
Abstract
Numerous Problems of Reactive Oxygen Species Detection
O
One of the most prominent biological parameters that may interfere with ROS detection is pH. ROS-generating complexes such as the mitochondrial electron transport chain or NADPH oxidases are intimately linked to substantial proton release and proton transport. It is therefore indispensable to consider the influence of pH on the ROS detection method. Obviously, such problems of interference occur in the detection of numerous other biological phenomena. For example, the dissociation constant of calcium indicators may depend on temperature, pH, ionic strength, and the presence of numerous other ions (58). Most indicators are well suited for physiological conditions in mammalian cells and much less for extreme conditions. Calibration of calcium measurements needs to be performed in situ because in vitro conditions rarely reflect the complex situation of a biological specimen (70). Thus, the history of intracellular calcium measurements may help us to improve ROS detection as well.
By definition, ROS are not stable especially in a biological environment offering numerous reaction partners. On the cellular level, they do not travel very far and the local concentration in subcellular compartments may be more relevant for the biological outcome than the overall level of ROS. However, some of these subcellular compartments represent a rather hostile environment, often quite different from the cytosol. In this review, we wish to highlight some of the difficulties that may arise when we attempt to measure ROS in such compartments. A prominent example for these difficulties is the phagosome, a site of strong ROS production in an environment that is designed to be cytotoxic for the internalized microorganisms.
Complex Biological Situations Where ROS Detection Is of Interest
The phagosome
Production of ROS in the phagosome of immune cells is triggered by the NADPH oxidase, NOX2. The NADPH oxidase is a multiprotein complex with membrane (gp91phox and p22phox) and regulatory cytosolic subunits (p47phox, p67phox, p40phox, and Rac) (47). On pathogen phagocytosis, these proteins assemble on the phagosomal membrane to form the active enzyme (51) that produces superoxide anion O2 •− (68). It is produced at high rates in the neutrophil phagosome and is the precursor of other ROS (83). ROS are essential for pathogen killing, and patients with NADPH oxidase deficiency face severe recurrent infections (25).
How much ROS are produced and how they react with the microorganism in the phagosome are still a matter of debate (28, 83). Winterbourn et al. modeled the reactions in the neutrophil phagosome, the immune cell, which produces the greatest amount of ROS, and estimated the superoxide anion concentration at steady state to 20 μM. Superoxide anions dismutate to hydrogen peroxide, H2O2. The level of hydrogen peroxide depends on the presence of myeloperoxidase (MPO) that catalyzes the formation of hypochlorous acid and consumes hydrogen peroxide. The H2O2 concentration can only increase modestly since it can diffuse out of the phagosome, where it encounters the cellular superoxide and peroxide, removal systems such as glutathione peroxidase and catalase (12).
The estimated maximal concentration of H2O2 with no MPO is 30 μM (84). It is in the same order of magnitude than the intracellular concentration measured in HeLa cells using ROS-sensitive nanoparticles after stimulation with epidermal growth factor (EGF) (11). It is still 1000-fold over the steady-state concentration in growing Escherichia coli (62), and ∼50 times the maximal intracellular concentration reported for mammalian cells (67).
Superoxide anion has been shown to damage bacterial dehydratase, which then releases iron. A possible scenario for bacterial killing would be that the iron released reacts with H2O2 to induce the formation of hydroxyl radicals (HO•) by the Fenton reaction. The hydroxyl radicals would then trigger DNA damage (30). Superoxide anion also reacts with nitric oxide radicals to form peroxynitrite anions, which are powerful oxidants (80). However, this generation of peroxynitrite anions is determined by the production of nitric oxide radicals, which depends on the physiological environment of the immune cells (28).
HOCl and pH in phagosomes and elsewhere
HOCl is formed in the neutrophil phagosome by the MPO, which is delivered to the phagosome by the fusion of granules containing MPO and proteases. More generally, HOCl is found in all sites of inflammation where phagocytes are present (35, 57, 85). In the latter case, the extracellular HOCl concentration is proposed to be in the micromolar range (27, 33). Furthermore, numerous organisms express haloperoxidases capable of producing HOCl for different purposes (5, 24). In all cases, HOCl quantities are mainly estimated or calculated and not measured, because appropriate tools to detect its production in live cells are still missing. HOCl is a strong oxidant and toxic to microorganisms (37). However, the amount of HOCl also depends on the presence of chloride.
The mechanism by which phagosomes acquire chloride is not clear; it probably involves different chloride channels that reach the phagosome on fusion with intracellular organelles such as secretory vesicles and endosomes/lysosomes (75). The chloride concentration of phagosomes was estimated to 70 mM at a steady-state level (54). Under such conditions and according to the model of Winterbourn, 90% of the oxygen consumed by NOX2 is converted to HOCl, which has a broad range of substrates (35, 84). However, HOCl first reacts with methionine, cysteine, or amines of the phagosomal proteins thereby inactivating phagosomal enzymes (38). Some HOCl may reach the bacteria; however, toxicity could also arise from the breakdown of phagosomal protein chloramines, generating diffusible and cytotoxic ammonia chloramines (84).
The fluorescence of the green fluorescent protein (GFP) is quenched by HOCl and not by H2O2 or chloramine (28). In two studies, bacteria expressing cytosolic GFP have their GFP bleached within 2 h after phagocytosis, suggesting that HOCl reaches the bacterial cytosol (55, 61). However, Schwartz et al. showed that the Staphylococci died before GFP bleaching (61).
Alternatively, the bleaching could be due to the drop of pH in the cytosol of the dead bacteria (28). This bacterial cytosolic acidification is consecutive to the acidification of the phagosome. However, neutrophil phagosomes acidify much slower than macrophage phagosomes, and several reports indicate neutral or even alkaline neutrophil phagosomes (19, 31, 40). These imaging studies also reveal considerable heterogeneity of pH between phagosomes even within the same cell. There is a special need for pH-insensitive fluorescent proteins (FPs) able to react quickly with HOCl.
In the neutrophils, ROS production could also arise at the plasma membrane on stimulation by soluble signals such as bacterial peptides or inflammatory mediators and also at intracellular sites other than the phagosome (18, 43, 48, 78, 80, 82). Neutrophil granules containing the membrane subunits of the NADPH oxidase are also a site of ROS production on activation of the neutrophil (13). Although there is no doubt about the importance of ROS production for phagosomal killing, the precise mechanisms remain elusive due to the complexity of the rapidly changing phagosomal chemistry. The development of time-resolved detection methods for ROS detection compatible with an accurate calibration in living cells is necessary to elucidate those mechanisms. Ideally, they will lead to the qualitative and quantitative characterization of the phagosomal chemistry.
In nonphagocytic cells, several intracellular compartments are sites of ROS production. Stimulation by cytokines may trigger ROS production after ligand and receptor endocytosis in the early endosome. This ROS production is necessary for NFκB activation (52). Although superoxide is initially produced in the lumen of the endosome, ROS need to cross the endosomal membrane to activate the signaling pathway. Superoxide may be transported out of the endosome through anion channels or dismutate to hydrogen peroxide, which diffuses through the lipid membrane (52). Certain aquaporins reportedly transport H2O2 across membranes (7). How much ROS are produced inside the redox-active endosome and how much diffuse in the cytosol are still unanswered questions.
Extreme extracellular environments for bacteria and archaea
The environmental conditions of the gastrointestinal tract are quite distinct from other tissues. As ROS are suspected to be involved in several gastrointestinal diseases such as ulcer, gastric cancer, and inflammatory bowel disease, it may become important to investigate ROS production in-situ (36). The pH of the stomach is below four and the intestine is rich in digestive enzymes and bile, which could affect ROS sensors when exposed to this environment.
An estimated 50% of the human population is infected by Helicobacter pylori, a microorganism capable of surviving in this extreme environment and is a major cause of gastric ulcer and cancer. Treatment of H. pylori infections with photodynamic therapy might be an alternative to classical antibiotics. The technique is based on light-induced ROS production with the help of photosensitizing material. If the latter is positioned close to the target such as infectious bacteria, selective ROS-mediated toxicity may be obtained (14). Thus, exploration of the redox homeostasis of H. pylori, preferentially in a physiological environment, would be helpful to characterize its sensitivity to ROS.
Certain archaea encounter and live in extreme conditions such as high salt (>1 mM NaCl), high temperature (>60°C), strong acidity (pH <4), or the presence of highly toxic compounds. In some cases, metal ions in the environment of archaea may catalyze the production of ROS by the Fenton reaction. Specific enzyme complexes for redox control as well as transcriptional control mechanism have been described for some archaea (42, 73). γ-radiation causes ROS production in cells, which is responsible for most of the radiation-induced DNA damage. ROS protection appears to be a major feature of certain archaea that are particularly resistant to radiation (16).
Furthermore, extremophiles are investigated for potential biotechnology applications such as bioremediation of polluted environments (53). Biomining, the extraction of metals with the help of biological systems, often involves oxidative reaction in environmental conditions quite different from mammalian physiology (32). Current ROS detection methods have not been tested under such diverse and harsh conditions and future experiments on ROS detection in extreme conditions will require numerous controls.
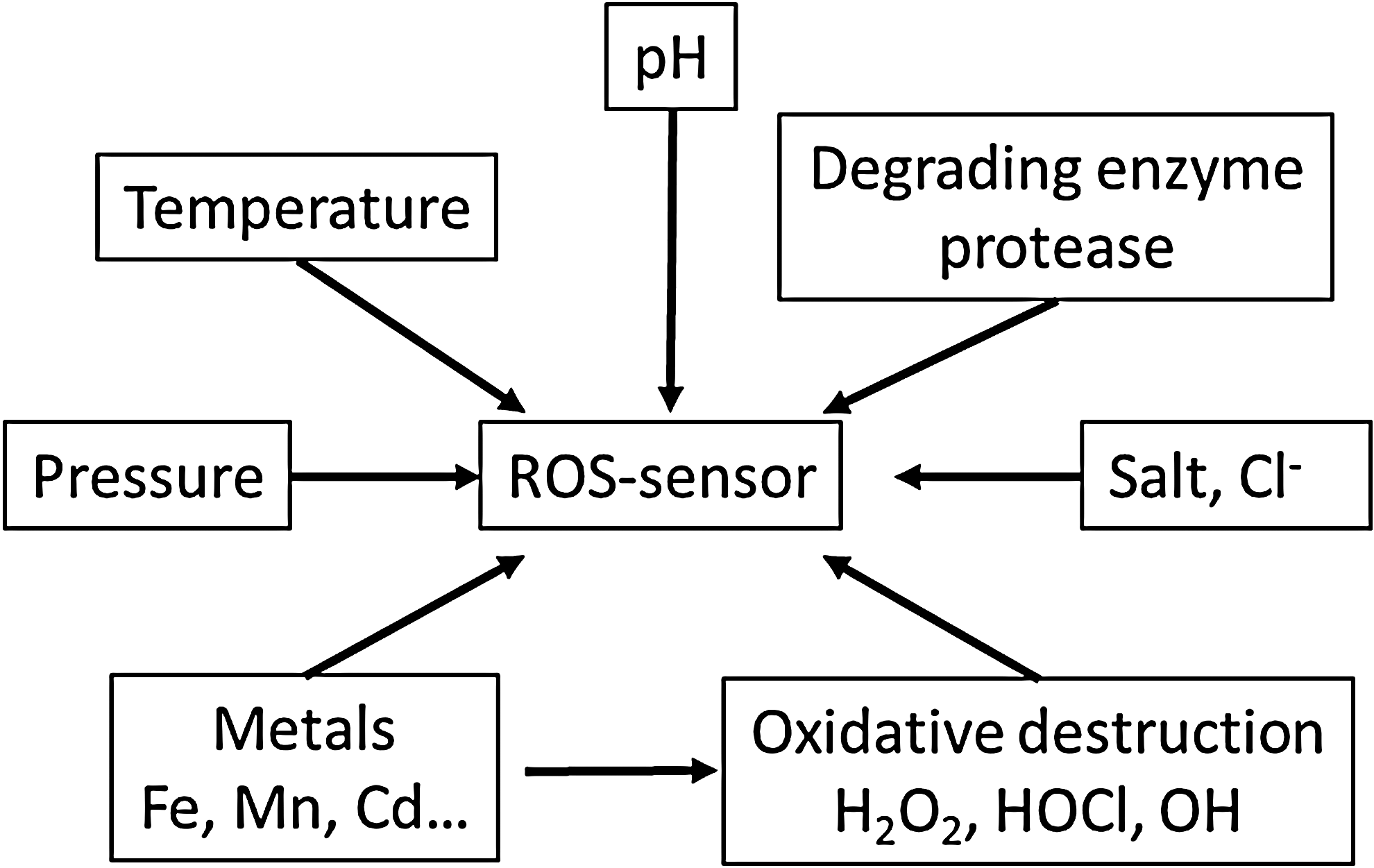
In conclusion, phagosomes, H. pylori, and archaea are examples of the harsh conditions under which oxidative stress is of fundamental importance. ROS detection (mainly O2 •−, H2O2, HOCl, HO•) under these conditions remains a challenge because the sensor has to resist to the ROS, proteases, low pH (pH 4 to 8 at least), temperature, and salt. The phagosome serves as a model system to illustrate the difficulties that maybe encountered and to point out potential solutions.
Features of a Hypothetical Quantitative Fluorescent Sensor for ROS
As described earlier, ROS are produced in the phagosome after internalization of the prey and some are transformed (O2 •−, H2O2), react with many targets (HO•, HOCl), or diffuse quickly into the cytosol (H2O2). Those fates are in competition with the reaction with the ROS probe that is either located on the prey or in the cytosol. Probably, only a fraction of the ROS produced is detected depending on the location and the relative reactivity of the probe. It is more likely to be able to measure in real time a relative concentration of ROS or variations of their concentration than absolute concentrations. The specificity of the probe is crucial to detect only one oxidant. The best approach to work in real time in live cell, taking into account the diversity of cellular behaviors, is to use fluorescent/luminescent probes combined with microscopy.
Probes can be classified in two main categories: the synthetic probes (from small organic molecules to particles) and genetically encoded FPs. Whatever the chemical nature of the probe, the analysis of a cellular parameter using fluorescence microscopy remains limited by the difficulty to quantify fluorescence intensities for each cell. The latter is proportional to the chromophore concentration inside the cell, which is difficult to control for synthetic dyes or transiently expressed genetically encoded fluorophores and to the cell geometry that may vary rapidly during highly dynamic events such as phagocytosis. In addition, the measurements may last for a few minutes and images could be subjected to small changes of focus that may also introduce artifactual fluctuation of intensities.
To record the variations of fluorescence intensities related only to the variations of ROS concentrations apart from all other sources of fluctuations, a common approach relies on the use of ratiometric sensors requiring the monitoring of two fluorescence intensities. In a first approach, one fluorescent dye should be insensitive to its environment and serve as a reference, its fluorescence intensity remaining constant during the whole experiment. The second dye allows the readout of the fluctuation of ROS concentration.
In the second approach, the biosensor can be exited or observed at two different wavelengths. The fluorescence intensities at those two wavelengths will both vary and their ratio will be used to monitor ROS concentration. The sensor may be a single organic molecule or a single FP similar to the classical sensors for calcium, fura-2, or pH, pHluorin, respectively (9). Alternatively, the sensor may also be composed of a sensing protein module either fused to a single FP or sandwiched between two FPs for biosensors based on Förster resonance energy transfer (FRET). For both approaches, the fluorescence ratio is independent from the probe concentration, the movements of the cell, or the fluctuations of the setup.
In the following paragraphs, we will focus on the difficulties raised by ROS detection in harsh environments in live cell microscopy either using organic dyes or genetically encoded biosensors based on fluorescent proteins.
Difficulties for ROS Detection on the (Sub)-Cellular Level
Probing ROS production using fluorescent organic dyes
Numerous synthetic fluorescent dyes for ROS detection have been developed in the past years, but most of them are unfortunately not yet commercially available (18, 43). In addition, only a few of them are functionalized with succinimidyl ester, maleimide, or any functional group involved in click chemistry reaction to stain covalently internalizable objects and thus locate the probe inside the phagosome.
We extensively worked with an old and well-characterized ROS probe, the dichlorodihydrofluorescein (DCFH2), which is commonly assumed to detect hydrogen peroxide in the presence of peroxidase. Its oxidation mechanism is complex and leads to a bright fluorescent dichlorofluorescein (DCF) from the nonfluorescent DCFH2 (84). In addition to DCF, we also identified a second and spectrally distinct oxidation product called Xfluo on reaction with HOCl (71). Xfluo is most probably a chlorinated version of DCF, and such a product has been already identified in the past on the closely related fluorescein (29). In the phagosome, the production of superoxide anion is large enough [20 μM (83)] to produce the micromolar concentration of hydrogen peroxide (84). Associated with a high content of MPO, this production gives rise to a very significant increase of fluorescence even in single phagosomes (17, 71, 72).
The succinimidyl ester version of the probe has been used to stain zymosan (17, 66), heat-inactivated yeast (71), or polystyrene beads (34). The amount of probes covalently linked to the object depends on the amount of active chemical sites and is quite different between various objects. We compared within the same cells the kinetic of the ROS production in their phagosome with yeast and zymosan stained with DCFH2 (Fig. 2). We clearly observed that the maximum fluorescence is reached earlier with zymosan than yeast. After 5 min, almost all the molecules of DCFH2 on the zymosan are oxidized and no more ROS production is detected after this period. In contrast, DCFH2-labeled yeast detects ROS production for at least 15 min. This comparison points out the importance of probe saturation that depends on the relative amount of probe and ROS molecules and on their ability to react together.
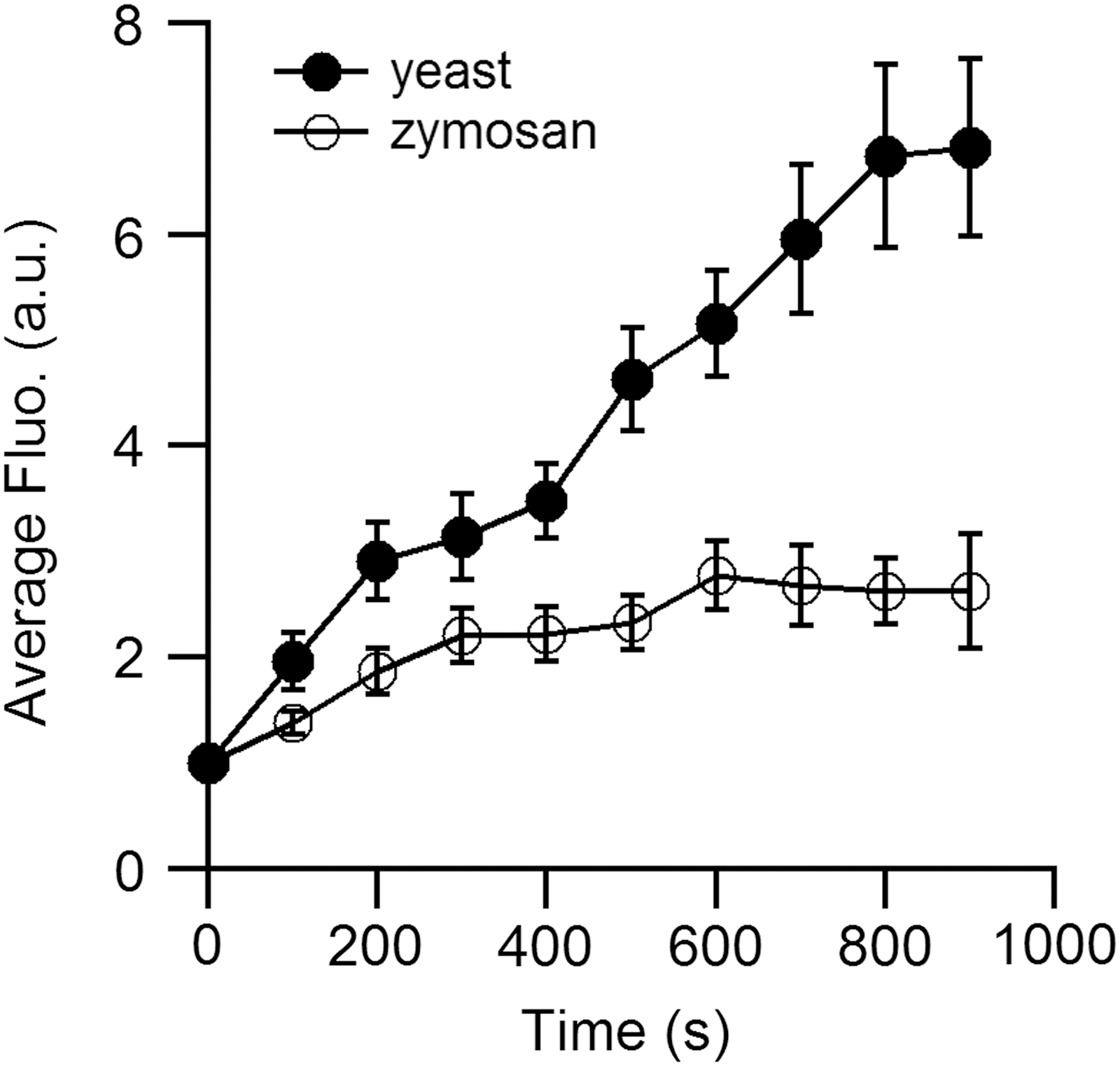
In the phagosome, where high amounts of ROS are produced, this limitation should be correctly addressed. For example, Steinckwich et al. analyzed the slope of the DCF fluorescence during the first 50s after phagosome closure (66). Kamen et al. also stressed the point that the fluorescence of DCFH2 covalently attached to beads saturates with a few minutes (34). ROS detection for periods beyond the saturation requires new probes that are currently not readily available.
The microscopic detection of phagosomal DCF is further complicated by the movement of the cells in and out of the focal plane. Flow cytometry is complementary to microscopy as it analyses a few thousand cells per sample. Since DCFH2 labeling is not uniform, the increase in DCF of individual cells cannot be assessed. Both problems can be partially addressed by double labeling with a ROS-insensitive reference dye. The reaction of heat-killed yeast with two succinimidyl ester dyes yields particles that are doubly labeled.
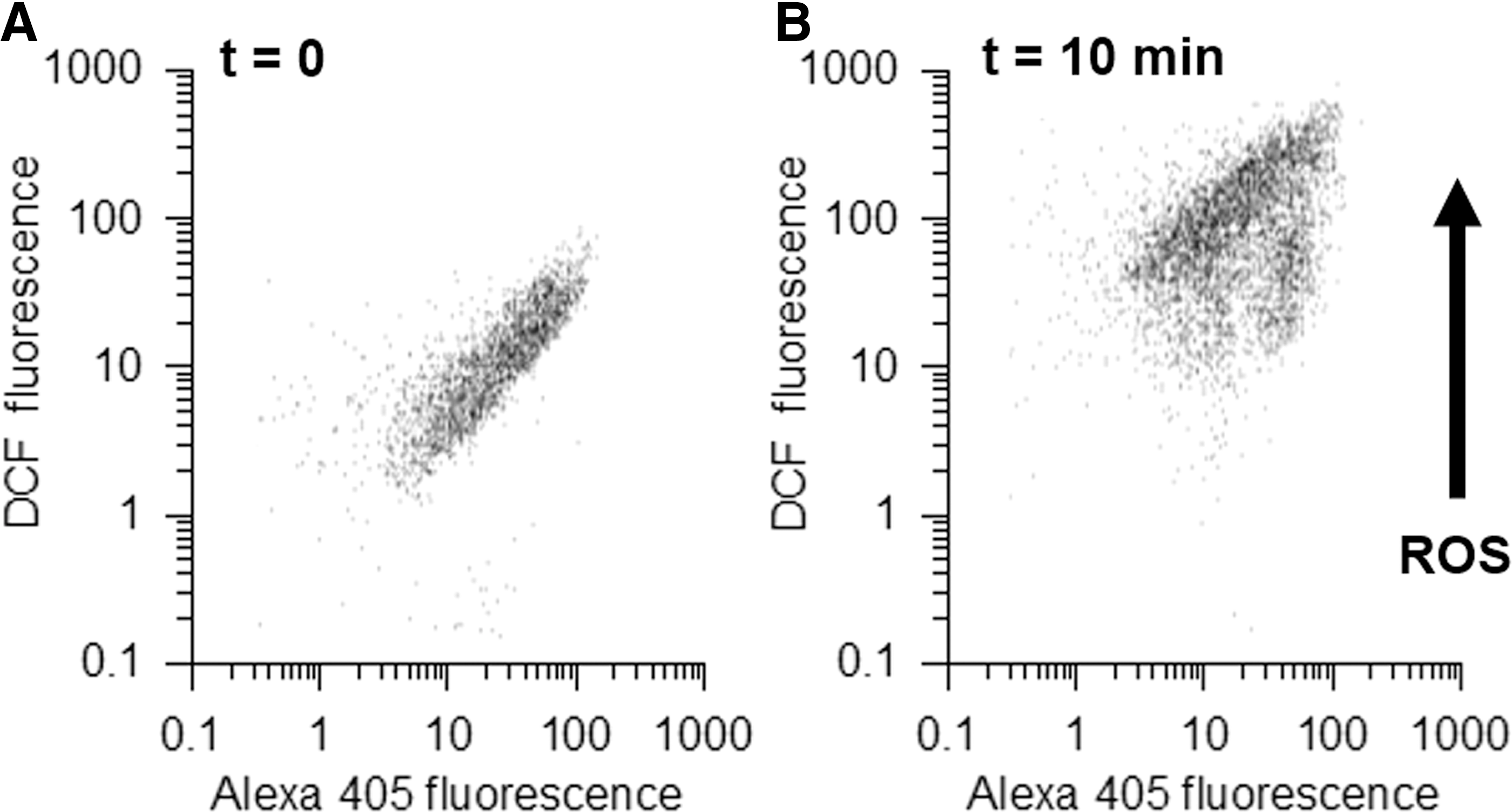
Although labeling with both dyes is heterogeneous, the labeling intensity for the two dyes is correlated so that particles that are intensely labeled for one dye are also intensely labeled for the other dye (Fig. 3A). In this study, 10 min after the beginning of phagocytosis, most of the internalized particles show much higher DCF fluorescence (Fig. 3B). The double labeling allows assessment of the level of phagocytosis and ROS production in the same sample. It also reveals that a small population of phagocytes has much less oxidized phagosomes. However, Alexa405 is insensitive to pH changes and H2O2 but reacts with HOCl. Inside the phagosomes of PLB985 cells, it loses about 30% of its fluorescence due to HOCl production. Other Alexa dyes may have different sensitivities, for example, Alexa647 is almost completely bleached inside phagosomes (data not shown).
Under the microscope, double labeling allows correction for changes in focus and labeling intensity. The example of Figure 4 shows an usual and rapid move out of focus that was rapidly corrected (around t = 450 s). While the traces of DCF and Alexa405 fluorescence were strongly affected by this move, the ratio DCF/Alexa405 remained almost stable. Thus, double labeling may not only overcome certain artifacts of ROS detection but may also illustrate the difficulty to find a reference dye that is completely insensitive to the condition of the phagosome. The chemical conditions in the phagosomes depend on the cell type, for example, neutrophils produce more ROS than macrophages, but their phagosomes acidify more slowly (50). Therefore, the potential interference with the ROS measurement is cell specific.

Biosensors based on FPs
FPs from the GFP family have given rise to a large variety of genetically encoded ROS biosensors. They can be expressed in the cell cytosol or specifically targeted to a cellular organelle. In addition, in conjunction with cell-specific promoters, these fluorescent reporters can be used to generate transgenic animals with cell- or tissue-specific expression.
The FP itself is composed of a highly stable 11-stranded β-barrel structure crossed by an α-helix, which harbors the chromophore (Fig. 5A) (15). The chromophore is the result of the autocatalytic cyclization of three residues in position 65, 66, 67 and is anchored in the β-barrel by an extended H-bond network. The residue 66 is always an aromatic residue. It is a tyrosine in GFPs, YFPs, and RFPs, respectively, the green, yellow, and red variants. In the cyan variants (cyan fluorescent proteins [CFPs]) derived from GFPs as Aquamarine, the aromatic residue is a tryptophan (Fig. 5A) (15, 44).
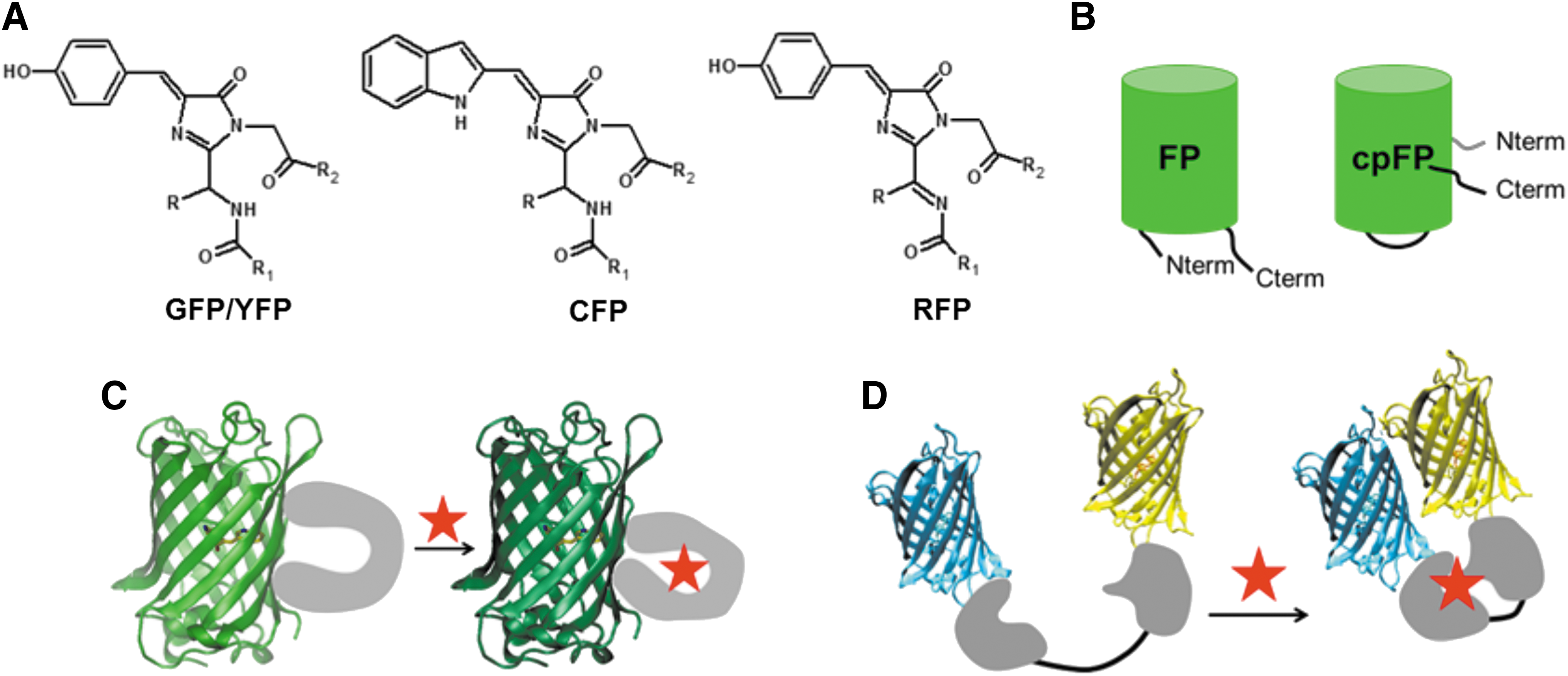
The photophysical and chemical properties of the chromophore can be influenced by modifications not only on the β-barrel in the chromophore pocket itself but also on more remote positions that may, for example, modify the overall protein flexibility or sensitivity to its environment.
The latter type of modification is illustrated by the circular permutation in the sequence of GFPs, YFPs, and RFPs (cpFPs) to displace the C and the N termini of the FP, which are initially on the same extremity of the β-barrel, just in front of the chromophore on the β-barrel side (Fig. 5B). The fusion of a sensing module at the C and the N termini of cpFPs leads to a large family of biosensors where the conformational changes induced in the sensing module during its activation directly promote rearrangements around the tyrosine-based chromophore (Fig. 5C). Such rearrangements ultimately shift the equilibrium between the two protonation states of the tyrosine moiety (neutral and protonated or anionic and deprotonated), which have different absorption spectra (49). The absorption shift of cpFPs allows a direct optical readout of the state of the sensing moiety. The equilibrium between the neutral and the anionic form of the chromophore is also responsible for the pH sensitivity of those FPs (9).
FP-based H2O2 biosensors and their pH sensitivity
This approach was successfully applied to create the ratiometric H2O2 biosensors of the Hyper family (41). The sensing part is the regulatory domain of E. coli's transcription regulator (OxyR-RD) that reacts specifically with H2O2. The reaction involves two Cys residues and leads to the formation of a disulfide bridge. The fusion of OxyR-RD with a cpYFP resulted in the first generation of Hyper. Hyper is a ratiometric probe that requires two excitation wavelengths at 420 nm (reduced form of the disulfide bridge and protonated chromophore) and 500 nm (oxidized form of the disulfide bridge and anionic chromophore) and one emission wavelength at 516 nm (4). The ratio
Hyper is a reversible probe because the disulfide bridge in the oxidized probe can be reduced by the cellular thiol reducing machinery involving glutaredoxin and glutathione (41). Whether this reduction would occur inside a phagosome is not clear. Hyper has been successively improved toward Hyper2 and Hyper3 (41). The latter presents a higher dynamic range combined with an optimization of the oxidation and reduction rates.
Hyper was used to detect H2O2 in the cytoplasm of RAW264.7 macrophages on phagocytosis of several zymosan particles during 60 min (46), and Hyper 3 allowed the direct visualization of the inflammation reaction at a wounded site on zebrafish larvae with imaging setups based either on fluorescence intensity or fluorescence lifetime measurements (8). A red version of Hyper (HyperRed) was recently engineered using a circularly permuted version of the red fluorescent protein mApple (22). HyperRed is excited at 575 nm and fluoresces at 605 nm leaving blue to yellow wavelengths available for the use of other reporters.
Two other fluorescent biosensors for H2O2, OxyFRET, and PerFRET (20) were built using as sensing part the cysteine-rich domain (CRD) of the yeast activator protein 1 (Yap1) involved in a redox relay of Saccharomyces cerevisiae. In PerFRET, one CRD is coupled with the oxidant receptor 1 (Orp1), which is indeed a peroxidase. The sensing part of the biosensor that undergoes conformational changes on oxidation is sandwiched between two FPs that form a FRET pair: Cerulean (Δ11) and cp173Venus (Fig. 5D). After its excitation, the donor, Cerulean, is susceptible to transfer part of its energy to the acceptor, cp173Venus, depending on their distance and their relative orientation, which are both modulated by the oxidation level of the sensing module.
The variation of FRET efficiency is monitored on excitation of the donor as the ratio between the sensitized fluorescence emission of the acceptor (
All the biosensors for H2O2 described above use pH-sensitive FPs characterized by their pKa, the pH value at which they have lost half of their fluorescence. In a pH range around pKa (typically pKa ± one pH unit), the variations of the fluorescence signals (fluorescence intensity, lifetime, or ratio) with pH have the greatest amplitude. At pHs lower than about two pH units below pKa, the fluorescence itself and its variations can be significantly decreased (4, 9, 21, 22).
Green and yellow FPs (GFP, YFP, Venus, Citrine) that bear a tyrosine in their chromophore have pKa values between pH 5.7 and 6.9 (64), which may increase in the presence of high chloride concentrations (6). The pKa is also increased for the circularly permutated version of a FP, whose barrel has been modified in front of the chromophore. For example, pKas of Citrine and cpCitrine are 5.7 and 7.7, respectively (26). For the Hyper family, pKa varies from pH 8.3 to 8.5; so, the fluorescence emission may vary strongly from pH 7 to 8 and could be very low at acidic pH below 6.5 (22). If the pH varies during H2O2 detection, H2O2-dependent variations of fluorescent signals of Hyper could be masked by those due to pH fluctuations (4, 79). In the case of OxyFRET and PerFRET, the detection relies on the use of Cerulean (pKa = 5.2) (23) and cp173Venus (pKa of Venus is 6, the one of cp173Venus should be slightly higher) (64).
We recently evaluated the influence of pH on donor fluorescence lifetime and ratiometric signals of FRET biosensors (6). The pH sensitivities of the donor and the acceptor both contribute to the variations of FRET efficiency with pH. Indeed, FRET efficiency is influenced not only by the relative orientation and distance between the chromophores but also by the spectral overlap between the fluorescence spectrum of the donor and the absorption spectrum of the acceptor.
When the pH decreases around pKa, the fluorescence intensity of the donor tends to decrease. In the meantime, the anionic band of the acceptor is shifted to its protonated form, which absorbs at lower wavelengths where the spectral overlap between both spectra vanished, rending FRET quenching inefficient. If the two events happen at similar pH ranges, they could compensate each other and lead to relatively stable fluorescence properties of the donor. In addition, if both intensities (
As a consequence, at low pH, the biosensor is unable to report any biochemical changes in its environment. Between pH 6.7 and 8, the emission ratio of OxyFRET is pH independent and the one of PerFRET decreases by 10%–15%. An extended pH range should be tested for their use in the phagosome and a detailed characterization with ratiometric- and also lifetime-based methods might be useful to dissect the behavior of OxyFRET and PerFRET at low pHs. Those biosensors might be improved by replacing the donor, Cerulean, with less pH-sensitive FPs such as Aquamarine or mTurquoise (44).
In the meantime, a useful advice is to combine the use of H2O2 biosensor with a pH sensor. Most of the pH biosensors are based on the green or yellow fluorescent protein, taking advantage of the variations of their absorption properties with the chromophore protonation level around their pKa (49). They need the same window of wavelengths than most of the H2O2 biosensors described previously and it is thus impossible to use them in the same experiment. Among all pH sensors, SypHer is a version of Hyper with a mutation in a key cysteine involved in the response to H2O2. It is particularly interesting since it requires exactly the same experimental settings as Hyper (59, 65).
There are only a few FP-based pH biosensors available at the edges of the visible spectrum: the cyan ECFP (60) and the red pHRed engineered from mKeima (69). The combined detection of pH with pHRed and calcium concentration with the ratiometric and slightly pH-sensitive GEM-GECO1 (pKa = 6.1) in the endosomal lumen of the same cells has been recently described (1). Besides the pH measurement itself, pHRed allowed the calibration and correction from pH variations of the signal measured by the GEM-GECO1 (1). This approach could be transferred for ROS sensing in acidic compartments.
Is it possible to use the sensitivity of pH-insensitive FPs to ROS for the detection of their production?
The reactivity of FPs toward ROS has been studied (2, 10). A potential role for detoxification in corals has been suggested (56) and FPs were also used to detect ROS (61, 74, 77). Indeed, using FPs as direct ROS biosensors is particularly tempting in compartments with a high protease concentration such as phagosomes and lysosomes; their high resistance to proteolysis due to their β-barrel folding pattern is in these cases a great advantage. This has been demonstrated during the identification of their chromopeptide or its chemical modifications with tandem mass spectrometry, which requires a proteolysis step to digest the protein in peptides. Proteolysis is achieved using either a tryptic cleavage combined with extremely harsh chemical conditions or a specific protease for residues present in the loops between the strands, which are more accessible than the residues involved in the β-barrel itself (3).
In 2008, it was first shown that cpYFP itself can be used to probe O2 •− flashes in mitochondria (77), but its fluorescence emission is also pH sensitive and its specificity for superoxide anion has been extensively discussed. An article of this Forum is dedicated to this biosensor and the controversy around its use (76). As shown in the previous example, ROS production is often coupled with pH variations. So, we investigated the reactivity of FPs that have a low pKa in vitro and thus are expected to be insensitive to the pH in most physiological situations and cell compartments with the ROS produced in the phagosome (O2 •−, HO•, H2O2, HOCl). Our aim was to explore their potentiality as biosensors in phagocytes.
We focused on the CFP, Aquamarine, and on the two RFPs mCherry and mStrawberry, which all have a pKa below 4.5 (Table 1) (21, 64). EGFP has been successfully used as a HOCl biosensor expressed in the cytosol of staphylococci bacteria trapped in the phagosome of neutrophils and has been added in our experiments as a reference (61). Aquamarine and EGFP are proteins derived from the GFP of the jellyfish Aequorea Victoria, while mCherry and mStrawberry were engineered from DsRed, a protein of the coral Discosoma sp. (Table 1) (15, 21). Even if they share the same three-dimensional structure, FPs derived from different species have low sequence identity and may exhibit different reactivity toward ROS.
For EGFP, ECFP, and Aquamarine, the chromophore is numbered 65, 66, and 67 and for RFPs, we kept numbers from the original publication (63).
From Shaner et al. (64).
From Merola et al. (44).
We followed the evolution of the fluorescence intensity in solutions of purified FPs on addition of ROS. FPs were expressed in E. coli and the purification was performed by affinity chromatography using conventional protocols already described previously (2). The concentration of FP transiently expressed in live cells varies usually from the high nanomolar range to some dozens of micromolars. Consequently, we choose an FP concentration of 10 μM, and ROS amounts are expressed as concentration ratios [ROS]/[FP]. Superoxide anion (O2 •−) and hydroxyl radical (HO•) were produced quantitatively using gamma irradiation directly in the protein solution (2), whereas H2O2 or HOCl was added as aqueous buffered solution.
During preliminary experiments with O2 •− and HO•, we observed an irreversible loss of fluorescence intensities (max. 20%) of FPs for concentration ratios [O2 •−+HO•]/[FP] between 10 to 15, consistently with our previous observations on ECFP (2). Aquamarine was the least reactive. This reaction is not specific to one FP and they might not be good candidates as biosensors for those species. Nonetheless, in the presence of a complex ROS mixture, part of the FP reactivity might be due to O2 •− and HO•, depending on their relative amount and this should be checked carefully with appropriate controls.
H2O2 results from the dismutation of O2 •− and its concentration in the phagosome was calculated in the low micromolar range (between 1 and 30 μM) depending on their reduction by phagosomal peroxidases (83). The fast and direct reaction of H2O2 with FPs is very unlikely due to its lack of reactivity in the absence of peroxidase (81). Indeed up to a concentration ratio [H2O2]/[FP] of 50, no variations of the fluorescence signals were observed for EGFP, ECFP, EYFP, and DsRed (74). We completed this study to a much higher concentration ratio of 1000 for Aquamarine, mCherry, mStrawberry, and EGFP with or without addition of commercial horseradish peroxidase (HRP at 40 U/ml).
For Aquamarine and EGFP, both derived from the GFP of A. victoria, we barely observed a decrease of the fluorescence emission after 24 h at a concentration ratio of 1000 (less than 10%) even after addition of a peroxidase (HRP) consistent with previous observations (74). In contrast, H2O2 reacts well with mCherry and mStrawberry, proteins derived from DsRed; a clear decrease of their fluorescence emission is observed after overnight incubation for concentration ratios [H2O2]/[FP] above 10 for mCherry and 50 for mStrawberry (Fig. 6). Interestingly, mCherry and mStrawberry have no cysteines, the main H2O2 target (81), whereas the insensitive EGFP shares with mStrawberry the same chromophore sequence, TYG. Thus, the mechanism for H2O2 oxidation might be complex and could involve not only the chromophore and its acylimine extension specific for the RFPs (Fig. 5A) but also other residues sensitive to oxidation by H2O2 such as methionines (81).
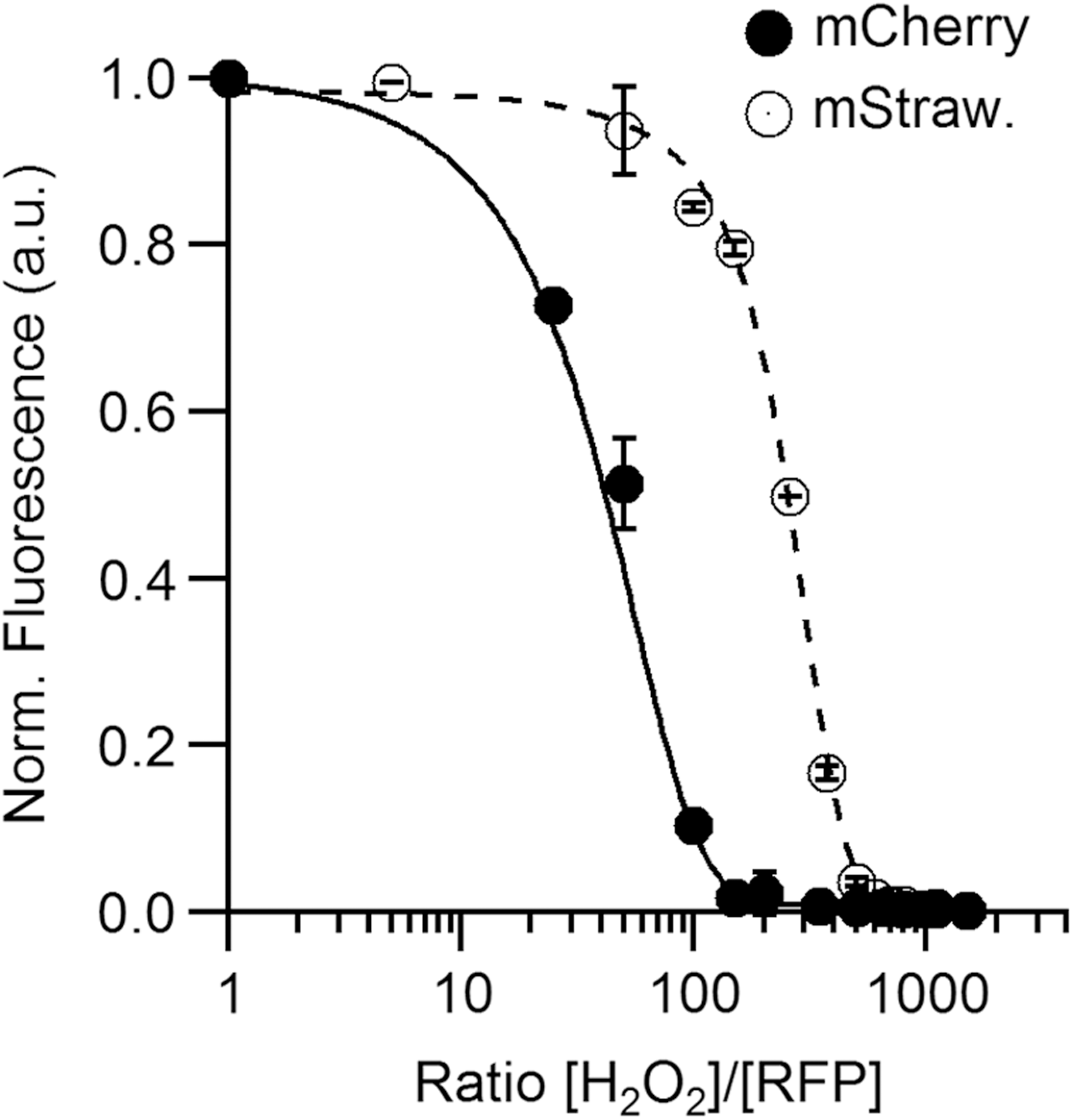
A next step will be to propose mutations to increase the reactivity of mCherry and thus improve its capacity to detect H2O2. In addition, in live cells, the long-term measurement of mCherry fluorescence intensity in environments, where significant amounts of H2O2 compared to FP concentration are produced, needs to be considered with caution as it may be bleached by H2O2 itself.
HOCl is produced in the presence of H2O2 by MPO in the phagosome of phagocytes at a very high rate (more than 100 mM·min−1 (83)). In our study, the complete quenching of the fluorescence emission of an EGFP solution is observed for a concentration ratio [HOCl]/[FP] of 100 (Fig. 7A), which is consistent with the reaction stoichiometry of ∼200 determined previously (55). Unfortunately, EGFP fluorescence properties are sensitive to pH variations below physiological pH (pKa = 5.5). Alternative solutions could be proposed by using Aquamarine, mCherry, or mStrawberry (RFPs). We observed rapidly (within seconds) a 50% quenching at a concentration ratio [HOCl]/[RFPs] of 65 for mCherry and mStrawberry (Fig. 7B) against a ratio of 40 for Aquamarine (Fig. 7B) and 35 for EGFP (Fig. 7A).

RFPs are less sensitive to oxidation by HOCl than other FPs, and Aquamarine appears to be the best candidate as HOCl biosensor in live cells. Indeed, the latter has a higher resistance to pH coupled to a mild reactivity with O2 •−/HO• and no reactivity toward H2O2. Typical concentrations of FPs expressed transiently in live cells should be in the 100 nM–10 μM range and on sites of inflammation, HOCl concentration is proposed to be much higher. With a production rate of more than 100 mM·min−1 in phagosomes (83), it appears, from our results, that Aquamarine should be rapidly 100% quenched and thus could be fully appropriate for HOCl probing.
Nevertheless, it should be kept in mind that in vivo, a large number of compounds other than the biosensor can react with the produced HOCl. In the phagosome, FPs should represent few ‰ of the total amount of proteins, decreasing the observed FP quenching kinetics. Taking advantage of the plasticity of FPs, Aquamarine variants with tuned reactivity adapted to various biological situations could be engineered in the future.
Conclusion
Numerous cells and tissues, such as the intestinal tract, phagocytes, or certain archaea, feature harsh environmental conditions that potentially interfere with many ROS detection methods. Nevertheless, the redox balance in these cells may be biologically or clinically relevant and require quantitative, localized, and time-resolved detection methods. For these cases, we recommend a set of basic controls that shall reduce the risk of artifacts due to interference with the environment (Fig. 8). The critical risk factors should be identified, which may not be trivial, because the actual chemical and physical conditions in these settings are not well known. Known risk factors shall be taken into account when choosing the probe. The sensitivity of the probe should be tested once it is localized in the appropriate cellular compartment.
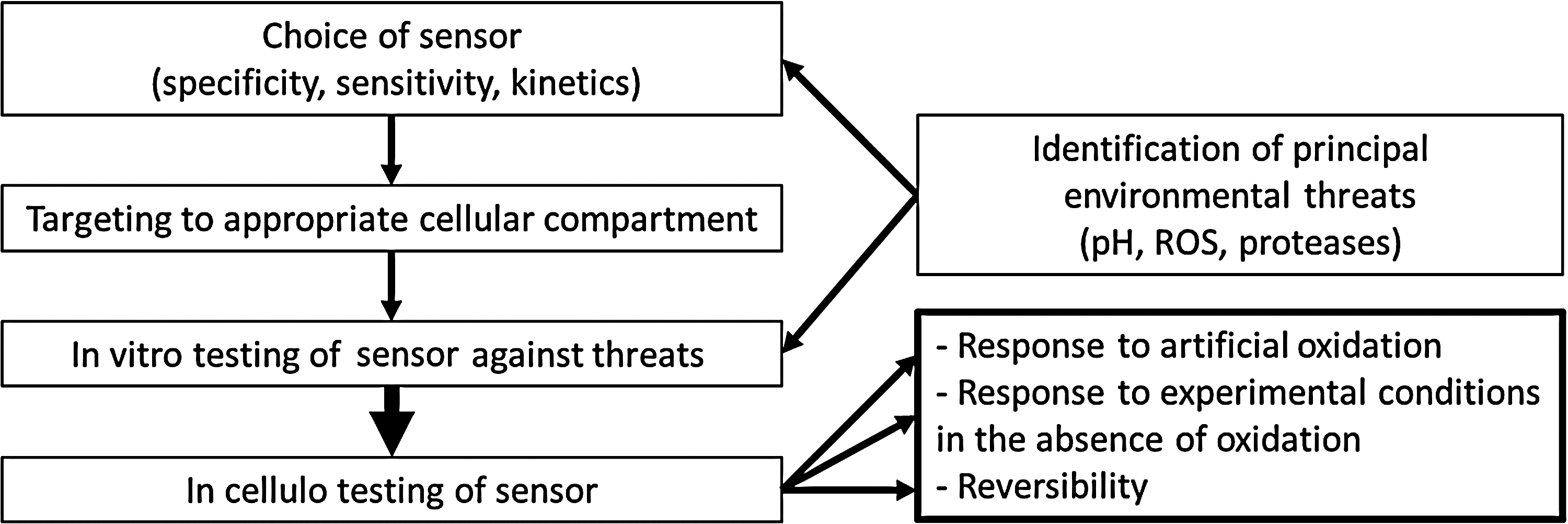
Control experiments for environmental factors such as pH, temperature, or salt conditions are straight forward. However, testing the sensitivity of an ROS sensor toward complex environments, such as the phagosome containing numerous enzymes, is more difficult, since the exact nature of this environment is not known. Artificial oxidation such as addition of H2O2 to cells will inform on the sensitivity of the experimental setup. However, due to limited diffusion, ROS scavengers and detoxifying enzymes such control experiments will most likely not be quantitative.
Another control consists of testing the probe in the experimental setup when oxidation is prevented. In the phagocyte, this may be achieved by blocking the NADPH oxidase or using cells that lack a functional oxidase. Finally, it is important to know, whether the probe is reversible such as Hyper for H2O2 or irreversible such as GFP with respect to HOCl. To develop new methods of ROS detection, the intrinsic ROS sensitivity of FPs maybe directly exploited but should also be considered when these proteins are integrated into complex biosensors.
Footnotes
Acknowledgments
The authors thank Yasmina Bousmah, Elodie Hudik, and Paul Machillot for technical assistance and Fabienne Mérola for fruitful discussions. They thank the Fondation pour la Recherche Médicale (DCM20121225747), UMR CNRS 8000, and INSERM UMR-S757 for financial support. L. Bouchab is the recipient of a doctoral grant from Region Ile de France (DIM Malinf). The authors declare that no competing financial interests exist.