their mass-to-charge and retention time, and selecting discriminatory peptides by matching peak area integration on extracted ion chromatograms with specific softwares: Progenesis QI (Nonlinear Dynamics), Mass Profiler Professional (Agilent Technologies), MaxQuant (Max Planck Institute of Biochemistry), and MassChroQ. The final step is identification of regulated peptides by targeted MS/MS. Label-free MS strategies have many advantages, including low cost, high throughput (no limit to the number of samples and conditions that can be compared), and time as sample preparation is easy [based on SDS-PAGE stacking gel purification/in-gel digestion (99) or in-filter digestion using FASP (101)].
Targeted proteomics
As part of a discovery strategy, SRM, also called MRM, is a method of absolute quantification in targeted proteomic analyses and usually carried out on triple-quadrupole mass spectrometers. In this method, isotopic absolute quantification (AQUA), peptides are spiked in complex samples and used as internal standards (41, 95). AQUA peptide is designed to be identical to the prototypic target peptide generated by sample digestion. Both peptides coelute in a known retention time window and are analyzed simultaneously. The concentration of the target peptide is then determined by measuring the signal response for the target peptide relative to that of the AQUA peptide. In the MRM mode, a particular pair of parent ion and several fragments are selected and detected to ensure the specific detection of low-abundance proteins in highly complex mixtures. The SRM method is specific and highly sensitive and a useful tool for high-throughput screening of biomarkers in a very large number of clinical samples.
Redox Arrays
We soon realized, in the course of our studies on redox proteomics of glutathionylated proteins, that some proteins are found over and over in proteomics studies. In fact, this is a very well-known bias of the proteomics approach. While MS is very sensitive, in a mixture of proteins, the abundant ones are identified more easily and can mask the less abundant ones (75, 78). For instance, PRDXs account for up to 1% of total cellular proteins (102). It is very likely that we would miss most of the cytokines or components that are present in the secretome at concentrations in the range of picogram per milliliter.
We have tried to circumvent this problem setting up a technology that we called “redox arrays” and that is based on commercially available antibody arrays (71). These are membranes or glass slides coated with antibodies to up to 1000 different proteins and are used to identify, in a semiquantitative manner, proteins that are differently expressed in different samples. We have adapted this approach to identify proteins undergoing glutathionylation in the inflammatory secretome. This strategy is outlined in Figure 6. The first part is based on the use of BioGEE to attach a biotin tag to glutathionylated proteins described above (Fig. 4). Then, the supernatant, containing biotinylated (glutathionylated) and nonbiotinylated ( nonglutathionylated) proteins, is applied to a membrane array (we used an antibody array consisting of 1000 antibodies immobilized on nitrocellulose membrane by RayBiotech); the membranes are washed as per the manufacturer's instruction by taking care of avoiding the use of any reducing agent (included in most protocols) that would remove the biotin tag. Detection with streptavidin–peroxidase followed by enhanced chemiluminescence will reveal a dark spot if a protein is released as glutathionylated. DTT-treated samples will allow identifying false-positive spots. If a protein is released but not glutathionylated, it will not show up.

Using this approach, both for the secretome and for the cell lysate, we could identify 80 potential glutathionylated proteins, most previously unrecognized to be glutathionylated. We use the word “potential” because the same caution should be used as in the MS identification and require validation as described above (section “studies specifically addressing the proteomic identification of the redox recretome”). In our methodological study, we validated two such proteins: soluble IL-1 receptor type II (IL-1sRII) in the secretome and the tyrosine-protein kinase Lyn in the intracellular fraction (71).
We have tried to establish if this method would allow the identification of low-abundance proteins and compared the proteins identified with this study (71) with those of our previous study using redox proteomics based on 2D gels (37), that of the study mentioned above based on the labeling with BioGEE followed by affinity purification on streptavidin and shotgun proteomics (23), as well as with a curated database, dbGSH, of known glutathionylated proteins identified so far (
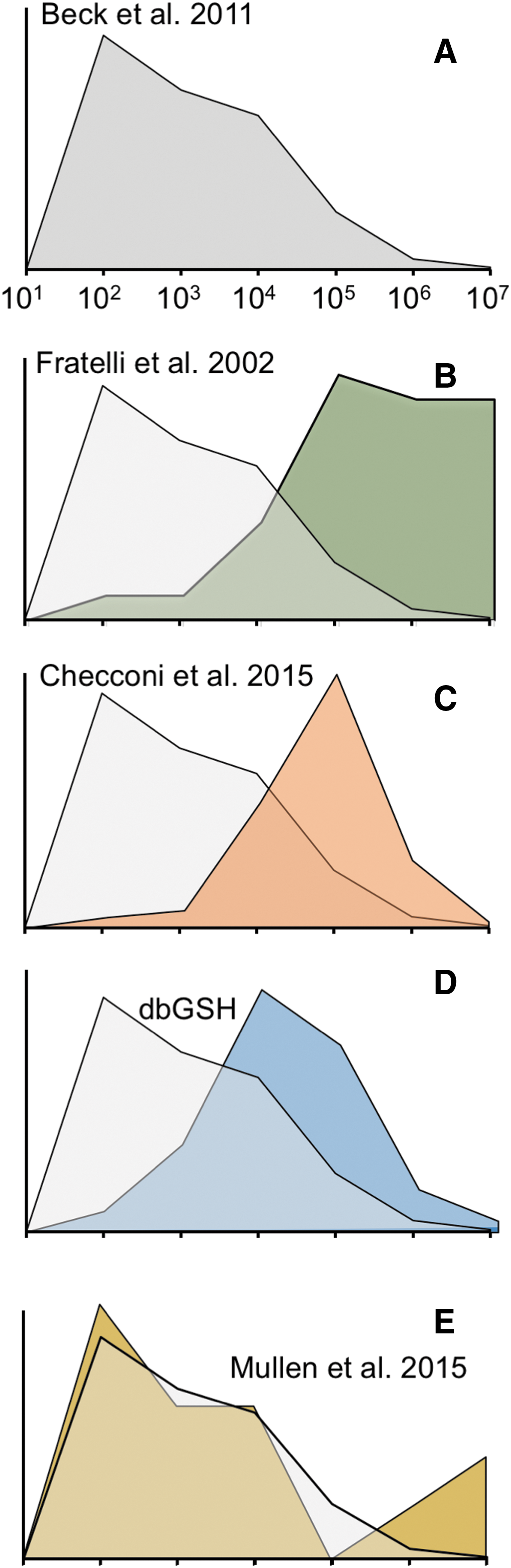
Figure 7B compares the distribution of glutathionylated proteins found with our first method based on radiolabeling of GSH with 35S, 2D electrophoresis followed by autoradiography, spot picking following Coomassie blue staining, and MS identification (37). It can be seen that the proteins identified are heavily skewed toward the high-abundance ones as one may expect from a study using Coomassie blue staining. The identification appears less skewed in our more recent approaches based on the use of BioGEE and MS identification (23, 81) (Fig. 7C). It is improved further in the curated database that clearly includes mainly not only proteins from proteomic studies but also proteins from studies aimed at specific proteins and not involving their identification by proteomics (Fig. 7D). The frequency distribution obtained with the redox array method [taken from (71)] is surprisingly close to the expected distribution for an unbiased method (Fig. 7E). Needless to say, this approach is far from being unbiased as the proteins for which the antibodies are spotted on the arrays are selected based on some criteria. For instance, if we were to use an antibody array for cytokines and growth factors, the frequency distribution would be skewed in favor of low-abundance proteins because most cytokines are not expressed constitutively and, even when induced, are in the picogram range.
We think that the strategy based on redox arrays can be tailored to identify other forms of oxidation or transient disulfides. For instance, it could be coupled to biotin switch approaches using reductants specific for S-nitrosylated proteins (52). We believe this technique might represent, for the field of redox biology, an equivalent to the arrays used to study the kinome and the phosphoproteome.
Biological Significance
Biomarkers of the redox state
The most obvious relevance of the redox secretome is in biomarker discovery. In fact, secreted proteins are likely to be detectable in circulation—an almost essential feature for a successful biomarker. Because reactive oxygen species are short lived, it is impossible to measure any of them in biological fluids, and we have to rely on biomarkers as proxy indicators. Oxidized proteins (such as carbonylated proteins or glutathionylated proteins) can be an indicator of the redox state of the organism provided they are prepared properly. In particular, for thiol-oxidized proteins, it is essential to treat the sample with NEM or another alkylating agent before storage to prevent thiol–disulfide exchange reaction with protein thiols or small molecular weight thiols.
Although not a secreted protein, many studies have measured glutathionylated hemoglobin in many disease conditions and found an association with some of them [reviewed in Ghezzi (45)]. The same approach could be used for secreted proteins. Oxidized proteins could be of the same use as glycated hemoglobin in diabetes that do not just reflect to the level of glucose at a given time point but really is an indicator of the exposure of the organism to high glucose (21). As mentioned earlier, the example of PTX3 suggests that the redox state of a biomarker may represent an additional dimension, improving its diagnostic or prognostic significance (29).
Functional significance
We think the study of the redox secretome can lead to the identification of novel soluble mediators (cytokines). Probably the best example of this is thioredoxin, which was discovered by Arne Holmgren in Stockholm as a reducing cofactor essential for the catalytic action of ribonucleotide reductase and Junyi Yodoi in Kyoto as an adult T-cell leukemia-derived factor; Yodoi was expecting to have identified an interleukin-1 gamma, but once the protein was sequenced, it was identified as TRX (104). In addition, PRDXs were first identified as natural killer (NK) cell activators even before their enzyme activity was established (85). The main cytokine-like activities of redoxins are summarized in Table 2 (24).
Redox arrays, Mullen et al. 2015 (70).
The identification of inflammatory cytokines has been a great success story: Tumor necrosis factor (TNF) was discovered as an inflammatory mediator in 1985 (7), and by 1998, anti-TNF antibodies were approved for use in inflammatory disease. Other cytokine inhibitors followed to block interleukins (ILs) IL-6, IL-1, and IL-17.
It may be worth noting that the most known cytokines have been discovered either by in silico mining or by biochemical purification of proteins with a biological activity. So far, we think that the only “cytokine” discovered by proteomics is HMGB1, which was observed as it was identified in Coomassie blue-stained gels as differentially expressed in the supernatant of macrophages stimulated with LPS compared to unstimulated ones (97). Thus, the study of the redox secretome may lead to the identification of previously unrecognized mediators and potential pharmacological targets.
As mentioned above, PRDX1, PRDX2, and FGF1 are secreted via disulfide-linked homodimerization. It will be interesting to investigate whether other proteins are secreted via formation of a disulfide-linked dimer as this may eventually describe a family of proteins whose secretion is dependent on the redox state of the cell and/or the extracellular environment.
Footnotes
Acknowledgments
We thank Professor Hubert Vaudry for his help in the article revision and Sonia Leach for proofreading and editing English. Supported by the EU, European Regional Development Fund, Interreg IVA France (Channel)—England, and project PeReNE (Peptide Research Network of Excellence).