
Abstract
Significance:
Haptoglobin (Hp) is an abundant human plasma protein that tightly captures hemoglobin (Hb) during hemolysis. The Hb-Hp complex formation reduces the oxidative properties of heme/Hb and promotes recognition by the macrophage scavenger receptor CD163. This leads to Hb-Hp breakdown and heme catabolism by heme oxygenase and biliverdin reductase. Gene duplications of a part of or the entire Hp gene in the primate evolution have led to variant Hp gene products that collectively may be designated “the haptoglobins (Hps)” as they all bind Hb. These variant products include the human-specific multimeric Hp phenotypes in individuals, which are hetero- or homozygous for an Hp2 gene allele. The Hp-related protein (Hpr) is another Hp duplication product in humans and other primates. Alternative functions of the variant Hps are indicated by numerous reports on association between Hp phenotypes and disease as well as the elucidation of a specific role of Hpr in the innate immune defense.
Recent Advances:
Recent functional and structural information on Hp and receptor systems for Hb removal now provides insight on how Hp carries out essential functions such as the Hb detoxification/removal, and how Hpr, by acting as an Hp-lookalike, can sneak a lethal toxin into trypanosome parasites that cause mammalian sleeping sickness.
Critical Issues and Future Directions:
The new structural insight may facilitate ongoing attempts of developing Hp derivatives for prevention of Hb toxicity in hemolytic diseases such as sickle cell disease and other hemoglobinopathies. Furthermore, the new structural knowledge may help identifying yet unknown functions based on other disease-relevant biological interactions involving Hps. Antioxid. Redox Signal. 26, 814–831.
Discovery
I
Expression and Regulation
Hp is expressed in many tissues and cell types, but, as other acute phase proteins, the liver is the quantitative major source. Hp is among the predominant plasma proteins next to the very abundant albumin and immunoglobins. The normal Hp plasma concentration is in the range of 0.5–3 g/L, which corresponds to Hb binding capacity of 0.3–1.8 g/L. The Hp concentration may increase substantially during acute inflammation in response to the acute phase mediator interleukin-6 (IL-6) and other cytokines (125, 140). Conversely, the concentration may drop to virtually zero during accelerated hemolysis owing to receptor-mediated removal of Hp in complex with Hb. In the clinic, Hp is therefore both a marker for the acute phase during infection (increased plasma Hp) and for hemolysis of any cause (decreased plasma Hp). Hpr is also expressed in the liver, but the plasma levels are more than 10-fold lower, probably because Hpr expression is attenuated by a retroviral-like element present in intron 1 (73). Unlike Hp, the Hpr plasma concentration is not affected by intravascular hemolysis because the Hb-Hpr complex does not bind the receptor CD163 (122). Due to the similarity of Hp and Hpr, immune-based assays in the routine clinic may not distinguish between Hp and Hpr, but rather provide a measure of the total concentration of the two Hps (122).
The Hp Genes and Evolution
The human gene encoding Hp exists as two major allelic forms, Hp1 and Hp2 (101, 139, 194). The Hp2 gene arose by a nonhomologous crossing-over event (101). This occurred after the divergence of humans in primate evolution. However, early human hominins such as Homo neanderthalensis and Homo denisova are also reported to have the Hp2 gene, and evidence is now provided that the present Hp1 gene variants in Homo sapiens arose from recurrent deletions in Hp2 (21).
Accordingly, three human major Hp genotypes exist, Hp1/Hp1 , Hp1/Hp2 , and Hp2/Hp2 (160, 161). The frequency of the Hp genotypes shows a high degree of geographical variation, with Hp1/Hp1 as the most prevailing genotype in South America, while Hp2/Hp2 is the major genotype in Southeast Asia (32). Outside these regions, the Hp1 and Hp2 alleles are more or less equally distributed. The differences in Hp gene allele frequencies may be a result of both genetic drift and natural selection (32). Both Hp1 and Hp2 have been linked to susceptibility to various diseases as discussed below and previously specifically reviewed (32, 93, 177). The molecular background for these associations is unknown. A similar but independent duplication event has given rise to an Hp2 allele in deer and cows (suborder Ruminantia) (190). The fact that this duplication event has occurred more than once suggests that the presence of the Hp2 allele may have been advantageous at some point in evolution.
The Hp gene encodes a polypeptide chain constituting a complement control protein (CCP) domain (also referred to as SUSHI) followed by a serine protease (SP) domain (Fig. 1) (62, 66, 90). This CCP-SP domain arrangement is also found in complement factors C1r and C1s which initiate the complement cascade through the proteolytic activity of the SP domains (42, 63). Hp has probably evolved from C1s by the loss of proteolytic activity and gain of a new function in Hb binding (4, 172). The duplicated region of the Hp2 allele corresponds exactly to the CCP domain. Consequently, the Hp2 gene encodes a peptide chain comprising two consecutive CCP domains followed by an SP domain, while the Hp1 gene product only contains a single CCP domain and an SP domain (Fig. 1).

The Hpr gene is located, 2.2 kb downstream of the human Hp gene (15, 100, 139). The Hp1 and Hpr genes are highly similar and encode proteins with >90% sequence identity (Fig. 1). The Hpr gene is only present in primates and appears to be the result of a gene triplication after the divergence of new world monkeys from other primates (105). Subsequently, one locus was deleted in humans leaving only two genes in the Hp gene cluster.
The Hp gene appears to be present only in mammals and bony fish. Birds and reptiles express a protein called Pit54, which is functionally related to Hp in the sense of a high affinity Hb binding and protection against heme-induced oxidative damage during intravascular hemolysis (78, 190). Surprisingly, the Pit54 gene encodes a protein composed of four scavenger receptor cysteine-rich (SRCR) domains, which are structurally related to the ectodomain of the Hb-Hp receptor CD163 (78).
Structure
Hp is expressed as a single polypeptide chain, including an 18 amino acid signal peptide, which is cleaved off in the endoplasmic reticulum (70). The core glycosylated prohaptoglobin is subsequently proteolytically cleaved into an α- and a β-chain by the C1r-like protein (189). The cleavage that occurs at the N-terminal side of Ile162 (numbering according to
Dimeric Hp (Hp1-1), which is the Hp1 gene product, has an overall structure resembling a barbell or a two-blade propeller that is ∼14 nm in length (Fig. 2). The Hp CCP domains constitute a central core that connects two oppositely positioned Hp SP domains (4, 187). Both electron micrographs and crystal structures indicate that the Hp dimer is a rigid structure possibly stabilized by intramolecular interactions between the CCP and SP domains. Hb dimers bind the Hp SP domains in the distal ends resulting in an Hb-Hp complex ∼18 nm in length.
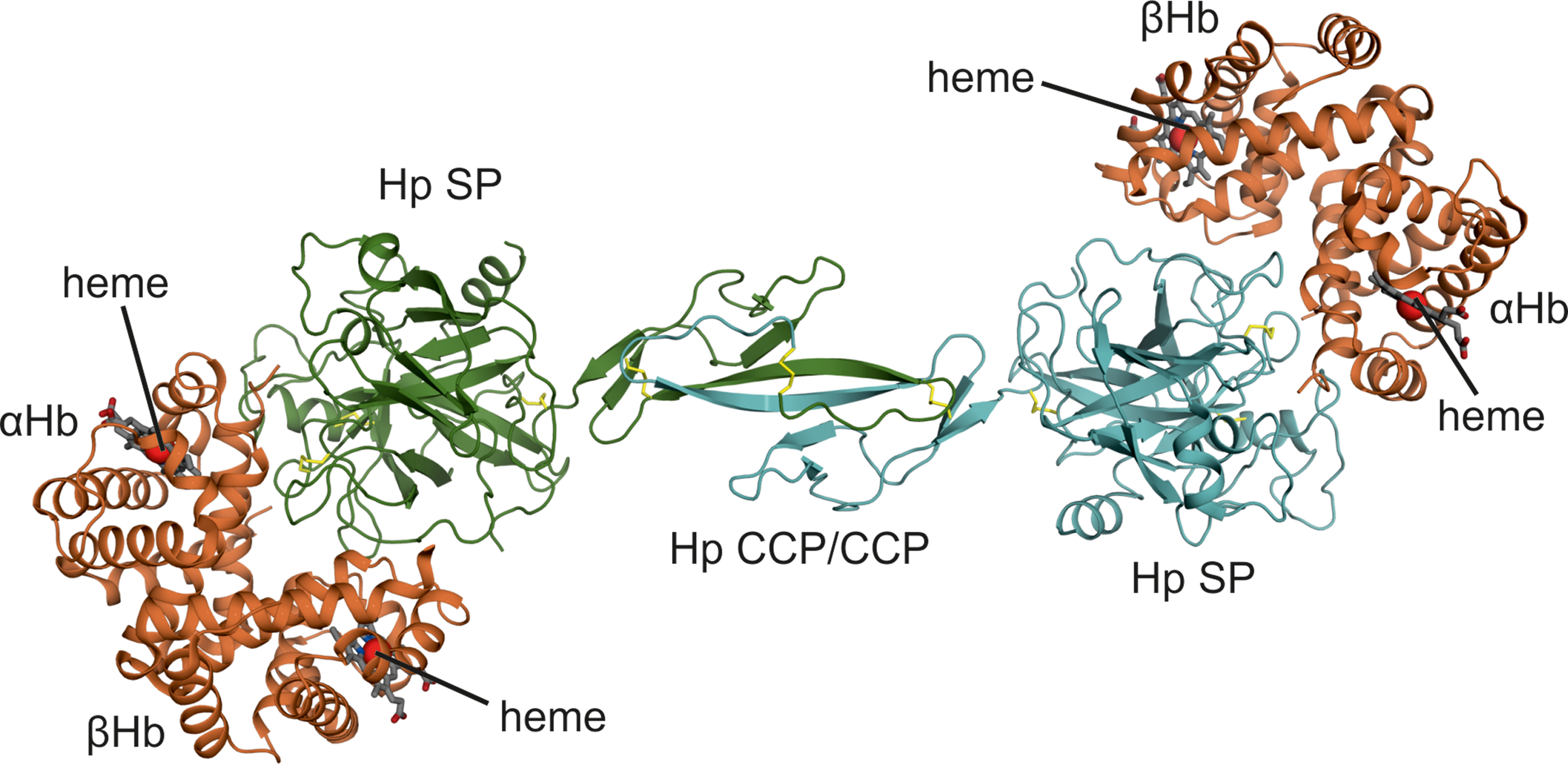
The Hp CCP domains have a β-sandwich arrangement similar to CCP domains in C1r/C1s (26, 27). However, Hp CCP domains dimerize through a beta-strand swap not previously observed for CCP domains (4, 167). The product is a fusion CCP domain structure with a central six-stranded beta-sheet.
The Hp SP domain has the typical fold of chymotrypsin-like SPs with two β-barrel subdomains each containing six β-strands and two or three α-helices (4, 92, 167). Several surface exposed loops differ in length and conformation compared to SPs. In particular, the region, designated loop 3, is significantly extended in Hp. This region protrudes from the surface of Hp and contains residues important for Hb-Hp recognition by the macrophage receptor CD163 (119, 123). The Hb binding site on Hp resides in several surface-exposed loops of the SP domain. Although Hp is not an active protease, the Hb-binding site is located in the regions responsible for substrate binding in other SPs, suggesting that the Hb-Hp interaction may originate from an enzyme–substrate complex (4).
Hp makes extensive interactions with both the α- and β-subunit of dimeric Hb, explaining the high-affinity interaction between the proteins. The Hp binding site involves many of the functionally critical residues surrounding the Hb heme-binding pocket. In addition, the Hb-Hp interface overlaps with the dimer–dimer interface in tetrameric Hb, which explains why Hp only binds dimeric Hb and not tetrameric Hb.
Hp Variants
The expression of the Hp2 gene in individuals with the Hp1/Hp2 and Hp2/Hp2 genotypes causes Hp to form higher order oligomers as originally observed by Smithies (160, 161). These oligomers are formed by pairwise joining of CCP domains via β-strand swapping. In addition, cysteine residues located in the N-terminus of the CCP domains covalently link the domains by a disulfide bond, giving rise to the characteristic ladder observed in nonreducing SDS-PAGE (Fig. 3). However, dimerization of CCP domains probably also occurs in the absence of the crosslinking disulfide bond because Hpr without this disulfide bond also dimerizes (4, 122). In the Hp2/Hp2 genotype, the tandem CCP domains in Hp2 allow each Hp2 unit to connect to two other Hp2 units resulting in a circular arrangement of CCP fusion domains (Fig. 4) (187). The most abundant Hp2 oligomer is a tetramer, but trimers and higher order oligomers are also present (Fig. 3). The presence of both Hp1 and Hp2 alleles (Hp1/Hp2 genotype) results in formation of both circular (Hp2-2) and linear Hp (Hp2-1) oligomers (Fig. 4). The linear Hp oligomers are formed by consecutive Hp2 units terminated at both ends by an Hp1 unit (75, 187). The minimal oligomer in the Hp1/Hp2 genotype is a dimer identical to the Hp1-1. Models of different Hp oligomers have been created based on the crystal structure of dimeric Hp (4) and validated by refined electron microscopy (Fig. 5) (Stødkilde et al., unpublished data) using more advanced analysis tools for high resolution imaging than available at the first original electron microscopic studies by Wejman et al. (187, 188). The model of the Hp2-2 trimer showing a three-bladed propeller structure (Fig. 5C) is in almost perfect agreement with single particle reconstruction of negative stain electron microscopy images of purified Hp2 from human plasma (Fig. 5B).



Hpr most likely has a structural fold similar to Hp1 and probably also dimerizes via fusion of two CCP domains. The Hb-binding region is almost completely conserved and so is the region for binding to a trypanosome parasite Hb-Hp receptor that binds this complex with the aim to obtain heme. In contrast, a high variation is seen in the Hp loop 3 region, which is involved in the binding of Hb-Hp to CD163. According to these structural features, Hpr binds Hb with high affinity (122) and the Hb-Hpr complex binds to the trypanosomal receptor (167). In contrast, Hb-Hpr does not bind to the scavenger receptor (SR) CD163, which precludes it from bloodstream clearance by CD163-expressing macrophages during hemolysis (119, 123). These differences enable Hpr to undertake an entirely different function in innate immunity toward infecting trypanosome parasites as discussed below. Hpr also differs from Hp by retaining its hydrophobic signal peptide after secretion. This allows Hpr to associate with a specialized high-density lipoprotein particle known as trypanosome lytic factor-1 (TLF-1) (71).
In addition to the genetic duplication of the ancestral Hp1 gene, different posttranslational modifications of Hp have also led to variant Hp proteins. One example is a hyperglycosylated form of Hp expressed in granulocytes, which store the protein in specific granules that are released upon activation (169). The role of the glycan moiety accounting for almost half of the mass of the Hp β-chain is so far unknown, but this hyperglycosylated variant binds Hb with high affinity. Another Hp variant is the protein designated “zonulin,” which is identified as preHp2. Zonulin/preHp2 differs from Hp2 by not being cleaved into α- and β-chains (60) and has been reported as a regulator of intestinal permeability by modulating tight junctions in the intestinal epithelium (59). According to ELISA measurements in 76 healthy individuals using antibodies that specifically recognize zonulin/preHp2, the protein is not detectable in some individuals (probably those with the Hp1/Hp1 genotype) and only present in μg/L levels in others (probably those with the Hp1/Hp2 or Hp2/Hp2 genotypes) (60). The fact that a large part of the human population has the Hp1/Hp1 genotype, and therefore cannot express Hp2, makes it in our view questionable whether zonulin/preHb2, has any essential physiological function.
Hp in the Defense Against Hb Toxicity
Hp is a detoxifier of Hb when red blood cells lyse within the vascular compartment (2). Under normal physiological conditions, Hb is bound and stabilized by Hp and subsequently cleared from circulation by the macrophage-specific receptor CD163 (89). However, in certain hemolytic conditions, such as sickle cell anemia, malaria, trauma, and severe infections, serum Hp is depleted leading to accumulation of free Hb in plasma.
The major problem connected to release of Hb from red blood cells is the oxidative properties of the Hb heme group, which can be oxidized in the presence of H2O2 (1, 115, 141). Ferric Hb (Fe3+) reacts with H2O2 to form an oxyferryl compound (Fe4+ = O2−) and a globin radical (81). The globin radical can be transferred to other molecules in close proximity, such as lipoproteins (e.g., low-density lipoprotein [LDL]) and cell membrane lipids. As a consequence this leads to cell damage and tissue injury (106). In addition, the globin radical can induce structural modifications within Hb [e.g., amino acid oxidation and globin crosslinking (81)]. This Hb-catalyzed self-destruction incapacitates the clearance mechanisms with an accumulation of toxic Hb degradation products as a result. Numerous oxidative modifications of the Hb globin chains have been identified and characterized (81, 134, 176). Of note, many of the H2O2-mediated oxidative changes of Hb are merely based on in vitro studies, and the physiological impact is unknown. However, such changes may occur close to activated granulocytes bursting H2O2 and other reactive oxygen species. Granulocytes also secrete Hp as described above. When Hb is bound by Hp, the oxidative damage in cells and tissues is prevented. However, binding of Hp to Hb does not appear to inhibit the oxidative properties of the Hb heme group and as a consequence radicals are still formed within the Hb-Hp complex (28, 176). Several of the Hb residues prone to oxidative modification are located within the Hb-Hp interface, and the protective role of Hp may be achieved through shielding and stabilization of the radicals on Hb (2, 4, 11, 35). Although it has been speculated that Hp acts as a radical sink that scavenges the free radicals from Hb (11, 28), no radicals have ever been detected on Hp, indicating that this may not be the case (176). It has also been speculated that Hb residues αTyr42 and βCys93 represent a pathway for migration of radicals from α1Hb to β2Hb (or from α2Hb to β1Hb) as these residues are located in close proximity in tetrameric Hb (134). Accordingly, Hp may serve as a restrictor of radical migration within Hb by preventing association of the Hb tetramer (4). Hp also protects against the H2O2-mediated destruction of Hb, which involves globin crosslinking, subunit dissociation, and heme release (28). This is probably achieved through the almost irreversible interaction of Hp with both Hb subunits. Furthermore, Hp binds close to the heme groups and it may stabilize this region of the globin moiety, which in turn may prevent heme release (4). This, for instance, reduces the transfer of the hydrophobic heme molecule from Hb to cell plasma membranes and the lipid layer of lipoprotein particles, where heme-mediated chain reactions can be triggered by lipid peroxides (80).
Plasma Hb also affects the bioavailability of nitric oxide (NO), a signaling molecule that controls many important functions, including smooth muscle relaxation and platelet aggregation (126, 150). NO reacts irreversibly with Hb to produce nitrate and methemoglobin. If the Hb-mediated consumption of NO is allowed to occur, it will disturb the NO homeostasis and cause vasoconstriction and hypertension (143). As with oxidation of Hb by H2O2, Hp-binding does not affect the Hb-mediated consumption of NO (9). On the contrary, the molecular mass of Hb has an impact on its vasoconstrictive activity, suggesting that extravasation and diffusion are factors that determine the vasoconstrictive effect of Hb (30, 114, 173). Hence, Hp probably functions as an attenuator of Hb-mediated NO consumption by increasing the mass of Hb. In general, the increased mass of the Hb-Hp complex may be an important contributor to Hp-mediated protection against toxic Hb, as it sequesters Hb within the intravascular space and prevents its filtration in the kidney and across the endothelial layers in other organs (58). In this way, Hp blocks Hb from entering areas, such as the kidneys, sensitive to oxidative damage, but instead keeps it accessible for clearance by macrophages.
The Hp-CD163-Heme Oxidase Axis for Hb Detoxification
The complex formation between Hp and Hb also promotes the removal of Hb from plasma via the macrophage SR CD163 that mediates a rapid endocytosis of the complex (Fig. 6) (89). This pathway parallels the phagocytosis of senescent red cells in M2-type macrophages, a process that ultimately also leads to lysosomal degradation of Hb. This process is followed by heme release into the cytosol and heme oxygenase-catalyzed conversion of heme to biliverdin that is further converted to bilirubin (56). The bilirubin is then exported from the macrophage and escorted by albumin for conjugation in the hepatocytes and subsequent biliary excretion. Owing to the high concentration of circulating Hp, the plasma compartment has a capacity for neutralization of many grams of Hb under a sudden hemolysis. The maximum clearance in two humans injected with Hp-saturating amounts of Hb has been measured to 0.13 g/h/L (94). Such a clearance rate, that corresponds to about 0.2 g Hp/h/L if fully saturated with Hb, may lead to complete consumption of Hb within hours (25). This explains that Hp is virtually absent in plasma in patients with severe hemolysis. The Hb removal under normal physiological hemolysis is far lower than the maximum capacity. About 0.5–1 g is removed by Hp under the assumption that a little less than 1% of red cells are exchanged per day and intravascular hemolysis accounts for 10% of the daily red cell destruction. The half-life of Hb-Hp complexes in the circulation may depend on the degree of hemolysis. In mice, the half-life of trace amount Hb-Hp has been measured to few minutes (54), and in humans, the half-life of Hb-Hp amounts below the maximum clearance capacity have been measured to 10–30 min (64, 86, 94). However, the injection of Hp-saturating amounts of Hb in humans (94) and dogs (22) shows an Hp half-life of several hours. Such a dose dependency of the clearance rate indicates that clearance of Hb-Hp occurs via a saturable system in accordance with uptake of Hb-Hp by receptor-mediated endocytosis.

There is no reason to believe that the catabolic fate of Hb is different, whether it is catabolized via CD163 or degraded as part of the process of erythrophagocytosis. However, CD163-mediated endocytosis may take place in a broader subset of macrophages than those responsible for red cell uptake (predominantly in spleen and bone marrow) because Hb-Hp complexes have better access to macrophages in the extravascular tissues. The crucial role of the macrophage for uptake of Hb-Hp is underscored by the finding that patients with drug-induced suppression of macrophages accumulate Hb-Hp in plasma (104). Furthermore, the specific role of CD163 in Hp removal is indicated by the fact that Hpr, which mainly differs from Hp by not binding to CD163, does not disappear from human plasma during hemolysis like Hp does (122).
The CD163 pathway (Fig. 6) for Hb degradation is upregulated during the acute phase response where IL-6 stimulates the expression of Hp, CD163, and heme oxygenase-1 (HO-1) (108). The expression of CD163 in humans is also regulated by cleavage by the metalloproteinase ADAM17, alias tumor necrosis activating enzyme that causes release of soluble CD163 (55, 57). These regulatory features are in accordance with a low or absent expression of CD163 in the “proinflammatory” M1 type macrophages and a high expression of CD163 in macrophages of the “anti-inflammatory”/alternatively activated M2 type macrophages important in tissue resolution in the late phase of inflammation. Macrophages have a plastic phenotype responding markedly on the microenvironment. For instance, clearance of Hb-Hp complexes stimulates the macrophages polarization into a special high-CD163-expressing macrophage Mhem phenotype with a suggested anti-inflammatory role (24). The Hb-Hp clearance itself also sparks an anti-inflammatory response, because the CD163 activity eliminates the proinflammatory Hb and leads to generation of anti-inflammatory metabolites (carbon monoxide and bilirubin) (185).
CD163 belongs to the class of membrane proteins with a single transmembrane segment and an extracellular domain consisting entirely of SRCR domains (56). This protein family also includes CD5, CD6, and the CD163 gene duplication product CD163 L-1/CD163b, which does not bind Hb-Hp (107). In a proposal of a new nomenclature of SRs, CD163 and CD163b are designated SR-I1 and SR-I2, respectively (136). CD163 has nine SRCR domains and Hb-Hp binds by a dual-point interaction with SRCR domains 2 and 3 in CD163. In each of these two domains a cluster of acidic amino acids is coordinated by a calcium ion. The two acidic clusters form the basis for electrostatic connection to Arg311 and Lys321 in the extended loop 3 of Hp (119). This is in analogy with other types of binding domains in other endocytic receptors such as the LDL receptor and the intrinsic factor-vitamin B12 receptor that also bind to their ligands in a calcium-dependent way (3). It is so far not known how CD163 recognizes Hb-Hp and not Hp alone. Free human Hb has been shown to bind to human CD163 with low affinity (153). However, there is so far no evidence that the Hb moiety in Hb-Hp directly participates in binding to CD163 and by this way reinforces the binding. In the mouse, free Hb seem to have a higher affinity for CD163, whereas the Hb-Hp complex has a lower affinity than in humans (54). The lower affinity for Hp can be explained by the fact that the mouse only has one of the two residues in Hp loop 3 needed for the dual-point interaction with CD163 (119).
This difference between mouse and man is only one of many subtle species difference in Hp, CD163, and related proteins as summarized in Table 1. Most striking are the many differences that have been introduced during primate evolution, which may suggest a strong evolutionary pressure caused by hemolytic and/or infectious disease. The need of combatting trypanosome infections causing sleeping sickness in Africa is a well-documented example that explains why the old world primates developed Hpr and apolipoprotein L-1 (ApoL-1) as weapons against this parasite. It is also possible that there are species differences in the Hb-Hp clearance, which in humans very much seem to rely on the CD163-mediated endocytosis by macrophages. Early studies have suggested uptake of Hb-Hp in hepatocytes via receptors of unknown origin (199). However, the studies on rodents have generally used human Hb-Hp, which may react and bind differently compared to rodent Hb-Hp complexes. Furthermore, the cross-species differences between rodents and humans may preclude preclinical studies in rodents when evaluating the use of Hp as a drug preventing Hb toxicity in humans (see below). However, dogs have shown a similar CD163-mediated handling of dog and human Hb-Hp and similar clearance rates as in humans (22). This suggests that dogs or other nonrodent species might be suitable for preclinical studies of Hp.
Hpr, haptoglobin-related protein.
The Hp and CD163 defense system against Hb and heme toxicity is backed up by other proteins that bind heme (121, 154, 159), if heme is released from Hb in plasma. Hemopexin strongly binds heme, and the heme–hemopexin complex is recognized by the multiligand receptor LDL-related protein/CD91 (76). The backup function of hemopexin in relation to Hp has been demonstrated by studies by Tolosano et al. who showed that intravascular hemolysis led to very severe pathological effects in mice with knockout of both the Hp and hemopexin genes compared to single gene knockouts (170). Heme binding to albumin and α1-microglobulin may also have a protective function (33).
Hp Phenotypes and Disease
Studies of SNPs in the Hp genome region indicate that the age of the Hp2 allele is less than 100,000 years, that is, before Homo sapien migration out of Africa (146). The Hp2 allele can, as mentioned, be found with varying frequency in all ethnic groups (32) and this rapid rise in Hp2 allele frequency has led to the hypothesis that it must have conferred a selective advantage early in human evolution, perhaps as protection against an infectious pathogen (23). Nevertheless, studies of diverse ethnic groups have shown that the Hp1 allele is overrepresented in elderly subjects, indicating that persons with the Hp1/Hp1 genotype have higher longevity (69, 116, 174, 198). Links between Hp genotype and a range of diseases have been described, but there are conflicting reports (32) and the general pattern does not point to either of the alleles being generally beneficial. Furthermore, it seems evident from several experimental in vitro and in vivo studies that there is no functional difference in the Hb-detoxification mediated by dimeric Hp1-1 versus multimeric Hp2-1 and Hp2-2 (97, 111). A similar mechanistic binding of Hb to the Hp SP domains of Hp2-1 and Hp2-2 is also highly suggested from a structural point of view (4).
It is, however, well established that the Hp concentration (g/L) is lower in the population with the Hp2/Hp2 genotype than in the Hp1/Hp1 population, which therefore may have higher overall protection against Hb-induced oxidative toxicity (177). The Hp2-Hp1 population has an intermediate Hp concentration. It has previously been proposed that these differences may contribute to the association between Hp genotype and risk of vascular disease (2, 34, 155). In line with this, studies of a human cohort of more than 20,000 individuals have shown that the Hp2 gene associates with a slightly higher cholesterol level, and this has been suggested to be driver for recurring deletions in the Hp2 gene leading to the present Hp1 gene variants (21). How Hp influences on the cholesterol level is not entirely clear, but it has been proposed to relate to apolipoprotein E-binding properties of Hp (21).
The reason for the difference in concentration of the Hp phenotypes is unknown, but we can tentatively explain this phenomenon by the difference in receptor-binding sites in the different types of Hb-Hp complexes. The hypothesis is as follows: when the Hp1-1 protein is saturated with Hb, the Hb-Hp1-1 complex exposes two CD163 binding sites. This allows the high avidity binding to two CD163s. The multimeric Hp2-2 and Hp2-1 have three or more Hp units and can therefore cross-link CD163 without being fully saturated with Hb as long as at least two Hp units are binding Hb. In short, Hp1-1 needs saturation with Hb to be effectively endocytosed, whereas multimeric Hp2-2 can be effectively endocytosed without being completely saturated with Hb. Multimeric Hp is therefore less “cost effective” in the removal of Hb, which implies a higher consumption of the multimeric forms compared to the dimeric form in the removal of the same amount of Hb. This has probably a limited importance during normal physiological hemolysis, but in cases of accelerated hemolysis and a high consumption of Hp, the higher Hp concentrations in individuals with the Hp1-1 phenotype may provide a stronger and longer protection against Hb toxicity.
The protective effect of Hp to oxidative damage by Hb is significantly impeded when Hb is glycated as seen in diabetes, where Hb-glucose is used as diagnostic biomarker. An increased abundance of glycosylated Hb, for which Hp has impaired antioxidative protection, has been reported in diabetic patients with the Hp2 /Hp2 genotype, leading to excessive oxidative damage in these patients (5). This finding, now confirmed in a large cohort (31), is proposed to contribute to cardiovascular complications in Hp2 /Hp2 diabetic patients. Several studies of complications in diabetes patients indicate that Hp2 /Hp2 is a risk factor (6). Furthermore, redox-active Hb is associated with LDL levels in diabetic patients leading to oxidated LDL (10). Vitamin E supplementation has been shown to decrease the risk of cardiovascular complications in Hp2 /Hp2 diabetics (180), which has led to the proposal that it should be a routine prophylactic treatment for Hp2 /Hp2 diabetics (180). However, it seems that the molecular background for Hp2 /Hp2 being a risk factor is still uncertain (145). The Hp concentration in the Hp2-Hp2 population may contribute but this is not known. It has also been reported that the endocytosis of Hb-Hp2-2 complexes is less efficient than of Hb-Hp1-1, but this is in contrast with results from other sides (6, 89). Actually, Hb-Hp2-2 is reported to have a higher functional affinity for the receptor than Hb-Hp1-1 (89). This is probably due to the multivalent binding of Hp2-2 to the receptors. Another explanation could be a different signaling mediated by the complex either via signaling by the receptor or the receptor cargo (heme). Recently, it has been reported that the Hb-Hp2-2 complex induces less anti-inflammatory signaling (involving phosphatidylinositol-3-kinase activation upstream of Akt phosphorylation) than Hb-Hp1-1 complexes (68, 91). A previous study (132) has also reported signaling, but it has not been confirmed by Schaer et al. (151), who raised the concern that many commercial Hb preparations contain contaminating endotoxin that is a strong toll-like receptor-4 stimulator. The problem is discussed in more detail elsewhere (120). Furthermore, when trying to elucidate the mechanistic role of Hp in disease, one should also consider other potential Hp ligands than Hb such as apolipoproteins. Very recently, it has also been shown that the proinflammatory cytokine HMGB1 binds to the Hp and the complex recognizes the receptor CD163 (195).
Many bacteria such as Neisseria meningitidis, Streptococcus pyogenes, and Staphylococcus aureus use Hb as an iron source (see also next section) by binding Hb-Hp complexes by various means and thereafter extracting the iron. Because of their polymeric structure, Hp2-1 and Hp2-2 can induce agglutination of bacteria. Hp2-1 and Hp2-2 have indeed been shown to inhibit the growth of S. pyogenes by agglutination (44). In patients, the Hp1/Hp1 genotype has been linked to increased risk of developing severe streptococcus infection (184). Furthermore, increased infection rates have been observed in Hp1/Hp1 cirrhosis patients (182).
On the contrary, Hp2/Hp2 individuals have been shown more prone to infection with Trypanosoma cruzi parasites causing Chagas disease, and they are reported to display more severe disease symptoms, including cardiac complications (82). The proposed mechanism behind this difference is based on less efficient clearance of the Hb-Hp complex in Hp2/Hp2 individuals—ensuring the availability of iron for the parasite, and on less efficient protection from oxidative stress and thus increased inflammation in the Hp2/Hp2 patients leading to cell damage and lowered cardiac function (112). Susceptibility to and severity of infections with Plasmodium falciparum have also been linked to Hp genotype, however, data are conflicting (41, 85, 130).
The increased free Hb concentration in Hp2/Hp2 individuals has also been linked to an increased risk of cerebral vasospasms following aneurysmal subarachnoid hemorrhage and, to a worse outcome, perceivably related to increased Hb concentration in the subarachnoid space (83, 96, 124).
Hp genotype has been linked to a range of other diseases, as summarized in Table 2, however, for many diseases, diverging results have been reported, or not linked to a functional explanation for the differences as previously reviewed (32, 38, 93, 177, 200).
Some diseases with few and conflicting reports on Hp genotype association are not included.
Even higher Hp2-2 rate in chronic renal failure patients with diabetes or hypertension.
Hp as a Carrier of Iron and Heme to Pathogenic Microorganisms
Iron is essential for the growth of most organisms, as its redox capabilities are required in numerous biological processes. To cope with the limited amount of available iron, pathogenic organisms use an array of strategies to snatch iron from the host. One route is to secrete siderophores or hemophores that can wrest iron or heme from host proteins (i.e., transferrin or hemoproteins) and then return to a specific receptor on the surface of the pathogen. Another route is the binding of Hb or Hb-Hp by bacterial wall-attached receptors followed by extraction of heme. Bacteria can boost the amount of available Hb/Hb-Hp by release of hemolysin, which triggers hemolysis (17). Some bacteria can only bind free Hb, whereas others, such as the gram-negative bacteria Neisseria meningitidis (149) and the gram-positive bacteria S. aureus (51), can bind and extract heme from both Hb and Hb-Hp. Thorough investigation of the proteins that orchestrate iron acquisition has revealed an elaborate yet elegant system of protein networks. One such system is that of S. aureus.
S. aureus expresses a range of iron-regulated surface determinant (Isd) proteins (IsdA to IsdI) involved in iron acquisition. IsdH is attached to the cell wall and binds Hb or Hb-Hp in the first step of iron acquisition. IsdH contains three NEAr transporter (NEAT) domains (IsdHN1, IsdHN2, and IsdHN3) (51, 52, 164). IsdHN1 binds with high affinity to the α-subunit of Hb and is probably essential for the initial recognition of Hb/Hb-Hp. Subsequently, IsdHN2-N3 extracts heme from Hb/Hb-Hp. IsdHN2 anchors to the globin moiety of Hb while IsdHN3 is placed over the heme pocket. By a combination of conformational strain and induced fit, heme translocates from Hb to a heme-binding site on IsdHN3 (49, 50, 88, 158). Another Isd-protein, IsdB, is also capable of heme extraction from Hb (171), however, this pathway has been less characterized in relation to the Hb-Hp complex. Once extracted from Hb/Hb-Hp, heme is transferred via IsdA and IsdC to the substrate-binding protein IsdE and transported into the cytoplasm by the ABC-transporter IsdF. In the final step of the pathway, intracellular heme is degraded by IsdG and IsdI and iron is released (67).
We have now observed that the interaction of Hb-Hp with IsdH blocks CD163 recognition and cellular uptake of Hb-Hp (150a). This may represent an S. aureus mechanism of obtaining heme from Hb-Hp without the risk of “loosing” the iron to macrophages. In addition to fuel the growth of the bacteria, the reduction of macrophage heme metabolism may reduce the anti-inflammatory response and thereby worsen a septic inflammation. Diagnostically, the mechanism may also skew the predictive value of using Hp as a marker for hemolysis.
The Hb-Hb complex is also an attractive target for heme auxotrophic microorganisms, that is, protozoan parasites of the species Trypanosoma brucei. These parasites infect mammals in large parts of Africa and cause the severe disease known as sleeping sickness (in humans) or nagana (in cattle). There are two T. brucei subspecies that are pathogenic to humans (T. b. gambiense and T. b. rhodesiense), whereas T. b. brucei primarily infects domestic animals. Trypanosomes require only a small amount of iron, which is met by receptor-mediated endocytosis of host transferrin (166). However, they lack all enzymes involved in heme biosynthesis (16). To obtain heme, they express an Hb-Hp receptor that enables binding and endocytosis of Hb-Hp (179). After endocytosis, the heme is subsequently liberated and incorporated into functionally important hemoproteins. The structure of the trypanosomal Hb-Hp receptor in complex with Hb-Hp shows that the receptor recognizes both Hp and the β-subunit of Hb, including the heme moiety (Fig. 7) (167).

Interestingly, it has turned out that a human innate immune defence mechanism has evolved from this heme scavenging pathway and it explains that humans, in contrast to some animals such as cattle, are resistant to infections with T. b. brucei. Hpr is an important component of this defence mechanism. Unlike Hp, Hpr is associated with other proteins in the specialized high-density lipoprotein subspecies designated TLF-1 (144). Within the TLF-1 particle, Hpr is still capable of binding Hb (122, 191) and the Hb-Hpr complex is also recognized by the trypanosomal Hb-Hp receptor (179). This leads to endocytosis of the entire TLF-1 particle, which also carries the primate-specific protein ApoL-1. ApoL-1 is capable of forming a pore in the lysosomal membrane, which ultimately leads to killing of the parasite (110, 178). To this date, Hpr has not been attributed other roles than mimicking Hp in this Trojan horse-like strategy. The human pathogenic trypanosome subspecies, T. b. gambiense and T. b. rhodesiense, have developed various mechanisms to avoid ApoL-1-mediated killing. Both subspecies express resistance proteins that bind and disarm apoL-1 and in this way prevent formation of lysosomal pores (43, 175, 193).
Therapeutic Properties
Heme/Hb toxicity is a serious complication of hemolysis as for instance seen in sickle cell disease and in other hemoglobinopathies, massive blood transfusion, autoimmune hemolytic anemia, and certain infections. These conditions may lead to consumption of plasma Hp, which expose the patient to severe Hb toxicity due to NO scavenging and oxidative damage. It seems therefore obvious to use Hp as a biological drug to supplement inadequate capacity of Hb neutralization during severe hemolysis as recently reviewed (152, 154). In fact, this is not a novel idea and plasma-derived Hp has since 1985 been on the market in Japan with hemolysis in conjunction to blood transfusion, extracorporal circulation, and thermal injury as primary indications. In addition, the product is used off-label in Japan for treatment of a range of hemolytic diseases. Several reports have documented a beneficial effect on reducing kidney intoxication (72, 74, 77).
There has also been a growing interest outside Japan to use Hp therapy in the treatment of sickle cell patients, who have recurrent attacks and hemolytic crises that strongly reduce length and quality of life (142). Preclinical studies have been done (97) or are ongoing, although the many differences between man and other species (Table 1) are a recurrent complication when studying animal models and in particular rodent models. Furthermore, it is discussed if also hemopexin, which to some extent backsup the function Hp, can be developed into a biological drug to be used in severe hemolysis (48). The binding and removal of heme by hemopexin might to a large extent substitute the effects of Hp. However, hemopexin–heme, which is recognized by CD91, must be taken up by a much wider span of cell types than macrophages, the “professional” cell type for Hb heme metabolism. It is unknown if this change of site for Hb heme metabolism may have any effects. From this point of view it seems most obvious to use Hp, the natural first line of defense against Hb toxicity, as the principal biological drug perhaps with hemopexin as an add on in combinatory therapy (48). Furthermore, instead of using plasma-derived Hp for development of Hp therapy against sickle cell disease and other indications, it may have biological and regulatory advantages to develop a recombinant Hp analogue for therapy. The present detailed insight into the Hp structure and structure–function relationship may facilitate the development of an optimal recombinant Hp analogue for use in biological therapy.
Footnotes
Acknowledgments
This review was supported by the TROJA ERC advanced grant 233312, the Danish Medical Research Council, The Danish Council for Independent Research (Sapere Aude program), and Novo Nordisk Foundation.