
Abstract
Significance:
Nonalcoholic fatty liver disease (NAFLD), characterized by liver triacylglycerol build-up, has been growing in the global world in concert with the raised prevalence of cardiometabolic disorders, including obesity, diabetes, and hyperlipemia. Redox imbalance has been suggested to be highly relevant to NAFLD pathogenesis.
Recent Advances:
As a major health problem, NAFLD progresses to the more severe nonalcoholic steatohepatitis (NASH) condition and predisposes susceptible individuals to liver and cardiovascular disease. Although NAFLD represents the predominant cause of chronic liver disorders, the mechanisms of its development and progression remain incompletely understood, even if various scientific groups ascribed them to the occurrence of insulin resistance, dyslipidemia, inflammation, and apoptosis. Nevertheless, oxidative stress (OxS) more and more appears as the most important pathological event during NAFLD development and the hallmark between simple steatosis and NASH manifestation.
Critical Issues:
The purpose of this article is to summarize recent developments in the understanding of NAFLD, essentially focusing on OxS as a major pathogenetic mechanism. Various attempts to translate reactive oxygen species (ROS) scavenging by antioxidants into experimental and clinical studies have yielded mostly encouraging results.
Future Directions:
Although augmented concentrations of ROS and faulty antioxidant defense have been associated to NAFLD and related complications, mechanisms of action and proofs of principle should be highlighted to support the causative role of OxS and to translate its concept into the clinic. Antioxid. Redox Signal. 26, 519–541.
Introduction
N
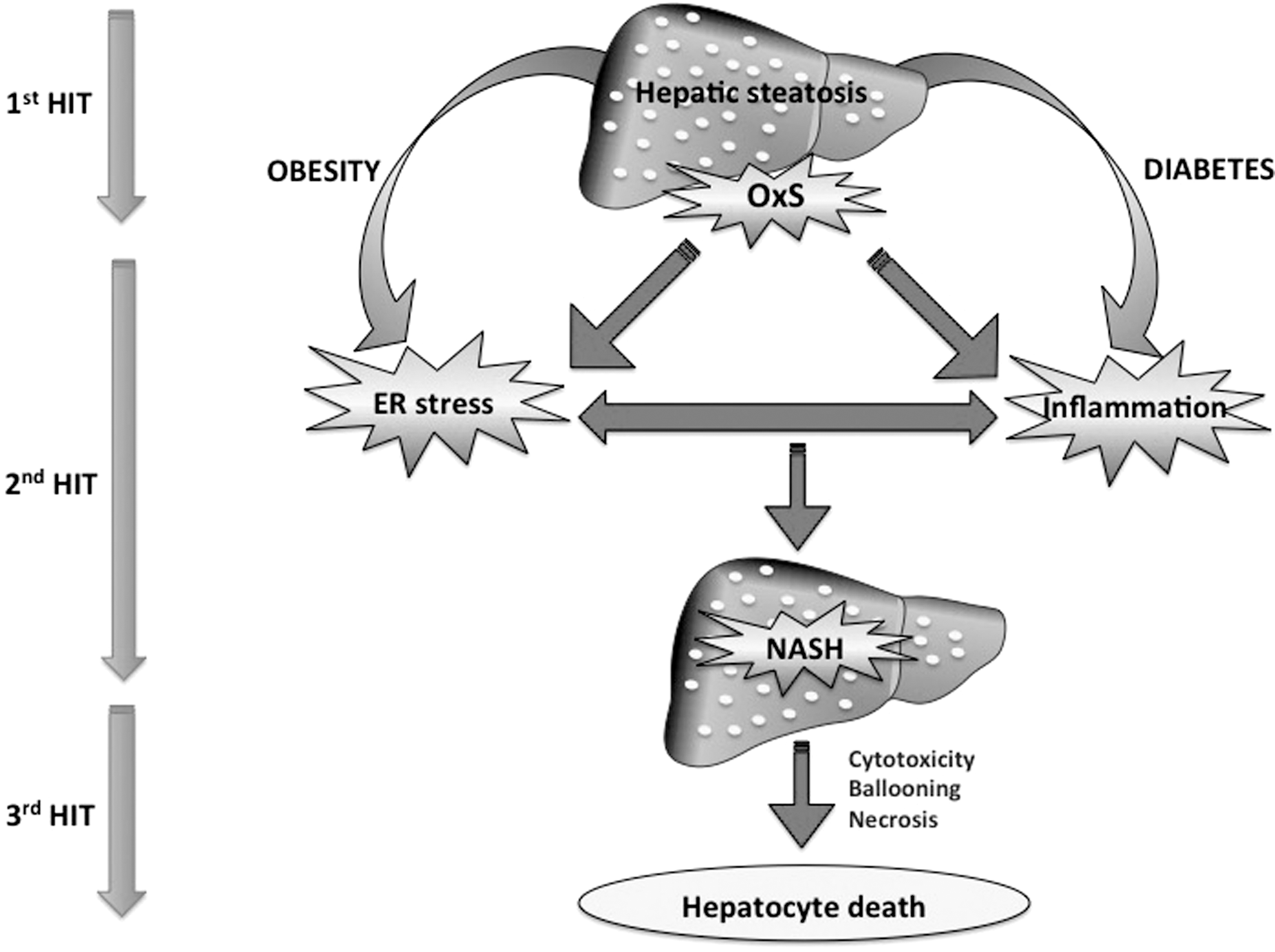
NAFLD presents a wide diversity of symptoms, ranging from a simple steatosis to the concurrent histological appearence of inflammation and ballooning, which defines nonalcoholic steatohepatitis (NASH) with a prevalence of 10–56% in the obese population (246). Up to 20% of the patients who develop NASH can progress to fibrosis, cirrhosis, and hepatocellular carcinoma (10). NAFLD and NASH are considered by the scientific community as hepatic expressions of the metabolic syndrome (MetS) since they are associated to obesity, insulin resistance (IR), and dyslipidaemia (24) along with chronic systemic oxidative stress (OxS) (48) and inflammation, especially in NASH (124). Although the mechanisms for their pathogenesis are still enigmatic, the “two hit hypothesis” is the widespread theory for the development of NAFLD (44). The first hit leading to steatosis results from IR, which leads to hepatic de novo lipogenesis and impaired fatty acid (FA) export. The “second hit” consists of elevated output of reactive oxygen species (ROS) that increase OxS capable of mediating inflammation and cytotoxicity, thereby culminating in NASH and fibrosis (8). A “third hit” has also been suggested, based on the fact that exaggerated OxS produces gradual hepatocyte death, lessens replication of mature hepatocytes, and favors progenitor cell expansion, ensuing in liver cirrhosis and hepatocellular carcinoma (55, 90). Overall, fat accumulation in the liver (first hit) augments vulnerability to OxS (second hit), which triggers inflammation, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, and the incapacity of hepathocytes to synthesize endogenous antioxidants (209) (Fig. 1). Hence, even if NAFLD and NASH are multifactorial diseases, OxS appears to be nearly the most significant mechanisms that convey liver injury in NAFLD (43, 222) with a branching of several intracellular events (e.g., mitochondrial dysfunction and ER stress) in combination with extracellular factors such as iron accretion and gut flora. Therefore, delineation of the OxS mechanisms is fundamental for understanding NAFLD development (174) and represents the main goal of this article. The interconnection with the dynamics of antioxidant actions will be discussed largely to glimpse the effective nutritional therapeutic avenues for NAFLD.
OxS in NAFLD and NASH
As aforementioned, OxS plays a key role in the initiation and progression of NAFLD from simple steatosis to NASH, even if the cause–effect relationship between OxS and the pathogenesis has not yet been robustly established (201). OxS occurs via elevated formation of ROS, which initiates lipid peroxidation by targeting the double bonds of polyunsaturated FA (PUFA). The subsequent formation of extremely reactive aldehyde components, namely 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA), causes intracellular damage. Many circulating biomarkers of lipid peroxidation were observed in NAFLD/NASH patients (86, 148, 165) (Table 1), and their high concentrations are associated with the intensity of liver disease (111). Concomitantly, a decline was noticed in antioxidant compounds such as catalase, glutathione (GSH), GSH S-transferase, superoxide dismutase (SOD), coenzyme Q (CoQ), and Cu-Zn SOD (60, 223) (Table 1). Other investigators emphasized the increase in urinary 8-iso-prostaglandin F2α and serum NOX2-derived peptide, a NADPH oxidase isoform (49, 50, 148). Besides, the expression of hemeoxygenase-1, acting as a cellular protector against OxS in normal conditions, augments in NASH patients, probably as an adapting response (120).
↑ increase; ↓ decrease; 4-HNE, 4-hydroxy-2-nonenal; 8-OHdG, 8-hydroxy-2-deoxyguanosine; 8-iso-PGF2α, 8-iso-prostaglandin F2α; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK-18, cytokeratin-18 tertiles; CoQ, coenzyme Q; GGT, gamma glutamyltransferase; GSH, glutathione; HAS, human serum albumin; HFD, high-fat diet; HFRD, high fructose diet; IR, insulin resistance; MCD, methionine-choline-deficient; MDA, malondialdehyde; MetS, metabolic syndrome; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; OSI, oxidative stress index; oxLDL, oxidized low-density lipoprotein; OxS, oxidative stress; PCC, protein carbonyl; sNOX-dp, NOX2-derived peptide; SOD, superoxide dismutase; TAS, total antioxidant status; TBARS, thiobarbituric acid-reacting substances; TOS, total oxidant status; T2D, type 2 diabetes; TG, triglycerides.
Various mechanisms have been reported to cause lipid peroxidation. Pro-oxidant systems such as cytochrome P450, lipoxygenase, and cyclooxygenase along with free radical products have been alone or synergitically implicated in the emergence of OxS in NAFLD (201). Similarly, raised hepatic expression of CYP2E (isoform of cytochrome P450) appears to be behind ROS production in NASH (13, 153). PUFA metabolic pathways are also singled out since their derived eicosanoids, including hydroxy-eicosatetraenoic, hydroxy-octadecadenoic, and oxo-octadecadenoic acids, have been found to be augmented in patients with NASH (67). Conversely, their inhibition contributes to the improvement of the disease (248). On the other hand, intensification of hepatic levels of 8-hydroxy-2-deoxyguanosine (8-OHdG), a reliable marker of mitochondrial oxidized DNA, is observed in patients with NAFLD and NASH (68), which suggests an overproduction of oxidative DNA damage-targetting mitochondia dysfunction. Intra-hepatocyte mechanisms for the development of OxS will be documented in the next few paragraphs.
Mitochondria Dysfunction, OxS, and Liver Steatosis
According to some groups, OxS is not only a primary cause of liver fat accumulation (161) but also implicated in fibrosis development (149, 193) in patients with NAFLD. The mitochondrion is a critical player in ROS overproduction (214), which results in abnormal respiration and dysfunction of mitochondria, culminating in NAFLD development and expansion (97, 189, 211). Some groups of researchers even say that mitochondrial ROS and dysfunction in patients with NASH are directly implicated in NAFLD and NASH pathogenesis (140). It is quite understandable to detect mitochondrial dysfunctions in patients with NAFLD since inflated ROS may damage lipids, mitochondrial proteins, and DNA. In addition to the reported impairment of the mitochondrial respiratory chain, ultrastructural mitochondrial alterations (29, 104), hepatic ATP synthesis deficiency (19, 41), and apoptosis (6, 202) are reported in NAFLD, probably as a consequence of massive OxS and inefficient antioxidant defense (79).
The mechanisms for redox imbalance-mediated mitochondrial dysfunctions can be summarized as follows (Fig. 2): (i) Excessive mitochondrial ROS are produced with enhanced electron leakage, which, in turn, elevates OxS and oxidative damage to mitochondria, thereby favoring a vicious cycle that leads to impairment of various defective mitochondrial (mt)DNA-encoded protein subunits and abnormal mitochondrial functions; (ii) increased MDA and 4-HNE levels, emanating from lipid oxidation, inhibit cytochrome c oxidase and induce uncoupling protein 2, leading to mitochondrial membrane damage (102, 160); (iii) increased ROS production and reduced hepatic ATP synthesis correlate positively with ultrastructural irregularities in hepatic subcellular mitochondria (e.g., distended and rounded, with deficiency of cristae and presence of paracristalline inclusions), thereby affecting mitochondrial functions (29, 171, 186); (iv) OxS is associated to the reduced expression of the transcription coactivator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), the most important regulator of mitochondrial biogenesis, in NAFLD/NASH with an obvious negative impact on the mitochondrial biogenesis process (1); (v) as PGC-1α coordinates numerous genes needed for mitochondrion production by mastering the activity of several nuclear transcription factors, its low expression in NAFLD/NASH disadvantages its interaction with both nuclear factor erythroid 1 and 2 (Nrf1 and Nrf2) to induce genes connected to the respiratory chain and mitochondrial import machinery, and transcription factors of mtDNA (TFAM, TFB1M, TFB2M), which govern mitochondrial functions (1, 133, 229); and (vi) the downregulation of PGC-1α in NAFLD/NASH may lead to the inactivation of antioxidant defenses, including SOD, catalase, and gluthatione peroxidase (197). Altogether, mtOxS is able to raise lipid peroxides, alter mitochondrial morphology, impair mitochondrial bioenergetics, and affect mitochondrial functions. These changes damage hepatocytes, trigger inflammation, and contribute to IR. However, efforts should be depoloyed to deepen the mechanisms of action using cellular and molecular sophisticated tools.

Oxidized Low-Density Lipoprotein, NAFLD/NASH, and Atherosclerosis
OxS is able to raise cardiovascular risks in patients with NAFLD via significant contribution to atherosclerosis risk factors and the induction of endothelial dysfunction (123). Elevated levels of ROS may lead to low-density lipoprotein oxidation (oxLDL) and macrophage transformation into foam cells, which represent the key stage in the development of the atherosclerotic damage (123). Interestingly, the postprandial increase in oxLDL independently predicted the severity of liver histology in subjects with NAFLD, thus providing a biological mechanism for the epidemiological association between liver disease and atherosclerosis in NASH (132, 207). Validation was obtained in high-fat diet (HFD)-fed mice that were administrated oxLDL (244). This experimental model could mimic human NASH, in which inflammatory events occur, including hepatocellular damage and inflammatory cell penetration (244).
oxLDL, as a product of ROS, is not only able to initiate intracellular OxS in Kupffer cells but also able to cause hepatic inflammation through the process of oxLDL uptake (21, 69). To examine whether oxLDL constitutes the basis for NASH pathogenesis, the recognition of oxLDL by Kupfer cells was inhibited by immunization of ldlr−/− (20). Consequently, a significant decline in hepatic inflammation was recorded as reflected by reduced macrophage, neutrophil and T-cell infiltration, and reduced gene expression of tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-1β, monocyte chemoattractant protein-1, and fibrosis-related genes. In an elegant study, oxLDL enhances lectin-like oxLDL receptor 1 expression and induces injury through ROS formation, nuclear factor kappaB (NF-κB) activation and endothelial nitric oxide synthases (eNOS) downregulation, endothelin 1 and caveolin 1 upregulation, and defenestration in human liver sinusoidal endothelial cells (251). Despite the relationship between oxLDL and liver steatosis, further studies are clearly needed to assess the clinical adequacy of plasma oxLDL as a noninvasive biomarker for NASH and to provide a basis for therapeutic strategies to improve NASH and related metabolic risk factors that conduct to T2D, cardiovascular diseases, and liver deterioration (225).
Myeloperoxidase, NAFLD/NASH, and Atherosclerosis
Various groups investigated the role of neutrophils in NAFLD since they are essential responders to infections and release the enzyme myeloperoxidase (MPO) that is endowed with substantial oxidizing potential in the liver (95). Increased MPO-positive Kupfer cells were observed in the liver of obese NASH patients in close link with raised plasma MPO levels (170). In fact, MPO activity may catalyze nitration of protein tyrosyl groups and may stimulate lipid peroxidation during hepatic steatosis (85) via accrement of HOCl-modified proteins and nitrated proteins in association with increased hepatic chemokine expression in NASH conditions (170). Conversely, MPO deficiency attenuates the development of NASH, supporting an important role for neutrophil MPO-mediated OxS in its pathogenesis (169).
Cross Talk Between OxS and ER Stress
Concretely, OxS and ER stress are two processes that are linked together. For example, protein folding in ER is connected to ROS since after accepting electrons from protein disulphide isomerase, ER oxidoreductase-1 transfers them to molecular oxygen and produces H2O2 to folding, modification, and trafficking of proteins (181). Additionally, RNA-dependent protein kinase-like ER eukaryotic initiation factor-2 α kinase phosphorylates Nrf2, which contributes to the dissociation of Nrf2–Keap 1 complex and promotes the gene expression containing antioxidant response element, reducing OxS by inducing the expression of antioxidant genes such as hemeoxygenase-1 (42). Conversely, the disturbance in GSH/GSSH ratio in ER lumen leads to misfolding of proteins and increased ROS generation (12, 31, 121). In fact, Ashraf's review summarizes the evidences and suggests that ER stress and OxS form a vicious cycle in the pathogenesis of NASH (12).
MicroRNA Regulation in NAFLD/NASH
microRNAs (miRNA) have been revealed to be differentially expressed in NASH patients. Their modulation seems to be beneficial to NAFLD. It has been described that the inhibition of the transcriptional activity of p53 via downregulation of miRNA34a in the liver of mice fed with HFD diminished OxS, apoptosis, and hepatic steatosis (53). An imbalance in nitric oxide levels potentiates a dysregulation in ROS metabolism, resulting in OxS. The enzyme NOS is a key player in cardiometabolic disorders and cardiovascular diseases and its expression is downregulated at the post-transcriptional level by miRNAs, including miR-222/221and miR-92a (235); whereas miR-21 upregulates eNOS activity (230). Currently, beneficial effect of miRNAs is proposed as an acceptable tool to lower OxS-related disorders, including NAFLD and MetS (11).
Genetic Mutations and OxS in NAFLD/NASH
To assess the role of cellular OxS in the development of NAFLD and NASH, numerous studies have focused on genetic variants (Table 2) that might reduce the ability of hepatocytes to protect against OxS. When the rs4880 polymorphism of SOD2 gene, coding for the mitochondrion-resident enzyme manganese-dependent SOD (MnSOD) was assessed, ineffective MnSOD mitochondrial targeting and reduced activity were recorded, suggesting that this variant is a good candidate modifier of NAFLD severity (4). In fact, a steady connection was observed between the functional SNP of SOD2 and fibrosis severity in NAFLD, which evidences that mitochondria-derived OxS is significant in the pathogenesis of advanced NAFLD. Similarly, moderate-to-severe steatosis and inflammation resulted from the −55CT variant of uncoupling protein 3 (UCP3) coding for the mitochondrial proton carrier uncoupling 3, a protein considered to be protective against OxS from β-oxidation of FA (7). A clear association was noted between NASH and the 129C/T polymorphism of glutamate-cystein ligase, the first and limiting enzyme in the formation of GSH (152). On the other hand, there was a protection against NAFLD by the 211G>A genotype of uridine-5′-diphosphoglucuronosyltransferase 1A1 coding for the enzyme synthesizing bilirubin (110), which exhibits potent antioxidant properties (233).
MnSOD, manganese-dependent superoxide dismutase.
Functional Evidence Illustrating the Causal Link Between OxS and NAFLD/NASH
In addition to environmental influences, the onset, incidence, and severity of the NAFLD pathological state are significantly influenced by different interacting genetic factors as noted earlier. As NAFLD is inextricably connected with OxS, unsurprisingly various researchers have first addressed the functional role of specific genes related to cellular redox activity. Over the past few years, genetic engineering technology in animals was used to improve molecular events, diagnostics, and therapeutics related to NAFLD and NASH.
For example, the development of SOD1 knockout (KO) mice shows a hepatic increase in FA synthesis (227) and a decrease in apolipoprotein B-100 levels (216), thereby leading to a lipid accumulation in the liver. In addition, SOD1 KO mice display an increment of iron in hepatocytes (187, 198), which raises OxS via Fenton reaction and the risk of liver disease progression, including fibrosis and NASH (177).
As Nrf2 empowers cells to tackle OxS by inducing cytoprotective genes, downregulates the expression of genes involved in FA synthesis (94, 234, 242), and counteracts inflammation (168), it is highly interesting to determine its connection to NAFLD/NASH. Global KO of Nrf2 strongly enhances the sensitivity of mice to develop NASH when they are administered a methionine- and choline-deficient regimen (38, 200). In contrast, genetic activation of Nrf2 by knockdown of Keap1 decreases their susceptibility to NASH caused by the choline-deficient diet (253). To approach the mechanisms, some researchers used mice with loss of Nrf2 and on HFDs. These animals develop a significant induction of lipogenic genes, a reduced expression of β-oxidation genes, a decline of AMP-activated protein kinase (AMPK) concentrations, and a low activity of acetyl coenzyme A carboxylase compared with the wild-type livers, which is compatible with a higher FA biogenesis and fat accumulation characterizing NAFLD (127).
The KO of the senescence marker protein-30 (SMP30) gene, located in the p11.3-q11.2 segment of the X chromosome, results in aberrantly broadened mitochondria with blurred cristae and unusual accumulations of neutral lipids and phospholipids (87). Similarly, SMP30 decline in response to aging, extreme lipid alimentation, or genetic deficiency leads to a failure of Ca(2+) pumping activity, which prejudices acute insulin discharge by pancreatic β-cells and exacerbates inflammation, OxS and ER stress in NASH (99). The repercussion of SMP30 reduction on OxS and liver steatosis is largely explained by the function of SMP30 as an active enzyme in the antioxidant vitamin C biosynthesis (98). SMP30 may, therefore, represent a new target to restore glucose metabolism and ameliorate NAFLD.
The renin-angiotensin system (angiotensin [Ang]-converting enzyme/Ang II/AT1) has been identified as an important pathway that contributes to the development of NAFLD (239). Indeed, Ang II is able to cause the expansion of NAFLD in the transgenic Ren2 rat model (with elevated tissue Ang II) by increasing hepatic ROS (231).
Other investigators have stressed the role of alcohol dehydrogenase 5 (class III), chi polypeptide (ADH3) in the production of OxS and liver steatosis. This enzyme is also known as GSH-dependent formaldehyde dehydrogenase, thereby contributing to the neutralization of exogenous formaldehyde (51) and protecting cells from the genotoxicity elicited by endogenous formaldehyde (175). In animals, the absence of ADH3 provoked the fast advancement of NASH, which illustrates that ADH3 favors a strong reaction against lipid accumulation by cooperating with the KEAP1-NRF2 system (73).
In the setting of NAFLD in mice with whole-body PEPCK knockdown (while being placed on a standard diet with 10% calories from fat), there is a constitutive oxidative metabolism that is capable of leading to collateral OxS and inflammation processes that intensify IR and hepatocellular injury (185).
On the other hand and contrary to expectations, mice with the deletion of glutamate cysteine ligase, the rate-limiting enzyme for de novo synthesis of GSH, did not exhibit an enhancement of the progression of NAFLD to NASH with severe hepatic injury (78). Instead, the authors found that despite their very low GSH levels, Gclm null mice were, to a great extent, protected against the development of NASH induced by the choline-deficient diet given their ability to develop compensatory mechanisms that protect the hepatocyte against metabolic abnormalities.
Although the identity of certain genes and impact on OxS certainly lead to significant pieces of information, functional studies are still required to delineate the mechanisms by which these risk factors cause the onset and worsening of NAFLD. Mouse models certainly constitute important tools for delivering novel understanding of the biology and genetics of NAFLD, which may clarify the mechanisms and means to develop and improve diagnostics and therapeutics.
Nutritional Antioxidant Interventions
Vitamin E
OxS has now been recognized as a central mechanism contributing to the improvment of liver lesions, as it accelerates the transition from simple steatosis to NASH (79). Therefore, different groups targeted OxS in animal and human studies using vitamin E (Table 3), a free radical scavenger, a chain-breaking antioxidant in the lipid peroxidation chain, and an anti-inflammatory agent (84). To examine the hepatoprotective activities of vitamin E, ob/ob mice were fed diets containing α- or γ-tocopherol before being injected lipopolysaccharides (LPS) to produce hepatic damage and NASH. The 5-week dietary supplementation of α- or γ-tocopherol significantly lowered hepatic LPS-elicited lipid peroxidation (MDA), inflammation (TNF-α), and liver injury, as noted by decreases in serum alanine aminotransferase (ALT) activity and serum lipids (free FA and triglycerides [TG]) without affecting body mass. Therefore, vitamin E preserved from pathogenic processes conducting to liver impairment and NASH in virtue of its antioxidant and anti-inflammatory efficiency, but not to any antiobesity effects. The authors also concluded that vitamin E actions were directed against the “second-hit” of NASH that is characterized by worsened hepatic injury, inflammation, and lipid peroxidation (45). This study with genetic ob/ob mice confirmed the data in rats and mice that were fed a methionine-choline deficient diet to provoke NASH, in which α-tocopherol supplementation reduced histologic signs of liver steatosis, inflammation, and OxS (138, 164). Likewise, subcutaneous administration of α-tocopherol to Zucker rats concomitantly raised α-tocopherol and lowered lipid peroxidation in mitochondria while improving liver injury (194).
↑ increase; ↓ decrease; ACC1, acetyl-CoA carboxylase 1; BW, body weight; CFU, colony-forming units; ChREBP, carbohydrate-responsive element-binding protein; DHA, docohexaenoique; EPA, eicosapentaenoic; FAS, fatty acid synthase; FSP27, fat-specific protein 27; GR, group; HDL, high-density lipoprotein; HFRD, high fructose diet; HMGB1, high-mobility group box 1; hs-CRP, high-sensitivity C-reactive protein; IL, interleukin; JNK, Jun N-terminal kinase; LPS, lipopolysaccharide; MCP-1, monocyte chemoattractant protein-1; NF-κB, nuclear factor kappaB; NKT, natural killer T cells; PPAR, peroxisome proliferator-activated receptor; PUFA, polyunsaturated FA; SREBP1c, sterol regulatory element binding protein-1c; TGF-β1, transforming growth; TNF-α, tumor necrosis factor-α; TLR, Toll-like receptors.
Data from numerous human clinical trials are consistent with animal observations. In a study with 10 subjects with NAFLD and 10 patients with NASH, a 1 year of α-tocopherol treatment at a dose of 300 IU/day improved serum transaminases (ALT, aspartate aminotransferase [AST], and gamma glutamyltransferase [GGT]) levels, hepatic inflammation histological grade, and fibrosis of only NASH patients (82). Confirmation was obtained by employing a more elevated dose (400 IU/day) of α-tocopherol in 11 subjects with NASH, suggesting that α-tocopherol was safe as no side effects were noted (182). Vitamin E (400–1200 IU/day during 2–4 months) could reduce transaminase levels in the serum of children with NAFLD regardless of changes in body mass index or the echogenicity of the liver observed by dual energy X-ray absorptiometry or ultrasound (103).
In the TONIC trial, vitamin E resulted in histologic improvement and NASH resolution along with amelioration in non-high-density lipoprotein (HDL)-cholesterol, LDL-, and total cholesterol levels in 56 children among the total 173 participants, suggesting an alleviation in cardiovascular risks in children with NASH (40). The PIVENS trial with 247 adults with NASH and without T2D showed that vitamin E at dose 800 IU daily during 96 weeks led to marked improvement in histologic features (43%) and significant resolution of NASH (47%) (182). Vitamin E with 1000 IU was more effective when combined with vitamin C (1000 mg/day) in improving fibrosis without changes in liver inflammatory factors over a period of 6 months (81). In contrast, a randomized, double-blind, placebo-controlled clinical trial reported that vitamin E (400 IU/day) combined with metformin (800 mg/day) was more effective in achieving the main objectif consisting of lessening ALT level and IR in pediatric NAFLD patients (3).
Overall, findings of human clinical trials have so far been inconsistent since most of them are retrospective studies, with insufficient sample size, and without control groups and liver biopsy documenting histological improvement. Even if current guidelines recommend vitamin E (at a dose of 800 IU/day) as the first-line pharmacotherapy for NASH (33), attention must be paid to the harmful concentrations of vitamin E reported in various studies (5). Therefore, the vitamin E therapeutic option in liver steatosis should be investigated in rigorously performed, randomized, controlled trials, especially since (i) vitamin E does not act against the oxidation elicited by endogenous enzymes and nonradical oxidants such as hypochlorite (143); (ii) the benefits of vitamin E have not been validated in patients with T2D as well as those with cirrhosis (184); and (iii) a meta-analysis revealed increased mortality with vitamin E administration (129).
Vitamin D
Growing evidence indicates the anti-inflammatory, immunomodulatory, and antifibrotic properties of vitamin D (93), as well as its inverse association with MetS components such as body mass index, body fat content, hypertension, IR, and T2D (37, 77, 199). These observations encouraged investigators to study the impact of vitamin D on fatty liver, especially given that (i) the liver is a major organ in vitamin D production; (ii) vitamin D deficiency is frequently reported in chronic liver disease (101); and (iii) vitamin D levels are associated to NAFLD in adults and children (208).
Earlier data in rats underlined the effectiveness of lipid transfer, metabolic proteins, and vitamin D3 levels as biomarkers for noninvasive diagnosis of steatosis progress and the usefulness of phototherapy as an appropriate supplementary treatment for NASH (135). Subsequently, it was reported that the development and progression of NAFLD by HFD and high fructose corn syrup diet (after 10 weeks of exposure) was exacerbated by vitamin D deficiency due to the induction of inflammation (IL-1β, IL-6 mRNA in the liver; IL-1β and IL-6 in the serum) and elevated OxS (HO-1 mRNA in the liver) (176). The authors proposed the activation of Toll-like receptors (TLR2 and TLR4) as the mechanism of action by way of CD14/LPS-binding protein with the induction of downstream inflammatory signaling molecules exaggerating steatosis and inflammation. Other investigators found that vitamin D may exhibit antiproliferative and antifibrotic effects in hepatic stellate cells, thereby contributing to liver fibrosis (15). They reached this conclusion in view of actions of vitamin D on: (i) inhibition of cyclin D1 expression and stellate cell proliferation; (ii) inhibition of collagen Iα1 promoter activity, mRNA and protein expression along with an antifibrotic effect; (iii) induction of matrix metalloproteinase enzymatic activity, which degraded extracellular matrix proteins; and (iv) inhibition of tissue inhibitor of metalloproteinase 1 mRNA expression levels, whereas vitamin D receptor (VDR) silencing impaired the effect of vitamin D on cyclin D1 and collagen Iα1 expression.
In a human cross-sectional design, scientists evaluated the VDR expression in the liver of patients with NASH (15, 163). VDR expression was negatively related to the significant worsening of liver histology and, once again, vitamin D/VDR system seemed to play a role in the progression of metabolic chronic liver injury in NASH. In other human studies, low serum 25(OH)D3 concentrations were recorded in subjects affected by the NAFLD (59), which probably promotes endotoxemia and thus activation of the innate immune system (122). Altogether, these observations provided a basis for designing clinical trials. When 27 patients with NAFLD in a parallel, double-blind, placebo-controlled study received either oral pearl consisting of 50,000 IU vitamin D3 every 14 day for 120 days, they displayed beneficial effects on serum MDA and high-sensitivity C-reactive protein (hs-CRP) levels without any changes in the serum levels of insulin resistance index (HOMA-IR) index, liver enzymes, and grades of hepatic lesions (192). Therefore, the authors suggested that supplementation with vitamin D might be considered an adjunctive therapy to reduce inflammation and lipid peroxidation alongside other treatments for NAFLD patients. Currently, thorough planned trials are needed to determine whether vitamin D supplementation may constitute a new therapeutic alternative in NASH care.
Coenzyme Q
CoQ is an endogenous lipid-soluble benzoquinone compound that exists in a reduced (ubiquinol, CoQ10H2) as well as in an oxidized (ubiquinone, CoQ10) form. The redox activity enables CoQ10 to behave as an antioxidant that is capable of preventing lipid peroxidation and peroxidative damage to membrane phospholipids (130). It acts as an essential cofactor in oxidative phosphorylation in mitochondria, because it operates in the mitochondrial inner membrane to transfer electrons from complexes I (NADH CoQ reductase) and II (succinate dehydrogenase) to complex III (cytochrome bc1 complex), which are closely involved in OXPHOS (80). It also functions as an inductor of ATP formation in tissues with high-energy turnover (heart, liver, and muscle) (23). It also serves as a cofactor and as a requisite cofactor of mitochondrial uncoupling proteins whose activation lessens free radical formation from mitochondria (17, 91). As OxS is closely linked to hepatic lipid accumulation and since IR impoverishes intracellular amounts of natural cellular antioxidants (74), CoQ10-mediated antioxidant activity is able to alleviate ROS concentrations, thereby allowing NAFLD alleviation. In HFD-induced rat model of NAFLD, CoQ administration led to an increase in apolipoprotein (apo) B synthesis, favoring the rise of the number and size of very low-density lipoprotein (VLDL) particles, which ultimately caused the secretion of larger VLDL and lessened hepatic steatosis (30). However, the changes in apo B48 concentrations elicited by dietary CoQ may also be due to diminished degradation of large VLDL that may represent atherogenic particles. Similarly, a diet containing 57% of energy from fat induces the characteristic features of NAFLD in rats, including IR, hypertriglyceridemia, hepatic steatosis, and liver damage, combined with altered CoQ10 metabolism (23). Besides, when 44 NAFLD patients were included in a randomized, double-blind, placebo-controlled trial and supplemented with 100 mg/day CoQ10 capsules, they improved various anthropometric and biochemical parameters in NAFLD, including serum AST and various adipose tissue-derived cytokines (64). Similarly, a randomized, double-blind, placebo-controlled trial showed that NAFLD participants receiving CoQ10 capsules (100 mg/day) exhibited a significant decrease in liver aminotransferases (AST, GGT, hs-CRP), inflammation (TNF-α), and the grades of liver steatosis, as well as raised levels of adiponectin (65).
Polyphenols
Since epidemiological investigations revealed that diets rich in vegetables and fruits could diminish the incidence of cardiometabolic disorders, their bioactive components are gaining more interest in improving health risks in view of their low toxicity and few side effects. Currently, polyphenol-rich plants and fruits are not only used to improve human health and promote quality life but also used to fight diseases, including obesity, T2D, cardiovascular disease, and cancer (156). For food experts and nutritionists, discovering appropriate functional nutrients and developing a dietary therapy are important strategies in avoiding NAFLD (Table 3). For example, blueberries that are highly recommended in view of the phenolic potential health benefits of their compounds have high anthocyanin content (112) that is beneficial in reducing the risk of MetS conditions, such as obesity, hyperglycemia, hyperinsulinemia, and hyperlipidemia (89, 190). Phenolic content of wild Chinese blueberries displays good efficacy in impeding steatosis in HepG2 cells (113).
In db/db mice, feeding a polyphenolic extract of lotus root for 3 weeks protects them from hepatic steatosis via the decline of lipogenic enzymes, such as FA synthase and malic enzyme (213). Red wine comprises several polyphenols with likely biological actions (108), particularly resveratrol and catechins (14), but only few other components from red wine have been tested. Similarly, ellagitannin blend extracted from European oak bark utilized in red wine maturation exhibits both cardiac and liver protection and markedly improves metabolic profile in high-carbohydrate, HFD-fed Wistar rats (155). Attenuation is observed in hepatic steatosis and fibrosis with infiltration of inflammatory cells in the liver, as well as in plasma activities of ALT and AST. As various investigations worldwide propose that the valuable actions of green tea are due to their capacity to regulate cell signaling and cell cycle (28), their epigallocatechin gallate is being used to treat diseases that are related to chronic inflammation. In Sprague–Dawley rats fed with HFD for 8 weeks to develop NAFLD, treatment with (−)-epigallocatechin-3-ga improves hepatic histology (given the decrease in fatty liver score as well as necrotic and inflammatory foci), reduces liver injury (e.g., ALT/AST ratio), and attenuates fibrosis (sirius red and synaptophysin-positive stain) along with downregulation in the expressions of key pathological oxidative (e.g., nitrotyrosine formation) and pro-inflammatory markers (e.g., inducible NOS, cyclooxygenase-2, and TNF-α) via transforming growth factors/SMAD, PI3K/Akt/forkhead box O1, and NF-κB pathways (236).
In human studies, the consumption of bayberries (250 ml twice daily for 4 weeks) containing high levels of polyphenols beneficially alters the levels of oxidative (protein carbonyl groups), inflammatory (TNF-α, IL-8, hs-CRP), apoptotic biomarkers (tissue polypeptide-specific antigen, cytokeratin-18 fragment M3), and liver enzymes (ALT, AST) in young individuals with features of NAFLD. Other researchers turn toward resveratrol because of its protection against OxS and inflammation that are involved in NAFLD pathogenesis. The design of placebo-controlled, double-blind, randomized clinical trials has permitted to document improvement of conditions (ALT, liver steatosis, markers of inflammation, and hepatocellular apoptosis) in patients with NAFLD (63) or NASH (224) pathogenesis.
Interestingly, flavonoids are often employed in NAFLD models, given their beneficial effects and especially their antioxidant power (173). The advantage of flavonoids may be related to their activation of peroxisome proliferator-activated receptor (PPAR)γ without serious side effects seen with the use of full agonists (34, 105, 128, 203). More clinical trials are required to explore whether the antioxidant and anti-inflammatory actions displayed by polyphenols are likely to make a significant contribution to the amelioration of NAFLD.
Berberine
Berberine is an isoquinoline alkaloid that is present in the bark, roots and stems, and plants of the genus Berberis. It displays various pharmacological activities and has been investigated for treatment of cancer, obesity, T2D, inflammation, atherosclerosis, Alzheimer's disease, rheumatoid arthritis, and cardiovascular diseases (2, 154, 228). This compound is able to fight MetS components such as abnormal glucose metabolism, dyslipidemia, and hypertension (100, 252). Since it has been able to suppress ROS production and to reduce inflammation (56, 144), it appeared reasonable to test its effectiveness in NAFLD. Using both in vitro and in vivo models, it has been shown that berberine is capable of inhibiting cholesterol and TG biogenesis by downregulating acetyl-CoA carboxylase via the activation of AMPK, thereby resulting in lower liver fat content (26).
Berberine is also capable of preventing liver fibrosis through the regulation of antioxidants (SOD) and lipid peroxidation (MDA and HNE) (249). In diabetic rats, berberine alleviates the pathological evolution of the liver and reverts the raised hepatic glycogen and TG to close the control levels by modulating PPARα/δ expression in the liver (255). Similarly, berberine exhibits the capacity to improve lipid dysregulation and fatty liver in db/db and ob/ob mice via peripheral AMPK activation and neural signaling (92). Another action on experimental NAFLD is the downregulation of liver UCP2 (mRNA and protein expression) in NAFLD rats. It can lessen hepatocyte fat accumulation while improving lipid metabolism disorder in the NAFLD rat model (241). Using berberine-containing Chinese medicine, Zucker rats display sustained glucose-lowering effects, attenuated IR, lowered fatty degeneration in association with activation of AMPK, Akt, and insulin-like growth factor-binding protein pathways, with downregulation of miR-29b and induction of a gene network implicated in cell cycle and NADPH metabolism (254). Importantly, mitochondrial dysfunctions (membrane potential, oxygen consumption, and ATP production) are improved in the liver in link with an increased activity of the mitochondrial sirtuin 3 in HFD-fed rats by berberin (210). Confirmation is obtained by demethyleneberberine, a natural mitochondria-targeted antioxidant, which exerts significant inhibition of mitochondrial dysfunction, OxS, and steatosis in the fatty liver disease mouse model (250).
A double-blind, crossover-designed trial versus placebo shows that 8 week treatment of a combined nutraceutical containing berberine, chlorogenic acid, and tocotrienols significantly improves anthropometric and biochemical parameters (39). In particular, total cholesterol, LDL-cholesterol, TGs, non-HDL cholesterol, fasting insulin, HOMA-IR, and steatosis are decreased in the nutraceutical group. In a randomized, parallel-controlled, open-label clinical trial, life style intervention in combination with berberine (0.5 g three time/day) has ameliorated NAFLD and related metabolic disorders (240). Given the limited number of human investigations, additional studies are needed to prove the effectiveness of berberine in preventing and treating NAFLD.
Resveratrol
Emerging evidence has pointed out the advantageous physiological actions of resveratrol in promoting health and treating diseases, including NAFLD (35, 106, 114, 247). This 3,5,4′-trihydroxy-trans-stilbene, derived mainly from plants, grapes, and berries, is a robust antioxidant and anti-inflammatory compound (158, 159). It is also capable of negatively modulating IR, glucose intolerance, and dyslipidemia (16). Through antilipidogenic effects, resveratrol may reverse steatosis by fighting de novo lipogenesis in various cell lines (72, 191, 226). In animal models, the administration of resveratrol resulted in preventive and therapeutic effects on hepatic steatosis (Table 3) via mechanisms leading to OxS reduction (27, 191). Despite the early promise of experimental studies with resveratrol, to date, very limited effort was made to uncover the impact of this natural polyphenolic compound on humans. In a small cohort of healthy obese men, resveratrol was able to decrease plasma ALT concentration and intrahepatic lipid content (212). However, additional thorough studies in human subjects are still needed over long periods to determine the efficiency of resveratrol in alleviating fatty liver diseases.
Silybin/sylimarin
Silymarin and silibinin are other natural polyphenolic flavonoids contained in the herb milk thistle (Silybum marianum). Silymarin represents the crude extract of milk thistle seeds and is a complex of biological compounds that include silibinin or silybin, which is endowed with potent antioxidant, immunomodulatory, antifibrotic, antiproliferative, and antiviral activities (70, 166). Silibinin is the most commonly employed for gastrointestinal clinics to treat liver disorders such as NASH and cirrhosis (115). When a 4-week daily dose (20 mg/kg i.p.) of silibinin is administrated to db/db mice that are fed a methionine-choline deficient diet, a model combining the features of the MetS with the histological pattern of NASH (Table 3), there is a marked antisteatotic effect accompanied by an antioxidant action of silybin, which is confirmed by decreased levels of hepatic OxS (isoprostanes, 8-OHdG, and nitrites/nitrate) as well as inflammation (TNF-α and IL-6 mRNA) via reduced JNK phosphorylation (179). In the same animal model, silibinin treatment counteracts the evolution of liver damage by controlling lipid homeostasis and overturning OxS-mediated lipotoxicity and NF-κB activation in experimental NASH (178). A clinical trial of 179 NAFLD patients reveals that 12 months of treatment with silibinin, combined with phosphatidylcholine and vitamin E, is associated with amelioration of IR, liver enzymes, and liver histology compared with placebo (115). Despite the huge number of findings collected from experimental models, one needs large human trials since the current translation of the evidence in clinical setting is far to be conclusive.
Polyunsaturated FAs
Scientific evidence showed that saturated lipids and fructose are more prospective to stimulate hepatic fat accumulation and progression of NAFLD, whereas unsaturated lipids seem to have a more preventive effect (46, 224). Especially, n-3 PUFA are capable of significantly reducing plasma and liver TG accumulation, which hampers NAFLD expansion via various metabolic pathways (126). Growing evidence, essentially based on cardioprotective properties (54, 71), suggests the effectiveness of n-3 PUFA to prevent or treat NAFLD. Eicosapentaenoic (EPA), docohexaenoique (DHA), and α-linolenic acids are the three main long-chain n-3 PUFAs. Given their sluggish biogenesis in humans, EPA and DHA are considered essential PUFA and must, therefore, be obtained from diet (22), mainly from fatty fish. Their number of double bonds, the length of the chain, and the presence of the first double bond provide n-3 PUFA with unique properties, which distinguish them from other unsaturated fats (47).
The n-3 PUFA affect several steps of the pathophysiological cascade and can prevent or inhibit the progression of NAFLD (Table 3). The n-3 PUFA lead to (i) decreased IR resulting from excessive caloric intake and lipid accumulation in adipose tissue; (ii) increased circulating adiponectin levels in response to obesity, which, in turn, diminishes fat accumulation in the liver; (iii) reduced adipose tissue-mediated exaggerated FA release, a condition contributing to dyslipidemia; (iv) lowering of blood lipid levels and raising n-3/n-6 ratio in cell membrane phospholipids; (v) enhancing the number of insulin receptors on hepatic cell membrane, a process improving insulin sensitivity; (vi) altering the transcription of liver genes involved in lipid metabolism regulation via PPARα, a major nuclear factor that controls enzymes involved in mitochondrial β-oxidation, promotes degradation of free FAs through stimulation of β-oxidation, thus preventing hepatic TG accumulation; (vii) downregulating expression of sterol regulatory element binding protein-1c (SREBP1c), a key transcription factor involved in TG synthesis that suppresses its activity, thereby averting liver TG accretion; (viii) impeding mitochondrial ROS production and OxS occurrence; (ix) limiting ER stress, which lessens hepatic lipotoxicity; (x) thwarting the synthesis of pro-inflammatory eicosanoids and cytokines in hepatocytes or downregulation of transcription of proinflammatory cytokines via their n-3 anti-inflammatory derivatives (protectins, resolvins); (xi) reducing hepatic fibrosis and NAFLD progression toward NASH and cirrhosis “second hits” (22); and (xii) controlling glucose homeostasis and impeding IR development, thereby slowing down NAFLD progression (131, 220) by negatively regulating carbohydrate-responsive element-binding protein expression (52).
The decrease in hepatic n-3 PUFA concentrations may contribute to exacerbating hepatic lesions. This hypothesis has first been tested in NAFLD animal models in which n-3 PUFA improves histological lesions, restores adiponectin secretion, induces PPARα expression, and hampers TNF-α and SREBP1c production (22, 205, 237). In this context, n-3 PUFA is able to decrease inflammation (36) via probably inactivation of the NF-κB pathway (36), resulting from inhibition of IκB-α phosphorylation/degradation and preventing NF-κB translocation to the nucleus for the induction of pro-inflammatory cytokines. Further evidence also shows that n-3 PUFA may attenuate the generation of free radicals (220) and the lipid peroxidation process (66).
In obese mice fed an HFD, the n-3 PUFA supplementation improves insulin sensitivity, while reducing OxS and inflammation associated with NAFLD (220). In a recent trial, the daily supplementation of NAFLD subjects with fish oil for 6 months ameliorates lipid profiles, decreases the discharge of pro-inflammatory cytokines from the liver, and inhibits the redox imbalance characterized by increased lipid peroxidation and free radical activity (220). In a study with young patients with NAFLD, the administration of different doses (250 and 500 mg/day) of DHA markedly decreases hepatic lipid content without apparent side effects, suggesting that n-3 PUFA doses are safe and effective (145, 146). Moreover, the increase of EPA to a dose of 2700 mg/day significantly improves levels of liver transaminases, free FAs, and cholesterol in patients with NAFLD (206). Although n-3 PUFA supplementation is a promising approach in the treatment of NAFLD patients, more randomized studies with large cohorts are necessary to validate their effectiveness and safety.
Pre-probiotics
Probiotic may restrain the role of bacterial pathogens in NAFLD in many animal models of HFD-induced NAFLD/NASH, as is largely shown with the most characterized probiotic VSL#3 mixture, (62, 109, 119). For example, treatment with the probiotic mixture VSL#3 displays a reduction of antioxidative and anti-inflammatory effects in NAFLD ob/ob mice (109), a model of NAFLD induced by an HFD (134), and a model of hepatic natural killer T-cell depletion in HFD-fed animals (119). Administration of VSL#3 to an experimental model of NAFLD/NASH attenuates fibrosis (Table 3) by reducing transforming growth factor-β, and collagen, α-smooth muscle actin, and matrix metalloproteinase-1 expression (221). Despite the numerous preclinical investigations carried out to evaluate the use of probiotics in the treatment of fatty liver disease, there are only a limited number of trials regarding their efficacy in human NAFLD. By testing a mixture of probiotics associated with prebiotics and vitamins in 10 patients with biopsy-proven NASH during 2 months of treatment, one can observe a substantial amelioration of hepatic injury and functional tests, as well as a partial sustained effect after the end of treatment (118). Another study carried out to evaluate the effects of VSL#3 probiotic therapy for 3 months in 22 NAFLD patients shows an amelioration of OxS and inflammation (116). Although these promising data are strongly indicative of a great potential for the use of probiotics in the prevention and treatment of NAFLD, there is a need of further clinical studies to better define this innovative strategy.
Conclusions
The prevalence of NAFLD raised rapidly in association with obesity and IR. As a metabolic stress involved in liver disease, NAFLD has become a worldwide health problem with increased risks of T2D, cardiovascular events, and mortality. Although the mechanisms remain poorly understood, OxS appears to play a major role in its pathogenesis and complications. The high production of ROS causes lipid peroxidation, followed by inflammation, activation of hepatic stellate cells leading to fibrogenesis, necrosis, cirrhosis, and carcinoma. This article summarizes the plausible evidences documenting the contribution of OxS originating from mitochondria to the progression of NAFLD and its aggravation to NASH. Various natural compounds represent potential therapeutic candidates, essentially due to their antioxidant, anti-inflammatory, and antifibrotic properties. In spite of the various data obtained in experimental models, further breakthrough related to OxS signaling and mechanisms are still warranted to understand the NAFLD pathogenesis. In addition, despite the available clinical studies, there is clearly a significant requirement for robust and well-controlled trials using large cohorts to translate the evidence in clinical setting and to prove intervention efficacy.
Footnotes
Acknowledgments
The current work was supported by research grants from the JA deSève Research Chair in nutrition (E.L.) and the FRQS doctoral Scholarship Award (S.S.).