Abstract
Significance:
Recently, chronic degenerative diseases have become one of the main health problems worldwide. That is the case of Alzheimer's disease (AD) and metabolic syndrome (MetS), whose expression can be influenced by different risk factors.
Recent Advances:
In recent decades, it has been widely described that MetS increases the risk of cognitive impairment and dementia. MetS pathogenesis involves several vascular risk factors such as diabetes, dyslipidemia, hypertension, and insulin resistance (I/R).
Critical Issues:
Reported evidence shows that vascular risk factors are associated with AD, particularly in the development of protein aggregation, inflammation, oxidative stress, neuronal dysfunction, and disturbances in signaling pathways, with insulin receptor signaling being a common alteration between MetS and AD.
Future Directions:
Insulin signaling has been involved in tau phosphorylation and amyloid β (Aβ) metabolism. However, it has also been demonstrated that Aβ oligomers can bind to insulin receptors, triggering their internalization, decreasing neuron responsiveness to insulin, and promoting insulin I/R. Thus, it could be argued that Aβ could be a convergent factor in the development of both pathologies. Antioxid. Redox Signal. 26, 542–560.
Introduction
A
In recent decades, it has been observed that diabetes and obesity could be risk factors for the development of neurodegenerative diseases. Recent epidemiological and clinical studies have indicated that the incidence of obesity and diabetes in young adults with cognitive impairment is increasing; hence, it is believed that dementia disorders—particularly Alzheimer's disease (AD)—will increase significantly in the coming years (47, 48, 54, 55).
In the 1990s, Ott et al. were pioneers in proposing a strong association between diabetes and AD. Vascular disorders and malfunction of the endothelium could represent the link because they are common characteristic features in both pathologies; also, it is believed that patients with cardiovascular disease and type 2 diabetes mellitus (T2DM) are more likely to develop cognitive impairment (154, 170). Currently, there are many studies that support this theory and explain how these two disorders may course together (55, 57, 165, 166).
In the same way, other pathological changes and risk factors that contribute to AD are insulin resistance (I/R), dyslipidemias, hypertension, inflammation, and endoplasmic reticulum (ER) stress. These factors are also associated with metabolic syndrome (MetS). Although there are some differences in the definition of MetS, it is clear that I/R and visceral obesity have an important role in its etiology (90).
In 2004 and 2007, Yaffe et al. conducted a study to determine the effect of MetS on cognition in an elderly population and assessed whether inflammation modifies this association. They observed that cognitive impairment could be the result of pathological processes such as MetS and inflammation in older people regardless of their backgrounds (224, 225). This association was also confirmed by Dik et al. in the Longitudinal Aging Study Amsterdam (LASA); however, the results obtained by these researchers showed that the main factor associated with MetS and cognition is hyperglycemia (66).
In the last years, plenty of evidence indicates that AD is a metabolic disease (57, 182, 211). In fact, it has been recently described that AD brains show increased microglial activation and increased expression of inflammatory mediators (21), as well as several markers of oxidative stress and I/R (37, 151, 190, 207). Thus, it is important to evaluate whether the central and peripheral inflammatory response may converge on a final common pathway leading to neurodegeneration associated with AD.
In this review, we describe the main aspects of AD and MetS, as well as the relationship between both pathologies, based on studies conducted and published in the literature. We also discuss the role of I/R and hypertension and its relationship to the presence of amyloid β (Aβ). Aβ toxicity and mechanisms for degradation are also discussed. Finally, we aim to discuss the role of Aβ in local and peripheral inflammation to establish its relationship with MetS, assuming that Aβ and I/R have an effect on the local and peripheral inflammation that leads to neurodegeneration.
Alzheimer's Disease
AD is the most common cause of dementia. It is a complex pathology that involves genetic and environmental factors, but the exact cause of its development is still unknown. It is a progressive neurodegenerative disease that accounts for ∼50% of all cases of dementia and it is located around the third and fourth place in leading causes of death in adults of industrialized countries. The incidence of AD increases by 5% in people over 65 years old and 20% in those over 80 years old (139, 150, 174).
Overall, AD has been classified into two types, depending on the age of onset: early-onset Alzheimer's disease (EOAD) and late-onset Alzheimer's disease (LOAD).
The manifestation of EOAD cases is very low; it is related to the presence of mutations in three genes: presenilin 1 (PSEN1), presenilin 2 (PSEN2), and the amyloid-beta precursor protein (APP), and it is inherited via an autosomal dominant pattern (24, 35, 71, 98, 137, 157, 194).
On the other hand, LOAD represents 90–95% of all AD cases and, although its etiology is not well understood, the main risk factor for this variant is aging. For many decades, it has been suggested that the neurodegeneration observed in the brains of patients with AD is the result of aggregation and accumulation of Aβ, hyperphosphorylated Tau, and inflammation. However, more recent studies indicate that AD is closely related with vascular and related factors, including total cholesterol and other lipid parameters, high blood pressure (BP), hypertension, and with metabolic alterations such as glucose utilization, impairments in brain insulin responsiveness, and energy metabolism (15, 117, 134, 169, 183, 198).
It is noteworthy that regardless of the different genetic and environmental factors, all patients (EOAD and LOAD) develop the same kind of protein aggregates in the brain: neuritic plaques (NPs) formed of Aβ (222) and neurofibrillary tangles (NFTs) formed of Tau protein (162).
Neurofibrillary tangles
The presence of NFTs in the brains of patients with AD is tightly related to the severity of dementia. NFTs are formed of paired helical filaments (PHFs) (131, 221), which are in turn formed by the association of several fragments of the microtubule-binding protein Tau (22, 26). The gene that encodes this protein is located on chromosome 17 and comprises 16 exons, from which exon 1 and exon 14 can be transcribed, but not translated (7, 162, 164). Tau protein has a role in the assembly and stability of microtubules; additionally, it is also related to the transport of vesicles and organelles.
In normal conditions, Tau protein is highly soluble, and it does not have an apparent secondary structure. However, in pathological conditions, Tau tends to self-assemble into insoluble filament structures (38, 99, 189). Inadequate Tau phosphorylation due to glycogen synthase kinase 3β (GSK3β) activation or inhibition of protein phosphatase 1 and 2A generates self-aggregation and formation of insoluble fibrillar aggregates known as PHFs and straight filaments that eventually produce NFTs (19, 119).
Lack of insulin signaling (in the brain of several animal models) may lead to changes in PKB/Akt, which has been demonstrated to normally dephosphorylate GSK3β, favoring Tau hyperphosphorylation and development of AD (60, 187, 188, 204). It has been observed that loss of sensitivity of insulin receptors (IRs) generates increased expression of Aβ and Tau (193, 211). These observations suggest that altered insulin signaling in the brain could lead to NFT and NP formation in AD, suggesting that I/R predisposes to neurodegeneration.
Neuritic plaques
NPs are extracellular deposits of 10–100 μm in diameter; they have an insoluble core surrounded by microglia, reactive astrocytes, and dystrophic neurites. The main component of NPs is a peptide of 39–42 amino acids with a molecular weight of 4 kDa, called Aβ (96, 97, 120, 191), which arises as a product of normal secretion of APP (126). APP is an integral membrane glycoprotein that is expressed primarily in the central nervous system (CNS) (185). The gene that encodes this protein is located on chromosome 21q21 and consists of 18 exons. Several isoforms are produced by alternative splicing with lengths varying between 365 and 770 amino acid residues.
The fraction known as Aβ is only present in the APP695, APP751, and APP770 isoforms. In neuronal cells, APP695 isoform is mainly expressed, unlike glial cells, which express mainly the isoforms, APP751 and APP770 (100, 168). APP can be processed by two types of proteolytic pathways through the activity of α-, β-, and γ-secretases (62). The nonamyloidogenic pathway is the main form of proteolytic cleavage of APP (Fig. 1) and it is performed by the α-secretase activity at residues 612–613, corresponding to the middle portion of the Aβ peptide, thereby preventing Aβ formation (75, 195).
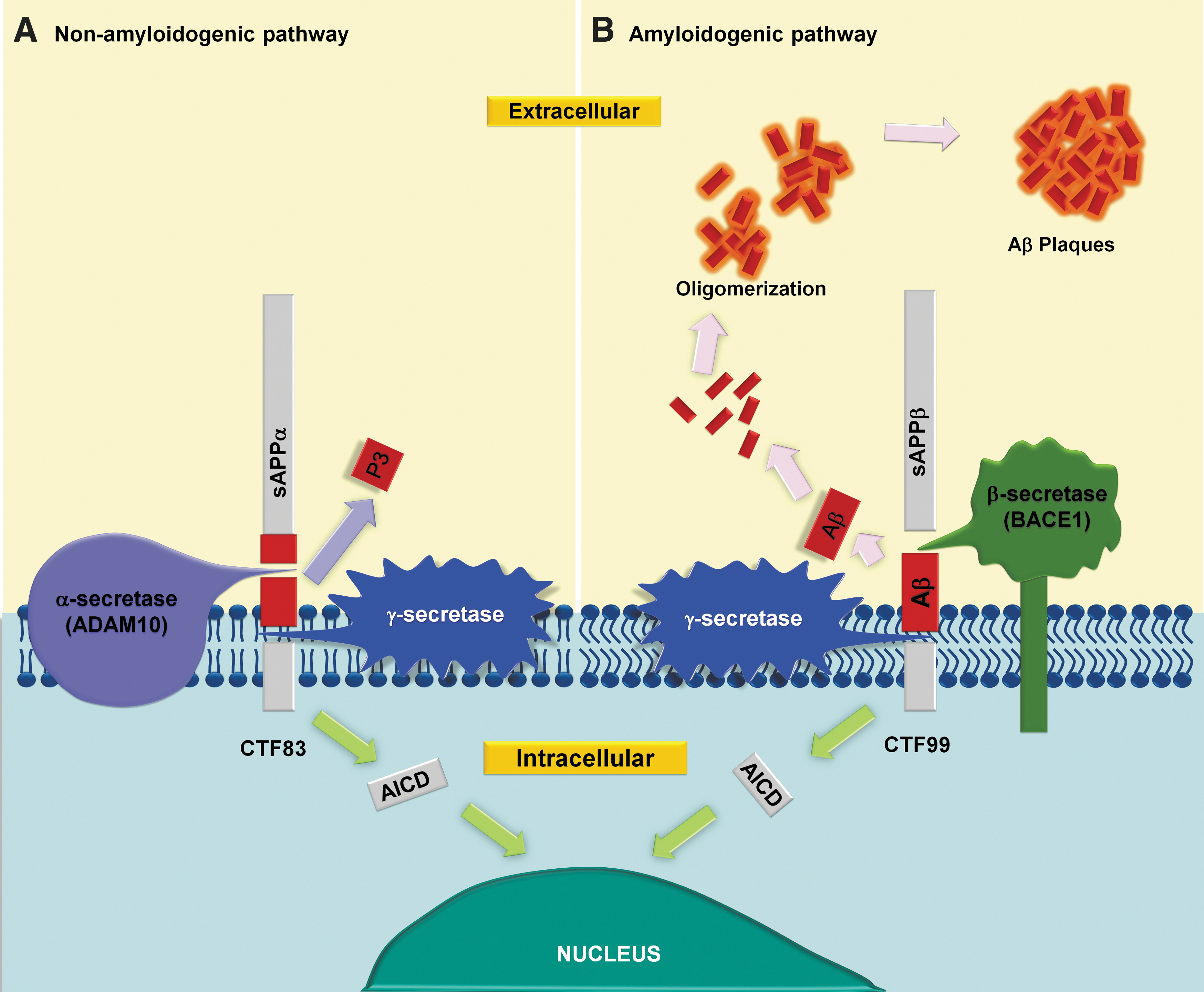
On the other hand, the amyloidogenic pathway is carried out by the β-secretase (BACE1) activity and results in the formation of a soluble product (sAβPP) and a carboxyl terminal membrane-anchored fragment (C99), which is processed by the enzyme γ-secretase, generating the neurotoxic fragment Aβ (Fig. 1) (69). Aβ peptides consist of 36–43 amino acid residues (Aβ36, Aβ39, Aβ40, Aβ42, Aβ43). The most abundant is Aβ40 peptide; however, Aβ42 is the most hydrophobic, therefore it constitutes a greater proportion of NPs observed in AD patients (121).
Aβ formation is a normal physiological process; however, in AD, there is an imbalance between formation and clearance, favoring the Aβ aggregation process in the brain of these patients (176). Damage caused by Aβ depends on the level of aggregation of the peptides, oligomeric forms being the most toxic (51, 128, 215). Progressive accumulation of these oligomers causes the formation of insoluble fibers that finally result in NP development. The physiological role of the Aβ peptide has not been fully elucidated, but it is known to participate in various processes of neuronal activation, including neuroprotective and cognitive functions (81, 102, 163, 208). Besides, APP processing can be altered by inhibition of energy metabolism and induction of amyloidogenic products (89).
Metabolic Syndrome
In recent decades, MetS has become a serious health problem worldwide. Although criteria for diagnosing MetS are divergent, the common denominator is central obesity or abdominal adiposity. When the storage capacity of adipose tissue is exceeded, fatty acids in the form of triglycerides start to ectopically deposit in organs such as heart muscles, liver, pancreatic islets, perivascular tissue, and skeletal muscle, promoting a reduction in the normal function of these organs and leading to a metabolic dysfunction (44, 179).
Not all overweight or obese people have metabolic abnormalities; the human body is capable of maintaining internal homeostasis through mechanisms of regulation of negative feedback. To maintain the stability of the internal environment, multiple body systems are involved, which are regulated by the limbic structures of the brain (prefrontal cortex, hippocampus, and the amygdala), which perform hormonal or metabolic regulatory responses (88, 210). Consequently, some authors have suggested that MetS is the result of a bad adaptation of the body to respond and suggest a potential link between metabolic disorders and brain action/dysfunction. In fact, several peptides and hormones produced in other organs outside of the CNS cross the blood–brain barrier (BBB) and exert their effects on the brain, and vice versa in a particular molecular cross talk between peripheral organs and CNS.
It has been documented that cortisol and estrogens, as well as insulin and leptin, could influence cognitive functions, while other neuropeptides such as orexin intervene regulating gastrointestinal motility and gastric secretion (130). Although little is known about the contribution of the brain to metabolic disorders, it is known that several neural circuits in the CNS may be involved in metabolic homeostasis. Through hormonal signaling and direct macronutrient sensing, the brain is continuously taking in external information about peripheral metabolism (78). Nutritional signaling integrated by cerebral structures such as the hypothalamus activates a chain of neurochemical events and elicits a relationship between metabolism and brain functions (87).
Metabolic syndrome and neurodegeneration
The predisposition to the development of metabolic diseases and neurodegenerative disorders is closely related to the presence of genetic and environmental factors as well as to the lifestyle of individuals. In general, MetS is a cluster of signs and symptoms that include several vascular risk factors such as I/R, abdominal obesity, arterial hypertension, and dyslipidemias. It is also an important risk factor for the development of vascular and cardiac diseases and, together with obesity and I/R, it increases the probability of development of AD and other neurodegenerative diseases in the elderly (88, 143, 153, 154, 199).
Several studies have shown that prediabetes and diabetes mellitus increase the risk of development of cognitive decline; they have also been found to be related to vascular dementia, and some authors support the idea of its relationship with AD (12, 34, 103, 105, 113, 193, 198). Recent studies in animal models have shown that sucrose-treated mice presented mitochondrial abnormalities, an oxidative imbalance, significant increase in Aβ levels, and slight increase in pTau levels, suggesting that metabolic alterations associated with sucrose intake could contribute to the development of AD-like pathological features (33, 34).
Afterward, they compared behavioral and cognitive functions, cerebral Aβ peptide levels, and vasculature integrity of 11-month-old T2DM and AD mice. Both models (DM2 and AD) have similar cognitive and behavioral disorders characterized by increased fear and anxiety and decreased learning and memory abilities, apart from increased cerebral vasculature permeability and plasma markers of endothelial/vascular dysfunction, all of this accompanied by a significant increase in Aβ levels. These results, along with others reported in literature, support the idea that T2DM increases the risk of developing neurodegenerative diseases, including AD (12, 34, 113, 193).
Both pathologies are characterized by the formation of deposits of insoluble protein aggregates, neuronal I/R, progressive I/R, desensitization and Aβ amyloidosis in the brain, neuroinflammation, and oxidative stress in addition to indirect ischemic effects (88).
Obesity is one of the most important features of MetS. It is partly responsible for the increase in free fatty acid (FFA) concentration that eventually leads to elevated very low-density lipoprotein concentration, and both of these constitute an important link between obesity and I/R (44, 152). Obesity-associated FFA elevations may influence AD pathogenesis. FFAs inhibit insulin-degrading enzyme (IDE), an important metalloprotease essential for normal insulin signaling that also participates in Aβ clearance (27, 49).
Similarly, inflammation in adipose tissue and obesity are closely linked, it is known that infiltration and expansion of macrophages can induce the production of inflammatory cytokines and they interfere with insulin signaling, generating an alteration in adipose tissue homeostasis (129). Therefore, we can say that inflammation may contribute significantly to the development of diabetes. A similar effect occurs in the AD brain, in which production and activation of inflammation-related proteins such as acute-phase proteins, complement factors, chemokines, and cytokines such as interleukin 6 (IL-6), interleukin 1 (IL-1), transforming growth factor β, and tumor necrosis factor α (TNF-α) (151) have been observed. Thus, we can say that in AD and diabetes, inflammation is an important feature and plays critical roles in the pathogenesis of both disorders (56, 82).
Insulin resistance
Although MetS is a complex condition, it is clear that a prevailing common denominator is disequilibrium between energy production and utilization, which in the long run, alters CNS function, favoring cognitive impairment and impaired executive function. Many of these alterations are related with I/R, and impaired insulin signaling has been involved in the progression and development of AD (28, 45). The term I/R refers to the fact that tissues do not respond efficiently to physiological concentrations of insulin. Therefore, much higher concentrations of insulin are required to maintain glucose homeostasis (220).
In the brain, insulin/insulin-like growth factor (IGF) signaling is important for synaptic maintenance, neuroprotection, and neuronal growth. Specifically, it has been observed that in the CNS, insulin could induce an increase in hippocampal long-term potentiation (LTP), promote neuronal outgrowth, increase glucose utilization in the entorhinal cortex and hippocampus, and affect neuronal activity in hippocampal pyramidal neurons (127, 171).
It is well known that insulin-like growth factor type 1 and type 2 (IGF-1 and IGF-2), insulin, and their receptors are expressed in glial and neuronal cells throughout the brain (61, 78, 110); however, the highest density expression is found in the hypothalamus (mainly in the arcuate nucleus), hippocampus, and olfactory bulbs (78, 116).
IRs and IGF receptor type-1 (IGFR-1) are tetrameric glycoproteins, comprising two α- and two β-subunits that belong to the receptor tyrosine kinase superfamily. In the mammalian brain, two IRs have been reported: a neuron-specific type and a peripheral-like type, expressed in glial cells. Both receptors (IR and IGF-1R) can be activated by insulin or IGF-1. When insulin binds the α-subunit of IR or IGF-1R, it promotes autophosphorylation of the β-subunit. As a result, the Tyr kinase activity of the β-subunit is triggered, and insulin receptor substrate proteins (IRS1–4) are phosphorylated at their Tyr residues (70, 156).
Subsequently, signaling molecules containing Src domains are recruited, which activate the catalytic subunit PI3K and the growth factor receptor-bound protein 2 (Grb-2), generating the activation of the PI3K/Akt/GSK-3β and the Ras/Raf-1/extracellular signal-regulated kinase (ERK1 and ERK2, ERK1/2) pathways (41, 70, 155, 209).
As we previously mentioned, activation of insulin/IGF signaling exerts neuroprotective effects and has been recently studied due to the importance of finding new therapies capable of modifying neurodegenerative diseases (95). In the case of AD, a direct link between the insulin pathway and Aβ metabolism has been described in neuroblastoma (SH-SY5Y) cell cultures, where exposure to insulin promoted the release of a soluble APP fragment derived from α-secretase activity (sAPPα) by the activation of tyrosine kinases and PI3-K (197). The sAPPα has been associated with beneficial functions in brain, such as neurite growth, modulation of neuronal excitability, synaptic plasticity, and synaptogenesis (149).
It was recently reported that the intracerebroventricular (icv) injection of sAPPα in aged mice ameliorated the decline in neurogenesis. This fact raises the possibility of an AD therapy in the future (64). Besides the relationship with sAPPα production, insulin is involved in Aβ trafficking from the intracellular to the extracellular space by activation of the tyrosine kinase/mitogen-activated protein kinase (MAPK) pathway (94). Meanwhile, IGF-1 seems to prevent the toxic effects of the extracellular peptide. Unfortunately, lower serum levels of this factor have been reported in Tg2576 mice and in AD patients (31, 217). Treatment of AD Tg2576 mice with IGF-1 induced Aβ clearance from the brain to the BBB by increasing albumin and transthyretin levels (31).
However, a posterior experiment revealed that IGF-1 activates IGF-1R at epithelial choroid plexus cells and promotes its transport to the brain by the multicargo transporter megalin/low-density lipoprotein receptor-related protein-2 (megalin/LRP2); the consequence of IGF-1 transport is the formation of complexes between Aβ and megalin/LRP2, which finally enhances Aβ clearance through the BBB (10, 30). As well as to insulin, exposure of human neuroblastoma cells to IGF-1 resulted in an increased sAPPα secretion by means of activation of different signaling pathways, such as PI3-K, MAPK, and cyclin-dependent kinase 5 (5). The evidence reassures the need to validate the therapies focused in promoting insulin/IGF-1 signaling due to its neuroprotective role.
I/R in the brain
Insulin has important effects on brain function. It is known that it facilitates memory; for example, insulin administration to rats results in better performance in passive avoidance tasks (171). It has been observed that when insulin is intranasally administered, brain atrophy is reduced, there is an improvement in memory processes, and neurodegeneration caused by apoptosis is reverted (86). Similarly, healthy people and patients with memory disorders treated with insulin (i.v. and intranasal application) showed improvement in memory processes (18, 50, 61, 127, 180).
It has been proposed that I/R significantly participates in AβPP metabolism, increasing Aβ accumulation and build-up of NPs (153). In a recent study, the icv injection of a subdiabetogenic dose of streptozotocin (icvSTZ) resulted in a significant increase in hippocampal Aβ42 levels as well as in cortical and hippocampal hyperphosphorylated tau protein levels; icvSTZ administration also induced a decrease in brain weight and cognitive decline (46). Other animal models have shown that I/R generates an obvious impairment in learning and memory.
Recently, an I/R rat model was implemented to assess pathological alterations and measure BACE1 brain expression; the results showed overexpression of this protease (138), suggesting that I/R participates in AD pathogenesis by promoting the metabolism of APP by β-secretase, thus eliciting the amyloidogenic pathway, which leads to Aβ release and amyloid plaque formation (29, 123, 138, 229). Previous studies have shown that Aβ oligomers bind to IRs, triggering their internalization and promoting I/R (58, 155, 230). This decreases neuron responsiveness to insulin, and it is likely to be associated with the learning and memory problems seen in AD patients (54).
It is not easy, in the presence of MetS, to establish a direct relationship between individual risk factors and dementia, where multiple risk factors, rather than one in particular, play a complex role in the pathogenesis of dementing disorders; however, the presence of amyloid appears to be a factor involved in the development of both AD and MetS.
Hypertension, MetS, and Aβ
Hypertension is one of the main risk factors for MetS. It causes functional and morphological changes in the cerebral vascular system that could be associated with cognitive impairment onset and progression. These changes include endothelial dysfunction, cerebral microbleeds, neuronal death, vessel remodeling, and BBB damage. Remarkably, these alterations are related to Aβ deposits in brain vasculature (77, 133), generating a reduction in blood flux and leading to a decreased supply of energy substrates such as glucose to neuronal cells, which in turn causes diminished metabolic energy production (172).
The molecular basis of the link between Aβ and vascular disruptions is poorly elucidated; however, it has been found that Aβ deposits lead to signaling pathway perturbations. In particular, the PKC cascade has been detected as a target leading to morphological vascular damage that prompts severe complications such as ischemia and stroke, which in turn generate the release of reactive oxygen species (ROS) and promote apoptosis (142). On the other hand, as mentioned before, Aβ deposits trigger oxidative stress, which may impact directly upon the sensitive layer of the endothelial brain vasculature. Endothelial dysfunction is undoubtedly the underlying event in the development of hypertension. Injured endothelial cells are a hub for inflammatory cells and further ROS generation and cause imbalance of vasoactive signals such as endothelin, angiotensin, and prostaglandins that increase vascular resistance (233).
Indeed, apart from the strong epidemiological link between hypertension and AD development, sufficient experimental evidence outlining the cellular mechanism that links hypertension and AD indicates a key participation of Aβ on this interaction. In this sense, it is important to consider that hypertension is a characteristic MetS component, which can be modified to prevent further complications, including neurodegeneration and vascular brain diseases.
Aβ Toxicity
As mentioned before, Aβ is a peptide formed of 39–42 amino acids derived from the proteolytic processing of a membrane-spanning glycoprotein called APP and it is detectable in cerebrospinal fluid (CSF) and plasma under normal physiological conditions (145). Aβ toxicity varies among different oligomeric amyloid species and their progressive accumulation causes NP formation (51, 128, 215). Some authors have suggested that Aβ fibers, generated by self-aggregation, are less toxic than oligomeric forms (161). This is supported by the fact that soluble Aβ oligomers correlate better with severity of cognitive impairment than NPs (Fig. 2) (108).
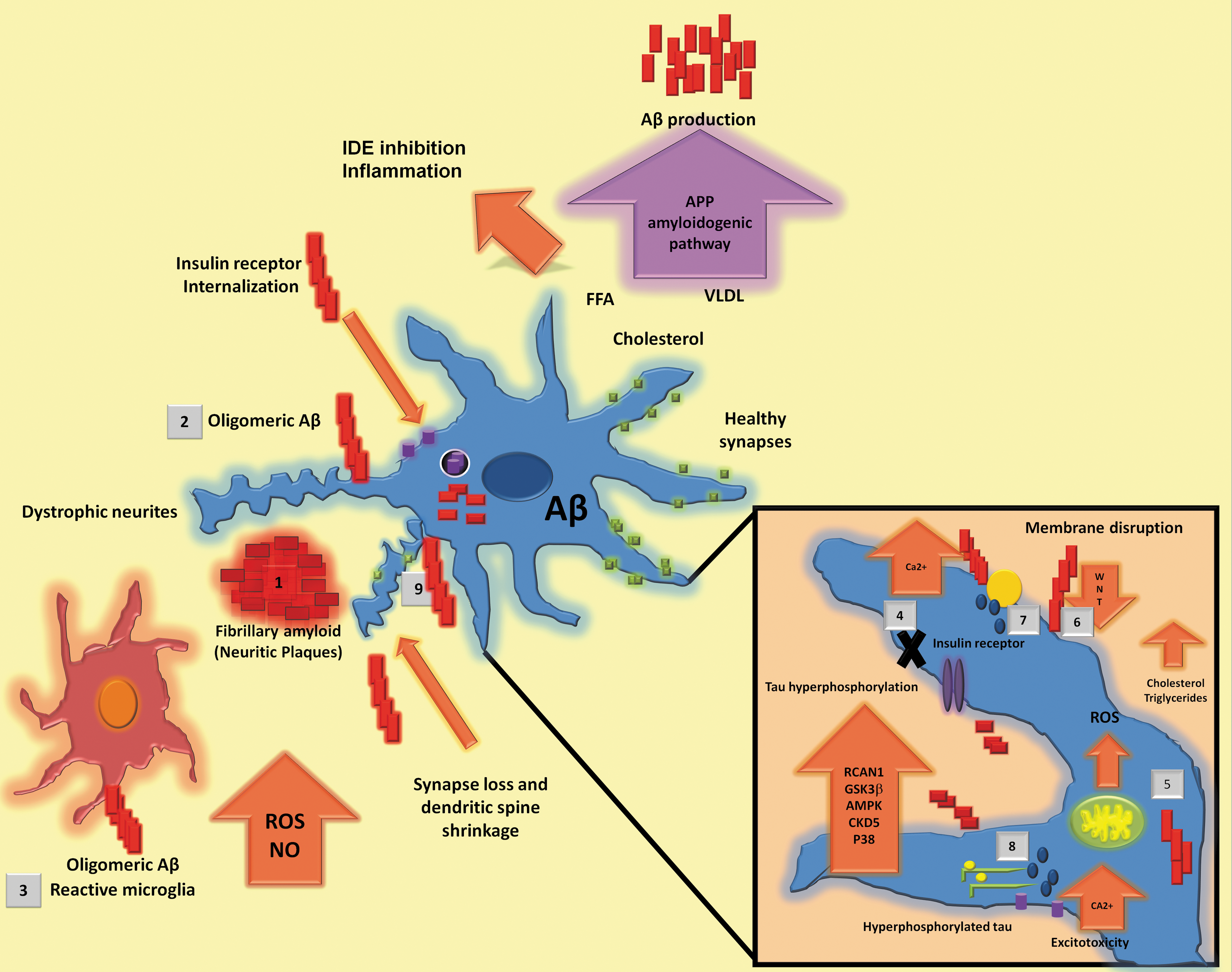
In vitro studies have demonstrated that direct administration of Aβ to cell cultures has a neurotoxic effect since it increases oxidative stress and induces apoptosis (85, 140). In cultured human neuronal cells, the presence of amyloid fibers generates a progressive damage in contrast with oligomers, which induce acute damage (65). In primary neuronal cell culture from embryonic rat, Aβ was shown to induce abnormal processes in the plaque vicinity or surrounding it and to cause neuronal dystrophy, although displaying a lesser effect in neuronal viability (101). Likewise, oxidative stress has been directly associated with Aβ accumulation and it has been observed in early stages of AD. In the same way, an increased inflammatory response has been associated with cognitive decline (67, 74, 186).
The presence of Aβ oligomers has been found to reduce spine density in a mechanism that involves NMDA-type glutamate receptors (NMDARs), calcineurin, and cofilin (192). Some reported effects of synthetic Aβ are a reduction of NMDAR cell surface expression and inhibition of LTP induction, apart from a decrease in dendritic spine density (135, 192, 196).
Furthermore, the presence of Aβ in cultured hippocampal neurons inhibits canonical Wnt signaling. In a study in a murine model mutant for APP/PS1 that favors the production of Aβ, lithium is capable of reverting cognitive impairment in spatial memory, gliosis, and neurodegeneration due to an increase in Wnt pathway activation, and it also diminishes plaque burden. (206). This was supported by another experiment in which addition of Wnt3a to cultured hippocampal rat neurons reduced neuronal death caused by Aβ (6). This group also found that in the same cultured hippocampal neurons, Aβ generates a reduction of β-catenin, a protein involved in the Wnt canonical pathway. By avoiding β-catenin reduction through Gsk3 inhibition, neuronal viability increased (91).
In relation to some features of MetS, it has been found that the Wnt pathway is reduced (52, 107, 231) and that Gsk3, which antagonizes the Wnt pathway, is increased (39). Additionally, there are studies in which restoration of this pathway avoids MetS features. In accordance with this, the Wnt pathway is downregulated during MetS, which might contribute to the neurodegeneration and cognitive impairment seen in the presence of Aβ (182) (Fig. 2).
Aβ Degradation
The physiological concentration of Aβ is the consequence of a balance among a number of factors, which include the regulation of AβPP cleavage that results in Aβ production, the transport of Aβ across the BBB, Aβ oligomerization, Aβ proteolytic degradation, and the capacity of Aβ-binding proteins, which in turn influence Aβ clearance and aggregation (13).
In the brain, microglial cells play an important role in Aβ clearance; they are able to engulf the peptides and also release proteolytic enzymes responsible for Aβ degradation. Studies have shown they are capable of internalizing and degrading both the fibrillar form of Aβ (fAβ) and soluble form of Aβ (sAβ) (36). The latter can be uptaken through two pathways: LDL receptor-related proteins (LRPs) and macropinocytosis. In contrast, fAβ forms are able to stimulate phagocytosis by their interaction with the cell surface innate immune receptor complex (Fig. 3) (136).

In the brain, amyloid can be removed in two ways: through proteolytic degradation of fibrillar and soluble forms and through the efflux of soluble Aβ to the peripheral circulation. Soluble Aβ can be cleared through its efflux across the BBB into the circulation by the low-density lipoprotein receptor-related protein 1 (LRP1), by its transport via the P-glycoprotein (Pg-P) across the BBB, or through the flow of interstitial fluid/CSF into the lymphatic system. These mechanisms are facilitated or inhibited by its binding to a transport protein, for example, Apolipoprotein E (ApoE), Apolipoprotein J, or albumin (63, 136).
LRP1 is a signaling receptor that functions as a scavenger and participates in cholesterol metabolism and transport associated with ApoE-containing lipoproteins. In addition, it functions as the main Aβ transporter, through the BBB (63, 132).
In the brain, ApoE regulates levels of Aβ peptide and participates in both its aggregation and clearance (115, 232); it is synthesized and secreted primarily in astrocytes and is the predominant brain apolipoprotein (122). Aβ degradation by ApoE depends not only on ApoE isoform but also on lipidation status. ApoE is lipidated mainly through the action of the ATP-binding cassette transporter ABCA1 (and related transporters) (63, 112, 214). ApoE lipidation status is an important factor determining whether the interaction of ApoE/Aβ leads to fibril formation and deposition into NPs or otherwise Aβ peptide efflux from the brain (17, 122). Studies in APP transgenic mice have demonstrated that ApoE3 and ApoE2 have higher affinity for Aβ, while ApoE4 binds the peptide with a lower affinity (2–3 times) (40, 114).
Aβ can suffer proteolytic degradation by a number of peptidases and proteinases known as Aβ-degrading proteases. Their activity relies upon their avidity to Aβ, optimal pH, and cellular localization. The specific proteases that degrade Aβ can be classified by enzymatic type (e.g., metalloprotease, serine protease, aspartyl protease, cysteine protease, threonine protease), by their subcellular localization (e.g., ER, extracellular space, mitochondria, endosomes, cytosol, lysosomes), and the aggregation state of the peptide substrate they hydrolyze (e.g., peptidases, oligopeptidases) (184). In microglia, the main intracellular and extracellular enzymes responsible for the proteolysis of Aβ are neprilysin (NEP) and IDE, respectively.
Neprilysin
NEP, also known as neutral endopeptidase or enkephalinase A, is an integral membrane zinc metalloendopeptidase with a molecular weight around 90 to 110 kDa. The gene that encodes this protein is located in the long arm of chromosome 3, contains 24 exons, and spans more than 80 kb (216). To date, there have been identified seven forms of this enzyme in humans, all of which have a short N-terminal cytoplasmic region, a membrane-spanning region, and an extracellular C-terminal catalytic domain that has an HExxH zinc-binding motif (32). This motif is responsible for the inactivation and degradation of a number of bioactive peptides, such as atrial natriuretic peptide, chemotactic peptides, bradykinin, angiotensins I and II, substance P, enkephalins, and endothelins; regulating lipid metabolism and BP.
NEP can also degrade Aβ monomers and oligomers (125). It is primarily expressed in kidneys and, to a lesser extent, in neuronal plasma membranes in the brain (159). It has a homolog, neprilysin 2 (NEP2), which is highly expressed in the pituitary, hypothalamus, and brain stem and also has the capacity to degrade Aβ (147). Mice deficient in the NEP2 gene showed elevations in total Aβ species, suggesting the importance of this NEP-like protease on the clearance and degradation of Aβ (109).
NEP has been indicated to be associated with lipid metabolism, BP, and I/R regulation. The suggestion that NEP contributes to I/R has been addressed by several studies. Arbin et al. (8, 9) found in obese I/R Zucker rats that the inhibition of NEP improves insulin-mediated glucose disposal (IMGD) and that the dual inhibition ACE/NEP improves IMGD more effectively than single inhibition. Moreover, Standeven et al. (203) tested the possible association of NEP with I/R and MetS. They carried out a study in 318 white males of European origin characterized by I/R and by the presence of MetS, finding that plasma NEP was elevated in subjects with MetS compared with those without and that its activity was correlated with insulin.
In addition, they found that NEP expression levels were increased during differentiation of human adipocytes and demonstrate in HFD-fed mice that the development of obesity and I/R increased with plasma NEP concentration. Furthermore, a study (14) identified NEP as a crucial player in the development of obesity. They observed that mice with genetic NEP deletion gain body weight, a feature often associated with obesity and related to deregulation of lipid metabolism. In regard to BP, Rice et al. (181) determined the heritability of circulating NEP and its role in BP in plasma from 534 subjects in the Leeds Family Study. They found that NEP was associated with systolic BP and diastolic BP and that it was correlated with body–mass index, cholesterol, and triglycerides, suggesting an association between NEP and MetS.
Insulin-degrading enzyme
IDE is a 110 kDa zinc-endopeptidase that requires a thiol (-SH) and the bivalent cation Zn2+ for its activity as a protease. The enzymatic activity of IDE is optimal at a pH range of 6 to 8.5. It belongs to the M16 family of zinc metalloendopeptidases (named inverzincins) (228), which present a zinc-binding motif, HxxEH, which is inverted compared with the conventional metalloprotease motif (160). Both histidines coordinate the union with the zinc atom, while glutamate has a role in catalysis.
The gene that encodes this protein is located in the long arm of chromosome 10 and comprises 120 kb (177). The enzyme has its highest expression in the muscle, liver, testes, and brain, whereas its subcellular location is in the cytosol, peroxisomes, endosomes, and the cell surface (80). IDE structure reveals that in its monomeric form, it is conformed by two equal size domains of ∼50 kDa, IDE-N and IDE-C, which are connected through a 28 amino acid loop (118).
In vivo IDE has multiple substrates with different Kms; its main function is degradation and clearance of insulin in vivo. The mutated form of IDE expressed in animal models increases insulin levels, due to a decrease in its degradation, and causes symptoms typical of human type 2 diabetes syndrome (76, 80, 177). Homozygous knockout mice for the IDE gene (IDE-/-) showed a decrease in insulin degradation in the liver and a decrease in amyloid degradation in primary neuronal cultures of up to 50% (79). Because progressive Aβ accumulation in the brain is an invariant feature in AD, its defective degradation is an important risk factor for the development of the pathology.
IDE is secreted by microglial cells at high levels and degrades Aβ in the extracellular space. Of all proteases secreted from these cells, only IDE has the ability to degrade monomeric amyloid (184); however, this action could be inhibited by insulin (175, 212). Considering the fact that insulin and Aβ compete for the union with the enzyme, insulin can inhibit the degradation of Aβ, resulting in an increase of Aβ extracellular levels (92). Insulin can regulate this action through the inhibition of extracellular degradation of Aβ by IDE or by promoting the release of Aβ (93). In this way, many lines of evidence suggest that AD pathology is related with IDE dysfunction; it is known that AD patients have low levels of IDE and also display impaired insulin clearance (43, 134, 173).
Aβ Toxicity: Inflammation and MetS
However, what is the relationship between amyloid toxicity and MetS? In recent years, several lines of evidence suggest that inhibition of energy metabolism may alter APP processing and induce Aβ accumulation (15, 89). Likewise, oligomeric Aβ causes internalization and cellular redistribution of IRs, blocking downstream hippocampal insulin signaling (23, 58, 144), and promotes hippocampal ER stress (37, 141). Recently, it has been demonstrated that icv injection of Aβ oligomers in mice triggers peripheral glucose intolerance. The accumulation of Aβ oligomers in the brain has an important effect on the hypothalamus and therefore affects peripheral metabolism through mechanisms similar to those of metabolic diseases such as type 2 diabetes (37).
In addition, icv-injected Aβ oligomers induce adipose tissue inflammation and impaired insulin-induced surface translocation of GLUT-4 in muscle cells (37). Finally, pharmacological inhibition of the inflammatory process in the brain and ER stress prevented glucose intolerance in treated mice, indicating that Aβ oligomers may act through a central pathway involving peripheral glucose homeostasis.
Aβ-induced local inflammation
Aβ aggregates trigger innate immune response. Under normal conditions, microglia are in a resting state; however, during the inflammatory response caused by amyloid, they are activated and release proinflammatory mediators such as cytokines (IL-6, IL-1β, TNFα), chemokines, prostaglandins, and free radicals whose aim is to raise an immune response that favors damage repair and homeostasis restoration; however, this response may also exacerbate damage and lead to a chronic inflammatory process that significantly contributes to cognitive impairment in patients with AD (200).
Microglial cells express cell surface receptors that recognize and internalize fAβ. These receptors include scavenger receptor type A (SR-AI/II), scavenger receptor type B (also known as CD36), receptor for advanced glycation end-products, Toll-like receptors, Fc receptors, and complement receptors (68). Microglial binding to fAβ is carried out by a cell surface receptor complex, which comprises scavenger receptor CD36, integrin-associated protein/CD47, and α6β1-integrin (11). Overall, Aβ degradation is important to maintain a balance between the rate of production and clearance. Otherwise, if this equilibrium is broken, it will lead to the accumulation of Aβ and therefore contribute to the occurrence of AD.
Moreover, microglial activation for the purpose of Aβ phagocytosis and degradation may lead to other consequences in AD pathogenesis. In this way, the stimulation of microglial cell cultures with fAβ induces TNF-α and glutamate release and both of them have been demonstrated to act in a synergistic mode on neurons to increase NMDAR activation and inducible nitric oxide (NO) synthase expression. This in turn leads to NO formation, which rapidly reacts with superoxide anion to produce peroxynitrite, a strong promoter of oxidative stress. For these reasons, chronic activation of microglia leads to excitotoxic damage and cell death (42, 84).
The most abundant glial cells are the astrocytes; they are in close contact with neurons to establish homeostasis and maintain BBB integrity (1, 16). For many years, it has been questioned if activation of astrocytes contributes to peptide clearance in parenchyma. The usage of primary cultures exposed to Aβ proved that they are capable of activating peptide uptake and presenting proteases to degrade it (20, 124, 223). However, it has been reported that astrocyte activation by Aβ causes an increase of intracellular Ca2+, ROS generation, and depletion of glutathione; this in turn promotes mitochondrial energy–redox dysfunction, leading to astrocyte apoptosis, and the oxidative stress generated by these cells impacts on neuronal viability (2 –4). In a recent study, it was noticed that Aβ causes hypometabolism of glucose, pyruvate, and lactate in human stem cell-derived cocultures of neurons and astrocytes; this could exacerbate energy dysfunction and oxidative damage (205).
Aβ-induced peripheral inflammation
Inflammation mediated by Aβ is not reduced only to a local response, but also the BBB has been extensively studied because of its relationship with peripheral inflammation (146). The barrier comprises a layer of endothelial cells linked by tight junctions and, surrounding them, the capillaries, the pericytes, and the end-feet of astrocytes give them support. Endothelial cells also have communication with microglia and neurons (219). Despite the fact that the BBB only allows diffusion of small and nonpolar molecules, it has been proposed that the proinflammatory mediators secreted as a consequence of glial cell activation could promote perivascular cell migration through the BBB (83, 201).
In general, disruption of the BBB favors the direct pass of Aβ and its deposition into blood vessels, originating cerebral amyloid angiopathy (CAA) in 80–90% of AD patients (226). The presence of Aβ vascular deposits induces an increase in systemic inflammation that has been linked with some clinical alterations in AD patients with CAA, such as hemorrhagic stroke and white matter abnormalities (73).
There is strong evidence of the fact that systemic inflammation represents a risk factor for the development of the cognitive decline associated with dementias, including AD (72). Chronic cardiovascular conditions, such as high LDL cholesterol, hypertension, diabetes, and low HDL cholesterol, have been linked with the rise of systemic inflammation and with the risk of AD development and progression (53, 202), which remarks the importance of focusing on chronic disorders that present these vascular alterations.
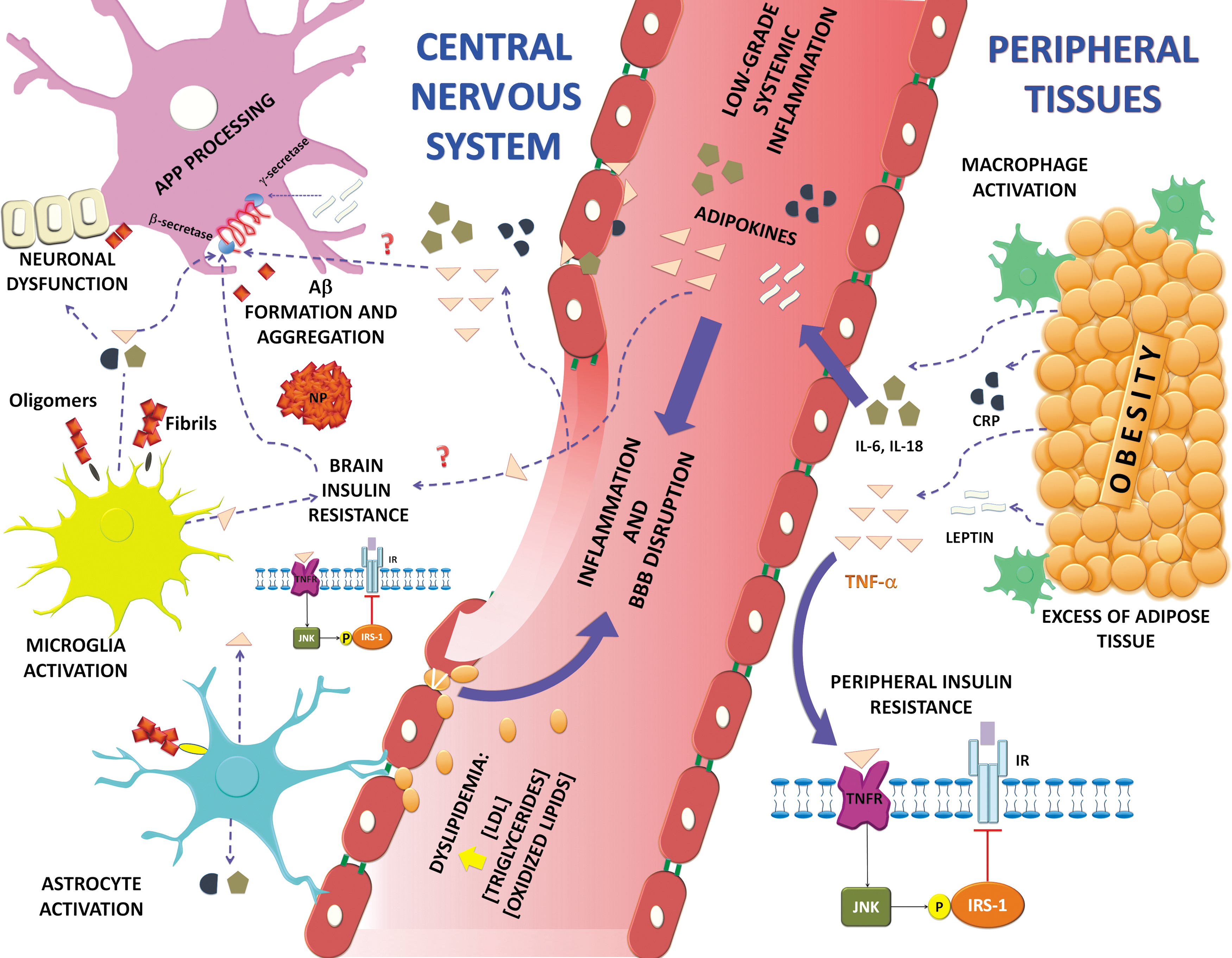
Link between Aβ-induced inflammation and metabolic syndrome
As we described above, MetS clusters a group of risk factors, such as abdominal obesity, dyslipidemia, hypertension, hyperglycemia, and I/R (106). The possible link between the presence of MetS factors and the susceptibility to develop AD and vascular dementia has been investigated (178, 218). Low-degree chronic inflammation could represent the linking point; in this way, a prospective cohort study in elderly people with MetS demonstrated a direct correlation within increased plasma or serum levels of C-reactive protein (CRP) and IL-6 with the presence of cognitive impairment (225).
It has been proposed that of all the MetS risk factors, obesity may play a major role in the development of low-degree systemic inflammation (148) and probably in the relationship between MCI and AD development (154). In conditions of obesity, the excess of adipose tissue promotes macrophage activation and subsequent release of adipokines into systemic circulation, including TNF-α, IL-6, IL-18, CRP, MCP-1, leptin, and resistin (167, 213).
Another feature that may be triggered by systemic inflammation originated from adipose tissue is I/R (104). Constant production of TNF-α in the periphery could induce the activation of inflammatory and stress kinases such as c-Jun N-terminal kinase, inhibitor κB kinases, and double-stranded RNA-dependent protein kinase. These kinases phosphorylate IRS-1 in the serine residues, which blocks downstream insulin signaling and promotes a decrease in glucose uptake favoring diabetes mellitus development (111, 158, 227), which is a component of MetS. I/R is also a pathological issue of AD that could impair neuronal survival and promote cognitive impairment (59). It has been reported that Aβ oligomers can activate TNF-α signaling and trigger subsequent activation of stress kinases that ultimately block insulin signaling (23).
Dyslipidemia is also an important component of I/R syndrome, and the major regulator of lipid metabolism is insulin. It reduces lipolysis and stimulates lipogenesis. It is the MetS condition that has been found in higher prevalence in AD subjects, who also present BBB impairment (25).
Conclusions
AD is a complex pathology that involves different aspects such as BBB disruption, local inflammation, oxidative stress, and impairments in brain insulin responsiveness, glucose utilization, and energy metabolism, as well as Aβ accumulation. In this way, the biochemical, molecular, and degenerative features of AD could correspond with the abnormalities that occur within metabolic disorders, suggesting a close relationship between both pathologies. Deregulating the insulin/IGF signaling pathway increases the formation of Aβ deposits, tau phosphorylation, and formation of reactive species; all features present in AD.
Aβ oligomers can bind to insulin receptors, triggering their internalization, decreasing neuron responsiveness to insulin, and promoting I/R. Furthermore, I/R alters AβPP metabolism, promoting its processing by β-secretase and thereby increasing Aβ formation. In addition, MetS risk factors confer chronic systemic low-grade inflammation that apparently could be linked with AD pathology. In the same way, Aβ oligomers could activate microglial cells and release proinflammatory mediators as cytokines (e.g., TNFα, IL-6, IL-1β, IL-18). Aβ oligomers can activate TNF-α signaling and trigger the posterior activation of stress kinases that finally block insulin signaling. As we can see, there are several evidences that demonstrate the fundamental role of amyloid and I/R in the development of both diseases.
Therefore, it can be assumed that both Aβ and I/R significantly influence the central and peripheral inflammatory responses that may eventually converge on a final common pathway leading to the neurodegenerative process associated with AD (Fig. 4).