
Abstract
Aims:
Many men endure immunosuppressive or anticancer treatments that contain alkylating agents before the age of sexual maturity, especially the increasing number of preadolescent males who undergo busulfan treatment for myeloablative conditioning before hematopoietic stem cell transplantation. Before sperm production, there are no sperm available for cryopreservation. Thus, it is necessary to identify a solution to ameliorate the busulfan-induced damage of spermatogonial stem cells (SSCs).
Results:
In this study, we demonstrated that melatonin relieved the previously described SSC loss and apoptosis in mouse testes. Melatonin increased the expression of manganese superoxide dismutase (MnSOD), which regulated the production of busulfan-induced reactive oxygen species (ROS). Moreover, melatonin promoted sirtuin type 1 (SIRT1) expression. SIRT1 participated in the deacetylation of p53, which promotes p53 ubiquitin degradation. Decreased concentrations of deacetylated p53 resulted in spermatogonial cell resistance to apoptosis. Acute T cell leukemia cell assay demonstrated that melatonin does not affect busulfan-induced cancer cell apoptosis and ROS.
Innovation:
The current evidence suggests that melatonin may alleviate the side effects of alkylating drugs, such as busulfan.
Conclusion:
Melatonin promoted MnSOD and SIRT1 expression, which successfully ameliorated busulfan-induced SSC apoptosis caused by high concentrations of ROS and p53. Antioxid. Redox Signal. 28, 385–400.
Introduction
A
We discovered that melatonin rescues busulfan-induced spermatogonial stem cell (SSC) oxidative apoptosis in mouse testes via the promotion of manganese superoxide dismutase (MnSOD) and sirtuin type 1 (SIRT1) expression in vivo and in vitro. In addition, this study provides experimental evidence in the potential reference that melatonin may alleviate the spermatogenic cell damage side effects of alkylating drugs, such as busulfan.
Melatonin (MT) has the potential to be effective in prevention of various ROS-related diseases, such as neurodegenerative disorders (34, 50). Melatonin comprises an indole neurohormone that is synthesized and secreted by the pineal gland; its concentrations are regulated by both dark–light and seasonal cycles in mammals (22, 45).
Melatonin is also one of the few antioxidants that eliminate ROS by providing electrons; moreover, its metabolites may amplify the antioxidant effect (29). It has been reported that melatonin increases the expression of antioxidant enzymes, such as manganese superoxide dismutase (MnSOD) and sirtuin type 1 (SIRT1) (15, 61, 62). Melatonin decreases the damage caused by ROS and other free radicals to mitochondria, as well as lipid membranes (44). Studies have demonstrated that melatonin may effectively reduce damaged side effects caused by radiation or chemotherapeutic drugs, such as cisplatin (18, 58).
The pineal gland comprises the part of the hypothalamic–pituitary–testicular axis that is important in mammalian testicular development and function (19, 46). Melatonin also plays a significant role in the regulation of self-renewal and differentiation via its antioxidant effects in various stem cells, such as mesenchymal stem cells (10) and spermatogenic cells (6). Moreover, melatonin prevents testicular damages caused by environmental toxins and drugs based on the characteristics of lipophilic and hydrophilic free radical scavengers (20, 23, 32).
Recent studies have demonstrated that in vitro melatonin strongly enhances cisplatin-induced cytotoxicity and apoptosis in HeLa cells (38). Thus, the use of melatonin may have potential as an auxiliary chemotherapy drug as a synergistic and protective agent. Studies have investigated the use of antioxidants to relieve the side effects of chemotherapy drugs; however, there is an urgent need to develop an evidence-based guidance on antioxidant supplementation to obtain the best possible care and avoid risky treatments for cancer patients (35). Specifically, few systematic studies have investigated the detailed mechanism by which melatonin ameliorates the effects of busulfan during spermatogenesis (30).
In this study, we investigated busulfan-induced damage and apoptosis in mouse testes; we demonstrated that melatonin ameliorated the previously described side effects and may be used as a potential drug with high potency and low side effects.
Results
Busulfan caused spermatogonial loss in mouse testes and in vitro
As described in the literature, the size and weight of mouse testes increased from 5 weeks of age until adulthood in normal mice (27). However, during busulfan treatment, the size and weight exhibited a clear downward trend. Twenty-one days after the busulfan injection, the testes in the busulfan-treated group were significantly smaller compared with the controls (Fig. 1A), and the testis/body ratio also exhibited a downward trend, which reached its lowest level at 35 days (Fig. 1B). These findings suggested that testis development may be suppressed in busulfan-injected mice.
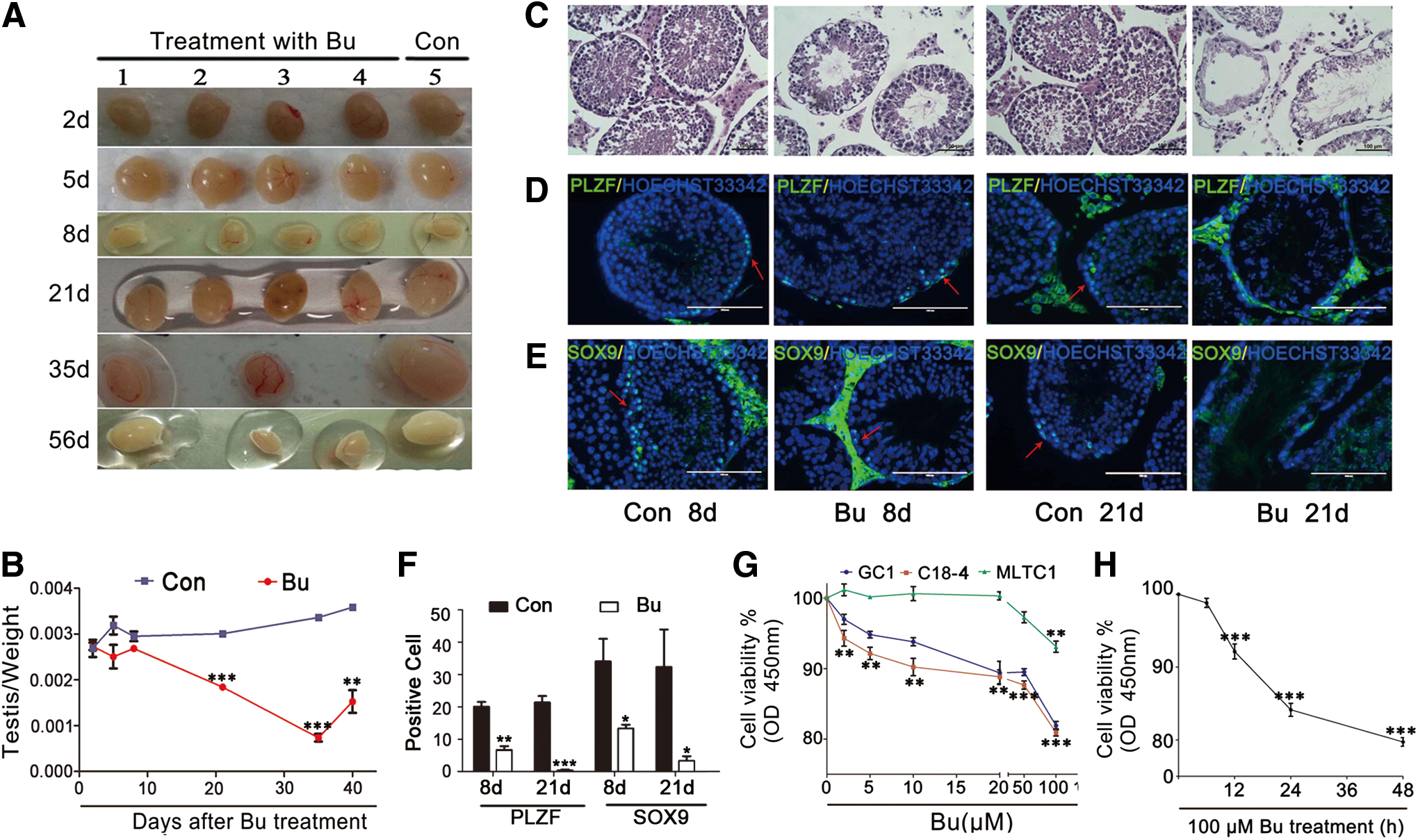
A histological analysis indicated a substantial number of vacuoles located in the seminiferous tubules of the busulfan-treated mouse testes (Fig. 1C). After 21 days, the seminiferous tubules exhibited a hollow tendency, and the numbers of spermatogenic cells, including spermatocytes, sperm, and spermatogonia, were significantly decreased. Compared with the control group, the morphology and location of the Sertoli and Leydig cells did not clearly change (Fig. 1C). To further analyze the type of cells that were lost in the testes, spermatogenic-specific markers were tested via immunofluorescence staining. The results demonstrated that PLZF, a self-renewal marker of spermatogonial stem cells (SSCs) (31), was decreased after busulfan treatment (Fig. 1D, F). However, SOX9, a marker of Sertoli cells (3), was also decreased (Fig. 1E, F).
To further confirm which cell type was more vulnerable to busulfan, the cell line viability was analyzed in cells treated with varying concentrations of busulfan for 24 h; these cell types included type A SSCs (C18-4) (51) and type B SSCs (GC1) (54), as well as Leydig cells (MLTC1) (17). The results demonstrated that cell viability for C18-4 and GC1 was more vulnerable compared with MLTC1 (Fig. 1G). We also demonstrated that cell vitality decreased with the treatment time for C18-4 cells (Fig. 1H).
In the beginning of this study, we analyzed busulfan-injected mice and identified spermatogonial loss and testis size suppression, which resulted from busulfan. Cytotoxicity tests also confirmed that busulfan caused spermatogonial loss rather than Sertoli and Leydig cell loss. This finding led us to further investigate the busulfan mechanism to understand how busulfan treatment caused SSC loss.
Busulfan-induced spermatogonial loss was caused by apoptosis
Evidence indicated busulfan-induced damage in spermatogenic cells in mice in a time and dose-dependent manner (Fig. 1B, F). Moreover, we analyzed the DNA content to confirm that busulfan induced cell death mainly in the G0/G1 phase rather than the S phase (Fig. 2A). We speculated that this finding was a result of cells undergoing cell cycle arrest. The results of our analysis indicated that cells with busulfan-induced damage remained at the G1/S checkpoint for repairing or undergoing apoptosis.

Thus, we used terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-X nicked end labeling (TUNEL) staining to analyze apoptosis in busulfan-treated mouse testes. In the control group, apoptosis was rare. However, there were significantly increased TUNEL-positive cells in the seminiferous tubules of the testes of the busulfan-treated animals compared with the controls (Fig. 2B). Two days after the busulfan injection, there were fewer apoptotic cells; after 8 days, the majority of apoptotic cells were spermatogonia and spermatocytes. After 21 days, the apoptotic cells comprised spermatocytes (Fig. 2B). Flow cytometry demonstrated that apoptosis was significantly increased in the busulfan-treated group compared with the control group (Fig. 2C). Similar to other alkylating agents, such as cyclophosphamide and acrolein, busulfan caused apoptosis in spermatogonia.
The mechanism of busulfan-induced spermatogenic apoptosis
We further examined the mechanism of apoptosis induced by busulfan. Thus, an activity assay or quantitative real-time polymerase chain reaction (qRT-PCR) was applied to examine the expression of apoptosis-related genes, such as Caspase3, MnSOD, p53, and SIRT1 (22, 43, 49). The data demonstrated that caspase3 activity, an apoptosis marker, was upregulated fivefold in the busulfan-treated animals compared with controls (Fig. 3A). Thus, we confirmed that apoptosis caused spermatogonial loss. We also demonstrated that the mRNA level of MnSOD increased compared with the control group, and the peak appeared 5 h after busulfan treatment (Fig. 3B); this finding was interpreted as being the result of a negative feedback regulator of busulfan-induced ROS.
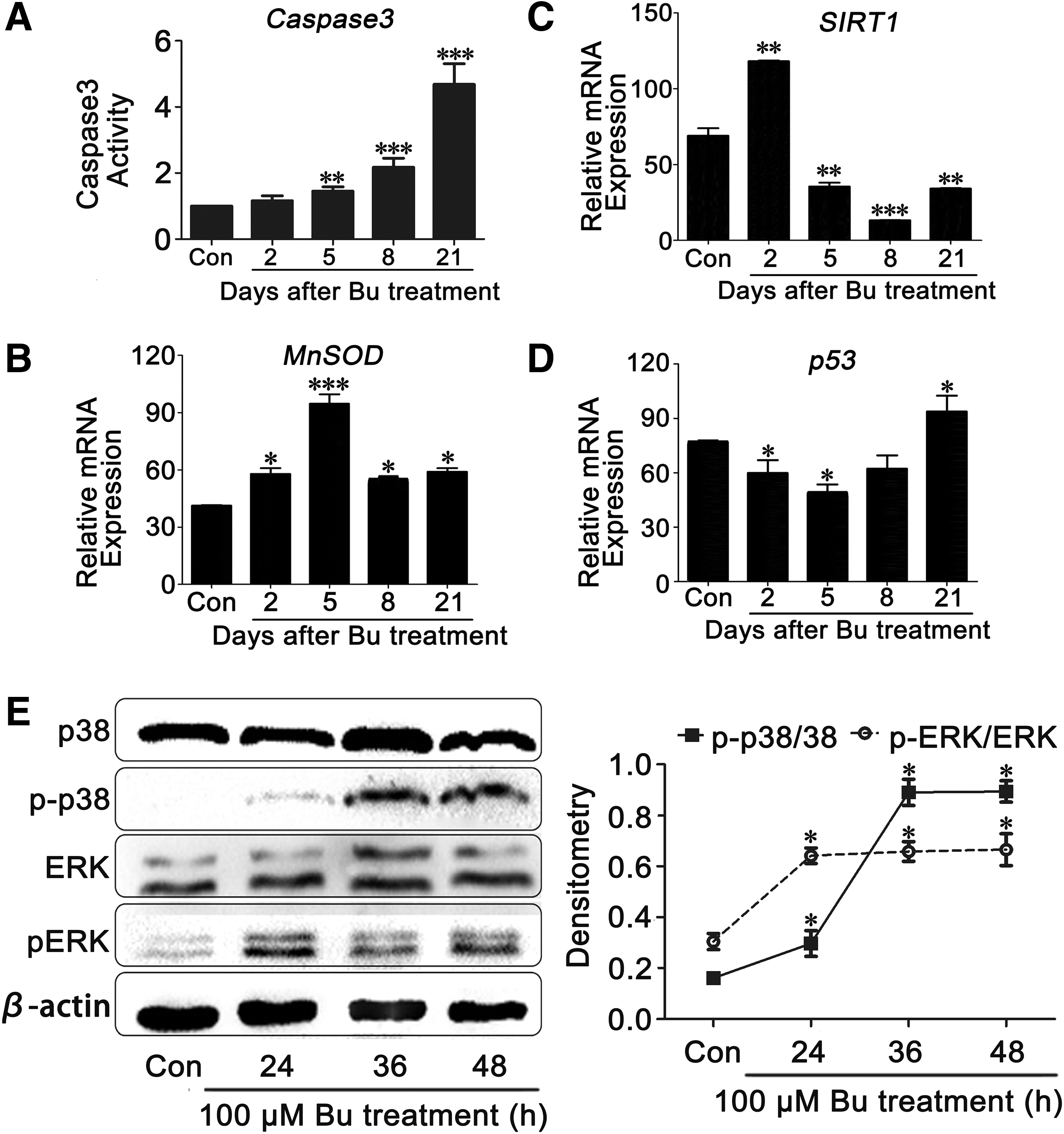
The mRNA level of SIRT1 exhibited the same trend as MnSOD; however, p53 had a completely opposite trend (Fig. 3C, D). These findings indicated that the expression levels of the two related genes were initially affected by the short-term stimulation and subsequently experienced long-term damage. We further investigated the stress-dependent extracellular signal-regulated kinase (ERK) and p38 signaling pathways and demonstrated that ERK and p38 phosphorylation was significantly increased in the testis samples obtained from the busulfan-induced group (Fig. 3E). Combined with previous toxicity studies of alkylating agents, we speculated that intracellular ROS may activate the p38 pathway and increase the transcription of MnSOD as a negative feedback signal.
Melatonin increased MT1 and MT2 expression, which increased SIRT1 expression
According to previous studies, the segmental physiological effects of melatonin are mediated by specific receptors, such as MT1 and MT2 (26, 52). This finding led us to investigate the expression of melatonin receptors in our population of samples. Immunohistochemistry demonstrated that MT1 and MT2 were widely expressed in SSCs, Leydig cells, and Sertoli cells of the mouse testes with or without busulfan or melatonin treatments (Fig. 4A, B). The mRNA results demonstrated that the MT1 and MT2 expression was increased in the melatonin-treated animals (Fig. 4C). To provide preliminary clues for further experiments, we examined receptor-mediated proteins. Western blot analysis indicated that SIRT1 decreased in the busulfan-treated mouse testes; however, melatonin inhibited the decrease.

We simultaneously used luzindole, an inhibitor of melatonin receptors, to authenticate the reversal of this effect. The results demonstrated that the SIRT1 expression was decreased in the testicular cell suspensions treated with luzindole; however, the data indicated that this effect was partially dependent on the melatonin receptor pathway (Fig. 4D).
Melatonin alleviated busulfan-induced spermatogonial cell loss
Melatonin receptors and their downstream targets were consistent with expectations in mouse testes; thus, we aimed to relieve the loss of SSCs caused by busulfan. One week after injection with 30 mg/kg busulfan, the mice were administered melatonin (10 mg/kg/day) for 2 weeks. The testis-to-weight ratio of the melatonin-injected group was significantly increased compared with the control group (Fig. 5A, B). Hematoxylin–eosin (HE) staining suggests that the melatonin-treated group did not exhibit the serious seminiferous tube vacuolization that was identified in busulfan-injected animals, which may indicate that melatonin functions to prevent the loss of spermatogonial cells (Fig. 5C).
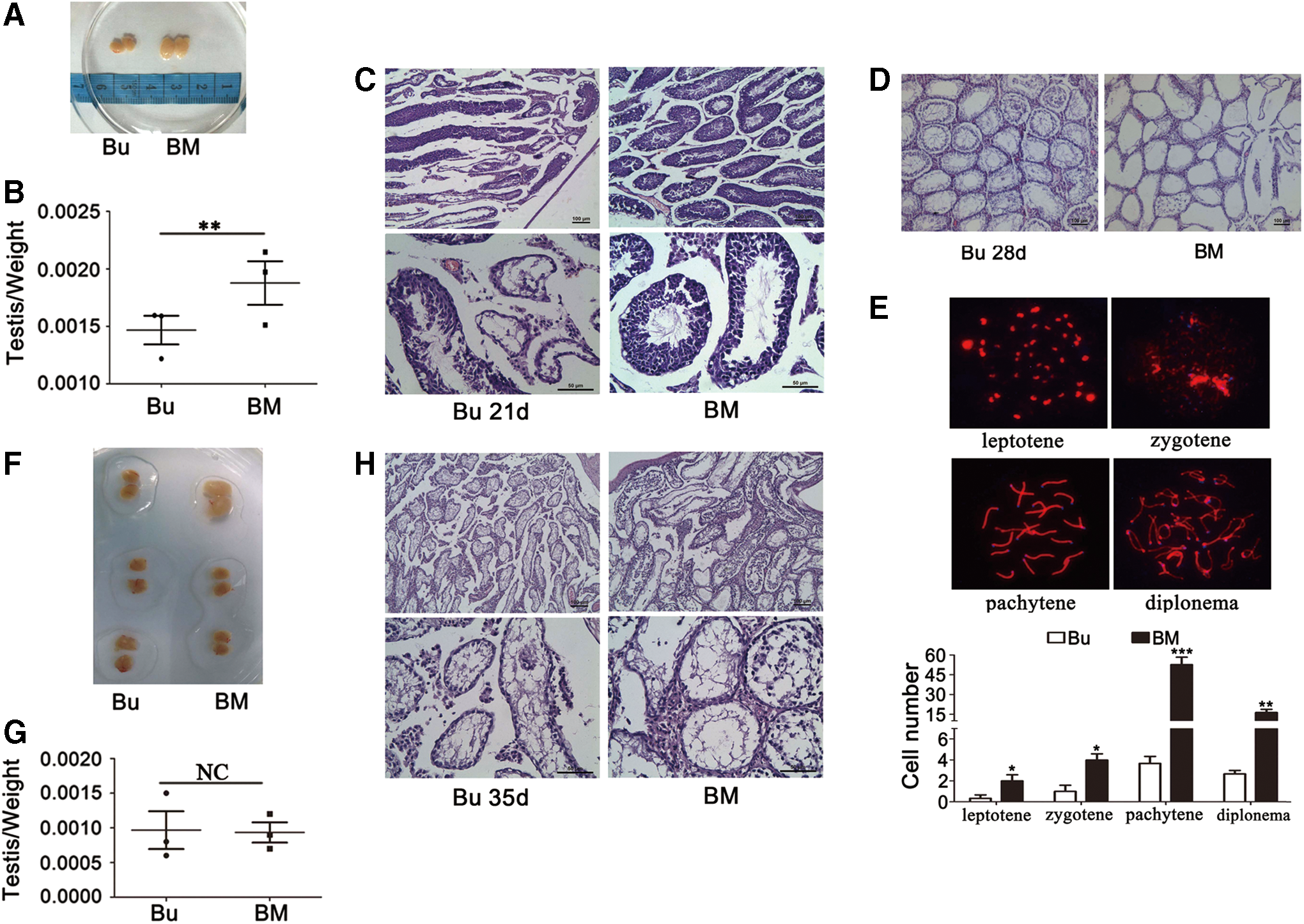
However, if melatonin was injected 2 weeks after busulfan treatment, curative effects of melatonin were significantly decreased. Specifically, SSCs were completely lost in all groups that received busulfan injections; however, spermatocytes exhibited partial survival in the mice that were administered melatonin (Fig. 5D).
These findings led us to use spermatocyte synaptonemal complex fluorescence immunostaining to examine meiosis in these cells, and there were few leptotene, zygotene, pachytene, and diplotene-stage spermatocytes at 28 days after busulfan treatment alone; however, these stages were identified in the melatonin-ameliorated groups (Fig. 5E). If we injected melatonin 21 days after busulfan treatment, the curative effect of melatonin was not observed. We identified no statistical differences in the testis/weight ratio (Fig. 5F, G) and HE staining (Fig. 5H).
Melatonin relieved busulfan-induced apoptosis in SSCs in contrast to cancer cells
Immunofluorescence staining demonstrated that PLZF-positive SSCs were increased in the BM group compared with the Bu group (Fig. 6A, B). We also demonstrated that melatonin cotreatment inhibited the activity of caspase3 (Fig. 6C). Moreover, Annexin V/propidium iodide (PI) staining was used to confirm the previous results, and busulfan induced 12.7% of SSC apoptosis, which was more than twofold higher than the control group. However, in the busulfan and melatonin groups, only 9.7% of cells were apoptotic (Fig. 6D). Taking into account that the percent of apoptosis was not substantially increased regardless of melatonin, we repeated the experiment three times; the statistical analysis indicated that there was a significant difference in apoptotic rates before and after melatonin treatment (Fig. 6E).

We used an acute T cell leukemia cell (ATLC) line referred to as J45.01 to determine whether melatonin affected the treatment effects of busulfan in cancer chemotherapy. In contrast to SSCs, melatonin did not ameliorate busulfan-induced apoptosis in the ATLC (Fig. 6F). To further determine why cancer cells were not sensitive to melatonin rescue, we used 2′,7′-dichlorofluorescein (DCF) staining to quantify the ROS levels of two kinds of cells. The data indicated that melatonin almost did not reduce the ROS in the ATLC (Fig. 6G).
Melatonin rescued busulfan-mediated ROS and mitochondrial dysfunction
According to our results (Figs. 3B and 6G), we hypothesized that ROS prevented apoptosis in busulfan-treated cells. Glutathione (GSH) is sensitive to intracellular ROS (48). The GSH/GSSG ratio indicates the status of the intracellular ROS balance. It was significantly decreased in the beginning of the busulfan treatment and gradually recovered to the normal level at 16 h after treatment; however, if we simultaneously added melatonin, the GSH/GSSG ratio was slightly decreased and promptly returned to the normal level within 1 h (Fig. 7A).
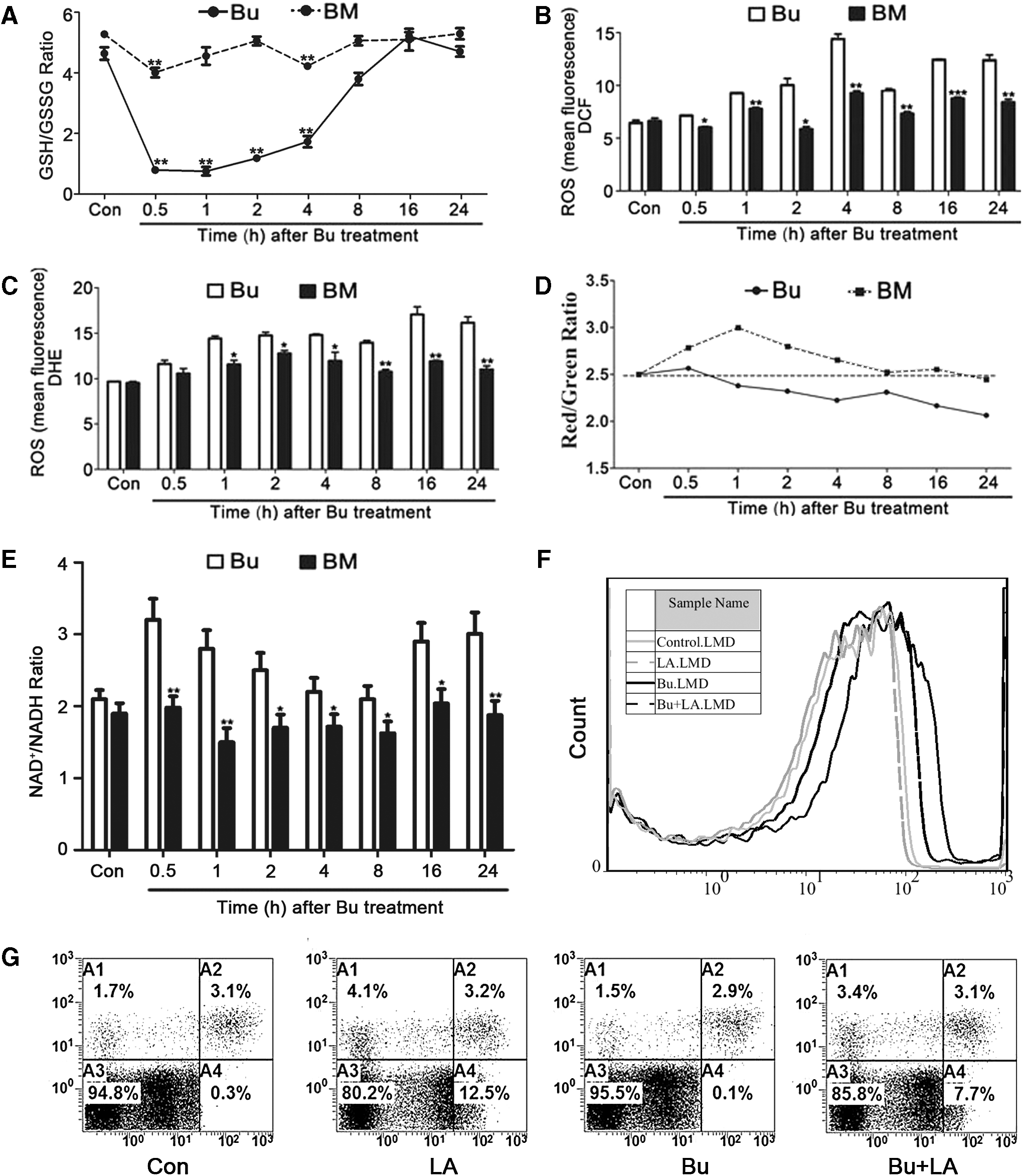
These findings suggest that GSH may compose the key factor in busulfan and melatonin treatment, which affects intracellular ROS. To assess the presence of ROS, the samples were stained with dichlorfluorescein diacetate (DCFH-DA) and dihydroethidium (DHE) to measure H2O2 and O2 •−, respectively, via flow cytometry. H2O2 and O2 •− are the main compositions of intracellular ROS. We demonstrated that intracellular ROS accumulation occurred in the busulfan treatment group and it was decreased in the melatonin-treated group (Fig. 7B, C).
In addition, we used cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazole-carbocyanine iodide (JC-1) to detect Ψm. The results indicated a trend of Ψm continuously decreasing after busulfan treatment; however, melatonin partially relieved this trend (Fig. 7D). The sustained reduction of Ψm is a marker of mitochondrial dysfunction.
The data demonstrated that melatonin rescued busulfan-mediated mitochondrial dysfunction. The NAD/NADH ratios in the Bu and BM groups are presented in Figure 7E. The ratio indicated a crosscurrent with intracellular ROS. We subsequently used alpha lipoic acid (LA) to confirm the involvement of ROS in busulfan-induced SSC apoptosis. Figure 7F indicated that LA treatment decreased the ROS levels in the busulfan and control groups. However, Figure 7G demonstrated that LA treatment only partially relived SSC apoptosis because ROS composed a critical mechanism to ameliorate busulfan-induced spermatogonial apoptosis. Compared with LA, melatonin activated the expression of SIRT1. Moreover, we determined that ROS and mitochondrial function were correlated in the presence of busulfan and melatonin treatments.
Melatonin increased MnSOD and SIRT1 expression; however, the expression of SIRT1 and acetylated p53 exhibited a strict reverse correlation
Based on the previous experiments and analyses, we focused on the mechanism of MnSOD and SIRT1. Western blot and qRT-PCR were applied to analyze the potential mechanism. The MnSOD expression was stimulated, transiently increased, and eventually returned to normal in the busulfan-treated groups; however, it constantly increased in the melatonin-treated groups (Fig. 8A, C–E). The SIRT1 expression was continuously suppressed; however, it significantly increased when busulfan was administered with melatonin compared with the group in which busulfan was administered alone (Fig. 8B–D, F). According to the classical theory, SIRT1 is a part of the p53 deacetylation complex (57).
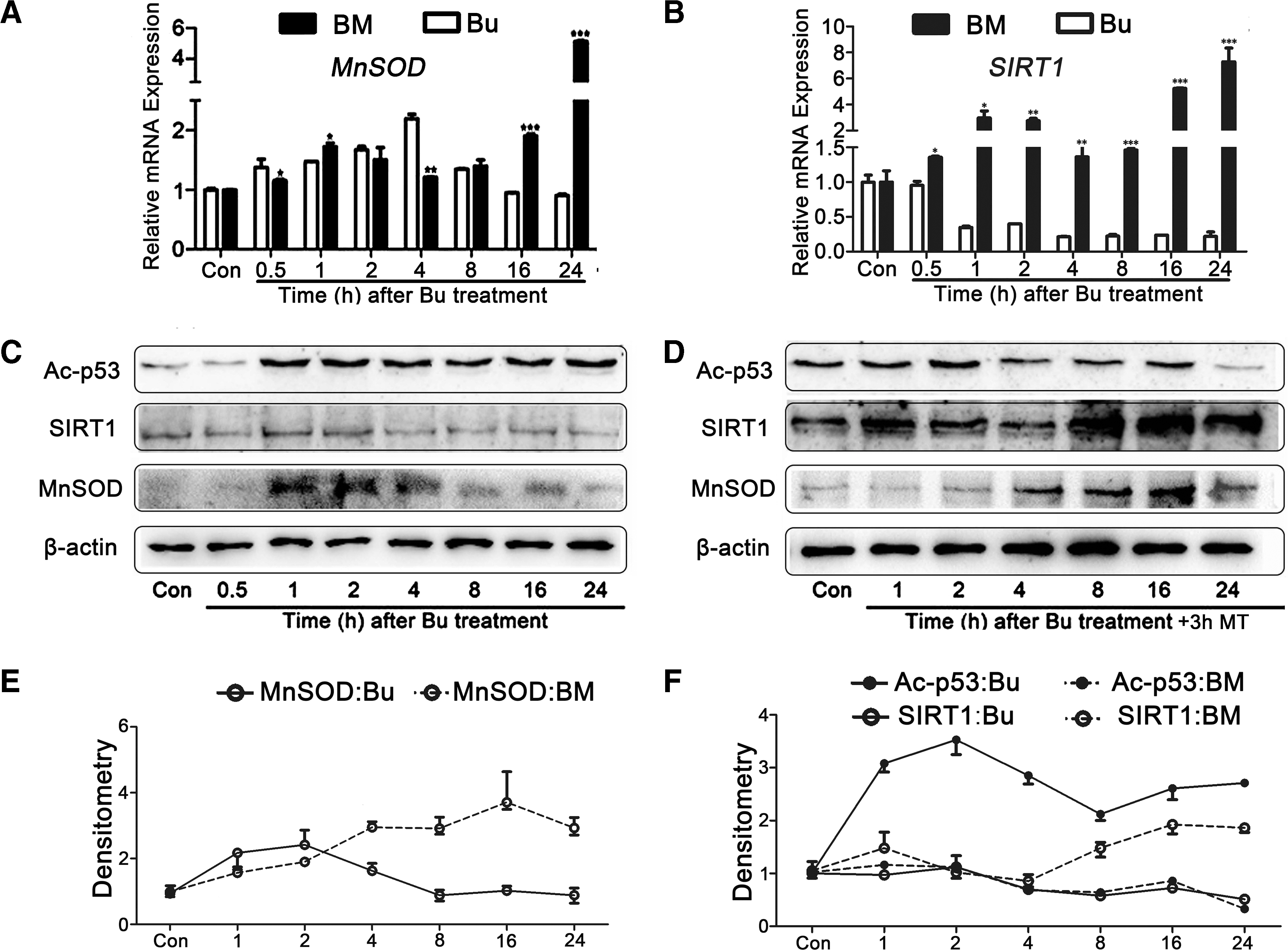
We also demonstrated that lower concentrations of acetylated p53 strictly appeared with increased SIRT1 expression, which demonstrated completely opposite trends (Fig. 8C, D, F). Therefore, SIRT1-mediated p53 deacetylation plays an important role in melatonin-relieved SSC apoptosis, which is caused by busulfan treatment.
Discussion
Alkylating chemotherapy agents, such as busulfan and temozolomide, do not have specific target proteins; however, they cause extensive ROS-mediated damage, which is the reason why they are used to treat middle and terminal cancers that exhibit multiple gene mutations (16, 40). This makes it difficult to alleviate their side effects because these drugs lead to extensive damage in their targets. Therefore, we measured melatonin-ameliorated effects in multiple directions, and the results indicated that melatonin effectively reduced spermatogenesis damage in busulfan treatment. The induction of cancer cell apoptosis is the fundamental reason to use chemotherapy drugs; thus, we should verify whether melatonin led the resistance to cancer cells in busulfan chemotherapy.
Busulfan is used in lymphoid and hematological tumors (53); our study also focused on adolescent males before sperm production and not sperm to be cryopreserved. Thus, we chose the J45.01 cell line, an ATLC line, which was established from the peripheral blood of a 14-year-old boy by Schneider et al. (47). We used this cell line as a parallel control experiment. In contrast to C18-4, an early apoptosis analysis indicated that melatonin could not ameliorate busulfan-induced J45.01 apoptosis (Fig. 6F).
According to previous literature, cancer cells have different ROS generation, elimination, and tolerance mechanisms compared with normal cells; it may be the reason why cancer cells were not sensitive to melatonin rescue. Therefore, we used dichlorfluorescein diacetate (DCFH-DA) staining to quantify the ROS levels of the two kinds of cells. The data exhibited a tendency consistent with previously described expectations (Fig. 6G).
Overall, the experimental results substantiate the feasibility of this research method. Thus, the current findings indicate that melatonin has the potential for use in the protection of spermatogonial damage in preadolescent male patients with chemotherapy in the future.
Evidence indicates that ROS levels are closely associated with increased levels of apoptotic events in busulfan-relieved SSC apoptosis. Moreover, melatonin effectively alleviated the extensive damage caused by busulfan via the elimination of ROS and repression of many apoptotic signaling pathways (Fig. 7). Our study demonstrated that busulfan caused extensive intracellular damage, which eventually led to apoptosis of spermatogonia.
Damages to different proteins or nucleic acid segments induce different results. However, damage to proteins that are essential for cell survival leads to serious consequences, such as apoptosis. For example, alkylating drugs may exhaust GSH as well as other antioxidants, which indicates that intracellular ROS will be out of control (12, 39). Excess ROS induced by mitochondrial damage results in the leakage of O2 •− from the respiratory chain; thus, the level of intracellular ROS is further increased (12, 14). This vicious circle eventually causes mitochondria-mediated apoptosis (56). Furthermore, we also demonstrated that the Erk/p38 signaling pathway and p53 led to significant changes in busulfan-treated animals, which may also represent the mechanism of apoptosis.
ROS were demonstrated to be the key mechanism of busulfan-induced damage to spermatogonia; thus, we investigated the use of melatonin, a certified ROS scavenger, to eliminate excessive production of ROS. The expression levels of MT1 and MT2 were increased in exogenous melatonin-treated mouse testes. In vivo experiments demonstrated that melatonin prevented the seminiferous tubule vacuolization and spermatogonial loss that results from apoptosis.
As ROS probes, DHE and DCF analyses indicated that melatonin significantly decreased intracellular O2 •− and H2O2, respectively, and the subsequent mitochondrial protection was confirmed via a JC-1 assay. Thus, we demonstrated that melatonin could break the vicious cycle of ROS production and mitochondrial damage. To assess the ROS scavenger effect of melatonin, we treated SSCs with melatonin alone. The results demonstrated that melatonin promoted MnSOD expression, which caused a decreased ROS concentration. In addition, the MnSOD expression was stimulated at a high level in the beginning of busulfan treatment, and melatonin ameliorated this effect and promoted MnSOD expression. Thus, we considered that melatonin-promoted MnSOD decreased the ROS caused by busulfan.
p53 also comprised a key protein in melatonin and prevented busulfan-induced apoptosis. In general, it is believed that mammalian cells degrade p53 through ubiquitination to maintain the appropriate p53 concentration (7, 24). P38 phosphorylates p53 to inhibit its ubiquitination, which increases the concentration of p53 (9, 55). SIRT1 is involved in the acetylation of p53, which changes the structure of the p53 protein so that p53 may be degraded via ubiquitination (25, 59). Melatonin was identified as a general inhibitor of the ubiquitin–proteasome system (42). Melatonin supplementation also enhances the efficiency of murine iPSC generation through inhibition of the p53-mediated apoptotic pathway (8).
Our study demonstrated that busulfan-induced ROS activated the Erk/p38 pathway, which increased the p53 concentration. However, the application of melatonin decreased the p53 concentration because it increased the SIRT1 expression. Therefore, p53 is an important protein in regard to the effects of melatonin and busulfan (Fig. 9).

Overall, we attempted to investigate the potential clinical function of melatonin and the related mechanism. Busulfan remains commonly used as a cytotoxic drug, particularly in anticancer and immunosuppressive therapies (5, 13, 21). Previous studies have demonstrated that melatonin exerted a protective effect against acrylamide-induced oxidative damage and alleviated acrylamide-induced lipid peroxidation and depressed the antioxidant capacity (1, 36). Therefore, these findings provide a reference to relieve the side effects of other alkylating drugs, such as cyclophosphamide.
Melatonin treatment modulated the stem cell properties of glioblastoma-initiating cells and induced cell death with ultrastructural features of autophagy (60). We demonstrated that melatonin prevented the spermatogonial loss caused by busulfan treatment as well as apoptosis through a complex mechanism. Animal and human studies have demonstrated that the short-term use of melatonin is safe, even in extreme doses (2). Thus, the study provided preclinical references for preadolescence males who suffer from exposure to alkylating agents during leukemia treatment.
In conclusion, melatonin ameliorated busulfan-induced SSC damage in mice in vivo and in vitro. Melatonin not only reduces the spermatogonial injury side effects of busulfan myeloablative pretreatment for preadolescence males before Hematopoietic stem cell transplantation (HSCT) but also provides a reference to relieve the side effects of other alkylating drugs.
Materials and Methods
Animals and animal experimental protocol
All experiments were performed on healthy, adult male Institute of Cancer Research (ICR) mice that weighed between 25 and 30 g. The mice were obtained from the animal center of the Fourth Military Medical University. The mice were housed in wire cages at 25°C under a 12-h light–12-h dark cycle at 70% humidity and fed ad libitum. The maintenance of animals and completion of experiments were in accordance with the Guide for the Care and Use of Laboratory Animals at Northwest A&F University. Eight-week-old ICR male mice received a single intraperitoneal injection of busulfan (30 mg/kg body weight) diluted in dimethyl sulfoxide (DMSO). Melatonin was dissolved in 1% ethanol (in normal saline), and 10 mg/kg/day of melatonin solution was intraperitoneally administered to the mice treated with busulfan for 0, 1, 2, and 3 weeks. Sixty mice were randomly assigned to three groups.
The Bu group
Twenty-four mice received a single intraperitoneal injection of busulfan as previously described and were subsequently randomized into six groups; each group comprised four mice. We collected samples at 2, 5, 8, 21, 35, and 56 days after injection.
The Con group
Six mice received a single intraperitoneal injection of normal DMSO.
The BM group
Thirty mice received a busulfan treatment and were then randomly allocated to six groups. Based on the previously described groups, the mice were injected with melatonin 0, 7, 14, and 21 days after busulfan treatment.
Cells and cell experimental protocol
C18-4 cells were cultured with Dulbecco's modified Eagle's medium/nutrient mixture F12 (DMEM/F12; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM L-glutamine (Invitrogen), and 100 U/ml penicillin and streptomycin (Invitrogen) (52). GC1 cells were cultured with DMEM (Gibco) supplemented with 10% FBS (Gibco), 2 mM L-glutamine (Invitrogen), and 100 U/ml penicillin and streptomycin (Invitrogen). MLTC1 cells were cultured with 1640 Medium (RPMI-1640; Gibco) supplemented with 10% FBS (Gibco), 2 mM L-glutamine (Invitrogen), and 100 U/ml penicillin and streptomycin (Invitrogen). J45.01 cells were cultured with 1640 medium (RPMI-1640; Gibco) supplemented with 10% FBS (Gibco) and 2 mM L-glutamine (Invitrogen).
Cells were subcultured every 3–4 days and maintained at 37°C in a humidified 5% CO2 incubator. Cells were dissociated via treatment with 0.25% trypsin-EDTA (Invitrogen) and reseeded into multiwell plates for the subsequent stage of the analysis. The medium was changed every 2 days. Cells were cultivated for 24 h before the experiments, and the cell density was ∼5 × 104/cm2.
The maintenance of animals and completion of experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals at Northwest A&F University.
The busulfan used in this step was dissolved in DMSO to 10−4 μM. Melatonin was dissolved in 1% ethanol in physiological saline to 10−7 M. Luzindole and PD0325901 were dissolved in DMSO to 10−5 M and 2.5 μM, respectively.
Immunofluorescence and immunohistochemical staining
Immunofluorescence staining was performed as previously described. Immunohistochemical staining was performed according to the manufacturer's instructions (DAB Detection Kit Streptavidin-Biotin; ZSGB-BIO). All information regarding the antibodies is listed in Table 1.
ERK, extracellular signal-regulated kinase; MnSOD, manganese superoxide dismutase; MT1 and MT2, melatonin receptor 1 and melatonin receptor 2; SIRT1, sirtuin type 1.
TUNEL assay
Testes were fixed in 4% (w/v) paraformaldehyde in 0.01 M phosphate-buffered saline (PBS) (pH 7.4), washed in PBS, dehydrated in ethanol (70%, 90%, and 100%), and embedded in paraffin wax. Testicular sections (5 μm) were rehydrated (xylene 5 min; ethanol 100%, 95%, 70%, 5 min each) and washed in distilled water before TUNEL staining. The sections were incubated for 15 min with proteinase K (20 μg/ml) at room temperature. The sections were rewashed three times with PBS (1 × ) and incubated for 60 min at 37°C in a moist chamber with 50 μl of TUNEL mix (5 μl of TdT and 45 μl of fluorescein-dUTP). The negative control only contained 50 μl of fluorescein-dUTP.
After three washes with PBS for 5 min each at room temperature, the sections were incubated with Hoechst 33342 for 10 min at room temperature. After three PBS washes, slides were mounted using a crystal mount (Biomeda).
Analysis of cell proliferation
Cell proliferation was assessed using a Cell Counting Kit-8 (CCK-8; Vazyme Biotech). Ten microliters of the CCK-8 solution was added to each well of a 96-well plate, and cells were incubated at 37°C for 1 h. The absorbance was determined at 450 nm using a microplate spectrophotometer (BioTek).
GSH and GSSG assay kits
The total GSH and oxidative GSH (GSSG) levels were measured using GSH and GSSG assay kits, respectively (S0053; Beyotime Institute of Biotechnology), via the colorimetric microplate assay and according to the manufacturer's protocol (12).
qRT-PCR analysis
Total RNA was extracted from cells using TRIzol reagent (Qiagen) according to the manufacturer's instructions. Total RNA was reverse transcribed into cDNA using an M-MLV Reverse Transcriptase Reagent kit (Thermo Scientific) according to the manufacturer's instructions. qRT-PCR analysis was conducted in triplicate on a CFX96 Real-time PCR system (Bio-Rad Ltd.) at a final volume of 15 μl. Each reaction contained 0.5 μl of cDNA (1:10 diluted), 7.5 μl of SYBR (Bioer Co. Ltd.), 6.3 μl of ddH2O, 0.3 μl of forward primer, 0.3 μl of reverse primer, and 0.1 μl of Taq DNA polymerase.
The β-actin expression was used as the housekeeping control. The qRT-PCR procedures were set as follows: 5 min at 94°C, followed by 40 cycles of amplification that consisted of denaturation for 20 s at 94°C, 30 s at 58°C for annealing, and 10 s at 70°C for elongation. The comparative CT method was used to measure the relative gene expression. The primers are listed in Table 2.
qRT-PCR, quantitative real-time polymerase chain reaction.
Western blot analysis
The testes or cell samples were collected, and protein extractions were obtained after surgery. The proteins were quantified using a BCA Protein Assay kit (Beyotime). The proteins were separated using SDS-PAGE and subsequently transferred to Immobilon NC membranes (Millipore). The membranes were blocked with 5% nonfat milk in TBST at room temperature and subsequently incubated with primary antibodies overnight at 4°C (Table 2), followed by three washes with TBST for 15 min each. The membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (1:3000) at room temperature for 1.5 h and washed with TBST three times for 15 min each time. Protein bands were detected using a Bio-Rad imaging system (Bio-Rad) and quantified using the Quantity One software package.
Cell cycle and apoptosis assay via flow cytometry
Cell cycle distribution was assessed via PI staining. Adherent cells were dissociated with 0.25% trypsinization, and detached cells were fixed in 70% ethanol at 4°C for 24 h. After being washed with PBS, the samples were stained with 50 μg/ml PI and 50 μg/ml RNase A at 37°C for 20 min and subsequently analyzed via flow cytometry (Altra; Beckman Co.). At least 5000 cells were collected per sample (11).
The distribution of cell apoptosis was assessed using Annexin V-FITC and PI staining. Cells were harvested and washed in cold PBS and cold binding buffer (1 × ). They were subsequently resuspended in cold 1 × binding buffer at 1 × 106 cells/ml. One-hundred microliters of cells (1 × 105 cells) was subsequently added to each labeled tube, and 5 μl of Annexin V-FITC was added to the appropriate tubes. Each tube was gently vortexed and incubated for 10 min at room temperature. Five milliliters of PI solution was subsequently added, and the tubes were kept still for 5 min at room temperature in the dark. Before the analysis, the cells were washed once with PBS and were subsequently resuspended in PBS. A flow cytometer was used to perform the experiment (Beckman Altra; Beckman Co.) (11).
Determination of ROS generation
C18-4 cells were washed in PBS before the addition of 50 μM DCFH-DA (ROS Assay Kit; Beyotime) in PBS; the cells were subsequently incubated in the dark for 20 min at 37°C. The cells were then washed twice with PBS and quantified using flow cytometry to measure mean fluorescence (Altra; Beckman Coulter) at excitation wavelengths of 488 and 530 nm emissions (12).
Mitochondrial membrane potential (Ψm) assay
The mitochondrial membrane potential was determined using the dual-emission, mitochondrion-specific lipophilic JC-1. Punctate red fluorescence (excitation 530 nm/emission 600 nm) represents the potential-dependent aggregate form of JC-1 in the mitochondria of healthy cells (polarized mitochondria). Diffuse green fluorescence (excitation 490 nm, emission 530 nm) represents the monomeric form of JC-1 in the cytosol of unhealthy cells (depolarized mitochondria).
Cells grown on coverslips were incubated in JC-1 (10 μg/ml) at 37°C for 15 min and washed in PBS; they were subsequently mounted on slides for observation with a Leica microscope equipped with an on-stage incubator (20/20 Technologies) for imaging. The TRITC and GFP filter sets (Semrock) were used to detect depolarized and repolarized mitochondria, respectively. Both color channels were overlaid in IPLab software (Becton Dickinson) to measure the distributions of repolarized and depolarized mitochondria in the field (12).
Immunohistochemistry and fluorescence immunostaining
Testicular tissues were shredded, and the released cells were spread evenly over microscope slides layered with a 1% paraformaldehyde solution (pH 9.2; Sangon) and Triton-X (Sangon). The slides were placed in a humid chamber for ∼24 h at room temperature and subsequently air-dried for ∼30 min. The dried slides were washed for 4 min in 0.04% Photo-Flo (Kodak), air-dried for 10 min, incubated for 30 min in DAB (Beijing Chinese Golden Bridge Biotechnology Co., Ltd.), drained but not allowed to dry, and overlaid with mixed primary antibodies: rabbit anti-MT1 (1:100; Bioss), rabbit anti-MT2 (1:100; Bioss), rabbit anti-SYCP3 (1:300; Abcam), rabbit anti-PLZF (1:100; Bioss), rabbit anti-SOX9 (1:100; Bioss), and human anti-CREST (1:800; Immuno Vision).
The primary antibodies were detected using secondary antibodies conjugated to the following fluorophores: Alexa555 donkey anti-rabbit (1:250; Molecular Probes) and 1-amino-4-methylcoumarin-3-acetic acid (AMCA) donkey anti-human (1:200; Jackson Immuno Research). The primary and secondary antibodies were incubated overnight at 37°C and for 90 min at 37°C, respectively. Twenty microliters of Vectorshield (Vector Laboratories) was applied per slide, and a coverslip was sealed onto the slide.
Caspase3 activity assay
Caspase3 activity was measured using a Caspase3 Activity Assay Kit (C1115; Beyotime) according to the manufacturer's instructions. The release of free p-nitroaniline (pNA) from caspase3 activity was colorimetrically monitored at 405 nm for 1 h using a microplate reader. Thus, caspase3 activity was detected by the pNA concentration (nmol)/protein (mg). The control group was set as 1 to measure the relative caspase3 activity of each group.
Statistical analysis
One-way analysis of variance (ANOVA) was used, and post-tests were conducted using Newman–Keuls multiple range test if the p-values were significant. Student's t-test was used when only two pairs of data were compared. All data are represented as the mean and SD, and the statistical significance is expressed as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data are representative of at least three different experiments and were analyzed using GraphPad Prism software.
Footnotes
Acknowledgments
This work was supported by grants from the China National Basic Research Program (JFYS 2016YFA0100203), the General Programs of Natural Science Foundation of China (31272518, 31572399), the National Major Project for Production of Transgenic Breeding (2014ZX08007-002), National High Technology Research and Development Program of China (SS2014AA021605), the Key Project of Chinese Ministry of Science and Technology (2013CB967401), and the Key Project of Shaanxi Province of Science and Technology (2015NY157).
Author Disclosure Statement
No competing financial interests exist.