
Abstract
Aims:
Circulating microparticles (MPs) from metabolic syndrome patients and those generated from apoptotic T cells induce endothelial dysfunction; however, the molecular and cellular mechanism(s) underlying in the effects of MPs remain to be elucidated.
Results:
Here, we show that both types of MPs increased expression of endoplasmic reticulum (ER) stress markers, X-box binding protein 1, p-eukaryotic translation initiation factor 2 α, and CHOP, and nuclear translocation of activating transcription factor 6 on human aortic endothelial cells (HAoECs). MPs decreased in vitro nitric oxide release by HAoECs, whereas in vivo MP injection into mice impaired the endothelium-dependent relaxation induced by acetylcholine. These effects were prevented when ER stress was inhibited, suggesting that ER stress is implicated in the endothelial effects induced by MPs. MPs affected mitochondrial function and evoked sequential increase of cytosolic and mitochondrial reactive oxygen species (ROS). Pharmacological inhibition of ER stress and silencing of neutral sphingomyelinase (SMase) with siRNA abrogated all MP-mediated effects. Neutralization of Fas ligand carried by MPs abolished effects induced by both MP types, whereas neutralization of low-density lipoprotein receptor on endothelial cells prevented T-lymphocyte MP-mediated effects.
Innovation and Conclusion:
Collectively, endothelial dysfunction triggered by MPs involves temporal cross talk between ER and mitochondria with respect to spatial regulation of ROS via the neutral SMase and interaction of MPs with Fas and/or low-density lipoprotein receptor. These results provide a novel molecular insight into the manner MPs mediate vascular dysfunction and allow identification of potential therapeutic targets to treat vascular complications associated with metabolic syndrome. Antioxid. Redox Signal. 26, 15–27.
Introduction
M
Among biological markers of endothelium injury, microparticles (MPs) have been involved in the pathogenesis and maintenance of cardiovascular, metabolic, and inflammatory diseases (9). Indeed, these small vesicles, released from plasma membrane of activated or apoptotic cells, carry proteins, nucleic acids, and lipids that can modify phenotype and function when delivered to target cells (21).
In the last years, an increasing interest on circulating microparticles (MPs), extracellular vesicles released from plasma membrane of cells, as players in the development and maintenance of cardiovascular diseases has been reported. Our study identifies a new pathway involving a spatiotemporal cross talk between the endoplasmic reticulum and mitochondria in the generation of oxidative stress accounting for the impairment of endothelial function induced by MPs from metabolic syndrome patients and allows identification of potential therapeutic targets to treat vascular complications associated with metabolic diseases.
In particular, we have previously described that MPs from both apoptotic T cells and from metabolic syndrome patients induced endothelial dysfunction characterized by a decrease of nitric oxide (NO•) release associated with the inhibition of endothelial nitric oxide synthase (eNOS) and an increase in oxidative and nitrative stresses in human endothelial cells (2, 25). In addition, when MPs from either apoptotic T cells or metabolic syndrome patients were injected into mice, an impairment of endothelium-dependent vasorelaxation in response to acetylcholine was observed illustrating their pathophysiological relevance (2, 25). However, the molecular and cellular mechanism(s) underlying the effects of MPs remain to be elucidated.
Several reports have shown that the activation of endoplasmic reticulum (ER) stress response plays an important role in the pathogenesis of metabolic disorders such as insulin resistance, type 2 diabetes, and obesity in animals (3, 26) as well as in humans (5). ER stress is induced by the accumulation of misfolded proteins in the ER lumen, which leads to the activation of unfolded protein response (UPR). UPR is initiated by three main ER transmembrane stress sensors: pancreatic endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 alpha (IRE1α), and activating transcription factor 6 (ATF6) (7). The UPR is initially activated as a prosurvival mechanism, however, prolonged UPR activation in conditions such as obesity and metabolic syndrome leads to apoptosis, oxidative stress, and inflammation and is referred to as the “ER stress response.”
Recently, Galán et al. (10) have demonstrated that chemical induction of ER stress with tunicamycin is associated with endothelial dysfunction and increased oxidative stress in the arterial wall. Under pathophysiological conditions such as hypertension, ER stress was reported to be involved in endothelial dysfunction induced by angiotensin II infusion (12).
Furthermore, several lines of evidence strongly support the link between mitochondrial dysfunction and cardiovascular complications associated with metabolic syndrome. In the skeletal muscle of type 2 diabetic patients, a downregulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha-responsive genes involved in oxidative phosphorylation has been described (24). Also, both basal and maximal adenosine diphosphate-stimulated respirations were decreased in type 2 patients (22, 28). Similar results have been observed in human cardiac muscle (23). Moreover, an important increase in ER-mitochondria contact sites via the mitochondria-associated membranes (MAMs) has been described during high-fat diet-induced obesity leading to a significant increase in hepatic lipid accumulation and decreased mitochondrial oxidative function (3).
In parallel, elevated reactive oxygen species (ROS) production in the cytosol, through the overexpression of the NADPH isoform NOX4, and in mitochondria, has been detected in smooth muscle aortic cells from atherosclerotic aged donors leading to impaired mitochondrial function (32).
The aim of the present study is to decipher how MPs transfer their message to induce endothelial dysfunction. Detailed analyses of the mechanisms involved with special interest in the cross talk between ER and mitochondria in the regulation of oxidative stress were conducted. This would help underpinning new molecular and cellular pathways governing the vascular effects of MPs and will pave the way for identifying novel potential targets against MP-induced endothelial dysfunction, which predisposes to the initial proatherosclerotic events transforming metabolic syndrome to different vascular diseases.
Results
T-lymphocyte MPs induce endothelial dysfunction through ER stress–response activation
In response to treatment by MPs from apoptotic lymphocytes, the UPR pathway activation was assessed using the ER stress inhibitor tauroursodeoxycholic acid (TUDCA), a chemical chaperone known to enhance adaptive response of ER. MPs activated the three canonical pathways associated with ER stress response: PERK, IRE1α, and ATF6. Indeed, MPs resulted in an increased phosphorylation of PERK and eukaryotic translation initiation factor 2 α (eIF2α), as well as mRNA (not shown) and protein CHOP expressions (Fig. 1A–C). In addition, MP treatment enhanced X-box binding protein 1 (XBP1) splicing downstream to IRE1α (Fig. 1D) and induced the nuclear translocation of ATF6 (Fig. 1E). Importantly, all these MP-evoked effects were prevented in the presence of the ER stress inhibitor TUDCA (Fig. 1A–E).
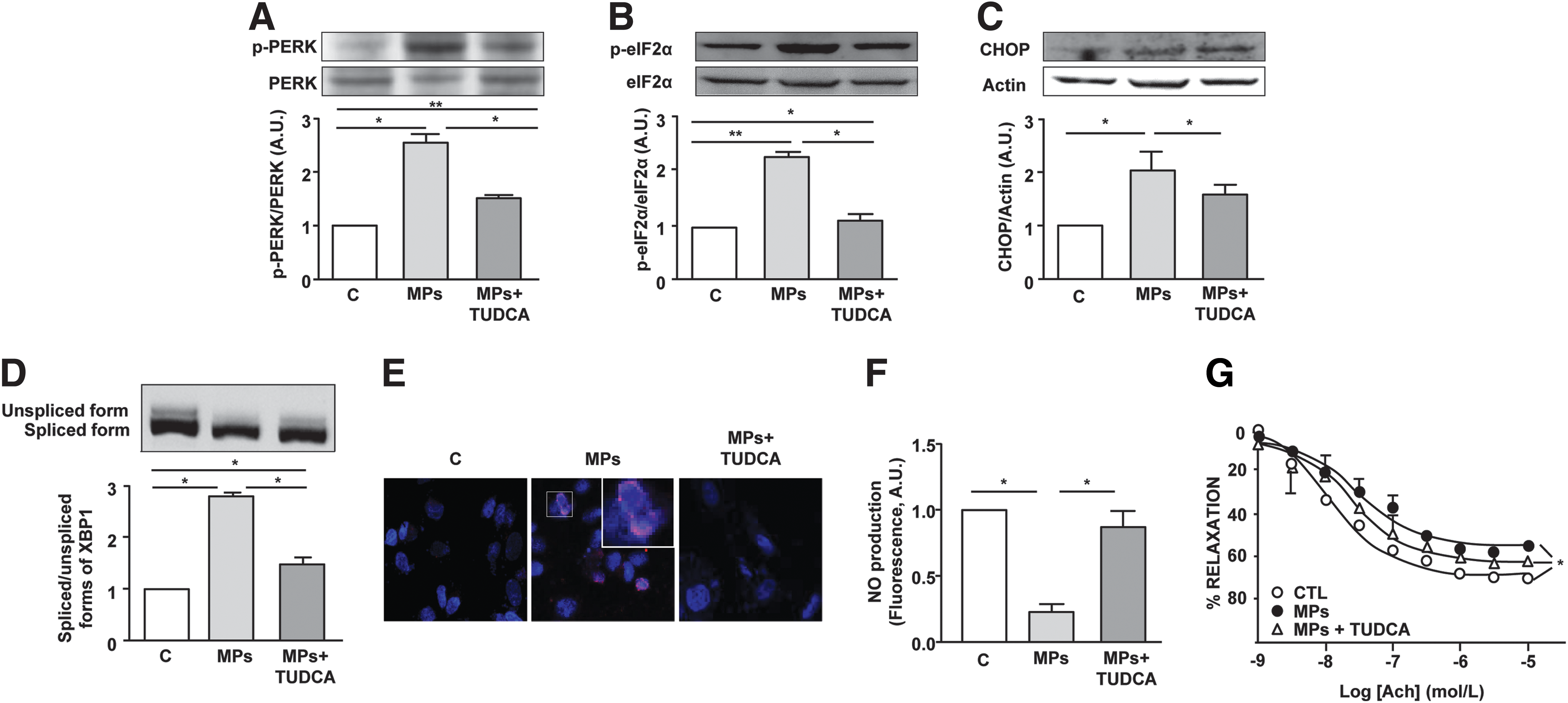
It should be noted that the ER stress inducer tunicamycin was able to activate the same three classical ER stress pathways, which were partially prevented by TUDCA (Supplementary Fig. S1; Supplementary Data are available online at
To verify whether ER stress–response activation accounts for the endothelial dysfunction induced by T-lymphocyte MPs (25), in vitro NO• production was evaluated in the absence or presence of TUDCA. As previously described, T-lymphocyte MPs significantly decreased NO• production in human aortic endothelial cells (HAoECs) (Fig. 1F). Interestingly, the inhibition of ER stress with TUDCA abolished the ability of MPs to reduce NO• production.
To establish the pathophysiological relevance of MPs, endothelium-dependent relaxation in response to acetylcholine was evaluated following MP incubation with mouse aortic rings in the absence or presence of TUDCA. MPs significantly reduced the maximal acetylcholine-evoked relaxation, which was improved by TUDCA (Fig. 1G). Finally, similarly to MPs, tunicamycin was able to reduce both in vitro NO• release by endothelial cells and acetylcholine-induced relaxation of mouse aortic rings. These effects were partially prevented in the presence of TUDCA (Supplementary Fig. S1).
Collectively, these results indicate that the activation of ER stress response is implicated in endothelial dysfunction induced by T-lymphocyte MPs.
Involvement of membrane receptors in T-lymphocyte MP-induced ER stress in endothelial cells
We have previously shown that T lymphocyte MPs, through their Fas ligand (FasL), induce vascular smooth muscle cell inflammation by directly interacting with Fas of the recipient cells (31). Also, Yang et al. (33) have reported the involvement of low-density lipoprotein receptor (LDL-R) in the effects induced by T-lymphocyte MPs on human retinal endothelial cells. To determine the mechanism(s) by which MPs would induce ER stress in HAoECs, the implication of both Fas/FasL interaction and LDL-R was analyzed on MP-induced eIF2α phosphorylation. Interestingly, we observed that neutralizing either FasL on MPs or LDL-R on endothelial cells significantly reduced the MP-induced phospho-eIF2α (Fig. 2A). Altogether, these data indicate that both Fas/FasL interaction and LDL-R pathway are involved in MP-mediated ER stress induction.
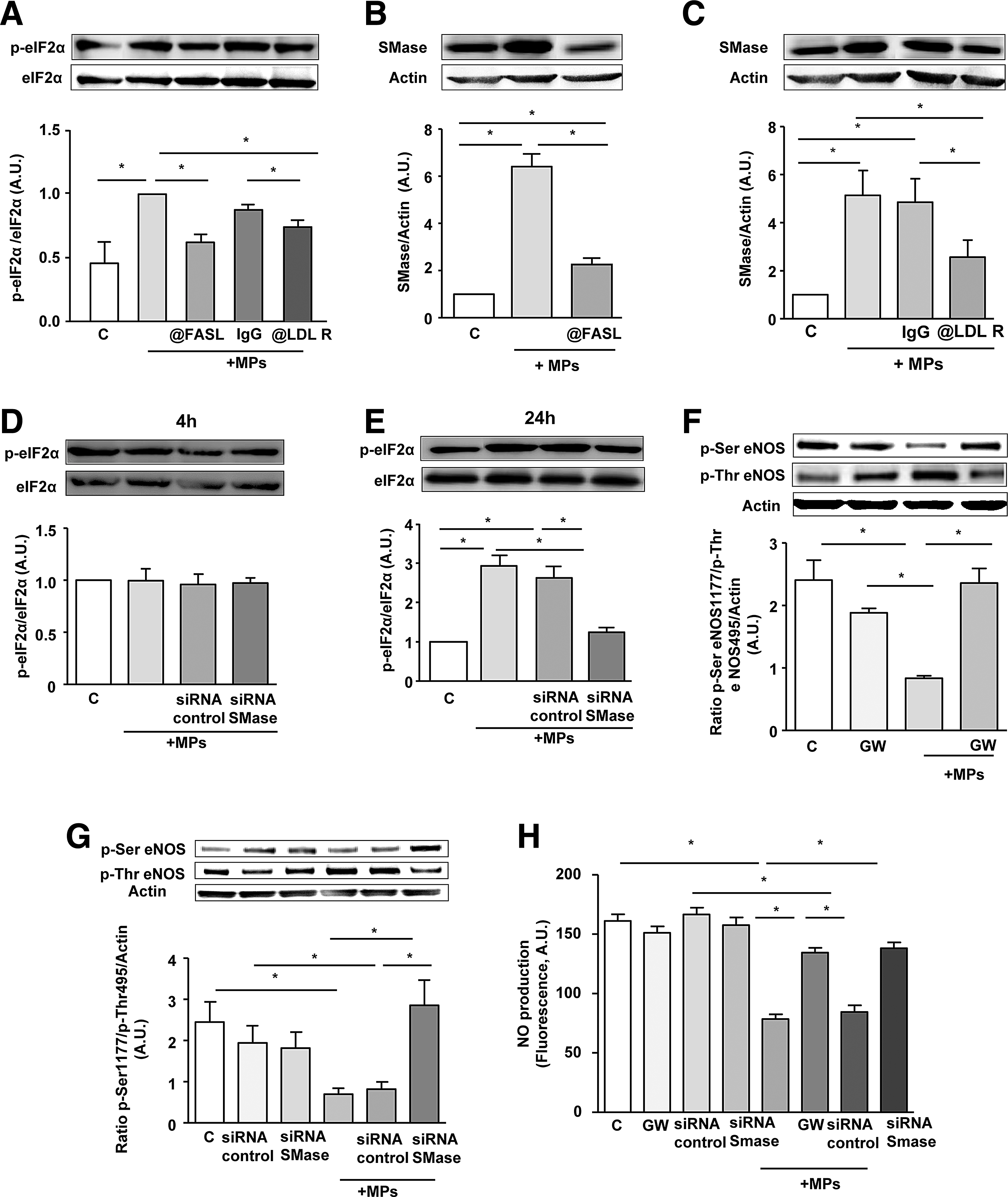
Neutral sphingomyelinase is the link between MP-activated receptor and ER stress–response induction
Since sphingomyelinase (SMase) cascade has been implicated in the regulation of ER stress response (15), we assessed the involvement of SMase on MP-induced effects. MP treatment induced an increase of neutral SMase expression, which was prevented after neutralizing FasL on MPs (Fig. 2B) or when LDL-R on endothelial cells was blocked (Fig. 2C). Treatment of endothelial cells by MPs for 24 h, but not for 4 h, increased phospho-eIF2α. Interestingly, silencing neutral SMase with a specific siRNA abolished MP-induced increase of eIF2α phosphorylation after 24 h of MP treatment (Fig. 2D, E).
Moreover, using either the specific neutral SMase inhibitor GW4869 or siRNA against neutral SMase completely prevented the MP-induced reduction of eNOS activity as reflected by the phospho-Ser eNOS/phospho-Thr eNOS ratio (Fig. 2F, G). Furthermore, MP-induced NO• reduction was abolished when neutral SMase was inhibited with GW4869 or silenced by siRNA (Fig. 2H). Altogether, these results suggest that the neutral SMase pathway controls the effects of MPs via Fas/FasL and LDL-R interaction upstream of ER stress, eNOS activation, and NO• production.
Cytosolic ROS production is upstream of T-lymphocyte MP-induced ER stress in endothelial cells
The possible involvement of oxidative stress in T-lymphocyte MP-induced ER stress activation was then assessed by incubating HAoECs with various inhibitors, the sources of ROS before MP stimulation. Indeed, intracellular enzymatic systems with the capacity to generate ROS were inhibited by allopurinol, apocynin, rotenone, or Nω-nitro-
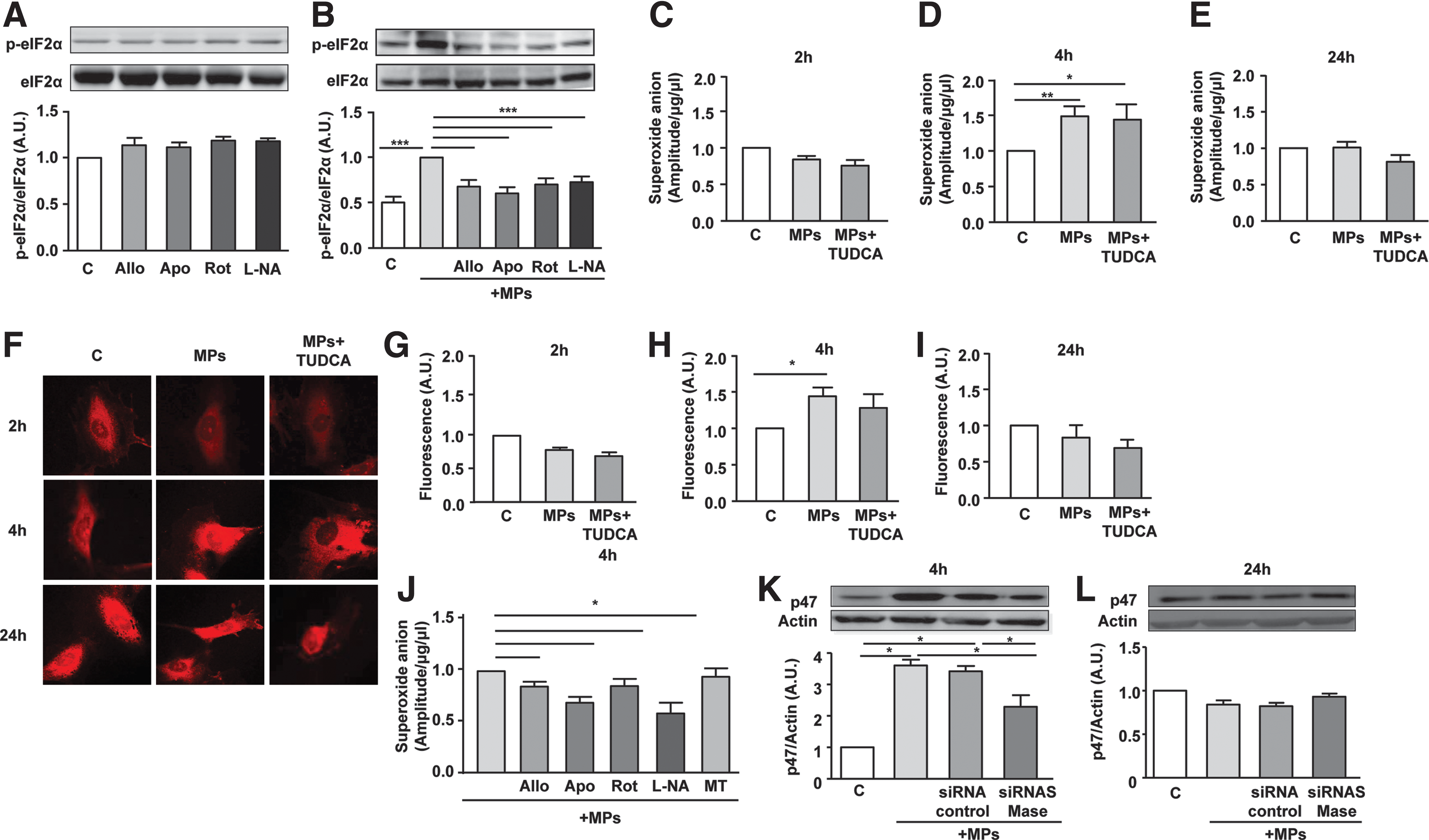
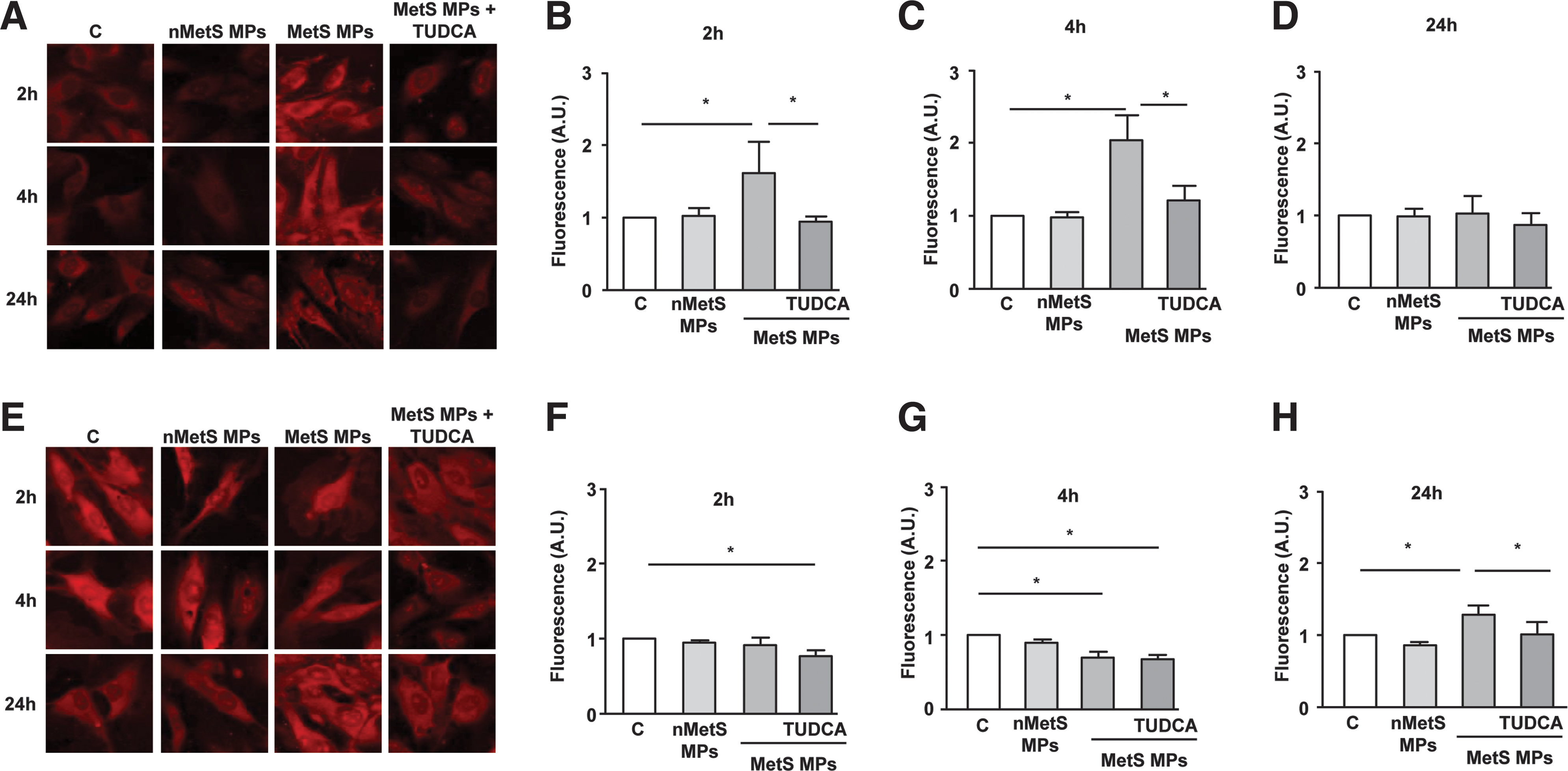
To establish the implication of cytoplasmic and mitochondrial ROS in the effects of MPs, time course (i.e., 2, 4, and 24 h) changes of ROS levels were measured. Analysis of ROS levels, using electronic paramagnetic resonance (EPR) and dihydroethidium (DHE) labeling, showed that T-lymphocyte MPs had no effect on ROS production at 2 h, increased at 4 h, and returned to basal levels at 24 h (Fig. 3C–I). This effect of T-lymphocyte MPs on ROS production was not affected by TUDCA. It is important to note that tunicamycin treatment induced similar patterns of ROS production as those obtained with MPs (Supplementary Fig. S2).
Interestingly, except for the mitochondrial ROS scavenger (mito-TEMPO), all of the ROS inhibitors tested (allopurinol, apocynin, rotenone, and
Altogether, lymphocyte MPs activate neutral SMase and induce an early increase of cytosolic ROS production, upstream of ER stress activation via at least the NADPH oxidase activation.
Cross talk between ER and mitochondria is involved in the effects induced by lymphocyte MPs in endothelial cells
To monitor mitochondrial ROS production, the MitoSOX probe was used. Quantification of confocal images showed that mitochondrial ROS fluorescence was reduced at 2 and 4 h, but was greatly enhanced after 24 h of lymphocyte MP treatment (Fig. 4A–D). Interestingly, the ER stress inhibitor TUDCA did not modify the reduction of MitoSOX fluorescence at 2 and 4 h but completely abolished the increase at 24 h. These results suggest that lymphocyte MPs induce a late increase of mitochondrial ROS by a mechanism downstream of ER stress activation. To confirm this, the mitochondrial ROS scavenger mito-TEMPO was used and did not show any effect on the increase of phospho-eIF2α induced by lymphocyte MPs (Fig. 4E).
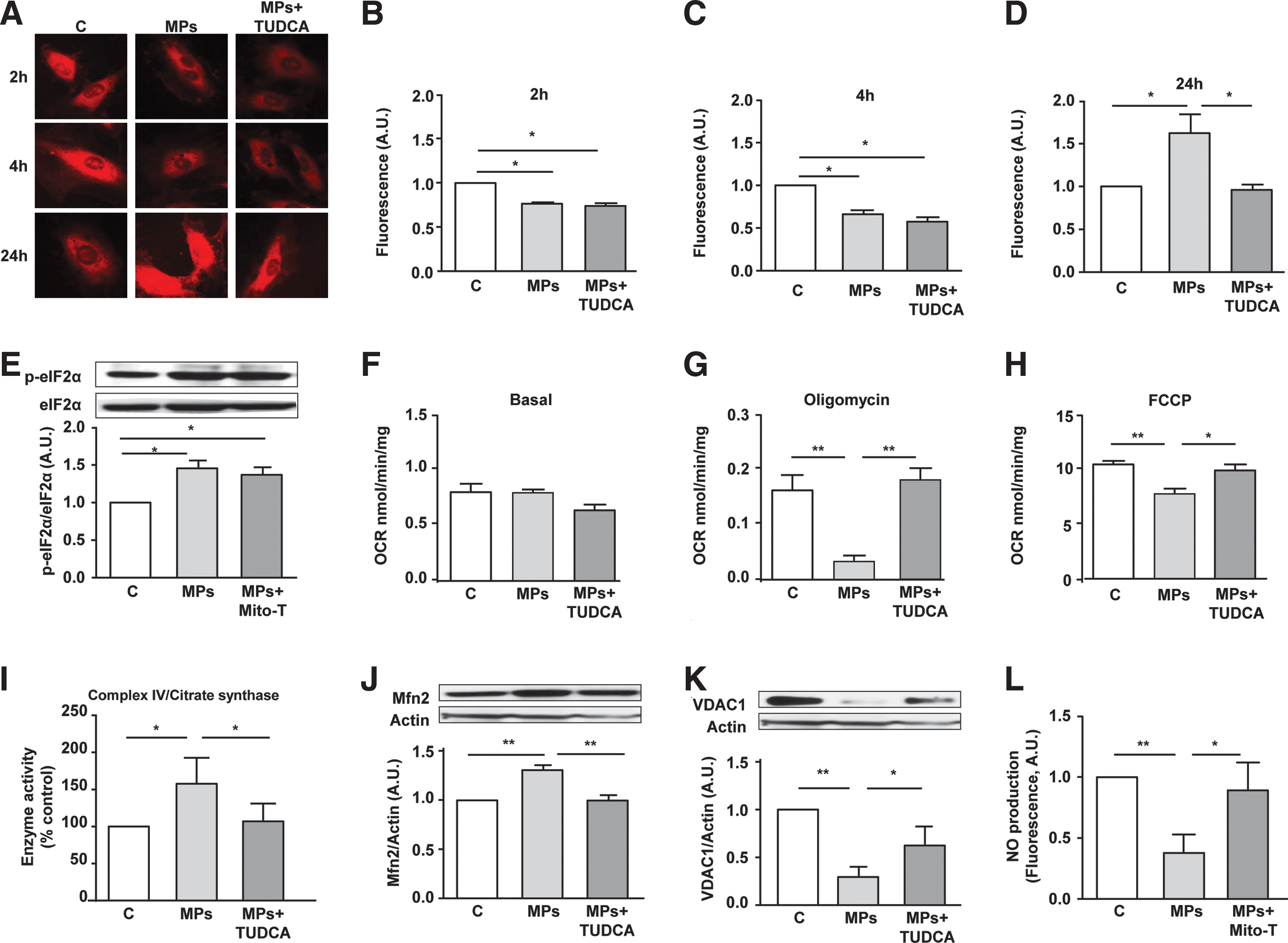
The effects of MPs on mitochondrial O2 consumption were then analyzed to decipher the role of mitochondria on the MP-induced endothelial dysfunction. Basal O2 consumption was not modified either by MPs or MPs + TUDCA (Fig. 4F). However, MP treatment significantly reduced both oligomycin and carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) mitochondrial respirations, which were abolished when ER stress was inhibited with TUDCA (Fig. 4G, H). These findings suggest that MPs are able to modify mitochondrial respiration by a mechanism implicating ER stress. Exposure to MPs resulted in an increase of complex IV activity (Fig. 4I) but had no effect on the activity of the other complexes of the electron transport chain (not shown).
To confirm the interaction between ER and mitochondria through the formation of tight physical junctions, MAM integrity was analyzed following MP treatment. As shown in Figure 4J and K, MPs induced MAM disruption characterized by an overexpression of mitofusin 2 (Mfn2) and a decreased expression of voltage-dependent anion channel 1 (VDAC1). Moreover, these effects were prevented in the presence of the ER stress inhibitor TUDCA, suggesting that MP-induced ER stress leads to MAM disruption on endothelial cells. Finally, to verify whether the MP effects on mitochondria lead to endothelial dysfunction, NO• production was measured in the presence of mito-TEMPO. Scavenging mitochondrial ROS with mito-TEMPO abolished the decrease in NO• release induced by MPs (Fig. 4L). This result suggests that alteration of mitochondrial function with respect to respiration and ROS production, as a consequence of ER stress, leads to endothelial dysfunction.
MPs from metabolic syndrome patients induce endothelial dysfunction via Fas/FasL pathway and neutral SMase activation leading to ER stress activation in vitro and ex vivo
We have previously described that MPs from metabolic syndrome patients, but not those of nonmetabolic syndrome subjects, induced endothelial dysfunction (2). In this study, we tested whether molecular pathways involved in the effects of MPs from metabolic syndrome required also the cross talk between ER and mitochondria and oxidative stress similarly to lymphocyte MPs. Therefore, MPs from metabolic syndrome patients or nonmetabolic syndrome subjects were isolated, for whom detailed clinical information is described in Supplementary Table S1. As expected, metabolic syndrome patients showed greater visceral obesity as given by waist circumference, enhanced triglyceridemia, and increased blood pressure.
The total number of circulating MPs was significantly increased in patients with metabolic syndrome compared to nonmetabolic syndrome subjects (Supplementary Table S2). Phenotypical characterization of cellular origin of MPs showed an increase of ∼6-fold (annexin V+), ∼3.3-fold (CD41+), and ∼9.6-fold (CD146+) of the circulating level of procoagulant-, platelet-, and endothelial-derived MPs in metabolic syndrome patients compared to nonmetabolic syndrome subjects, respectively. Also, levels of erythrocyte- and leukocyte-derived MPs (CD235a+, CD45+) were elevated (∼9- and ∼2.7-fold, respectively) in these patients.
As shown in Figure 5A and B, MPs from metabolic syndrome patients, but not from nonmetabolic syndrome individuals, induced an increase in phosphorylation of eIF2α and nuclear translocation of ATF6, which were prevented in the presence of TUDCA. Similarly and as expected, MPs from metabolic patients, in comparison to nonmetabolic syndrome subjects, caused a reduction in NO• release from endothelial cells, which was abolished when ER stress was inhibited by TUDCA (Fig. 5C). Moreover, incubation of mouse aortic rings with MPs from metabolic syndrome patients, but not from nonmetabolic syndrome subjects, was associated to a reduced endothelium-dependent relaxation in response to acetylcholine (Fig. 5D). This effect was prevented when vessels were incubated in the presence of TUDCA.
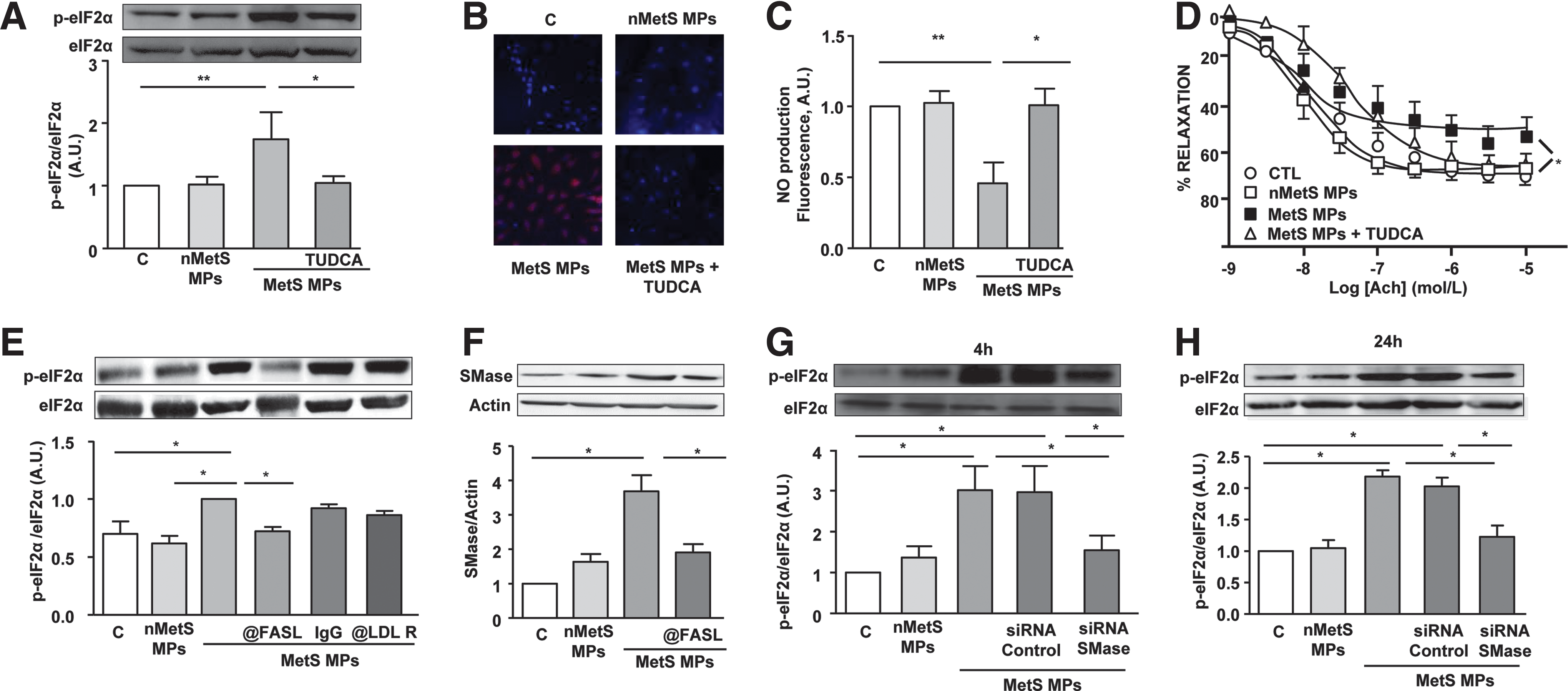
Taken together, these results indicate that ER stress plays a key role in the induction of endothelial dysfunction induced by MPs from metabolic syndrome patients.
To establish the receptor involved in the effects of MPs from metabolic syndrome patients on endothelial cells, we neutralized either FasL on MPs or LDL-R on HAoECs. Interestingly, neutralization of FasL but not LDL-R on HAoECs, prevented the phosphorylation of eIF2α by metabolic syndrome MPs (Fig. 5E) and abolished their ability to enhance the expression of SMase (Fig. 5F). Finally, it is important to note that silencing neutral SMase with siRNA significantly abolished the increase of eIF2α phosphorylation by metabolic syndrome MPs both at early (4 h) and late stage (24 h) of treatment (Fig. 5G, H), in contrast to T-lymphocyte MPs that were able to increase eIF2α at 24 h only (Fig. 2E).
Cytosolic and mitochondrial ROS productions are downstream of metabolic syndrome MP-induced ER stress in endothelial cells
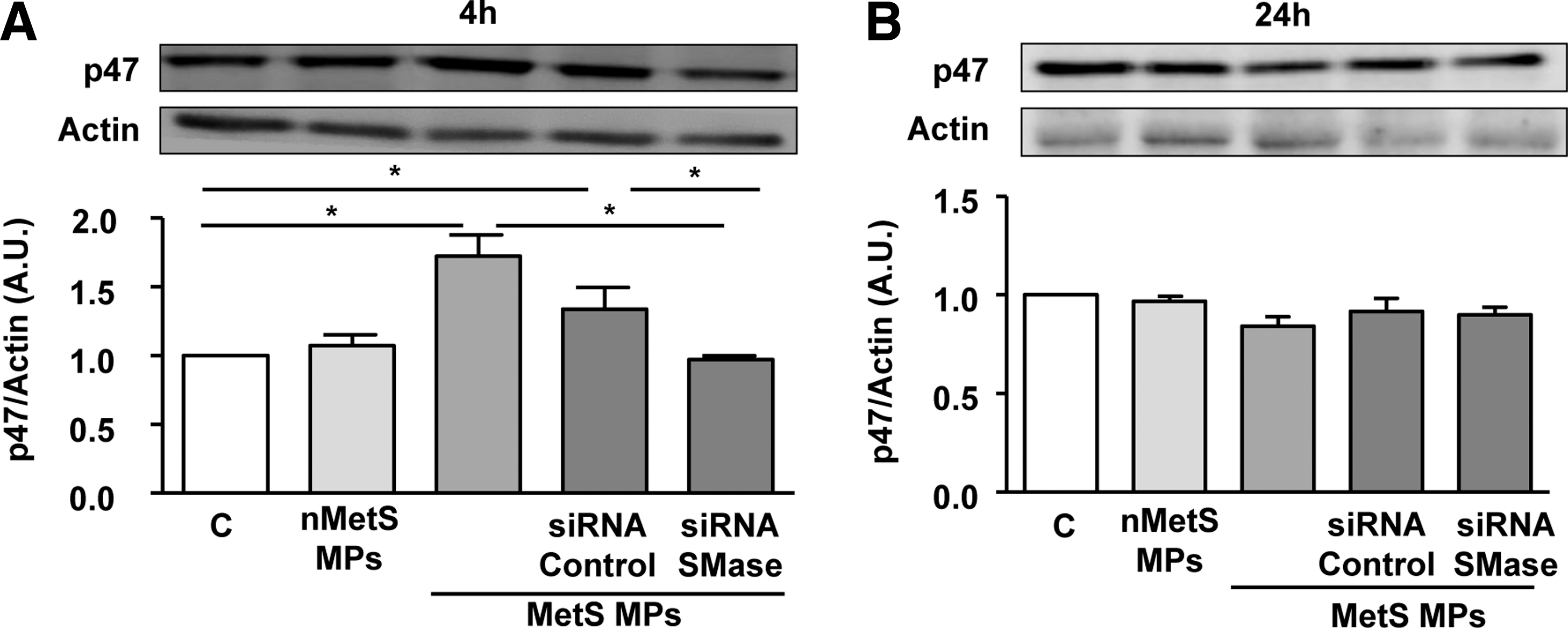
As illustrated in Figure 6A–H, MPs from metabolic syndrome induced oxidative stress by increasing both cytosolic and mitochondrial ROS. DHE staining showed that MPs from metabolic syndrome patients, in comparison to nonmetabolic syndrome subjects, enhanced cytosolic ROS at 2 and 4 h, but not at 24 h (Fig. 6A–D). Also, MPs from metabolic syndrome patients but not those from nonmetabolic syndrome subjects increased MitoSOX fluorescence at 24 h, but not at 2 or 4 h of treatment of HAoECs (Fig. 6E–H). Cytosolic ROS, but not mitochondrial ROS production, was abolished in the presence of ER stress inhibitor TUDCA. Interestingly, MPs induced p47phox expression at 4 h, but not at 24 h, which was abolished after SMase silencing (Fig. 7A, B). These results suggest that MPs from metabolic syndrome patients activate neutral SMase and induce a spatiotemporal increase of cytosolic ROS at early stage via the NADPH oxidase that concur to ER stress and mitochondrial ROS at late stages of stimulation. All of these effects participate in metabolic syndrome MP-induced endothelial dysfunction.
Discussion
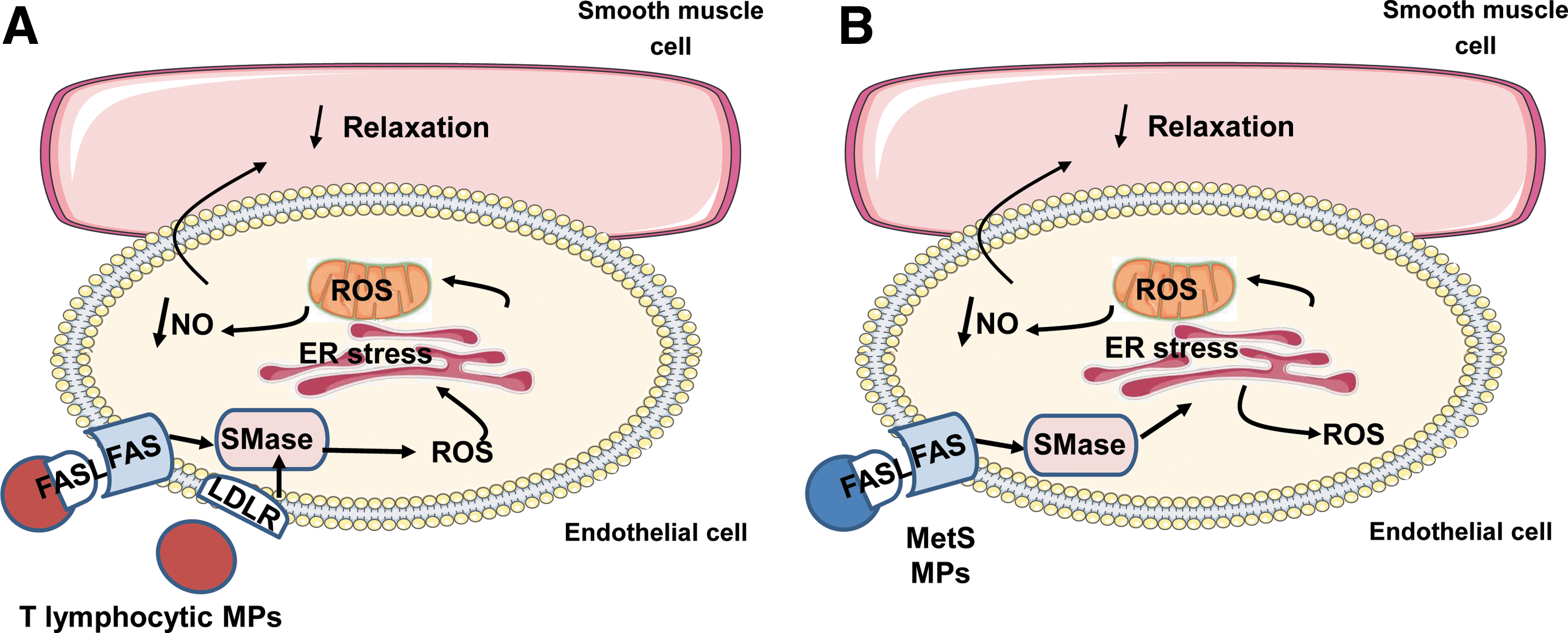
In the present study, we provide evidence to support the crucial role of ER and mitochondria interaction in the deleterious effects of human MPs on endothelial function. In detail, MPs from apoptotic T cells act on Fas and LDL-R; then, via neutral SMase cascade induce cytosolic ROS production that activates ER stress. Through the interaction between ER and mitochondria, mitochondrial ROS are increased and contribute to decreased bioavailability of NO• and the subsequent impairment of endothelium-dependent vasorelaxation. Concerning MPs from metabolic syndrome patients, Fas/FasL, but not LDL-R, participates in the response induced by MPs. In addition, neutral SMase activation induces directly ER stress which, in turn, increases both cytosolic and mitochondrial ROS. All of these events lead to reduction of NO• release and the subsequent impairment of endothelium-dependent vasorelaxation.
MPs have been described as cargo to deliver biological information between cells. We have previously described that MPs from either apoptotic T cells or metabolic syndrome patients induce endothelial dysfunction through the inhibition of eNOS (2, 25). In the present study, we show that both types of MPs induce endothelial dysfunction by ER stress activation. Thus, in a similar manner to tunicamycin, MPs activate the three canonical UPR pathways, as illustrated by the increased phosphorylation of PERK and eIF2α, XBP1 splicing, and nuclear ATF6 translocation. ER stress activation has been described in endothelial cells of obese patients (11) and after an intralipid infusion in endothelial cells from healthy subjects (30), suggesting that metabolic disorders can lead to ER stress that may be relevant to vascular diseases.
In the present study, the involved mechanisms in the ER induction of MPs from T cells and those from metabolic syndrome patients were found to be slightly different, probably due to the differences between MP origins. Indeed, we show that metabolic syndrome patients display elevated levels of platelet- and nonplatelet-derived MPs, mainly from leukocytes, erythrocytes, and endothelial cells. In this respect, we have previously shown that the effects of metabolic syndrome MPs on endothelial cells are probably supported by nonplatelet-derived MPs, since this subset of MPs reduces NO• production (2). In addition, Chironi et al. (6) have found that circulating levels of leukocyte-derived MPs were higher in metabolic syndrome patients than in healthy individuals and that these levels positively correlate with the number of metabolic syndrome components.
MPs carry proteins, lipids, and nucleic acids, mainly mRNA and miRNA. Since RNAse treatment has no effect in MP-induced responses on HAoECs (not shown), we decided to analyze the direct interaction between MPs and target cells via the ligand–receptor binding. We have previously shown that interaction of FasL carried by MPs with Fas at the surface of target cells mediates the induction of proinflammatory proteins in the vascular wall (1, 31), whereas Yang et al. (33) demonstrated the involvement of LDL-R in the MP-induced effects.
In this study, we show that both mechanisms participate to the effects of MPs from T cells, but only Fas/FasL interaction is involved in the effects generated by MPs from metabolic syndrome patients. Indeed, neutralization of LDL-R on endothelial cells does not affect metabolic syndrome MP-induced ER stress. Nevertheless, both types of MPs are able to activate neutral SMase, which has been directly related to endothelial dysfunction [for review see ref. (27)]. In addition, we provide proof that inhibition of neutral SMase, or silencing its expression, strongly blocks MP-induced ER stress and improves both eNOS activity and NO• production, illustrating the implication of neutral SMase pathway on MP-induced endothelial dysfunction.
Nonetheless, intracellular pathways involved in the effects of both types of MPs show differential spatial and temporal regulation with respect to cytosolic and mitochondrial pools of ROS. Activation of ER stress by MPs from T cells is dependent on ROS production from different origins since inhibitors of NADPH oxidase, xanthine oxidase, and mitochondrial complex I prevented phosphorylation of eIF2α, which suggests a link between the different ROS sources. Thus, it has been proposed that NADPH oxidase plays a central role in orchestrating the activation and dysfunction of other enzymes generating ROS [for review see ref. (14)]. In addition, the early increase (at 4 h of treatment) on cytosolic ROS production evoked by MPs from apoptotic T cells was not inhibited by TUDCA, indicating that the enhancement of cytosolic ROS is upstream to ER stress.
Interestingly, silencing SMase partially prevented the increase of p47phox subunit of NADPH oxidase induced by MPs at 4 h, suggesting that SMase activation precedes NADPH oxidase-dependent deleterious effects of MPs in endothelial cells. The SMase-p47phox-ROS cascade has been described in vascular smooth muscle cells under hypoxia conditions (8) and in neurons exposed to tumor necrosis factor-alpha (4), however, to the best of our knowledge, the relationship of this cascade with ER stress has never been studied. Furthermore, several studies showed that cellular toxics such as arsenic or ischemia/reperfusion injury exert their cytotoxicity by inducing apoptosis through ROS-induced ER stress, which is associated to a mitochondrial dysfunction (29, 34).
In the present study, we provide evidence that mitochondrial ROS increase, changes on mitochondrial respiration and complex IV activity are detected at late (24 h) but not early (4 h) MP treatment, and are abolished when ER stress is inhibited, indicating that ER stress activation is mandatory for the generation of mitochondrial ROS and perturbation of mitochondrial function. Remarkably, scavenging of mitochondrial ROS by mito-TEMPO restores NO• production, illustrating the involvement of mitochondrial ROS on the MP-induced endothelial dysfunction. Moreover, the enhanced MAM alteration, which suggests an interaction between ER stress and mitochondria, could account for the subsequent modifications of the mitochondrial function. It is worth noting that ER and mitochondrial cross talk have been described in neurodegenerative (13) and metabolic disorders (3, 18), suggesting that rescuing mitochondrial and ER stress impairments may identify new targets against these pathologies.
In this respect, activation of SMase pathway by MPs from metabolic syndrome patients directly induces ER stress, which in turn evokes an increase of both cytosolic and mitochondrial ROS. It is important to note that the spatiotemporal regulations of ROS induced by MPs from metabolic syndrome patients are different from those of T-lymphocytic MPs. In fact, with respect to the increase of cytosolic ROS, ER stress activation is downstream for the former and upstream for the latter. This differential response probably results from the fact that a mixture of MP subsets triggers the effects of metabolic syndrome MPs although we previously reported that nonplatelet MPs are responsible for endothelial dysfunction. Indeed, it is well known that the content of MPs depends on the cells they originate from, the stimulus of production, and the mechanism of vesicle generation (9).
The difference in the mechanism involved in ER stress response induced by T-lymphocytic MPs and those from metabolic syndrome patients might explain the different level of endothelial dysfunction induced by both types of MPs. We have no evidence of any NOS uncoupling nor diminished levels of tetrahydrobiopterin in the present study, but it is most likely that reduced acetylcholine-induced relaxation is due to spatiotemporal increase of oxidative stress that leads to reduced NO• production. However, we cannot exclude that mechanisms other than NO• pathway, such as cyclooxygenase pathways, might be involved.
Nevertheless, the present study emphasizes the importance of ER stress as a key control of oxidative stress leading to endothelial dysfunction by both types of MPs. In this respect, it has been described that the presence of unfolded proteins in the ER lumen is sufficient to activate oxidative stress [for review see ref. (19)]. This process may be related to the fact that ER stress induction by MPs from metabolic syndrome patients takes place early, only after 4 h of MP treatment.
Collectively, we demonstrate that endothelial dysfunction triggered by MPs involves temporal cross talk between ER and mitochondria with respect to spatial regulation of oxidative stress via the SMase route (Fig. 8). These events occur via interaction of MPs on endothelial cells with Fas and/or LDL-R. These results highlight novel potential targets to fight against the pivotal role of MPs on endothelial dysfunction leading to the increase of cardiovascular complications, including those associated with metabolic syndrome.
Materials and Methods
MP production from T cells
The human lymphoid CEM T-cell line was used for MP production. Cells were seeded at 106 cells/ml and cultured in serum-free X-VIVO 15 medium (Lonza). MPs were produced as previously described (20, 25). Briefly, cells were treated with actinomycin D (1 μg/ml; Sigma-Aldrich) for 24 h. The supernatant was obtained by centrifugation at 750 g for 15 min and then at 1500 g for 5 min to remove cells and large debris, respectively. MPs from the supernatant were washed after three centrifugation steps (45 min, 14,000 g) and recovered in 400 μl of sterile NaCl 0.9%. Last, a washing medium was used as vehicle. The determination of the amount of MPs was conducted by measuring MP-associated proteins using the DC Protein Assay (Bio-Rad). MPs were used at 10 μg/ml corresponding to 1330 ± 312 × 103 MPs. This concentration of lymphocyte MPs is found in plasma from patients undergoing carotid endarterectomy (0–2 × 106 MPs/ml plasma) (16).
Metabolic syndrome patients
This study was approved by the Ethics Committee of the University Hospital of Angers (France; NCT: 00997165). A total of 30 patients were included with metabolic syndrome from the METABOL cohort at the Department of Endocrinology and Nutrition of the University Hospital of Angers. Patients were eligible for inclusion, according to the National Cholesterol Education Program-Adult Treatment Panel III, when they had at least three of the following five criteria: (i) waist circumference >102 or 88 cm for men and women, respectively; (ii) systolic and diastolic pressures ≥130/85 mmHg; (iii) fasting glycemia ≥1.1 g/l; (iv) triglycerides ≥1.5 g/l; and (v) high-density cholesterol lipoprotein <0.4 g/l in men or <0.5 g/l in women (Supplementary Table S1). Patients with a history of cardiovascular diseases, preexistent chronic inflammatory disease, and cancer were excluded. Normal controls consisted of 20 subjects who met less than two of the metabolic syndrome criteria (70% without any component of metabolic syndrome).
MP collection from patients
Peripheral blood (20 ml) from nonmetabolic syndrome or metabolic syndrome subjects was collected in ethylenediaminetetraacetic acid tubes (Vacutainers; Becton Dickinson) from a peripheral vein using a 21-gauge needle to minimize platelet activation and was processed within 2 h. After a 20-min centrifugation at 270 g, platelet-rich plasma was separated from whole blood, which was then centrifuged for 20 min at 1500 g to obtain platelet-free plasma (PFP). Two hundred microliters of PFP was frozen and stored at −80°C until subsequent use for MP characterization by flow cytometry. Remaining PFP was subjected to three series of centrifugations each at 21,000 g for 45 min to pellet MPs for in vitro studies, and the supernatant was replaced by 200 μl of sterile 0.9% NaCl and stored at 4°C until subsequent use. MPs from nonmetabolic syndrome or metabolic syndrome subjects were used at the circulating concentration detected for each individual, as previously described (1, 2).
Characterization of MP phenotype
Membrane MP subpopulations were discriminated in PFP according to the expression of membrane-specific antigens (1, 2). Irrelevant human immunoglobulin G (IgG) was used as an isotype-matched negative control for each sample. For numeration studies, 10 μl of PFP was incubated with 5 μl of specific antibody (Beckman Coulter). Annexin V binding was used to numerate phosphatidylserine-expressing circulating MPs (2 μl of annexin V/5 μl PFP). After 30 min at room temperature, samples were diluted in 300 μl of sterile 0.9% NaCl or annexin-V labeling buffer, respectively. Then, an equal volume of sample and Flow-Count beads was added and samples were analyzed in a flow cytometer 500 MPL system (Beckman Coulter).
Cell culture
HAoECs were maintained in culture in an endothelial cell growth medium MV2 (PromoCell) supplemented with 1% streptomycin/penicillin (Lonza). Cells were treated for 24 h with ER stress inducer tunicamycin (0.8 μg/ml; Sigma-Aldrich), 10 μg/ml of T-lymphocyte MPs, or those from nonmetabolic syndrome or metabolic syndrome subjects, in the absence or presence of the ER stress inhibitor, TUDCA (100 μmol/l; Sigma-Aldrich). In another set of experiments, 30 min before MP treatment, cells were pretreated with the NADPH oxidase inhibitor, apocynin (Apo, 100 μmol/l; Calbiochem), the inhibitor of xanthine oxidase, allopurinol (Allo, 50 μmol/l; Sigma-Aldrich), the inhibitor of mitochondrial complex I NADH dehydrogenase, rotenone (Rot, 5 μmol/l; Sigma-Aldrich), the NO• synthase inhibitor,
Western blot
After treatment, cells were homogenized and lysed. Proteins (40 μg) were separated on 4–12% NuPAGE gels (ThermoFisher Scientific). Blots were probed with antibodies against PERK, phospho-PERK, eIF2α, phospho-eIF2α, CHOP, phospho-Thr495 eNOS (Cell Signaling), Mfn2, VDAC1α (Abcam), p47-phox, and phospho-Ser1177 eNOS (BD Biosciences). A polyclonal mouse anti-human β-actin antibody (Sigma-Aldrich) was used for standardization of protein gel loading.
In another set of experiments, cells were pretreated with the LDL-R antibody (5 μg/ml; R&D Systems) or its control IgG antibody for 30 min before MP treatment. Also, FasL carried by MPs was preincubated with human anti-FasL (5 μg; BD Biosciences) for 30 min at 4°C to allow neutralization of MP FasL, after being washed two times with NaCl 0.9% to remove unbound anti-FasL antibody.
Nuclear translocation of ATF6 by confocal microscopy
Cells were seeded on glass slides (Ibidi) for 24 h and then treated with either tunicamycin or MPs for 24 h in the absence or presence of TUDCA. Cells were then washed and fixed with 4% paraformaldehyde. Then, cells were treated with 5% bovine serum albumin, permeabilized by 0.1% Triton, incubated with anti-ATF6 (Abcam) for 1 h, followed by a 1-h incubation with the Alexa fluor-546 secondary antibody (ThermoFisher Scientific) at room temperature. Finally, 4′,6-diamidino-2-phenylindole (DAPI, 300 mmol/l; Santa Cruz Biotechnology) was added for 5 min. Cells were washed and fluorescence was measured with a confocal microscopy (CLMS 700; Zeiss, ZEN software). All images were acquired using 40× objective.
Quantitative and semiquantitative polymerase chain reaction
Cells were treated for 6 h as described above, and then, frozen cell pellets were used to investigate the expression of mRNA for CHOP by real time (RT)-polymerase chain reaction (RT-PCR; human forward primer GAACGGCTCAAGCAGGAAAT, human reverse primer TTCACCATTCGGTCAATCAGAG). Total RNA was isolated using the miRNeasy Qiagen MicroKit. cDNA was generated using the PrimeScript TM RT Reagent Kit (Takara/Clontech) from 500 ng of RNA with random hexameric primers. RT-PCR analyses were performed using a CFX96™ Real-Time PCR Detection System and SYBR Green detection (Bio-Rad). Semiquantitative PCR was made for the expression of XBP-1 (human forward primer AAACAGAGTAGCAGCTCAGACTGC, human reverse primer TCCTTCTGGGTAGACCTCTGGGAG) by using PTC-200 (MJ Research). β-Tubulin was used as the housekeeping reference gene.
siRNA transfection
Transient transfection of HAoECs was done according to the manufacturer's protocol (Santa Cruz Biotechnology). Briefly, HAoECs were treated with either control siRNA or neutral SMase siRNA for 6 h in a serum- and antibiotic-free medium. Cells were then washed and normal fresh medium was added for 24 h before treatment with MPs for 24 h.
NO• measurement
After treatment for 24 h, as described above, cells were loaded with 4, 5-diaminofluorescein diacetate (10 μmol/l; Santa Cruz Biotechnology) for 30 min at 37°C, and fluorescence was measured.
Endothelial function
Aortic rings were obtained from male Swiss mice bred at the animal facility at the University of Angers. After 24 h of MP incubation, aortic rings were mounted on a wire myograph filled with physiological salt solution, as previously described (20). Endothelium-dependent vasodilatation was studied by cumulative application of acetylcholine (1 nmol/l–10 μmol/l; Sigma-Aldrich) in aortas with functional endothelium precontracted with U46619 (Sigma-Aldrich).
Anion superoxide determination by EPR studies
The detection of anion superoxide production was performed by the EPR technique using 1-hydroxy-3-methylcarbonyl-2, 2, 5, 5 tetramethyl pyrrolidine (CMH; Noxygen) as spin trap. Briefly, after 24 h of treatment, the medium was replaced with 500 μl of deferoxamine-chelated Krebs–Hepes solution containing CMH (500 μmol/l), deferoxamine (25 μmol/l; Sigma-Aldrich), and sodium diethyldithiocarbamate (5 μmol/l; Sigma-Aldrich), and incubated for 45 min at 37°C. ROS measurement was performed on a tabletop x-band spectrometer Miniscope (MS200; Magnettech). Recordings were made at 77°K, using a Dewar flask. Instrument settings were 10 mW of microwave power, 1 mT of amplitude modulation, 100 kHz of modulation frequency, 150 s of sweep time, and three scans. The quantitative measurement of the O2 •− signal amplitude was reported to the relative units for protein concentration (amplitude/μg/μl).
ROS measurement by DHE or MitoSOX staining
Endothelial cells were grown on glass slides (Ibidi), and after different treatments, they were washed and then incubated with either the oxidative fluorescent dye DHE (5 μmol/l; Sigma-Aldrich) for 30 min at 37°C or MitoSOX red (5 μmol/l; ThermoFisher Scientific) for 10 min at 37°C. Then, cells were fixed by 4% paraformaldehyde for 20 min at room temperature. DAPI was added for 5 min. Finally, cells were visualized with confocal microscopy (CLMS 700; Zeiss, ZEN software). All images were acquired using a 63× objective.
Measurement of whole-cell respiration
Respiration rates were measured in treated HAoECs. The cells were collected by trypsinization, washed once in culture medium, and centrifuged (500 g, 5 min). The pellet was resuspended in a respiratory medium, corresponding to normal culture medium without fetal bovine serum and antibiotics, both reagents known to interfere with mitochondrial respiration. Mitochondrial oxygen consumption was measured at 37°C using a high-resolution Oxygraph-2K respirometer (OROBOROS). Oxygraphy experiments were performed on cells cultured in T75 flasks, corresponding to an average of 5 × 106 cells per flask. The basal respiration rate of cells was determined by measuring the linear rate of oxygen consumption. Oligomycin (1 μg/ml) was then added to determine the nonphosphorylating respiration rate. The uncoupling respiration rate was also recorded by stepwise addition of FCCP (0.2–1 μmol/l) up to the optimal concentration representing the maximal capacity of the respiratory chain without the regulation of ATP synthase. Finally, the nonmitochondrial oxygen consumption rate was determined by adding antimycin A (1 μg/ml). At the end of the experiment, a standard volume of respiratory medium is collected and centrifuged for 2 min at 16,000 g. The cell pellet was then resuspended in NaCl (0.9%) and protein concentration was determined spectrophotometrically. Respiration rates were expressed in nmoles of O2 consumed per minute and per mg protein.
Mitochondrial enzyme activity measurements
The activity of the different mitochondrial respiratory chain complexes was measured on cell homogenates, at 37°C, using an SAFAS spectrophotometer (SAFAS).
Statistics
Data are expressed as mean ± standard error of the mean. Statistical analyses were performed by one-way analysis of variance (ANOVA), then the Mann–Whitney U test or ANOVA for repeated measures, and subsequently, the Bonferroni post hoc test. p < 0.05 was considered to be statistically significant.
Footnotes
Acknowledgments
We thank M. Wertheimer and SCAHU staff (Université d'Angers) for taking care of animals, G. Hilairet for expert technical assistance for confocal microscopy, and the staff of Centre Hospitalo-Universitaire d'Angers for analysis of clinical data of METABOL cohort. This work was supported by Institut National de la Santé et de la Recherche Médicale, Université d'Angers and Centre Hospitalo-Universitaire d'Angers. Z.S. is the recipient of a doctoral fellowship from the “Association de Spécialisation et d'Orientation Scientifique” from Lebanon.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.