
Abstract
Significance:
Metabolic syndrome (MetS) is associated with a greater risk of diabetes and cardiovascular diseases. It is estimated that this multifactorial condition affects 20%–30% of the world's population. A detailed understanding of MetS mechanisms is crucial for the development of effective prevention strategies and adequate intervention tools that could curb its increasing prevalence and limit its comorbidities, particularly in younger age groups. With advances in basic redox biology, oxidative stress (OxS) involvement in the complex pathophysiology of MetS has become widely accepted. Nevertheless, its clear association with and causative effects on MetS require further elucidation.
Recent Advances:
Although a better understanding of the causes, risks, and effects of MetS is essential, studies suggest that oxidant/antioxidant imbalance is a key contributor to this condition. OxS is now understood to be a major underlying mechanism for mitochondrial dysfunction, ectopic lipid accumulation, and gut microbiota impairment.
Critical Issues:
Further studies, particularly in the field of translational research, are clearly required to understand and control the production of reactive oxygen species (ROS) levels, especially in the mitochondria, since the various therapeutic trials conducted to date have not targeted this major ROS-generating system, aimed to delay MetS onset, or prevent its progression.
Future Directions:
Multiple relevant markers need to be identified to clarify the role of ROS in the etiology of MetS. Future clinical trials should provide important proof of concept for the effectiveness of antioxidants as useful therapeutic approaches to simultaneously counteract mitochondrial OxS, alleviate MetS symptoms, and prevent complications. Antioxid. Redox Signal. 26, 445–461.
Introduction
M
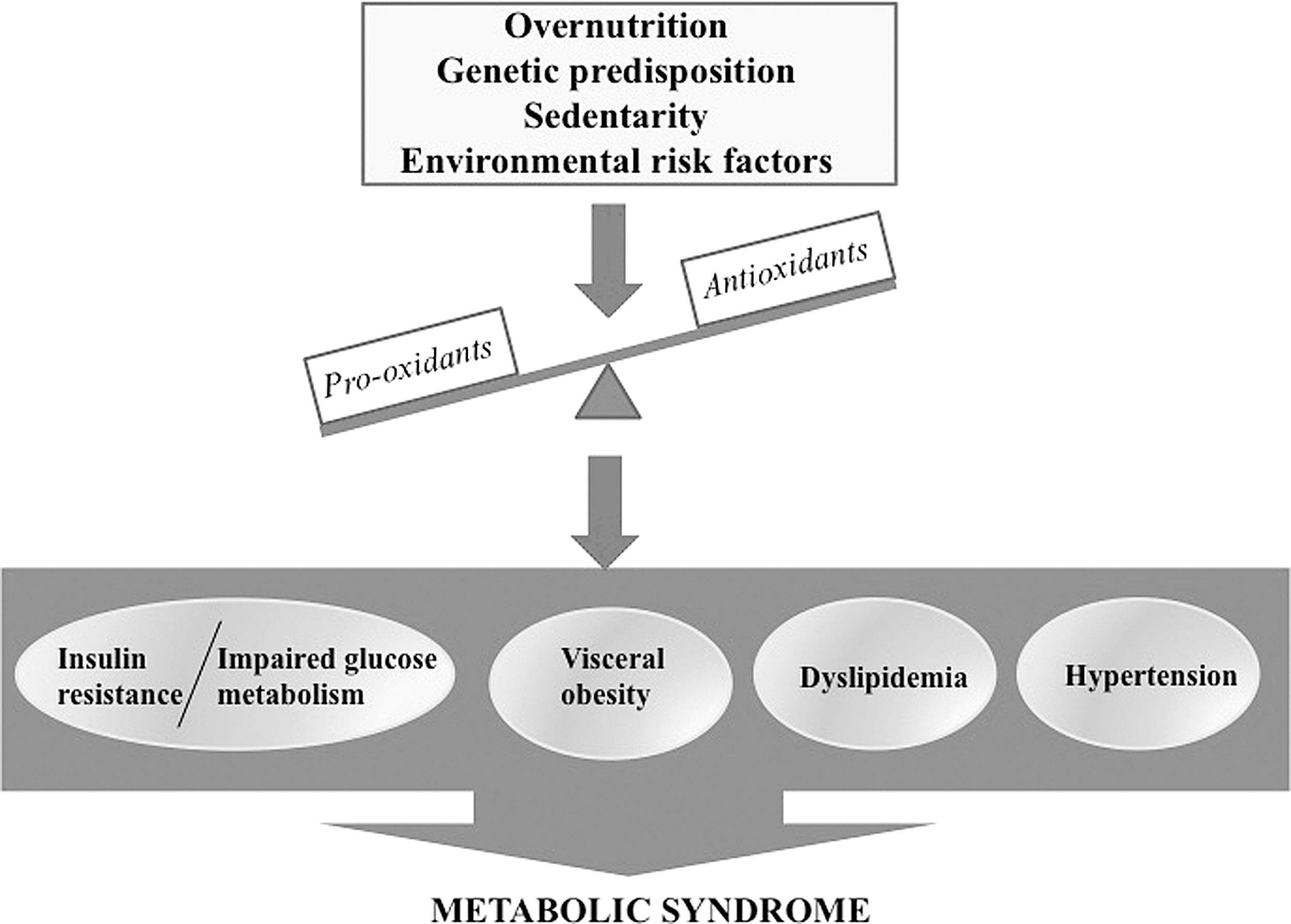
Growing evidence suggests a dominant pathogenic role for oxidative stress (OxS), a dominant event in cellular damage and dysfunction, given its strong relationship with various MetS combinations and resulting clinical complications (137, 166). In fact, it has been proposed that MetS-induced OxS is an early event in all metabolic manifestations in view of the association of the end products of free radical-mediated OxS with body mass index, insulin resistance (IR) state, hyperlipidemia, and hypertension (56, 88, 218).
In this review, we will focus on the state-of-the-art knowledge pertaining to the role of OxS in the pathogenesis of dysregulated MetS parameters. A link with mitochondrial dysfunction and metabolic signaling pathway derangements will be emphasized. In particular, the impact of OxS-associated MetS on the insulin sensitivity of metabolic organs and cross talk with low-grade inflammation will be evidenced, since OxS promotes inflammatory agents that conversely participate in reactive oxygen species (ROS) generation, thereby creating a vicious pathogenic circle that amplifies oxidative processes. Critical cellular and molecular determinants will be discussed to determine whether OxS is causal or simply related to MetS. Finally, whether OxS lowering represents an interesting target for MetS prevention and complications will be considered with particular focus on the impact of therapies aimed at restoring redox homeostasis and fighting MetS.
OxS: A Brief Overview
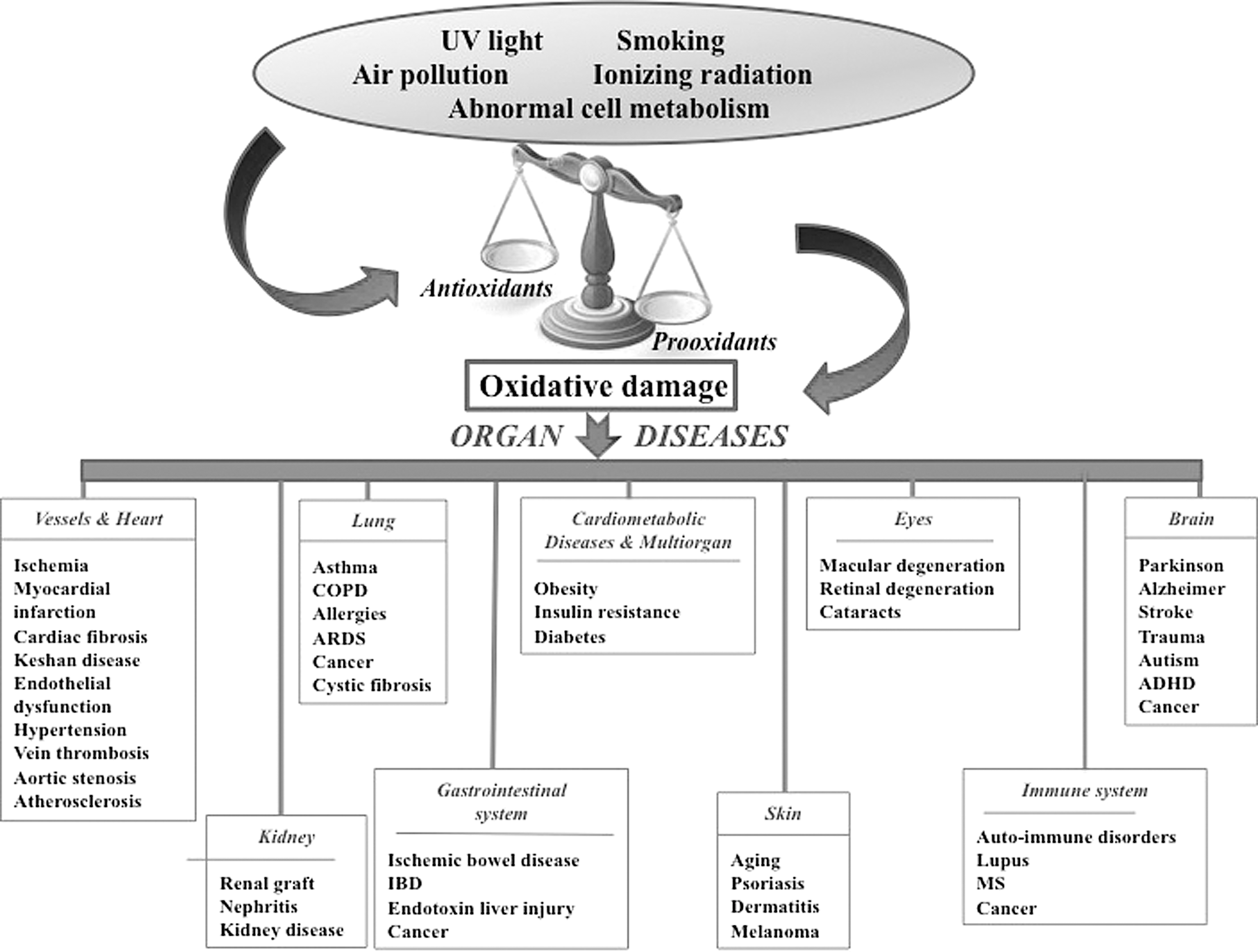
OxS results from a state of disequilibrium between exaggerated ROS output and a limited biological capability to neutralize free radicals in living organisms. The discrepancy between excessive reactive molecules and weak endogenous defense results in damaged lipids, proteins, DNA, and cellular structures (Fig. 2), ultimately culminating in the pathogenesis of a broad range of diseases (Fig. 3). OxS is generated in various dynamic intracellular organelles such as the endoplasmic reticulum, lysosomes, and mitochondria as by-products of oxidative protein folding, dysfunctional autophagy, and mitochondrial respiration and detoxification. The main ROS resulting from chemical reduction of O2 include superoxide anion (O2 −•), hydroxyl radical (•OH), singlet oxygen (1O2), hydrogen peroxide (H2O2), hypochlorous radical, peroxynitrite radical (ONOO−), ozone, and nitric oxide (NO) (166). The relevant sources of ROS are mitochondrial electron transport system and the homologs of nicotinamide adenine dinucleotide phosphate oxidase (NOX1, NOX2, and NOX3), as well as cyclooxygenases, xanthine oxidase, lipooxygenases, and uncoupled nitric oxide synthase (NOS) (137). The reactive molecules are formed by reduction–oxidation (redox) reactions in response to the activation of the several types of oxidases. In a first step, O2 is reduced with the combination of additional electrons, thereby producing the powerful O2 −• that reacts with other endogenous molecules to generate secondary oxidizing molecules, either directly or via reactions catalyzed by free metals (e.g., Cu, Fe, Cd, and Pb) or metals containing components (88). Thereafter, the reduction of O2 −• by spontaneous or enzymatic dismutation reactions leads to the output of the stable by-product H2O2, which actively participates in the formation of the highly reactive •OH and thiol functional groups. Moreover, O2 −• may react with NO to form the potent nitrating and oxidizing ONOO−. In contrast to the short-lived reactive species O2 −• due to different reactions (transformation into quick and spontaneous dismutation to H2O2 or via rapid catalysis by superoxide dismutase [SOD]) (59), H2O2 is characterized by its long life span, relative stability, easy intra- and intercellular diffusion, and tight regulation by endogenous and exogenous enzymes, which convert it into water and O2 or possibly into different metabolites (22). It is important to mention that ROS signaling actions depend on the subcellular site of production, type of species generated, proximity to antioxidants, species half-life, cell membrane permeability, and local concentration of ROS (69, 94). While H2O2 is particularly important in cellular signaling, both O2 −• and H2O2 are able to oxidatively modify thiols on protein cysteine residues and, thereby, induce structural changes and trigger the signaling activation/inactivation of various molecules such as mitogen-activated protein kinases (modulating the activation of defense mechanisms following exposure to OxS), tyrosine kinases (central players in the post-translational activation of antioxidant enzymes), and transcription factors (controlling several biological processes such as cell growth, cell migration, the expression of proinflammatory genes, and the biogenesis of extracellular matrix proteins (50, 58, 112). Importantly, the biological activity of a wide variety of proteins may be compromised not only by thiol oxidation but also by carbonylation, side-chain oxidation, fragmentation, unfolding, and misfolding. In addition to damaging ROS during redox homeostasis instability, cells have to tackle organic analogs, including reactive nitrogen species, nitrous oxide, ONOO−, peroxynitrous acid, nitryl anion, nitrosyl chloride, nitrosyl cation, nitrogen dioxide, dinitrogen trioxide, and nitrous acid. The ROS responsible for protein modifications in plasma are as follows: radical species produced by the activities of NOX, NOS, and oxygenase, in addition to reactive nitrogen species from myeloperoxidase (MPO) and NOS activities, and hypochlorous acid from MPO.
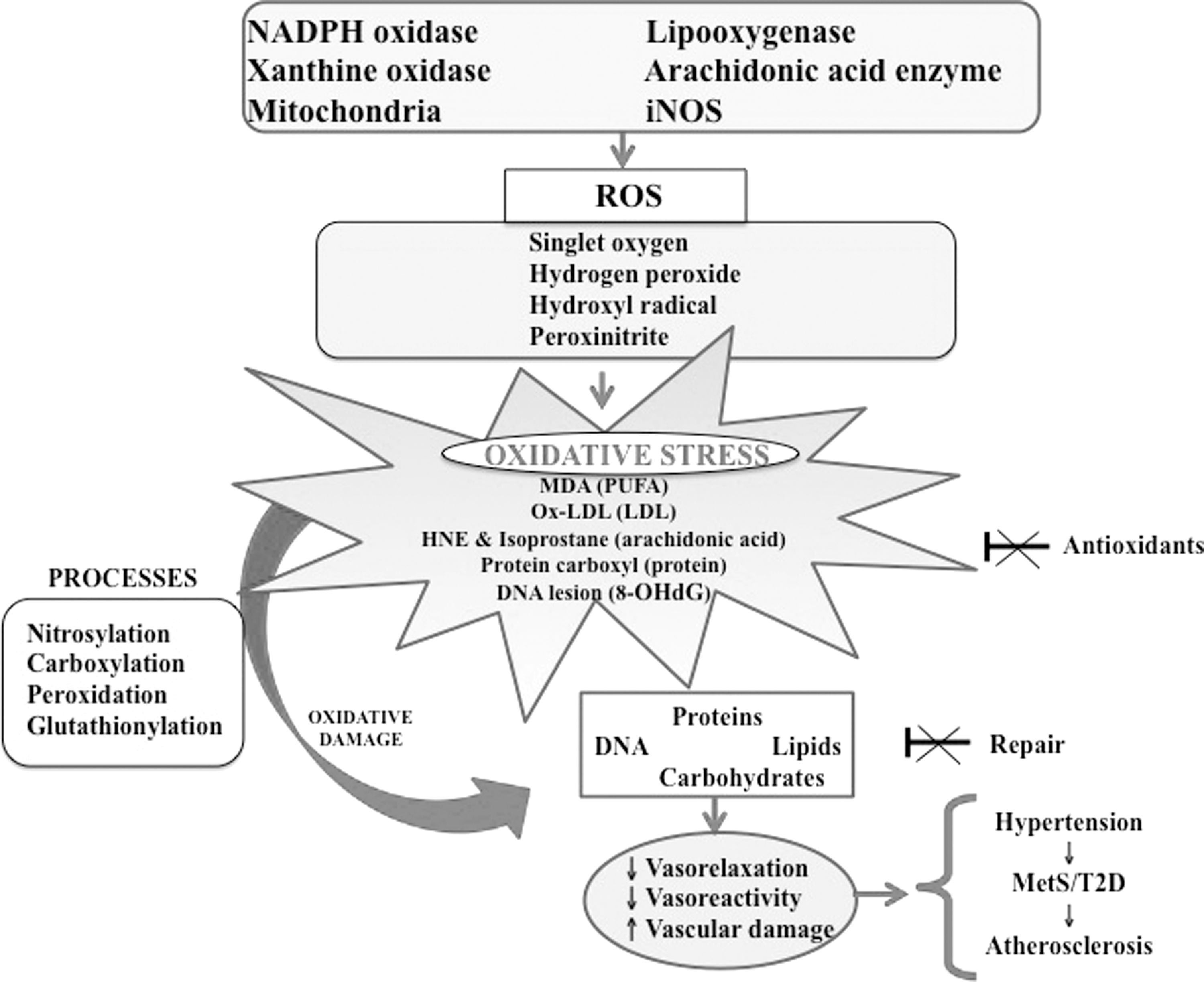
Antioxidants provide a critical defense against OxS. The antioxidant defense system consists of several endogenous enzymes (catalase [CAT], glutathione peroxidase [GPX], thioredoxin reductase, Cu Zn SOD [SOD1], Mn SOD [SOD2], extracellular SOD [SOD3], and glutathione reductase [GR]) and various circulating biomolecules (bilirubin, coenzyme Q10, N-acetycysteine, melatonin, uric acid, glutathione, coenzyme, NO, pyruvate, albumin, and ceruloplasmin), as well as dietary components (vitamins A, C, E, folic acid, flavonoids, polyphenols, Zinc, and selenium) (37, 97, 133, 157, 204).
OxS and MetS
MetS is characterized by an inappropriate rise of various factors, but particularly plasma-free fatty acids (FFAs) (74) that activate ROS production (86). Even though the mechanism is still poorly understood, FFA increase has been shown to enhance •OH, O2 −•, and H2O2 in endothelial and vascular smooth muscle cells (VSMC) (86). Moreover, fat accumulation constitutes a trigger for ROS augmentation in adipose tissue, likely by stimulating NOX and lowering antioxidative enzymes (62). In contrast, the exposure of adipose tissues to OxS under MetS conditions results in a decrease in anti-inflammatory adiponectin and an increase in inflammatory cytokines (154, 188), both of which lead to compromised insulin signaling through the induction of insulin receptor phosphorylation and deterioration of glucose transporter 4 (GLUT4) translocation and gene transcription (15).
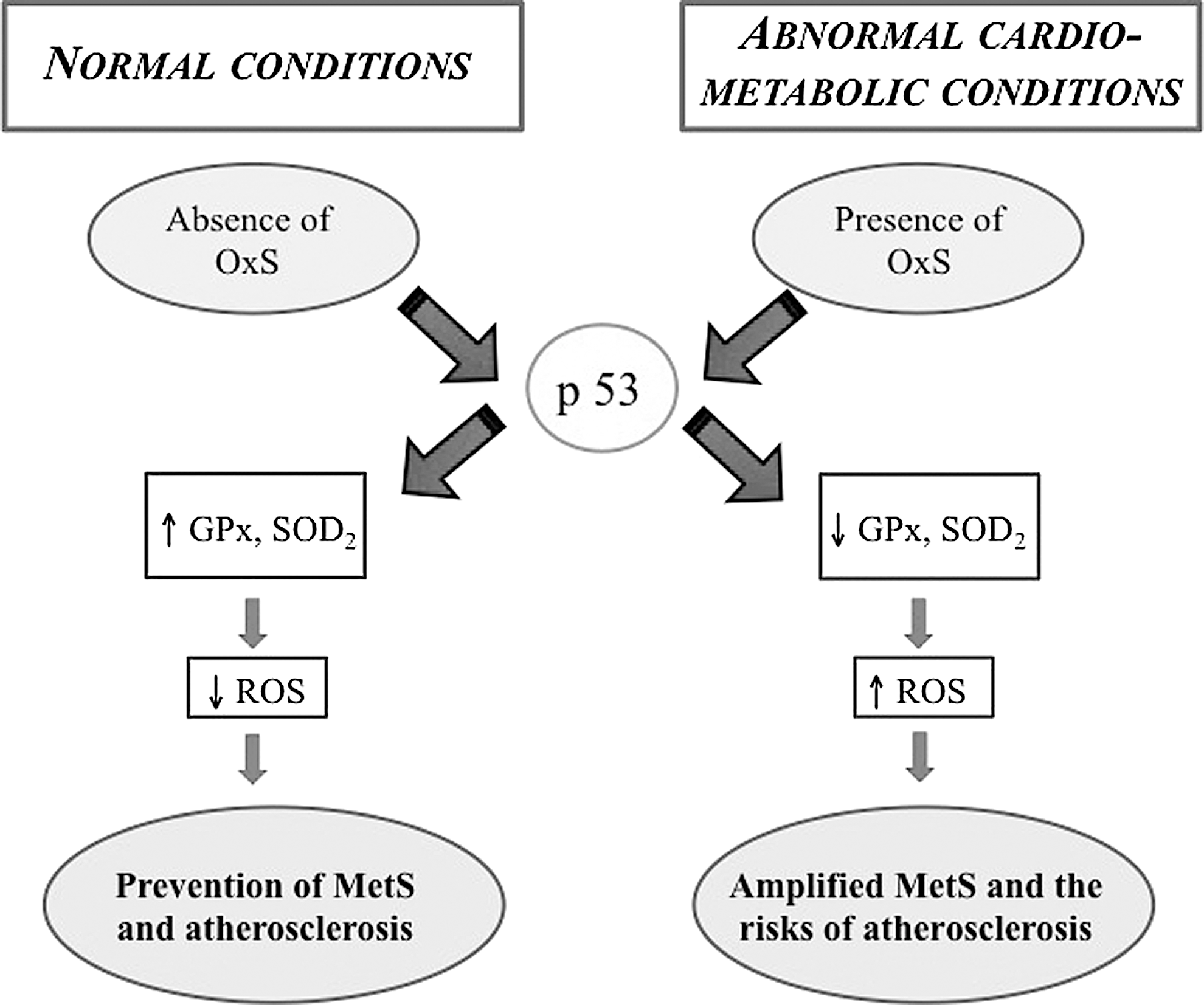
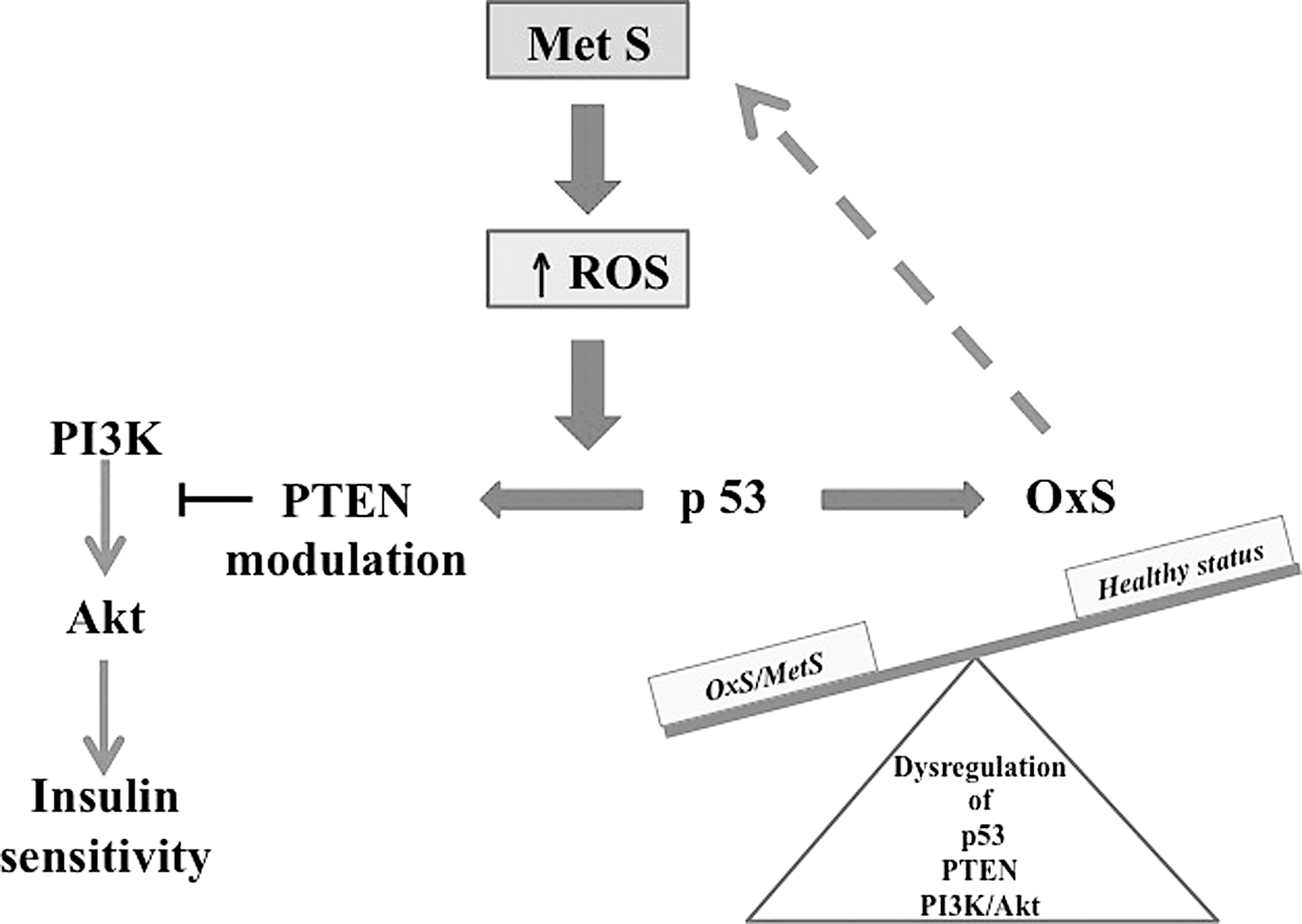
Additional studies have reported the regulatory role of transcription factor p53, a critical tumor suppressor and protector of the genome in the expression of GPX1, the most profuse isoform of the GPX family capable of detoxifying H2O2 (195). Under pathophysiological conditions such as MetS, suppression of p53 downregulates basal GPX1 and SOD2 expression, which triggers cellular ROS production, leading afterward to oxidative damage, whereas reestablishing physiological levels of p53 enhances the antioxidant enzymes and lowers ROS formation (108, 117, 174) (Fig. 4). It is important to note that elevated ROS concentrations could inactivate tumor suppressor phosphatase and tensin homolog (PTEN) deleted on chromosome 10, a member of the protein tyrosine phosphatase family, either in a forthright manner by oxidizing its cysteine residue or indirectly by stimulating its phosphorylation, thereby interfering with PTEN presence on the plasma membrane and resulting in the activation of the PI3K/Akt signaling cascade (98) (Fig. 5). Conversely, PTEN upregulation modulates PI3K/AKT signaling, which reduces cellular ROS generation (216). Collectively, these findings suggest the existence of multiple mechanisms of OxS elaboration by ROS in MetS. However, further research is needed to define the role of OxS-mediated PTEN and p53 in insulin target tissues and IR (Fig. 5).
OxS and obesity
Obesity, an excess accretion of adipose tissue, is a global pandemic (57). Its ever-increasing prevalence is related to the dysregulation of caloric intake and energy expenditure. A positive energy balance promotes OxS that is likely a mechanistic connexion between obesity and related complications, including IR. For example, FFAs that result from high fat and carbohydrate intake in obese rodents and humans may favor ROS production because of the saturation of the electron transport chain in the mitochondria (3, 62). In turn, OxS may amplify fat accumulation by stimulating preadipocyte proliferation, adipocyte differentiation, and mature adipocyte size (62, 79, 113). It has also been observed that OxS controls food intake and body weight by upholding various effects on hypothalamic neurons with a concomitant impact on satiety and hunger (82). When obesity sets in, adipose tissue becomes an ideal site capable of producing ROS by upregulating NOX activity (62, 129).
Low levels of antioxidant molecules (146, 207) along with high plasma advanced oxidation protein products, oxidized low-density lipoprotein (oxLDL), thiobarbituric acid reactive substances (42), and additional cellular/systemic OxS indicators have been reported in obese subjects. The activities of antioxidant enzymes, including GPX and CAT, have been found to be markedly decreased in obesity (155, 173). Antioxidant defenses, which normally protect against infectious processes and contribute to normal functions such as proliferation, differentiation, and signaling (19, 32, 61, 92, 171, 194, 215), when diminished, are unable to scavenge ROS. Although an inverse relationship between total antioxidant capacity and visceral fat was noted, regardless of other variables (35), an inverse relationship between body mass index/total body fat percentage could not be clearly demonstrated (23). One possible explanation is that different tissues attempt to increase their antioxidant defenses to counteract OxS.
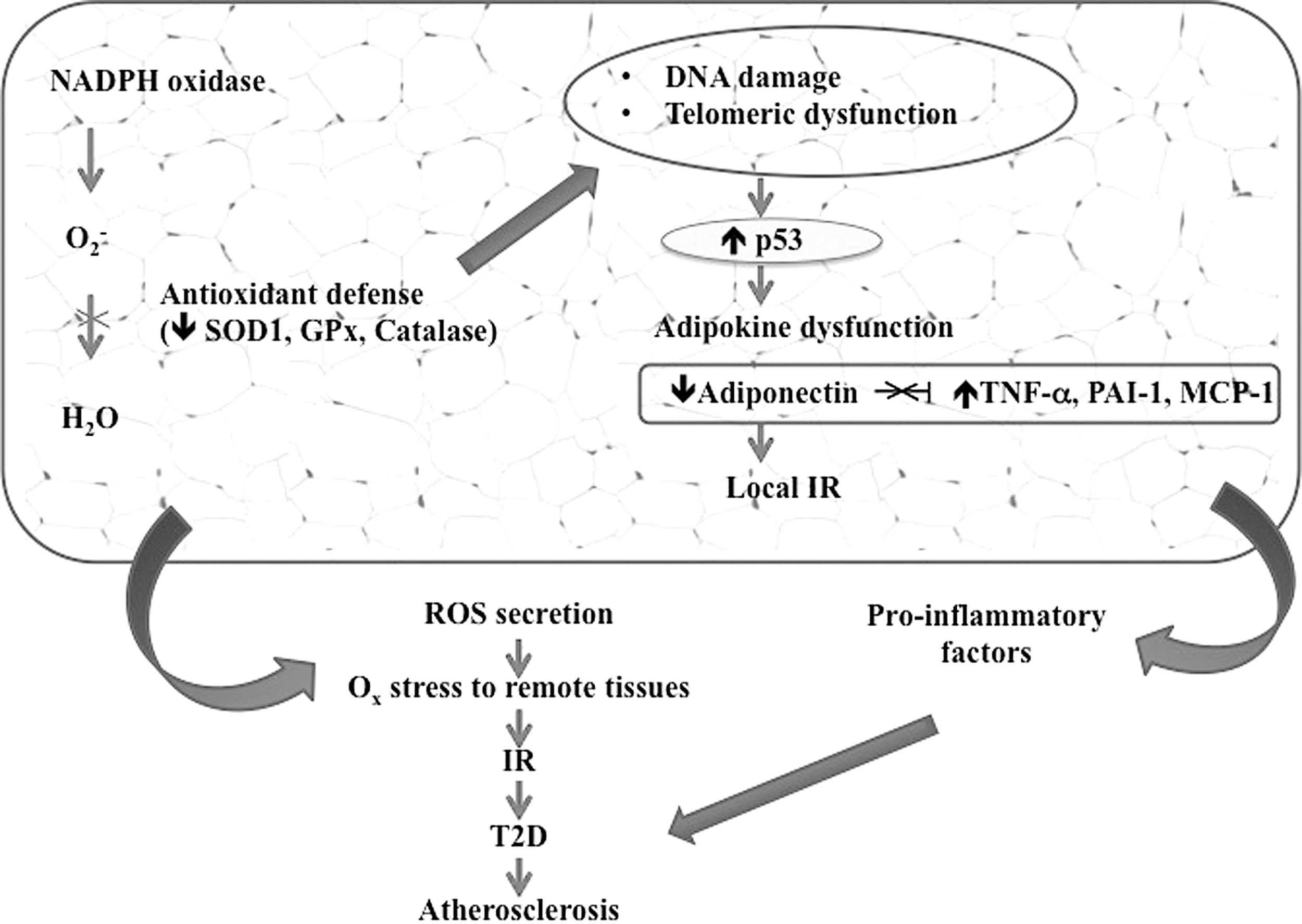
NOXs in vascular cells constitute a major ROS site that can set off downstream components such as NOS and the mitochondria (136). Interestingly, elevated vascular ROS in mice overexpressing p22phox and under a high-fat regimen trigger obesity and even a MetS phenotype by raising inflammation, promoting adipogenesis, and developing exercise intolerance (221). Fat accumulation-induced OxS also takes part in the pathogenesis of obesity-associated MetS through the deregulated synthesis of adipocytokines such as PAI-1, leptin, resistin, visfatin, adiponectin, tumor necrosis factor-α, and interleukin-6 (71) (Fig. 6). While several groups agree that obesity can cause metabolic derangements and that it precedes and predisposes to the development of MetS via leptin resistance (221), the induction of leptin resistance by vascular ROS is able to disrupt the inhibition of insulin biogenesis and delivery from pancreatic β-cells occurring under normal conditions, thereby constituting a feed-forward cycle forcing weight gain (106). Further support of this concept may be reflected by the diffusion of NOX-generated ROS to the mitochondria to activate OxS, thereby causing mitochondrial dysfunction, limiting muscle exercise capacity, and favoring weight gain (26, 221). Accordingly, Mn-SOD knockout mice exhibited severe exercise disturbances (109).
OxS and impaired glucose metabolism/IR
IR plays a crucial role in interlinking the various constituents of the MetS cluster and in fortifying the syndrome's evolution. OxS has been shown to be associated with IR (54), a key feature of MetS (3). This association is not surprising, given the insulin action of glucose metabolism and cascade signaling (143), the abnormalities of which contribute to MetS (223). Indeed, insulin insensitivity leads to increase circulating glucose levels (as frequently noticed in the postprandial state) because of the inability to coordinately suppress hepatic glucose production and glycogen degradation while enhancing glucose uptake into the muscle and adipose tissue via the mobilization of insulin-dependent GLUT4 from intracellular storage membrane vesicles to the cell surface (7, 192). To better understand this aspect, investigators exposed skeletal muscle cells to fructose and noted diminished glucose uptake, sluggish GLUT4 translocation, and disturbed insulin signaling in response to the ROS-interceded activation of intracellular c-Jun N-terminal kinase and extracellular signal-regulated kinase 1/2 (ERK1/2) pathways (89). Antioxidant mechanisms to shut off ROS-initiated signals prevented IR and its metabolic consequences. Similarly, the administration of antioxidants to fructose-fed rats lowered ROS production and precluded IR (189). Therefore, excessive ROS concentrations were capable of damaging cells by oxidizing biomolecules and stimulating various stress-sensitive intracellular pathways such as c-Jun N-terminal kinase, ERK1/2, and NF-κB, resulting in the development of IR through transcription factor and stress kinase-mediated, chronic low-grade inflammation (104, 162). It is worth noting that IR affects GLUT1 (the transporter of glucose) expression and dehydroascorbic acid (the oxidized form of ascorbate), both of which function in antioxidant defense (75, 177). This is important, since GLUT1 is largely abundant in glycolytically active cells and has been snapped up by vitamin C auxotrophs to preserve hepatic redox states through the transport of dehydroascorbate. Generation of the electron carrier nicotinamide adenine dinucleotide phosphate in the pentose phosphate pathway and the conversion of dehydroascorbic acid to ascorbate depress ROS levels: the former by donating electrons to antioxidant pathways such as GR and thioredoxin reductase and the latter by serving as an antioxidant.
ROS overbuilding and restricted antioxidant enzyme concentrations not only increase IR, but also affect the secretory capacity and viability of β-cells (96), a major component of MetS. Accordingly, IR-associated hyperglycemia is linked to the excessive generation of free radicals and superoxides via the proton electrochemical gradient, which overwhelms the antioxidant defense capacity of the various tissues (147, 204). Persistence of hyperglycemia is thought to favor glucose autoxidation and NOS activation that collectively enhance OxS (45, 67), and culminate in polyol, hexosamine, advanced glycosylation end products, and protein kinase C pathways (24), which collectively exacerbate the complications of T2D (14, 38, 53, 102, 122). Furthermore, elevated circulating and cellular OxS markers in patients with nascent MetS are associated with increased levels of oxLDL, nitrotyrosine, monocyte superoxide, and NOX activity along with a decrease in the nuclear factor erythroid-derived 2 like 2 (Nrf2) antioxidant defense system (91). A decline in transcription factor function in response to the disequilibrium in the redox system results in imperfectly weak antioxidant defense, since Nrf2 cannot be freed from kelch-like ECH-associated protein, thereby preventing its translocation into the nucleus to bind to the antioxidant reactive element in the genes that encode antioxidant enzymes such as GR, aldo-keto reductase, heme oxygenase-1, and γ-glutamylcysteine synthetase (31).
OxS and dyslipidemia
Dyslipidemia is characterized by increased plasma triglycerides (TGs), ascribed to augmented synthesis of very low-density lipoproteins (VLDLs), FFAs, small dense LDL particles, and reduced high-density lipoprotein cholesterol (HDL-C) in MetS (52, 72, 170). It is now thought that small, dense LDL particles, non-HDL-C, and apolipoprotein B represent valuable markers for the diagnosis and severity of MetS (65, 95, 128, 135). Interestingly, elevated OxS has been simultaneously associated with hypertriglyceridemia, deficient HDL antioxidant capacity, and a chronic inflammatory state in patients with MetS (19). A link between OxS and lipid disorders in MetS has been reinforced by the administration of statins that improved not only atherogenic dyslipidemia but also the oxidant/antioxidant status in subjects with MetS (19). Similarly, inhibition of intestinal cholesterol absorption by ezetimibe was accompanied with an amelioration of IR by lowering ROS in obese mice (193). Additional evidence and potential mechanisms explain the relationship between OxS and lipoprotein levels in the context of dyslipidemia: (i) increased production of O2 −• via the NADP oxidase pathway is detected during hypertriglyceridemia, hypertension, and obesity (21, 222); (ii) the susceptibility of small, dense LDL particles to oxidation is a function of hypertriglyceridemia degree (105); (iii) LDL oxidation intensifies with augmented waist circumference, C reactive protein concentration, and the number of LDL particles (29, 44, 60, 200, 205, 210); (iv) the lowering of bilirubin levels by MetS contributes to increased LDL oxidation and cardiovascular risk (140), whereas normal bilirubin concentrations can prevent lipid oxidation (215); (v) the aberrant expression of small noncoding RNAs disturbs mitochondrial and endoplasmic reticulum metabolic homeostasis, thereby impacting on lipid and lipoprotein metabolism in the MetS (34); and (vi) red meat consumption is associated with the occurrence of OxS, hypertriglyceridemia, and MetS (36).
OxS and hypertension
The endothelium, interfacing with the blood stream, is not only a physical barrier tightly controlling vascular permeability such as the passage of luminal macromolecules into the tissue but it also ensures vessel homeostasis via modulation of vascular tone, platelet aggregation and coagulation, inflammation, and growth factors. For instance, barrier dysfunction occurs with high levels of angiopoietin 2 and vascular endothelial growth factor. On the contrary, NO as an endothelium-derived relaxing factor inhibits platelet aggregation, neutrophil attachment to endothelial cells, adhesion molecule expression, and smooth muscle cell proliferation (64, 138). Under NO deficiency, the onset of atherosclerosis can be observed. Interestingly, a controlled production of low concentrations of ROS, as signaling molecules, maintains vessel integrity by orchestrating endothelial function and vascular contraction/dilatation (199). However, an aberrant balance between free radicals and antioxidants results in endothelial dysfunction, enhanced contractility, VSMC growth, monocyte intrusion, lipid peroxidation, inflammation, and marked deposition of extracellular matrix proteins, key components in hypertensive vascular injury (49, 197) (Fig. 2). Available conclusive reports underscore the critical role of ROS in the development of vascular dysfunction and hypertension (130). This link between OxS and deep alterations in the vasculature can be due to rich providers of NOX in endothelial, adventitial, and VSMC (111). Indeed, O2 −• produced in high concentrations interacts with NO to produce the highly reactive intermediate ONOO− that acts as a vasoconstrictor and a cytotoxic molecule with the potential to cause oxidative damage to cellular constituents (219). ONOO− is also an inhibitor of prostacyclin synthase and endothelial NOS activities, which restrain the bioavailability of antioxidants and endothelial mediators with protective functions (12, 110). This radical species, closely implicated in experimental and human hypertension, is now recognized as a mediator of vascular injury in cardiovascular pathologies (78, 116, 158, 196). For instance, genetic animal models such as the spontaneously hypertensive rat (SHR) exhibit a number of polymorphisms in the gene promoter region of the NOX p22phox subunit with augmented protein expression and enhanced NOX activity (39). The resulting abundance of vascular O2 −• generation reduced NO bioavailability and raised OxS, whereas the administration of antioxidant vitamins, NOX inhibitors, SOD mimetics, BH4, and Ang II type-1 receptor blockers concomitantly reduced vascular O2 −• formation and markedly alleviated hypertension in these animals (158, 171, 184), which suggests that NOX upregulation and OxS increase precede the development of hypertension. This concept is strengthened by the elevated expression of p47phox, another key component of NOX endowed with the ability to stimulate O2 −• production (190) in the renal vasculature, macula densa, and distal nephron from SHRs (61, 103). Human studies also provide ROS by-product accumulation in hypertensive patients (40, 167, 217), probably arising from decreased SOD and CAT antioxidant activity in combination with low concentrations of ROS scavengers (vitamin E, glutathione) (167, 209). Interestingly, the renin–angiotensin system appears as a potent contributor of NOX activation and ROS generation (49, 111, 198). In this context, it is appropriate to recall that the therapeutic blood pressure (BP)-lowering actions of Ang II type-1 receptor blockers and angiotensin-converting enzyme inhibitors have been ascribed to NOX inhibition and decreased ROS production (66, 220).
Currently, hypertension is considered a primary risk factor for MetS. The association between hypertension and MetS highlights the production of ROS and the development of OxS as common causative links. It is possible that OxS produced in hypertension may lead to other components of the MetS cluster. IR per se or via an increased plasma concentration of FFAs, a prominent attribute of the MetS, could increase ROS production through NOX activation, deteriorate vascular reactivity, alter the smooth muscle cell phenotype, and enhance vascular growth (86, 99, 212, 222), thereby partially accounting for the phenotypic modifications that result in hypertension. For example, IR in rats caused by a high-fructose feeding was closely linked to OxS characterized by an upsurge in the generation of O2 −•, culminating in raised plasma levels of lipid peroxidation markers. Elevated cardiac O2 −• production is associated with an overexpression of p22phox with the parallel occurrence of IR and near the start of development of cardiac hypertrophy, mild hypertension, and vascular alteration as pointed out by an elevation in the media/lumen ratio of mesenteric arteries (47). Therefore, amplification loops may take part in the intensification of OxS in MetS. The evidence indicating that antioxidant treatment ameliorates OxS and reduces BP in various animal models, including the SHR (144, 211), fortifies not only the perception that OxS is undeniably a fundamental element in defining the stage of endothelial dysfunction and BP but also the prognosis of patients with conventional MetS closely determined by the status of the pro-oxidant/antioxidant balance, which necessitates powerful therapeutic agents able to synergistically scavenge reactive-free radicals from every MetS component.
Mitochondria-Derived OxS
As mentioned before, ROS are adversely implicated in the deviant signaling and tissue harm detected in the pathophysiology of MetS. The failure to regulate properly redox-sensitive signaling pathways and the OxS-mediated abnormal changes in cellular components are major contributors to tissue dysfunctions. Mitochondria, as major sources of ROS, promote OxS and are clearly implicated in the development of MetS components and complications. Furthermore, mitochondria are targets of cellular ROS that severely affect mitochondrial functions and result in defects in mitochondrial electron transport, complex enzyme activities, adenosine triphosphate (ATP) diminution, caspase 3 liberation, and mitochondrial DNA lessening (43, 77, 101, 131, 163), thereby contributing to MetS progression. In turn, mitochondrial dysfunction may have repercussions on its regulation of cellular functions, such as intermediary metabolism (77), redox signaling (43), calcium (Ca2+) homeostasis (27, 169), cell proliferation (6, 153), development (134, 178), and cell death (68, 208).
Mitochondria-derived OxS and obesity
A number of studies have stated that obesity causes mitochondrial OxS and dysfunction in different tissues. The vicious cycle between muscle depletion and the accretion of ectopic fat, termed sarcopenia, may result from an intricate interplay of various factors such as OxS and mitochondrial dysfunction. Apparently, high caloric intake raises plasma FFAs and glucose levels, which are closely associated with high ROS generation and obesity exacerbation (149). Conversely, calorie restriction alleviates sarcopenia by reducing mitochondrial abnormalities (124), which highlights the mechanistic implication of mitochondrial OxS and dysfunctions in obesity comorbidities. The mitochondria of subcutaneous white adipose tissue from obese patients with or without T2D displayed a pronounced increase in protein carbonyls and lipid peroxidation products, reflecting an enhanced mitochondrial ROS production rate along with decreased activities of mitochondrial antioxidant enzymes like SOD and GPX, a phenomenon that is likely to cause metabolic abnormalities in adipose tissue during obesity (30).
In young patients, excess body weight impairs mitochondrial function in cardiomyocytes even in the absence of heart failure and diabetes (145). Mitochondrial dysfunction is characterized by proapoptotic Bax and Bcl-xS, reduced antiapoptotic Bcl-xL expression, cytochrome C release and partially activated caspase cascade, high protein carbonyl content, and 8-hydroxy-2′-deoxyguanosine in cardiomyocytes. In addition, the use of several experimental animal models of hyperglycemia and overweight/obesity allowed for the detection of mitochondrial ROS-induced OxS, uncoupling, and lessened ATP production in the heart of rodent models, whereas the pharmacological approach targeting mitochondria could effectively scavenge mitochondrial ROS, improve cardiac hypertrophy, and normalize mitochondrial energetics and insulin-stimulated glucose utilization, thus indicating that obesity and IR are linked to heart mitochondrial dysfunction and OxS (84). Obesity also results in mitochondrial dysfunction in skeletal muscle and adipose tissue in humans and mouse models (101). Given the decline in the number of mitochondria and the decrease in the expression of genes related to mitochondrial biogenesis in adipocytes, which occur concomitantly with aberrant mitochondrial morphology and oxidative phosphorylation functions (16, 182), mitochondrial dysfunction may be considered a key factor in deleterious intra-adipocyte metabolism with a negative impact on nonadipose tissues. For example, it has been shown that adipose tissue dysfunction in perilipin-1 null mice, which affects heart health and contributes to the development of metabolic cardiomyopathy through mitochondrial swollen forms, disrupted cristae and OxS in cardiomyocytes (118). Among the molecular mechanisms behind cardiac dysfunction in obesity, there are alterations in intracellular Ca2+-mediated regulation, which significantly impact on contraction and relaxation frequency (13, 90). Obesity is also a significant risk for hepatic lipid accumulation caused by the uptake of circulating FFA and induced de novo lipogenesis, forcing out lipotoxicity, mitochondrial dysfunction, OxS, and IR (107), thereby culminating in the development of nonalcoholic fatty liver disease (73, 141). Therefore, the alleviation of mitochondrial OxS and dysfunctions in excessive adipose tissue may be useful in shielding from obesity-induced hepatosteatosis and lipid accumulation in nonadipose tissues.
Mitochondria-derived OxS and IR
Mitochondrial dysfunction related to mitochondrial biogenesis, reduced mitochondrial content, and/or decreased protein content, along with oxidative protein activity, can affect insulin sensitivity (41). The resulting decrease in substrate oxidation (e.g., FFA) leads to lipid accumulation and the accretion of metabolically active diacylglycerols and ceramides, which inhibit insulin receptors and protein kinase AKT (25, 176, 179). An aggravating factor is the reduced fuel oxidation that disturbs electron flow through the electron transport chain, leading to more electron leakage toward oxygen and the production of superoxide that, in combination with mitochondrial DNA lesions, protein aggregations, and lipid peroxidation, gives rise to either mitophagy or apoptosis (131). Therefore, it is not surprising that dysfunctional mitochondria are observed in the context of glucose intolerance (100), largely accompanied by abnormally low mitochondrial NADH:O2 oxidoreductase activity, small size, downregulated biogenesis, and oxidative phosphorylation pathways (101, 132). The mechanism responsible for most of these defects was linked to the declined expression of Peroxisome proliferator-activated receptor gamma coactivator 1 α (PGC1 α), the master regulator of mitochondrial metabolism (101, 132) as illustrated in human studies (76, 83, 168). Nevertheless, these investigations remain inconclusive as to whether the primary cause of these abnormalities is due to the mitochondrial number or actual metabolic changes within the mitochondria (11, 20, 81, 161). Moreover, impaired mitochondrial function in IR is not unanimously accepted, since various groups have reported no correlation or even the absence of alterations in the markers of mitochondrial content and functions, while others have insisted on the offsetting augmentation of mitochondrial oxidative ability with raised lipid provision (46, 63, 87, 175, 191, 201, 202). For now, despite the associative studies of IR and mitochondrial dysfunction, we cannot definitively conclude whether modifications in mitochondrial function are a cause or consequence of IR, particularly in view of various differences in methodology and experimental protocols. Although several groups have attempted to tackle this critical issue using transgenic approaches such as posttranslational modification of mitochondrial transcription factor A (controlling mitochondrial transcription), PGC1 α (regulating mitochondrial biogenesis and interacting with a great number of transcription factors and nuclear hormone receptors that are associated with mitochondrial function), and acetyl-CoA carboxylase 2 (the inhibition of which stimulates the access of long-chain fatty acids into mitochondria for β-oxidation), their findings are still unable to clearly demonstrate functional defects in mitochondria as upstream faults causing IR (1, 33, 80, 114, 151, 181, 206, 213, 214).
Mitochondria-derived OxS and dyslipidemia
The cause that links hyperlipidemia to the pathogenesis of MetS is frequently attributed to the toxic action of lipids in various target tissues. Indeed, it is assumed that lipid overflow causes ectopic fat deposition, which impairs the function of various organs (48). Multiple biochemical mechanisms have been proposed indicating that lipid excess induces cell OxS and damage (180). Free cholesterol, oxLDL, and glycated HDL are additional potential causes of mitochondrial dysfunction and/or apoptosis (164). For example, oxLDL affects mitochondrial functions, specifically oxidative phosphorylation in vascular endothelial cells, and may increase ROS, resulting in mitochondrial-specific OxS, 8-oxo-deoxyguanine (183).
Mitochondria-derived OxS and hypertension
Mitochondrial-dependent apoptotic pathway actively participates in vascular endothelium disruption triggered by oxLDL accumulation (120). In fact, oxLDL accretion promotes ROS production in endothelial cells, which leads to proapoptotic effects in association with substantial perturbations in mitochondrial ATP production and abnormally high permeability, culminating in cytochrome C discharge and the subsequent activation of executioner caspases (172). In turn, the deterioration of mitochondrial function raises ROS generation and exacerbates OxS in the atherogenic process (8). By acting on the underlining mechanisms (i.e., oxLDL-induced cellular apoptosis and mitochondrial dysfunction) through the mitochondrial-dependent apoptotic pathway and glycogen synthase kinase 3β/β-catenin signaling pathways, it is possible to fight vascular dysfunctions and atherosclerosis (115). Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) is a key protein to control to prevent severe damage to the endothelium. It represents the major receptor responsible for the binding, internalization, and degradation of oxLDL. LOX-1 is implicated in various pathophysiological events, including endothelial dysfunction, hypertension, and atherosclerosis (51). It is upregulated not only by oxLDL but also by ROS and angiotensin II (119) and results in mitochondrial dysfunction (126). Importantly, full control of LOX-1 may suppress the induction of oxidative damage to mitochondrial DNA and proteins and may improve clinical conditions recognized to undergo vascular remodeling and tissue dysfunction, such as T2D, hypertension, and dyslipidemia (51).
Microbiota and MetS Traits
Despite the importance of intestinal microbiota, there are only few data concerning its extraintestinal metabolic role, namely in MetS. Currently, it is well established that this gut ecosystem not only influences intestinal functioning but it may also confer metabolic endotoxemia and low-grade chronic inflammation because of the transfer of microbial metabolites into systemic circulation via an abnormally permeable intestinal barrier (142), which eventuates MetS (28). Our recent studies clearly showed that improvement of OxS, endotoxemia, inflammation, and MetS features by polyphenols is associated with a robust modulation in the relative abundance of Akkermansia spp. in high-fat/high-sucrose-fed mice (4, 5). Conversely, uncontrolled prolonged generation of short chain fatty acids by dysbiotic microbiota promotes MetS in Toll-like receptor 5 in knockout mice (185). Apparently, modification of the gut microbiota by nutritional status, improvement of intestinal integrity, and induction of microbial metabolic output could positively impact on host health and may represent beneficial avenues for the management of MetS. So far, it has not been established whether microbiota produces beneficial effects on MetS components by regulating mitochondrial events.
Effects of Antioxidants on MetS Traits
Many investigators believe that implementation of lifestyle changes (e.g., healthy diets and physical activity) is of the utmost importance for the treatment of MetS and its consequent complications (150, 152). MetS is a constellation of interrelated conditions, including hypertension, dyslipidemia, IR, T2D, and central obesity. Treatments that target individual components may fight MetS at the forefront of T2D and CVD. Therefore, the current therapy for obesity includes Orlistat, an inhibitor of gastrointestinal lipase activity for weight reduction, lorcaserin, phentermine–topiramate, bupropion/naltrexone, and liraglutide in addition to lifestyle modification and bariatric surgery. The pharmacologic agents for abnormal glucose metabolism/IR and T2D are metformin and glucagon-like peptide 1 receptor agonists. The first line medication for elevated BP includes amiloride and thiazide diuretics for unproblematic patients, angiotensin-converting enzyme inhibitors, or angiotensin receptor blockers for subjects with T2D, congestive heart failure, or chronic kidney disease, or beta blockers for persons with angina (18, 156, 160). Finally, dyslipidemia is treated with HMG Co-A reductase statin inhibitors (LDL-cholesterol), fibrates (TG), and niacins (to raise HDL-cholesterol and lower TG and LDL-cholesterol). Interestingly, short-term fenofibrate treatment may ameliorate not only dyslipidemia but also IR, hypertension, inflammation, and OxS markers in individuals with MetS, signifying that this drug can efficiently decrease the risk of arteriosclerosis via different pathways (203).
Nevertheless, lifestyle modifications and better diet control seem to be more effective than medications (123, 139). Several clinical investigations have corroborated the benefits of consuming bioactive nutraceuticals from functional foods for the amelioration of hyperglycemia (9, 10, 32). Various nutritional antioxidants such as flavonoids, arginine, vitamin C, vitamin E, carotenoids, resveratrol, and selenium can be used to prevent and treat health complications resulting from MetS (70). However, the underlying mechanisms are not fully elucidated for most of these natural compounds. Flavonoids and other phenolic compounds are widely used in view of their antioxidant content capable of neutralizing OxS or activating cytoprotective gene transcription, which keeps out oxidative impairment of macromolecules (2). Indeed, several natural compounds can stimulate the endogenous defense system by activating the nuclear factor Nrf2, a critical modulator of antioxidant response (194). Various studies reported its preventive effects on the development of MetS and related complications, including the transition from pre-T2D to T2D (93). In view of the significant contribution of mitochondria to ROS production, energy metabolism, and MetS development, several therapeutic strategies have recently been designed to prevent mitochondrial dysfunction (55, 121, 186, 187). For example, targeting the antioxidant ubiquinone MitoQ to mitochondria led to enhanced defense against pathological oxidative repercussions in MetS (127, 165).
Conclusions
OxS is increasingly perceived as the root cause of the initiation and evolution of MetS and may constitute the major unifying mechanism of the obesity, dyslipidemia, IR, and hypertension components associated with MetS. Although mitochondrial redox state and dysfunction are closely implicated in the pathogenesis of MetS, it remains unclear whether they may be the cause or consequence. Substantial evidence indicates ROS scavenging potential in normalizing mitochondrial biogenesis, energetics, and functional capacity while preventing MetS complications and severity, which provides additional mechanistic insight into the role of OxS in MetS development and subsequent complications. However, the therapeutic utility of ROS scavenging and particularly mitochondria-targeted antioxidants as a therapy for MetS-related disorders requires further investigation, given the controversial findings. Further studies should be instrumental in determining whether mitochondria-targeted approaches improve mitochondrial structure, DNA integrity, ROS production, dynamics, mitophagy, and function, before turning to the wide range of effects and mechanisms of action in MetS features.
Footnotes
Acknowledgments
The current work was supported by research grants from the JA deSève Research Chair in nutrition (E.L.) and FRQS doctoral Scholarship Award (S.S.). The authors thank D. St-Cyr-Huot for her technical assistance.