
Abstract
Introduction
T
Because numerous diseases reflect an imbalance between positive and negative ROS effects (34), researchers also need to measure this electron deregulation and understand which redox-active components are present and at what concentrations. Most research on redox sensing has focused on direct investigation and detection of oxidants that are actively involved in redox processes. Oxidation can be non-catalytic, mediated by ROS, and catalytic, performed by enzymes. However, the majority of studies have focused on ROS. It should be noted that “ROS” does not refer to a single entity; these species can be divided into non-radical oxidants, such as hydrogen peroxide (H2O2) and hypochlorous acid (HOCl), among others, and radical oxidants with an unpaired electron, such as superoxide anion (free radical) (O2 −) and nitric oxide (NO). The latter are also called free radicals.
Having an efficient and reliable technique to directly detect changes in intracellular ROS production is a fundamental need in many research fields. Experimental culture conditions (e.g., substrates in the medium, oxygen tension) will greatly affect the sources and magnitude of cellular ROS production. In addition, owing to the high reactivity and extremely short half-life of ROS, quantification of specific ROS has always been challenging and prone to artifacts. Several techniques are currently employed to measure ROS in cultured cells and given the massive expansion of the ROS field, rigorous recommendations have been proposed (29). The purpose of this article is to discuss synthetic sensors. The growing field of protein sensors, such as genetically encoded redox probes, though very important, is outside the scope of this article and will not be addressed.
On a cellular level, sensor-based fluorescence detection is superior because of its high sensitivity, experimental convenience, and non-invasiveness. Fluorescent sensors can be measured with spectrophotometers or microplate readers and using wide-field and confocal microscopy, and flow cytometry. Only the last two approaches allow evaluation at the single-cell level. A large number of fluorescent synthetic probes capable of being introduced into living cells and organisms have been developed. These probes exhibit a change in their fluorescence intensity in response to oxidation. In most applications, fluorescent probes are generated in situ by the reaction of an inactive form of the probe with ROS. Thus, the probes are stable in a reduced state, but they can be oxidized by the species of interest. Two types of probes are employed: chemiluminescent probes, such as luminol and lucigenin (28, 51), and fluorescent probes, such as the extensively used reagents, dihydroethidium (DHE) (107), dichlorofluorescein derivatives (65, 81), dihydrorhodamine (82), and Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine [AmR]) (55, 56). However, the fact that the detection principle is based on a redox reaction will directly affect selectivity. Even if the sensor is stable in the reduced state, it must be sufficiently reactive and easily oxidized; moreover, the identity of all the molecules responsible for the oxidation is often difficult to determine. Recently, it was demonstrated that reactive sulfide species can even be detected with high sensitivity by some of these fluorescent probes (19). The sensor undergoes two electron oxidation steps, usually through formation of a radical intermediate. Consequently, these probes are associated with several common pitfalls: They have limited resistance against oxidation, the products of probe reactions with ROS may be highly unstable, and they may also be subject to redox cycling that generates additional ROS (5, 31, 42).
After a brief introduction to these common probes, we describe practical experiences that not only highlight difficulties associated with their use but also indicate how these problems can be solved. A number of alternatives have been developed to circumvent problems encountered in using these probes. Some include sensors based on a non-redox mechanism, such as deprotection of a fluorescent molecule by ROS (21, 52), that still uses changes in fluorescence intensity as the detection principle; these are reviewed in Maghzal et al. (53) and for boronate-protected sensors in Lin et al. (49) and Zielonka et al. (109). Others include analytical measurements to quantify the products of oxidation by high-performance liquid chromatography (HPLC) or mass spectroscopy (42). There is also a high interest in protein sensors, such as genetically encoded redox probes (24), that are discussed in another article of this Forum. In addition to considering these approaches, this article will discuss two other types of detection techniques: electron paramagnetic resonance (EPR) (46) and fluorescent lifetime-based methods (76). Both techniques directly detect free radicals based on their paramagnetic properties. We will then address the question of location, focusing on one subcellular compartment—the mitochondria. We will describe possibilities of targeting synthetic probes to mitochondria, and we will then evaluate contributions from different techniques to the field of ROS detection.
Intensity-Based Fluorescent Synthetic Sensors—Oxidant-Sensitive Probes
“Oxidation as a common activation route for probes” from Wardman (99).
In addition to the book by Halliwell and Gutteridge (34), there are numerous reviews on intracellular ROS detection. During the past 10 years, several reviews were published that focused on its various aspects. In 2006, Bartosz proposed an interesting description of the problems that might arise due to the limited stability and high reactivity of fluorescent sensors (5). Soh's discussion included singlet oxygen 1O2 (88). In 2007, Dikalov et al. presented the major assays with their strengths and weaknesses (23). Wardman completed some unexplored aspects of these earlier reviews (99). In 2010, Rhee et al. focused on H2O2 (72) and in 2014, Fan and Li concentrated on anion superoxide (27). Chen et al. presented unconventional sensors (14). In 2013, Woolley et al. drew up a list of ideal criteria that sensors for ROS detection must fulfill and designed a logic diagram for deciding which sensors should be used for which experimental design (100). Kalyanaraman and Zielonka continue to warn about the most used sensors (18). All authors advise the reader about the complexities associated with ROS detection and the difficulty, if not impossibility, of using well-known synthetic probes for this purpose. They recommend that these probes be considered redox indicators. However, it is understandable that biologists who need to quantify ROS will be unable to resist using commercially available probes, especially since they are cited in thousands of publications. In addition to reiterating concerns that have been well covered in the aforementioned reviews, we will also focus here on difficulties, usually not published, that can be encountered in attempting to apply these sensors in the laboratory.
DHE (hydroethidine)
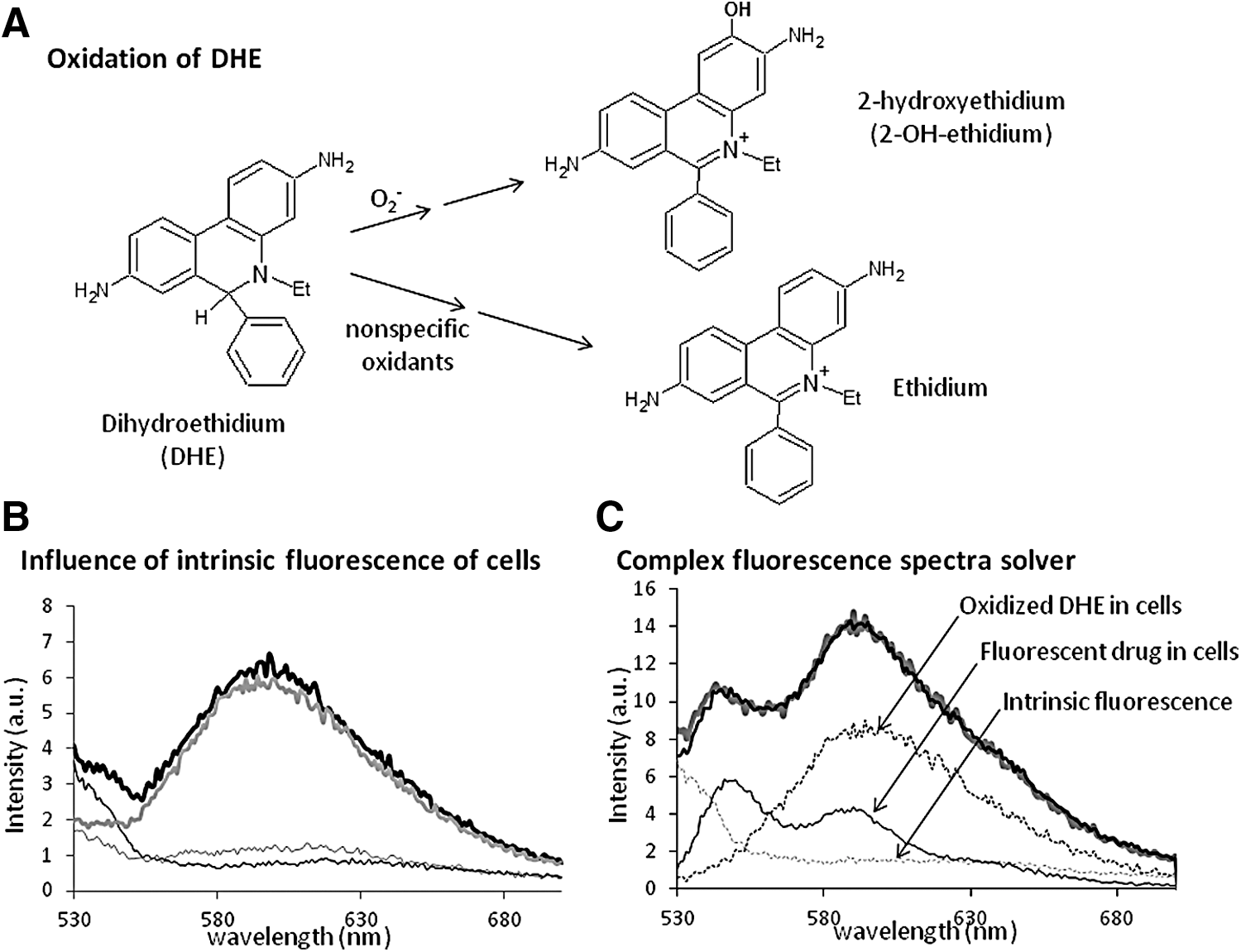
The usefulness of DHE, a cell-permeable compound, as an indicator of redox components has been reviewed by Zielonka and Kalyanaraman (107). Intracellularly, DHE is oxidized to the fluorescent products, ethidium and 2-OH-ethidium (2-hydroxyethidium; Fig. 1A). For several years, ethidium was considered the sole product of DHE oxidation (94). Both oxidized compounds can intercalate into nuclear DNA, which increases the quantum yield of their fluorescence. Oxidation of DHE is a superoxide-dependent process in which 2-OH-ethidium is formed by superoxide anion, although the reaction of DHE with other ROS or oxidant species and subsequent formation of ethidium may interfere. Because the fluorescence of 2-OH-ethidium overlaps with that of ethidium, it has been suggested that HPLC be performed to quantify 2-OH-ethidium and thus superoxide anion (107).
Fluorescence-based techniques
The traditional excitation wavelength is based on the maximum emission of ethidium at 518 nm. The two forms of oxidized DHE, ethidium and 2-OH-ethidium, fluoresce as single bands at 593 and 567 nm, respectively (104). The relative intensity of the two compounds differs slightly among published reports (79, 105, 110); however, they are of the same order of magnitude. After excitation above 510 nm, the fluorescence spectrum of different DHE-loaded cell lines shows a maximum at ∼587 nm (ethidium fluorescence), but not at 568 nm (Fig. 1B). As calculated by HPLC (110), the level of ethidium detected in biological systems is nearly 10-fold higher than that of 2-OH-ethidium, making ethidium fluorescence the major contributor. Robinson et al. proposed that excitation at 396 nm would preferentially excite 2-OH-ethidium, which would thus exhibit more intense fluorescence than ethidium (79). These authors published a general protocol using two excitation wavelengths (78). It should be noted that autofluorescence of cells (Fig. 1B), and in some case fluorescence of incorporated drugs (Fig. 1C), can interfere with the fluorescence of oxidized products. An example is the interaction with doxorubicin, described by us (69). In such cases, we recommend working with the entire spectrum. The complex fluorescence spectra of cells treated with drugs and loaded with DHE can be analyzed to identify and quantify all fluorescent components that contribute to the complex experimental spectra. The method was previously described (10) and uses the spectra of all fluorescent components obtained individually: drugs in cells, ethidium in cells, and the intrinsic fluorescence of cells (Fig. 1C). A computer tool allows the reconstruction of a theoretical spectrum by combining the characteristic fluorescence spectra obtained beforehand. The theoretical and experimental spectra are compared using graphical or numerical estimators.
Stability of DHE
Because DHE is also fluorescent (excitation at 350 nm, emission at 400 nm), a ratiometric approach was initially proposed for DHE-based assays using the intensity ratio, 640/420 nm, between oxidized and reduced forms of DHE (13). Subsequently, a single-wavelength, cytometry-based application of DHE has come to be the classical method for assessing ROS (59). We and others (9, 91) have found that DHE is not stable when irradiated by ultraviolet light; after irradiation at 355 nm, the fluorescence of DHE disappears within a few minutes. Because compound stability is a prerequisite for using the ratiometric configuration of the assay, DHE intensity must be recorded shortly after preparation of the compound and using extremely low levels of irradiation. However, although we have found that application of these rather drastic conditions to ratiometric assays seems to increase the reproducibility of our data, we consider that the benefit does not justify the considerable experimental effort.
Practical recommendations
The literature seldom mentions the blue color characteristic of solutions containing reduced DHE. When working solutions (1 mM) are prepared immediately before use by diluting stock solutions into degassed medium under a nitrogen flow, the resulting solution is pale pink rather than blue owing to contamination by the oxidized form of DHE. Zielonka et al. recommend preparing stock solutions in an anaerobic chamber (110). In this case, a stock solution of DHE prepared in degassed dimethyl sulfoxide at 10 mM can be stored under N2 at −20°C for <1 month. Longer storage might require keeping DHE at −80°C. Working solutions prepared after this point take on a dark pink coloration owing to partial or total oxidation and must be discarded. Robinson et al. proposed that if contamination does not exceed 10%, DHE can be chemically reduced or purified by HPLC (79). Because the photo-oxidation of DHE is oxygen dependent, solutions are prepared under subdued light in degassed solutions. Extracellular molecules that fluoresce at wavelengths above 530 nm, such as Phenol Red, must be removed from the medium by washing. Consequently, suspensions of cells are usually incubated with 20 μM DHE in phosphate-buffered saline (PBS) in the dark at 37°C for 15–30 min. After that, the preparation can be used with or without washing. Fluorescence values must be normalized to the number of cells in each sample. In the literature, the formation of oxidative species by DHE owing to redox cycling is described as being less important than is the case for other superoxide sensors, such as luminol and nitroblue tetrazolium.
Amplex Red
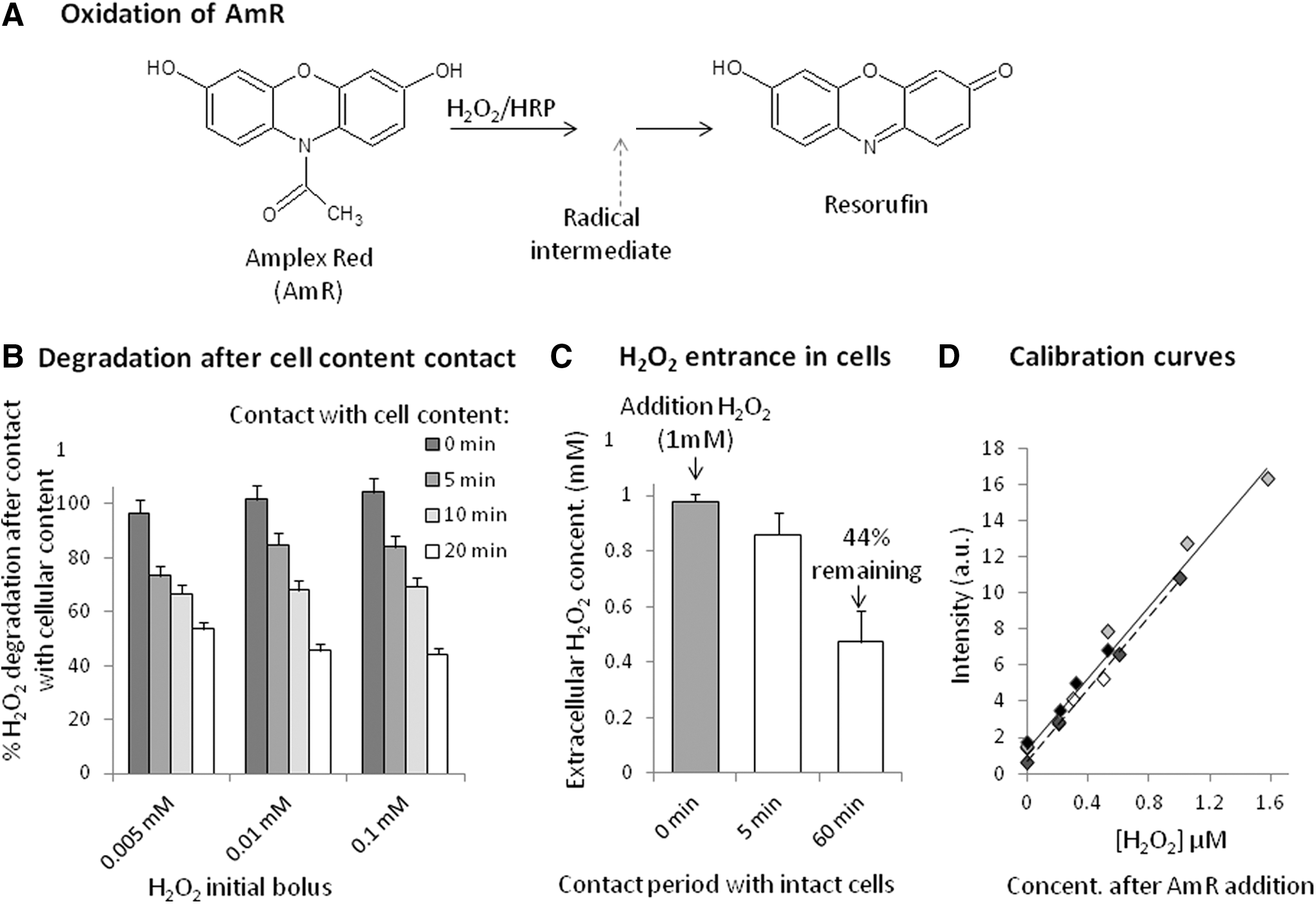
The AmR assay is based on the enzymatic oxidation of the colorless, non-fluorescent compound AmR through a radical intermediate (32). AmR reacts with H2O2 in the presence of horseradish peroxidase to generate the stable colored and fluorescent product, resorufin (106) (Fig. 2A). AmR is not permeable, and it thus measures extracellular H2O2. Because H2O2 diffuses through lipid membranes, this assay gives an indication of cellular production, provided cells release enough H2O2. Production and secretion of H2O2 by cells has been reported for several cell types, including hemocytes (57), activated human leukocytes (56), neutrophils (106), endothelial cells (89), synaptosomes (3), and podocytes (45). Most other cell lines do not release detectable concentrations of H2O2 into the extracellular medium. Kits are commercially available and easy to use. However, as is the case for all synthetic sensors, AmR presents several pitfalls (18, 103). Notably, exposure of AmR to light also results in the formation of resorufin (103); thus, experiments with AmR should be conducted under subdued lighting.
Cells that do not release H2O2
Because some cell lines do not release detectable concentrations of extracellular H2O2, several teams have modified the technique to include a cell lysis step to release intracellular H2O2 into the external medium. Some preliminary adjustments for lysis conditions (time, concentration, medium) are necessary, and we perform them using the inexpensive FOX-2 (ferrous oxidation in xylenol orange, version 2) method (87), which allows for the determination of hydroperoxides by measuring absorbance. We have found that experiments cannot be performed in culture medium, because H2O2 is eliminated by cell culture components, possibly sodium pyruvate (30); thus, these experiments are performed after washing cells three times and resuspending them in PBS. Cell lysate components are also effective in eliminating extracellular H2O2, as shown in Figure 2B, which presents the decrease in H2O2 concentration over time caused by contact with cell lysate. Accordingly, we recommend performing cell lysis and measurements within 5 min. Alternatively, for cells that release H2O2 slowly, the duration of AmR loading was increased up to 60–120 min to accumulate AmR signal (23, 58). For such a long experiment, one has to be careful about photo-oxidation of the sensors. Noteworthy, protocols to generate oxidative stress often propose the addition of exogenous H2O2 solution at 1 mM to cells. After 1 h, ∼56% of added H2O2 enters into the cells (Fig. 2C). Obviously, H2O2 is also converted inside the cells during the 1-h treatment, as evidenced by the fact that <1 μM of H2O2 equivalents are released after lysis; in contrast, non-lysed cells do not release H2O2 (70).
Practical recommendations
The proposed protocol was not only developed for cardiac cells but can also be performed on other adherent and detached cells. After washing and trypsin treatment (if necessary), cells are suspended in PBS or KRPG (AmR kit), and then 5% Triton-X (without H2O2) is added to a final concentration of 0.25% (v/v). Cells are lysed for 5 min (including 2 min of centrifugation, if needed) before addition of the reagent. The lysate is suspended in half the volume of the micro-wells. Identical volumes of AmR reaction mixtures are added, avoiding exposure to light, and spectra are recorded between 560 and 600 nm after excitation at 550 nm (max: 580 nm) for 1 h. Fluorescence changes can be converted to H2O2 equivalents (μM released) using a standard curve. H2O2 standard curves must be prepared daily by adding equal amounts of AmR reaction mixture to different dilutions of H2O2 in KRPG (AmR kit). Fluorescence readings become stable within 10–20 min. If the slope of the standard curve is stable and ranges from 9 to 11 μM −1, the intercept varies depending on the time between AmR preparation and measurement (Fig. 2D). We strongly recommend preparing blank and H2O2 solutions for each set of experiments. According to several authors, AmR assays are reported to be able to detect concentrations as low as 0.05 μM (106), although in our hands, with a conventional fluorimeter, this technique requires concentrations >0.1 μM. Because resorufin is subject to autoproduction of ROS and other artifacts, it is doubtful that biologically realistic H2O2 production can be quantified using this approach.
Chemiluminescent sensors
Lucigenin (bis-N-methyl-acridinium) and luminol (5-amino-2,3-dihydro-1.4-phthalazinedione) have frequently been used for the detection of superoxide anion (Fig. 3A). The complex chemistry of both compounds was already described in a review in 1993 (28). Proceeding through free radical formation, an electronically excited molecule is produced that emits a photon when returning to its ground state. The luminescence can be detected by a scintillation counter or a luminometer (2). The main disadvantage consists of production of O2 − during the process (a phenomenon known as redox cycling). Because of redox cycling, strong pH dependence, and absence of specificity, luminol was abandoned for this application (44). L-012 (8-amino-5-chloro-7-phenyl-pyrido[3,4-d]pyridazine-1,4(2H,3H)dione), a brighter analog, was developed (65, 108). The other compound, lucigenin is also prone to redox cycling. The use of low concentrations of this compound minimizes this problem (98), but its use remains controversial (23, 99). Therefore, other compounds, not prone to redox cycling, were proposed. Based on the central structure of coelenterazine and cypridina luciferin (Fig. 3B), various structural analogs were developed. Among them, MCLA [2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidazo(1,2-α)pyrazin-3-one] is described as being a useful probe for O2 − detection, even if its specificity is not entirely sure (43). New analogs are still under development that have various emitting wavelengths (41). It should be noted that the localization of these sensors is disputed: extracellular (54), intracellular, or even mitochondrial localization (68). One explanation of this discrepancy could be that localization depends on concentration and cell line. Bioluminescence was also used to detect H2O2 (33). PCL-1 [peroxy caged luciferin 1 ((4S)-2-[6-[(4-Boronophenyl)methoxy]-2-benzothiazolyl]-4,5-dihydro-4-thiazolecarboxylic acid)] is a boronate-protected analog of firefly luciferin peroxynitrile (Fig. 3C). After being deprotected by H2O2 (95) and/or peroxinitrile (85) and a reaction with luciferase, photons will be emitted.

Alternative Techniques for Detection of Free Radicals
Two detection methods based on the paramagnetic properties of free radicals have been developed. The first, EPR (or ESR) spectroscopy allows the direct detection and characterization of molecules or a paramagnetic center having unpaired electrons (46); however, it cannot directly detect short-lived, intracellular free radicals. Instead, detection of these species is based on the formation of persistent radicals by spin trapping or spin probing (92, 97). The second method is based on fluorescence lifetime measurements (6, 83). Sensors with long fluorescence lifetimes—the time required for a population of excited sensors to return to their non-excited state by emitting light—are sensitive to collisions with paramagnetic species (20, 66). This phenomenon, called dynamic quenching, decreases the fluorescence intensity and lifetime of the sensors, with the decrease being proportional to quencher concentration.
Electron paramagnetic resonance
The fundamental physical concepts of EPR are analogous to those of nuclear magnetic resonance, except that electron spin, rather than nuclear spin, is excited. An EPR spectrum is usually obtained by varying magnetic field strength at a fixed microwave frequency. This technique has historically required expensive equipment and skilled operators; however, a number of manufacturers have worked to produce EPR spectrometers that are easy to operate. Each free radical exhibits a specific EPR spectrum, and the intensity of an EPR signal is proportional to its concentration, allowing for identification and quantitative measurement of free radicals. In principle, this technique is ideal, but it has limitations, namely that it is not sensitive enough to directly detect highly reactive free radicals in cells. Increasing the signal requires the addition of spin traps or spin probes—diamagnetic molecules that react with intracellular free radicals to form new, more stable radicals with half-lives that are long enough to accumulate and be EPR detectable (5, 23).
Spin traps form covalent bonds with the radical by an addition reaction that yields a long-lived spin adduct. The reaction creates a unique adduct for each free radical that can be specifically identified (Fig. 4A). The most common traps are nitroso, nitrone (12), and cyclic nitrone (1, 90), which yield nitroxide adducts (accurately termed aminoxyl radicals) that can be detected by EPR. Among conditions that spin traps must fulfill to be considered efficient free radical detectors, the most crucial are kinetic criteria; specifically, they must trap free radicals rapidly. Besides low spin-trapping rates, spontaneous and cell-enhanced decomposition to products not detectable by EPR and the need for high concentrations of spin traps are potential pitfalls that limit the applications of spin traps in biological systems (25).

Over time, research has improved spin adduct stability and reduced its toxicity through structural modifications to the trap (37). Hydroxylamide, a diamagnetic product, is formed by direct reduction of nitroxide adduct, a reaction that competes with free radical quantification. However, this drawback can be usefully exploited to differentiate between normal and pathological redox states status by using the reduction of cell-permeable nitroxides to hydroxylamines (35). Cyclic hydroxylamine-based ROS sensors that are oxidized by ROS without binding have also been developed (Fig. 4B). Because they do not “trap” the radicals, the terms “spin label” or “spin probe” are preferred. A redox reaction involving one-electron oxidation by a free radical leads to the recovery of the nitroxide radical, which is detected by EPR (25); consequently, hydroxylamines yield the same nitroxide radical regardless of the free radical being detected. Among the advantages of hydroxylamine spin probes is their suitability for use at very low concentrations (sub-millimolar range) compared with spin traps; they also show better stability and faster kinetics. However, free radical identification is complicated by the fact that hydroxylamines can be potentially oxidized by other oxidants. It should be noted that the term “spin labeling” is also used for an entirely different method in which nitroxide radicals are used for probing local dynamics of protein or biological membranes, a nomenclature overlap that might lead to confusion for the unprepared reader (7, 38).
Time-resolved microfluorimetry
The change in the fluorescence lifetime of a given sensor can be used to detect and quantify free radicals. Dynamic quenching, which forms the basis of this technique, is in some ways similar to Förster resonance energy transfer (FRET), a more common process. In both cases, the excited fluorescent sensor loses its energy without emitting light. In FRET, this energy is transferred to a second molecule in close vicinity; whereas in dynamic quenching, this energy is lost via collision with quenchers. In the latter case, the fluorescence intensity and lifetime of the sensor decrease as a result of multiple processes that occur at the moment of collision. Paramagnetic species, such as oxygen and free radicals, have the property of quenching long-lived fluorophores, such as metal complexes and aromatic compounds, that have long lifetimes (hundreds of nanoseconds). Use of lifetime-based fluorescent sensors circumvents a certain number of problems associated with intensity-based sensors, since the measurement does not depend on variations in sample thickness, cellular uptake, or sensor concentrations. Several additional advantages include stability of the sensor, which does not need to react with free radicals, and the reversibility of the process, since cessation of free radical production would be immediately observed. Measured values are not attributable to the accumulation of free radicals, but they instead reflect their production at the exact time of measurement.
Successful applications
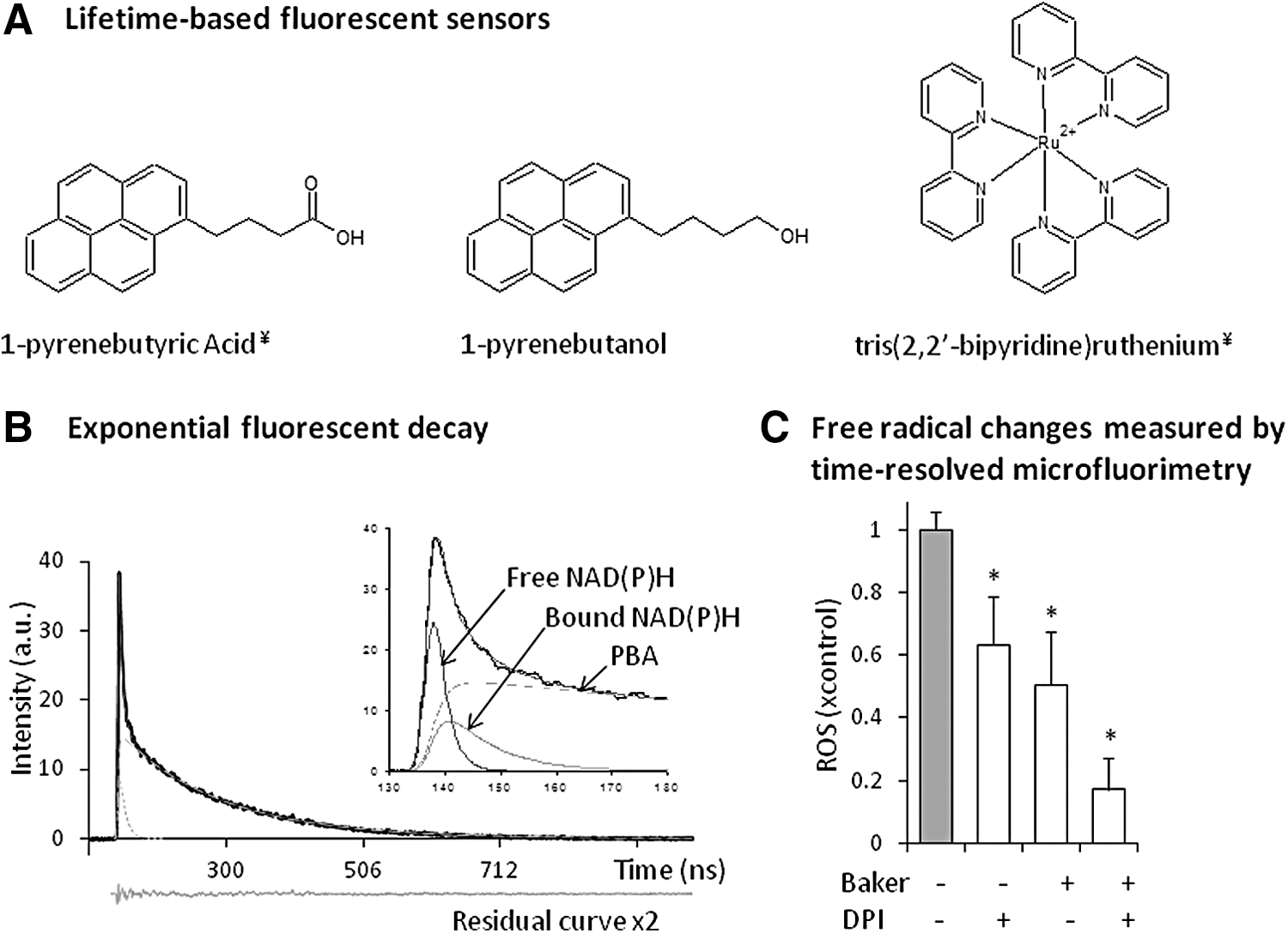
Ten years ago, we started using 1-pyrene butyric acid (PBA) in living cells, and by following its lifetime variation, we were able to quantify free radicals. Dynamic quenching plays an important role in measuring oxygen, and in the 1970s, Vaughan and Weber were the first to demonstrate that PBA could be used as an oxygen probe (96). More recently, it has been shown that fluorescence quenching of PBA can also be used to measure free radicals (16, 20, 66). The concept has been validated for PBA and other probes (Fig. 5A) in solution (66) and in loaded primary cells (57, 67, 73), cultured cells (69, 71, 84), and microalgae (8). We have shown that this sensor is sensitive to small, mobile radical molecules, but not to non-radical ROS such as H2O2.
Fluorescence-based techniques
Pulsed-lifetime measurements are recorded using a previously described apparatus (74, 76). Fluorescence decay of PBA in PBA-loaded cells is recorded and analyzed for each individual cell. Application of a 404-nm filter to the emissions path after excitation with a nitrogen laser at 337 nm allows decay to be resolved into three exponential curves (Fig. 5B). Time constants (i.e., lifetimes) and amplitude values of each exponential curve in the decay are obtained using the downhill simplex method (63). The two short time constants are attributable to free and bound NAD(P)H, and they provide information about cellular activity, a property that will not be further discussed here. The relationship between the long time constant (>100 ns), which is characteristic of pyrene derivatives, and free radical concentration is described by the Stern–Volmer equation. One can, therefore, calculate intracellular free radical production in treated cells by a comparison with control cells (expressed as fold change) using the following equation:
where τ is the fluorescence lifetime measured for treated cells, τc is the mean of the lifetime measured for non-treated cells, and τ0 is the mean of the fluorescence lifetime in the absence of free radicals.
Practical recommendations
Cells must be rinsed once with Hanks Balanced salt solution (HBBS) to remove protein that can bind PBA and prevent loading. Then, cells must be incubated at 37°C for 5–15 min in HBSS containing 0.2–1.6 μM PBA, depending on the cell line. Removal of all extracellular PBA is a key point; cells are rinsed three times and resuspended in HBBS, after which the cell suspension is placed on the objective. A total of 12–20 cells should be observed in a single recording session, reducing the time required to complete the entire process to ∼30 min. During the recording procedure, the cells remain physiologically intact, and ROS formation is stable.
One challenge in applying the lifetime-based method is that quantification of free radical changes requires obtaining intact cells that no longer produce reactive oxygen. Generally, placing cells in paraformaldehyde (Baker solution) stops mitochondrial activity and ROS generation (70), while preventing a loss of membrane integrity and probe reorganization. However, for other cell lines such as cardiac cells (69), which are known to contain membrane-bound NOX, fixation is not sufficient to stop all free radical production, and additional treatment with the NOX inhibitor diphenyleneiodonium chloride (DPI) is necessary (Fig. 5C). DPI is extensively used as anion superoxide inhibitor. It is a flavin inhibitor that blocks not only NOX but also mitochondrial production of superoxide (50). The method is sensitive to substantial variations in intracellular oxygen. However, several experiments have shown no impact of oxygen (70, 76), probably owing to the rapid diffusion of oxygen inside cells. It is important to bear in mind that fluorescence lifetime (as intensity) is highly sensitive to changes in the environment, which must be adequately controlled.
Mitochondrial Targeting Sensors
Mitochondria play a central role in ROS biology. They are an important source of ROS within cells, are involved in cell survival and death, and are promising therapeutic targets. Different carriers and vectors have been developed to reach mitochondria. To serve these functions, these probes need to cross not only the cell membrane but also outer and inner mitochondrial membranes to access the organelle. Although some research has focused on detecting redox-active components in isolated mitochondria (4, 60), there is considerable interest in performing measurements in living cells. As is the case for intracellular ROS detection, mitochondrial ROS detection has been the subject of a number of reviews (22, 24, 102), which describe problems similar to those encountered with cellular sensors (107).
Lipophilic cationic compounds
The families of mitochondrial vectors are presented in Figure 6A. These vectors exploit the higher membrane potential maintained by mitochondria, which offers a unique chemical opportunity to selectively target the organelle. Structurally, these vectors include positive charges and lipophilic groups (40, 77, 86). The lipophilic part facilitates movement across the membrane, whereas the positive charge takes advantage of the proton gradient and subsequent electrochemical potential generated within the matrix of mitochondria. However, good ROS sensors must enter mitochondria regardless of their polarization status and should not change the potential gradient on entering the organelle. Since fluorescence signals are directly dependent on the number of mitochondria per cell, mitochondria number must be determined simultaneously. Finally, sensors should not interfere with the biological system. It is possible that loading sensors in mitochondria could disturb cells and influence both membrane potential and ROS. Designing properly working sensors for mitochondria is, therefore, not trivial.

Triphenylphosphonium-based probes
The triphenylphosphonium (TPP) group has a positive charge on the phosphonium and is surrounded by three lipophilic phenyl groups (80). There are several sensors that carry the TPP group (Fig. 6B–F). These allow ROS detection by various methods, including fluorescence intensity as well as mass spectroscopy and spin trap. Some are already commercially available, such as the widely used Mito-HE, better known as mitochondrion-targeted DHE (MitoSOX). MitoSOX (Fig. 6B) is based on DHE linked to TPP by a hexyl carbon chain. Designed for anion superoxide detection, it behaves similarly to its parent compound DHE (79, 107). Not unexpectedly, MitoSOX is also preferentially oxidized by superoxide to a hydroxylated product by the same mechanism as DHE. Among other TPP-based sensors is MitoPY1 (triphenylphosphonium conjugate with boronate-based peroxy-yellow 1; Fig. 6C), which was designed for H2O2 detection (21). The protecting boranate moiety reacts with H2O2; removal of the boronate moiety allows light emission. Notably, it also reacts with peroxynitrite. To avoid difficulties associated with fluorescence-based detection, several teams have developed mitochondria-targeted sensors that use alternative detection methods. A new sensor, MitoB [(3-boronophenyl)methyl]triphenyl-phosphonium (Fig. 6D), detected by mass spectroscopy, was recently described (15). Also in the area of EPR, several nitrone spin traps (26, 36) (Fig. 6E) and hydroxylamine spin probes (25) (Fig. 6F) have been modified by addition of a TPP group.
Lipophilic cations
Rhodamine and rosamine sensors share the property of encompassing a cationic functionality in an otherwise nonpolar framework. Their accumulation is usually strongly dependent on membrane potential. Rh123 ([6-amino-9-(2-methoxycarbonylphenyl)xanthen-3-ylidene]azanium chloride) and Mito Tracker series probes are extensively used for mitochondrial imaging and membrane potential evaluation. A number of fluorescent ROS sensors derived from these parent compounds have been developed (Fig. 6G–J). One of these, DHR [dihydrorhodamine 123 (2-(3,6-Diamino-9H-xanthene-9-yl)-benzoic acid methyl ester; Fig. 6G], is transformed by oxidation into the fluorescent Rh123, but can react with various ROS and oxidizing species (17). However, DHR cannot correctly quantify ROS in the mitochondria, because it is cationic and only localizes in mitochondria after oxidation. More recently synthesized rhodamine-like sensors include MitoHR and MitoAR (Fig. 6H) (47). Among the best-known sensors in the Mito Tracker series is Mito Tracker Red CM-H2XRos (Fig. 6I). This reduced, non-fluorescent version of MitoTracker Red that fluoresces on oxidation was characterized in a comparative study by Kuznetsov et al. (48). Our group has used a rhodamine carrier to synthesize the pyrene-based sensors, PRE-4 [(1"-pyrene butyl)-2-rhodamine ester] (75) and PRE-6 [(1"-pyrene hexyl)-2-rhodamine ester] (Fig. 6J), which successfully reach mitochondria. However, folding of these two chromophores and subsequent energy transfer between them has so far prevented their utilization.
Peptidic delivery agents
Peptides containing both naturally occurring and non-natural amino acids designed by alternating lipophilic and positively charged moieties constitute the third group of probe types (39, 101). Shiller and Szeto peptides were designed to target mitochondria (93), they present intrinsic antioxidant activity. Subsequently, the Kelley laboratory created mitochondria-penetrating peptides (MPP) combining synthetic and natural residues that are either cationic (e.g., arginine) or hydrophobic (e.g., cyclohexylalanine). The solid-phase synthesis of such peptides is relatively straightforward, and sensors can be added through peptide bond formation. A variety of cargos have been proposed (101), but to our knowledge, no ROS sensor has been designed using this technique. Our group recently used this approach to synthesize Mito-PB [pyrene-FXrFXrFXr (FX = cyclohexylalanine, r = darginine)], a pyrene moiety carried to mitochondria by this peptide vector (Fig. 6K). Because fluorescent lifetime is used for detection, changes in mitochondrial membrane potential or mitochondria numbers will not influence quenching by free radicals and, thus, do not affect free radical quantification. However, our initial studies showed that the presence of the sensor in mitochondria can disturb cellular activity in a concentration- and cell line-dependent manner.
Conclusions
There is a vast literature on the role of ROS in biological systems, how they cause damage, and how they are involved in cell regulatory and signaling pathways. To continue and broaden the progress in these areas, researchers need robust methods. Consequently, the challenges presented by difficulties associated with measuring redox species, especially intracellular ROS, require solutions. As summarized during the conference, “Chemistry and Biology of Reactive Oxygen Species” (61), two points warrant particular emphasis: Available knowledge must be transmitted to a wider audience, and better approaches for ROS detection need to be developed. These approaches must include the design of sensors with improved selectivity, reversibility, and compartment-targeting properties. Among the numerous approaches under development are fluorescence-based sensors that can be used with in vivo imaging technology to visualize redox-active components at the level of intact cells, whole organs or tissues, and even living organisms.