
Abstract
Significance:
Redox signaling is one of the key elements involved in cardiovascular diseases. Two important molecules are the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) and the oxidoreductase thioredoxin-1 (Trx-1).
Recent Advances:
During the previous years, a lot of studies investigated Nrf2 and Trx-1 as protective proteins in cardiovascular disorders. Moreover, post-translational modifications of those molecules were identified that play an important role in the cardiovascular system. This review will summarize changes in the vasculature in atherosclerosis and ischemia reperfusion injury of the heart and the newest findings achieved with Nrf2 and Trx-1 therein. Interestingly, Nrf2 and Trx-1 can act together as well as independently of each other in protection against atherosclerosis and ischemia and reperfusion injury.
Critical Issues:
In principle, pharmacological activation of a transcription factor-like Nrf2 can be dangerous, since a transcription regulator has multiple targets and the pleiotropic effects of such activation should not be ignored. Moreover, overactivation of Nrf2 as well as long-term treatment with Trx-1 could be deleterious for the cardiovascular system.
Future Directions:
Therefore, the length of treatment with Nrf2 activators and/or Trx-1 has first to be studied in more detail in cardiovascular disorders. Moreover, a combination of Nrf2 activators and Trx-1 should be investigated and taken into consideration. Antioxid. Redox Signal. 26, 630–644.
Introduction
S
In the cardiovascular system, important antioxidative molecules are catalase, superoxide dismutases (SOD), glutathione, glutathione S-transferases, glutathione peroxidases, heme oxygenases, thioredoxin reductases, and thioredoxins. The expression of those antioxidative molecules can be regulated by the transcription factor Nuclear factor erythroid 2-related factor 2 (Nrf2). Genetic variants in Nrf2 and Nrf2 regulated genes have been associated with cardiovascular diseases (86). Therefore, activation of Nrf2 can be one potential therapeutic option in cardiovascular diseases. Over the previous years, it had become clear that thioredoxin-1 (Trx-1) was not only regulated by Nrf2 via the two antioxidant response elements (AREs) in its promoter but also that Trx-1 regulated Nrf2 by keeping Nrf2 in its reduced state and thus allowing Nrf2 to increase expression of antioxidative enzymes (63). Moreover, Trx-1 has been shown to regulate proteins by direct interaction in the cardiovascular system (114) and is, therefore, not solely an antioxidative molecule and could be of therapeutic interest in cardiovascular disorders. A combination of activation of Nrf2 and an increase in Trx-1 could be considered.
Thus, this review will first introduce Nrf2 and Trx-1 and then the effects of Nrf2 and Trx-1 in atherosclerosis and in ischemia reperfusion injury will be described.
Nuclear Factor Erythroid 2-Related Factor 2
Nrf2 is a transcription factor that binds to AREs in promoter regions of target genes to coordinate transcription of genes in response to oxidative stress. Nrf2 belongs to the transcription factor family of cap ‘n’ collar basic region leucine zipper (CNC-bZIP) proteins. Under basal conditions, Nrf2 remains sequestered in the cytoplasm by the Kelch-like ECH-associated protein 1 (Keap1) (Fig. 1). Keap1 is a cysteine rich protein that binds Nrf2 through cysteine residues, thereby inhibiting nuclear translocation of Nrf2 (56, 103). Protein levels of Nrf2 are moderate, because association with Keap1 leads not only to cytoplasmic retention but also to ubiquitination and proteasomal degradation of Nrf2 in a Cullin3-dependent manner, preventing basal expression of Nrf2 target genes (68). Oxidants and electrophiles serve as Nrf2 activators; however, the exact mechanism of activation remains unclear. Experimental evidence suggests that electrophilic molecules or oxidants induce the release of Nrf2 from the cytosolic complex by oxidation of cysteines within Keap1.
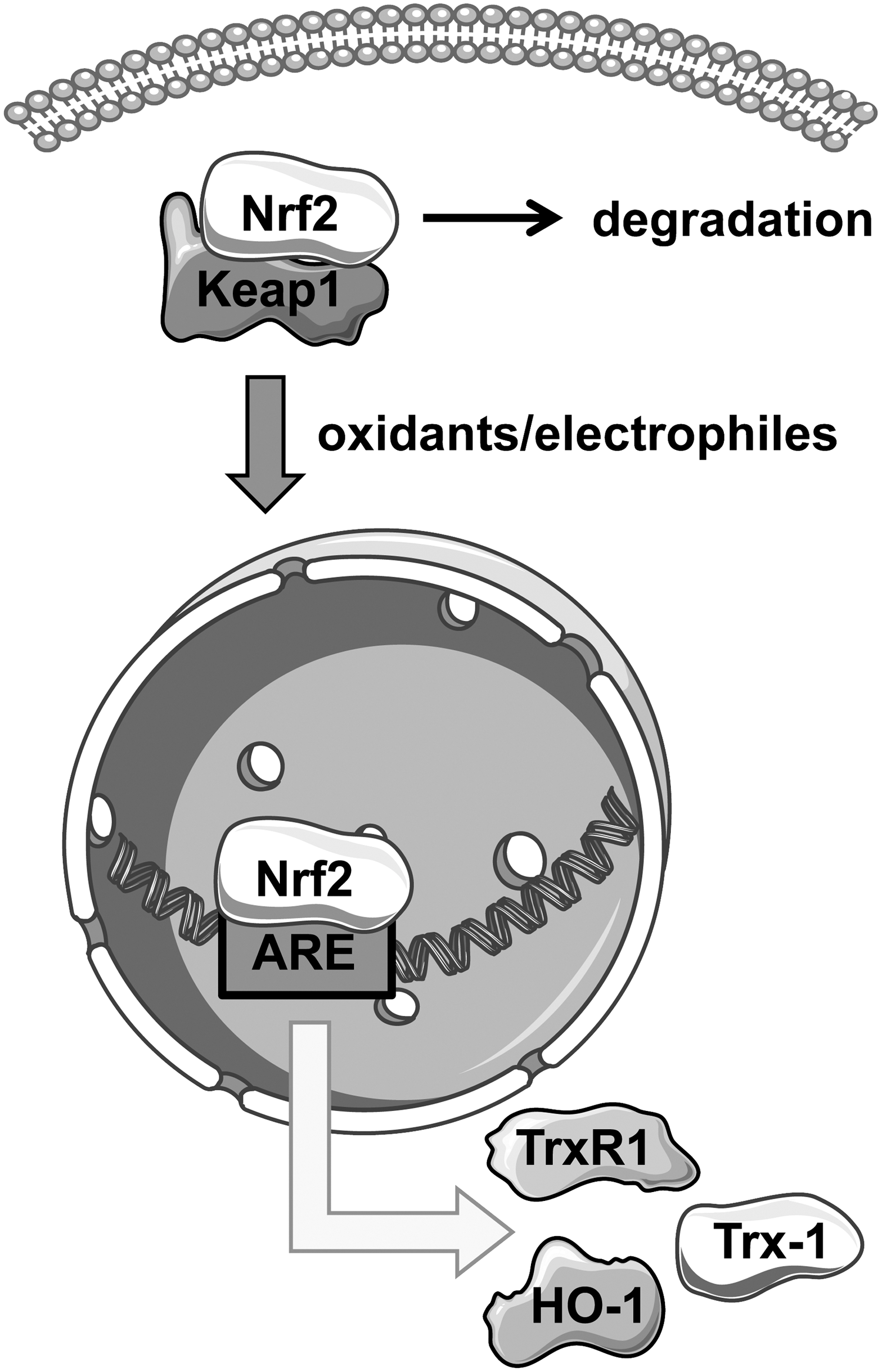
Regarding expression of Nrf2 itself, analyses with luciferase reporter systems demonstrated that activity of the Nrf2 promoter was enhanced by treatment with 3H-1,2-dithiole-3-thione (59). Moreover, 3H-1,2-dithiole-3-thione also increased Nrf2 messenger RNA (mRNA) and protein levels in the rat cardiac H9c2 cell line, which was accompanied with reduced intracellular reactive oxygen species (ROS) levels (11).
During vascular development, enhanced expression of Nrf2 has been reported in the tip cells of retinal vessels. Two models, one involving Nrf2 deficiency and another involving an endothelial-specific Nrf2 knockout, showed reduced angiogenic sprouting in the retina without changes in vascular endothelial growth factor (VEGF), implying that Nrf2 is a cell-autonomous regulator of angiogenesis (106).
Target genes of Nrf2 regulate drug metabolism, redox homeostasis, DNA repair, and inflammatory processes. The most prominent examples are SOD, Glutathione-S-transferase (GST), heme oxygenase-1 (HO-1), and thioredoxin reductase 1 (TrxR1). Besides increased sensitivity against oxidative stress, Nrf2-deficient mice show disturbances in lipid metabolism, which will be addressed in more detail in the sections: “Role of Nrf2 in Atherosclerosis, Especially in the Endothelium” and “Role of Nrf2 and Trx-1 in Atherosclerosis, Especially in Macrophages.” The importance for Keap1 in regulation of Nrf2 has also been demonstrated by the deletion of Keap1 in hepatocytes of mice. This leads to a constitutive activation of Nrf2. In models of steatohepatitis, improved hepatic steatosis and reduced apoptosis have been observed in those Keap1-deficient mice. However, chronic liver inflammation and fibrosis progression were not inhibited by loss of Keap1 (85), suggesting that sustained activation of Nrf2 could be detrimental.
Another important target of Nrf2 is the ubiquitin-proteasome complex, which has also been discussed as a therapeutic target in cardiovascular diseases (77). In mouse embryonic fibroblasts, the 20S proteasome is regulated by Nrf2 in response to oxidative stress. One hour of hydrogen peroxide (H2O2) treatment resulted in elevated proteasomal activity after 24 h and small interfering RNA (siRNA) knockdown of Nrf2 prevented this activation. Thus, Nrf2 serves to limit the levels of damaged or oxidized proteins by promoting their degradation (81).
Since Nrf2 activation induces the expression of protective enzymes against oxidative stress, modulation of Nrf2 activity arose as a potential therapeutic approach. Interestingly, many Nrf2 inducers are plant-derived natural compounds (6, 42). Nrf2 signaling is important in various diseases such as cancers, respiratory disorders, neuronal diseases, diabetes, and cardiovascular diseases [reviewed in Magesh et al. (64)]. A critical consideration in targeting Nrf2 therapeutically is that this transcription factor is believed to regulate the expression of up to 10% of all mammalian genes (98); therefore, one has to bear in mind that modulation of Nrf2 may lead to unexpected outcomes. A phase 3 randomized trial revealed an increased risk for heart failure in patients with type 2 diabetes treated once daily with bardoxolone, a potent Nrf2 activator (15), highlighting the aforementioned problem. On the other hand, brusatol has been reported to enhance the efficiency of chemotherapy by inhibiting Nrf2 (87). However, it was also shown that brusatol can have adverse effects in non-tumorous cells and enhances the sensitivity to chemical stress induced by bardoxolone, further supporting how critical it is to tamper with Nrf2 using inhibitors and activators (76).
Post-Translational Modifications of Nrf2
Besides the regulation of Nrf2 expression and its interaction with Keap1 as described earlier, there is recent evidence that Nrf2 is also modified post-translationally, which results in changes in Nrf2 activity and/or localization.
Protein kinase C (PKC)-dependent phosphorylation of Nrf2 at serine 40 has been described in oxidative stress in a tumor cell line, leading to increased nuclear localization of Nrf2 (41). Similar observations were made in rat pheochromocytoma (PC12) cells. Treatment with the nitric oxide (NO) donor S-Nitroso-N-acetyl-DL-penicillamine (SNAP) induced serine phosphorylation of Nrf2, analyzed by immunoprecipitation and use of a phospho-serine antibody. Interestingly, this study not only reported enhanced phosphorylation of Nrf2 but also enhanced nitrosation of Keap1. The abrogation of nuclear localization of Nrf2 by concomitant use of a pharmacological PKC inhibitor did not affect nitrosation of Keap1. Thus, nitrosation of Keap1 seems not to be responsible for the localization of Nrf2 (100). Enhanced nuclear Nrf2 activity was also observed after acetylation of lysines 588 and 591, which was dependent on cAMP responsive element binding protein (CREB). In this study, the authors showed that enhanced activity of the deacetylase NAD-dependent deacetylase sirtuin-1 (SIRT1) or mutation of both lysines (588 and 591) in Nrf2 promoted cytosolic accumulation of Nrf2 (50).
Interplay Between Nrf2 and Trx-1
It has been demonstrated that parts of the Trx-1 system are regulated by the transcription factor Nrf2, because Trx-1 and its reductase, TrxR1, each harbor AREs in their promoter region (43, 53, 89), leading to increased Trx-1 expression on nuclear Nrf2 activation under several conditions (Fig. 1). Reciprocally, it has been shown that overexpression of Trx-1 enhanced Nrf2 activity and that binding to AREs was increased, as analyzed by a protein-DNA binding assay after immunoprecipitation of nuclear Trx-1 (34, 91). This suggests a potential direct interaction of both proteins as well as a crosstalk between Nrf2 and Trx-1. A recent study describes the influence of Trx-1 on nuclear localization of SKN-1, the ortholog of Nrf2 in Caenorhabditis elegans. It was observed that loss of Trx-1 in the whole organism led to enhanced nuclear localization of SKN-1, demonstrating that interaction and crosstalk of Nrf2 signaling and the Trx-1 system contribute to inter- and intra-organ communication (67).
The cross-regulation between Nrf2 and Trx-1 either leads to protection from organ failure and maintenance of cellular homeostasis or, in the case of loss of functionality of these proteins, promotes pathophysiological complications. It is necessary to understand the underlying molecular mechanisms of the crosstalk between Nrf2 and Trx-1 to be able to develop new kinds of treatment of cardiovascular diseases or to improve the already established therapies. Moreover, the Nrf2-independent functions of Trx-1 seem to play a role in cardioprotection and will be introduced later.
Thioredoxin-1
The Trx-1 system consists of the two major proteins, Trx-1 and TrxR1. The conserved redox motif, with amino acids—CGPC—at positions 32–35 in human Trx-1, serves as a regulator of the cellular redox-steady-state equilibrium. Because of the proximity of both cysteines, a rapid oxidation accompanied by formation of an internal disulfide bridge is possible. This reversible oxidation of Trx-1 is recovered under nicotinamide adenine dinucleotide phosphate (NADPH) consumption through TrxR1, resulting in restoration of Trx-1 in its reduced form. Trx-1 contains three more cysteine residues at positions 62, 69, and 73; it has been shown that, in addition to intramolecular disulfide bridge formation between C32 and C35, a second S-S bridge can form between C62 and C69 under oxidative conditions with distinct impact on the three-dimensional structure of Trx-1 (Fig. 2) (35, 105).

As a thiol-disulfide-oxidoreductase, Trx-1 acts as a reducing agent for oxidized proteins. The two-step mechanism, by which the target molecule is reduced with concomitant oxidation of Trx-1 at C32 and/or C35, has been extensively reviewed elsewhere (61). The sensitivity of the Trx-1 system has been demonstrated by kinetic modeling, which showed that changes of concentrations or activities of single members of this crucial redox cycle affect kinetics of other members (82).
Post-Translational Modifications of Trx-1
Oxidation of cysteines is not the only post-translational modification (PTM) that occurs within Trx-1. For example, another fundamental PTM of Trx-1 in the endothelium is S-nitrosation (Fig. 2). Binding of NO to cysteine 69 elevated the redox activity of Trx-1 and enhanced the resistance to apoptosis (31). Nitrosation of Trx-1 under physiological conditions seems to occur in a selective manner. When Trx-1 was oxidized, nitrosation induced by incubation with nitrosated glutathione (GSNO) was observed mainly at C73 (3) (Fig. 2). However, it is unclear whether these findings are due to the oxidation at the historically considered active site at C32 and C35, or whether they are due to disulfide bridge formation between C62 and C69, which also occurs under oxidative stress. The most likely explanation is that oxidation of C62 and C69 prevents S-nitrosation of C69.
Besides nitrosation, Trx-1 is also nitrated, which is a modification of mostly tyrosines by peroxynitrite (ONOO−). In various studies, nitration of Trx-1 in the cardiovascular system was investigated. Tao et al. demonstrated a decade ago inactivation of Trx-1 by 3-N-morpholinosydnonimine (SIN-1), which is often used to generate superoxide radical anions and NO at the same time to yield ONOO-, in vitro.
Moreover, it has been shown that ischemia/reperfusion (I/R) reduced the activity of Trx-1 and enhanced its nitration. This was paralleled with reduced interaction of apoptosis signal-regulating kinase 1 (ASK-1) (96). Similar findings were observed in old mice compared with young mice, indicating that nitration of Trx-1 in the heart is even more pronounced in old mice (112), suggesting that the inability of the old heart to cope with an insult is due to reduced Trx-1 activity. In high-glucose-treated cardiomyocytes, inactivation of Trx-1 due to a drastic increase of nitration was observed without changes in expression levels. This was even more pronounced after sham-simulated ischemia reperfusion, suggesting that high-glucose sensitization of cardiomyocytes is dependent on nitration and, thus, inactivation of Trx-1 (62).
Regulation of Proteins by Trx-1
Interaction of Trx-1 with other proteins often depends on its redox status. This is reasonable, because Trx-1 reduces oxidized proteins to restore their function. For example, interaction of Trx-1 with the energy-sensing protein and cardioprotective 5′ AMP-activated protein kinase (AMPK) is required to maintain cysteines within AMPK in a reduced state, which is crucial for its activation (92). However, interaction that is dependent on cysteines in the active site may also inhibit the function of the target molecule. In the endothelium, apoptosis is a crucial event, since endothelial cells (EC) serve as a barrier organ and maintenance of the functional monolayer integrity is essential for blood vessel homeostasis. It has been demonstrated that Trx-1 is able to inhibit apoptosis by interacting with the N-terminus of ASK-1. This interaction results in decreased ASK-1 activity and was abrogated under oxidative stress conditions or mutation of cysteines 32 and 35 (88). Another study demonstrated that overexpressed Trx-1 led to degradation of ASK-1 in EC.
Furthermore, usage of Trx-1C32S and Trx-1C35S mutants revealed that a single cysteine is sufficient to inhibit ASK-1 activity, which was even seen in the presence of H2O2. However, the responsible cysteine—if it is only one—in ASK-1 remains to be investigated. The interaction between ASK-1 and Trx-1 can be inhibited by Trx binding protein-2 (TBP-2), also referred to as vitamin D3 upregulated protein 1 (VDUP1), or thioredoxin interacting protein (TXNIP) (46). Therefore, TXNIP is a negative regulator of Trx-1 and counteracts its protective properties.
In addition, Trx-1 possesses anti-inflammatory capacities. Overexpression of Trx-1 in EC downregulates the release of monocyte chemoattractant protein 1 (MCP-1), an inducible cytokine that recruits monocytes and macrophages. This was shown to be accompanied by suppressing nuclear translocation of apurinic/apyrimidinic endonuclease redox effector factor-1 (APEX-1) and reduced DNA-binding activity of activator protein 1 (AP-1) (12). This study lacks experiments regarding involvement of interaction between Trx-1 and APEX-1, but it has been shown that direct interaction of Trx-1 with APEX-1 leads to transcriptome changes due to modulation of AP-1 activity in HeLa cells (38).
Modulation of activity or functionality of other proteins in a covalent or indirect manner (75) by Trx-1 is not restricted to the cytoplasm (114). Trx-1 is able to shuttle to the nucleus in a karyopherin-α-dependent manner (91). Nuclear interaction partners of Trx-1 include hypoxia-inducible factor-1 α (HIF-1α), glucocorticoid receptor, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and, as already mentioned, Nrf2 (23, 61, 65).
Atherosclerosis
The onset of atherosclerosis has been associated with the common lifestyle of industrial nations, which includes lack of exercise, overconsumption of sugars, fats, and cholesterol. Each of these habits can result in a damaged vasculature (19, 26). Multiple layers of the vasculature can be damaged and lead to the formation of atherosclerotic lesions and plaques. Distinct cell types, which become dysfunctional during atherogenesis, include EC, smooth muscle cells (SMCs), and macrophages. The endothelium, formed by a monolayer of EC, is located closest to the blood flow and forms a semi-permeable barrier between blood contents and the media and adventitia. As a consequence, EC need to be sensitive to chemical and physical changes in the body to induce relaxation or contraction of blood vessels [for review, see Mehta and Malik (69)].
Loss of EC or injury to the endothelial monolayer can result in remodeling of the vessel wall, which is marked by invasion of inflammatory cells, proliferation of SMCs, and integrin-dependent recruitment of platelets as early events in the development of atherosclerotic plaques [for review, see Bui et al. (8)]. Endothelial dysfunction contributes to the development of atherosclerosis. As an initial event, EC become activated and start to express leukocyte adhesion molecules. After recruitment of monocytes, oxidized low-density lipoprotein (OxLDL) through scavenger receptors such as CD36 leads to the formation of foam cells (25), a hallmark of atherosclerotic lesions. A recent study has shown that SMCs in atherosclerotic lesions may be derived from proliferating progenitor cells in the adventitia (94).
Role of Nrf2 in Atherosclerosis, Especially in the Endothelium
In EC, knockdown of Nrf2, or overexpression of its repressor Keap1, led to a reduction of EC migration and tube formation. Moreover, it was demonstrated that Caspase-3/7 activity was increased under basal conditions and was even more pronounced after treatment with 3 μM H2O2 for 18 h (101). Knockdown of Keap1 also protected EC from tumor necrosis factor α (TNF-α)-induced ROS formation and monocyte adhesion. In addition, an injection of Keap1 siRNA before lipopolysaccharide (LPS) injection reduced the NOx content and TNF-α plasma levels in mice (52). Further evidence supporting the benefit of an intact Nrf2-ARE system is provided in a study by Zakkar et al., which shows that absence of Nrf2 leads to vascular cell adhesion molecule-1 (VCAM-1) expression in EC whereas activation of Nrf2 leads to the opposite (111).
To address the hypothesis that activation of Nrf2 at atheroprone sites (commonly at branch points with low shear stress) will prevent EC activation and atherogenesis, the investigators pharmacologically activated Nrf2 using sulforaphane. Sulforaphane treatment was shown to reduce VCAM-1 expression in sites of low shear stress in wild-type (WT) but not Nrf2-null mice (111). This study provides evidence that Nrf2 regulation in EC is intimately linked with initiation and progression of atherosclerosis.
In addition to oxidants, it has been demonstrated that shear stress induces Nrf2 target gene expression, which was abrogated after Nrf2 knockdown. These expression patterns were antagonized by preincubation of human EC with oxypurinol or diphenyleneiodonium (DPI; NADPH oxidase and Xanthine oxidase inhibitors) (104). This study found that both oscillatory and laminar flow enhanced Nrf2 nuclear localization, whereas only laminar flow induced the expression of Nrf2 target genes. This suggests that Nrf2 has anti-atherogenic properties, since the pro-atherogenic oscillatory flow did not enhance binding to the ARE (40).
Surprisingly, various studies observed that Nrf2 acts in a pro-atherogenic manner. Sussan et al. reported that aortic plaques are reduced in young and old Nrf2−/− ApoE−/− mice, and that this was gender independent (93). Yet another study found that Nrf2-related gene expression was lower in aged of LDLR−/− mice placed on a high-fat diet (18). As a possible reason for these discrepancies, the authors discuss methodological differences to the earlier study, as well as differences in mouse models used (apolipoprotein E [ApoE] vs. LDL receptor [LDLR] deficiency). Reduction in lesion size was reported in Nrf2-deficient mice, and as a molecular mechanism, this is believed to be due to induction of interleukin (IL)-1 in cholesterol crystals via the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, activated by Nrf2 (27). These various contradictory findings underline the complexity of both modulating the signaling of a transcription factor-like Nrf2 and cardiovascular diseases such as atherosclerosis.
Studies show that disruption of Nrf2 activity allows EC to succumb to oxidative stress and leads to the development of coronary artery disease (21). The presence of SOD in the endothelium is reduced in patients with coronary artery disease, implying a deficiency in the Nrf2-mediated antioxidant response (60). Conversely, it has also been shown that reducing superoxide production by decreasing NADPH oxidase expression results in smaller lesions in a mouse model of atherosclerosis (4). Successful attenuation of atherosclerosis was also achieved in aged high-fat-diet-fed mice when subjected to antioxidant treatment (18). However, the use of exogenous antioxidants in humans to combat atherosclerosis and cardiovascular disease, in general, has not been very successful (73), stressing the importance to understand the intracellular redox homeostasis and defense mechanisms in more detail.
In addition to the oxidized lipids and pro-inflammatory cytokines, EC proximal to atherosclerotic lesions are also exposed to highly oxidative molecules, such as hypochlorous acid (HOCl). HOCl is formed by myeloperoxidase, which is present both intercellularly in EC and extracellularly at lesion sites (107). Treating EC with HOCl will induce expression of HO-1, an Nrf2-dependent antioxidant enzyme promoting cell viability. Inhibition of HO-1 by protoporphyrin-IX results in apoptosis of HOCl-stimulated EC. In the case of atherosclerosis, hypoxia is another factor that comes into play. It was found that intermittent hypoxia, an inducer of atherosclerosis and vascular inflammation, leads to a decrease of reduced glutathione (GSH) and an increase of modified glutathione (GSSG) (33). Sustained hypoxia was also found to induce pro-inflammatory gene expression in EC, while concurrently inducing Nrf2 and Nrf2-dependent transcription of HO-1 (83).
Another key function of EC is the production of NO by endothelial nitric oxide synthase (eNOS) to combat coagulation and regulate vascular tone (16). Dysfunction of the EC in atherosclerosis develops on a few different levels, ranging from cell viability to promotion of inflammation. These problems arise from the high level of oxidative stress associated with atherosclerosis. An essential factor that distinguishes the atheroprotective and atherogenic sites along the endothelium is expression of Nrf2. A study that investigated atherosclerotic susceptibility of sites within the mouse vasculature found that lesion development inversely correlated with shear stress experienced by the EC (111). EC located at branch points in the vasculature were noted to have a higher likelihood of forming atherosclerotic lesions compared with cells found in areas of high shear stress and laminar flow.
The production of VCAM-1 in EC has been associated with atherosclerosis (111). During the development of atherosclerosis, OxLDL particles gather within the arterial intima. The bioactive components of these particles (oxidized phospholipids [OxPL]) induce VCAM-1 production in neighboring EC. VCAM-1 promotes monocyte binding to lesions in the endothelium. The accumulation of immune cells, which adopt a pro-inflammatory state, provides an environment filled with inflammatory stimuli (MCP-1, IL-1β, TNF-α) and subsequent oxidative stress. It was also found that VCAM-1 is induced in a p38-dependent manner, and Nrf2 activation suppresses this signaling (111). Sites of high shear stress dampen the effect of pro-inflammatory stimuli on EC. The converse was found in sites of low shear stress where they were observed to be highly vulnerable to plaque formation and greatly increased levels of VCAM-1 expression (111).
Thus, Nrf2 plays an important role in protecting the endothelium from damage and inhibiting the onset of endothelial dysfunction and, subsequently, atherosclerosis.
Role of Trx-1 in Atherosclerosis, Especially in the Endothelium
The function of Trx-1 in atherosclerosis and especially in the endothelium has not been studied in depth, since regulation of Nrf2 results in an increase in Thioredoxin-1 Reductase and, thus, in increased Trx-1 activity. In a few studies, it has been demonstrated that overexpression of Trx-1 enhanced Smad3 phosphorylation and inhibited the OxLDL-induced expression of adhesion molecules VCAM and intercellular adhesion molecule (ICAM) in human EC. Moreover, Smad3 was identified as an interaction partner of Trx-1, as was shown by immunoprecipitation of Trx-1 in EC lysates after adenoviral transduction with either Trx-1 WT or Trx-1 C32/35S (13).
One important player in the inflammasome is the TXNIP. TXNIP interacts with Trx-1 and is, therefore, inhibited. Trx-1 activity was investigated in vascular smooth muscle cells (VSMCs) isolated from WT and TXNIP ApoE-deficient mice. TXNIP-deficient mice show increased Trx-1 activity and reduced ROS levels. TXNIP ablation also leads to reduced expression of VCAM, ICAM, and MCP-1 (9). Changes of the TXNIP/Trx-1 interaction have been found in another in vivo study. Atherosclerosis, as an age-associated disease, was analyzed in old mice (25 months), which were fed with resveratrol for 3 months. These authors demonstrated a reduction of TXNIP expression and an improvement of aortic distensibility, a parameter reflecting elasticity. Although they found no changes in Trx-1 mRNA levels, it is likely that the release of Trx-1 from the complex with TXNIP is responsible for the improved outcome (5).
Given the fact that Trx-1 is one of the most important anti-apoptotic proteins in the endothelium and loss of EC importantly contributes to the onset of atherosclerosis and associated events, for example, MI, it is reasonable to conclude that Trx-1 is a protective molecule in the vascular wall.
Role of Nrf2 and Trx-1 in Atherosclerosis, Especially in Macrophages
A consequence of atherogenic VCAM-1 and proinflammatory gene expression in EC is the infiltration of macrophages and monocytes into the lesions, as VCAM-1 induction promotes binding of monocytes to the endothelium. The migration of monocytes through the endothelium is an important event in the development of atherosclerosis, as blocking monocyte recruitment significantly slows disease development (70). These monocytes are exposed to many inflammatory mediators, including, but not limited to, MCP-1, TNF-α, and IL-1β. Details of monocyte interaction with the endothelium in atherosclerosis have been reviewed by Moore and Tabas (71). Nrf2 and Trx-1 play critical roles in controlling inflammation and immune system function. Disruption of Nrf2 results in increased mortality of endotoxin- and cecal ligation-challenged mice (97). The intracellular role of Nrf2 and Trx-1 in macrophages remains the same as in other cells, a defense mechanism against oxidative stress (45). At the intracellular level, Nrf2 and Trx-1 are believed to protect immune cells, namely macrophages, from ROS generated during inflammation (57).
High-cholesterol diets dramatically increase the presence of LDL in the circulation, and atherosclerotic lesions are burdened with high levels of oxidative stress, resulting in an environment filled with oxidized peptides and lipids. OxLDL, namely the phospholipid surface of the lipoprotein, produces OxPL. OxPL comprise one of the primary bioactive components of OxLDL, and OxPL constitute the active component of minimally modified low-density lipoprotein (mmLDL). OxPL promote monocyte adhesion to the endothelium (28) and an inflammatory response in macrophages. The hallmark of OxPL-dependent macrophage polarization is the induction of Nrf2-dependent gene expression, as represented by the markers HO-1 and TrxR1, to name a few (47).
OxPL inhibit phagocytosis, one of the principle functions of macrophages, promoting further inflammation and plaque accumulation. OxPL induce a novel macrophage phenotype, known as Mox, which has a unique gene expression profile as compared with the pro-inflammatory (M1) and anti-inflammatory (M2) macrophages. Mox is characterized as having mild (in comparison to M1) pro-inflammatory gene expression as well as significant Nrf2-dependent gene expression (Fig. 3). The Mox macrophages make up one third of the macrophages present in atherosclerotic lesions (Fig. 3) (47).
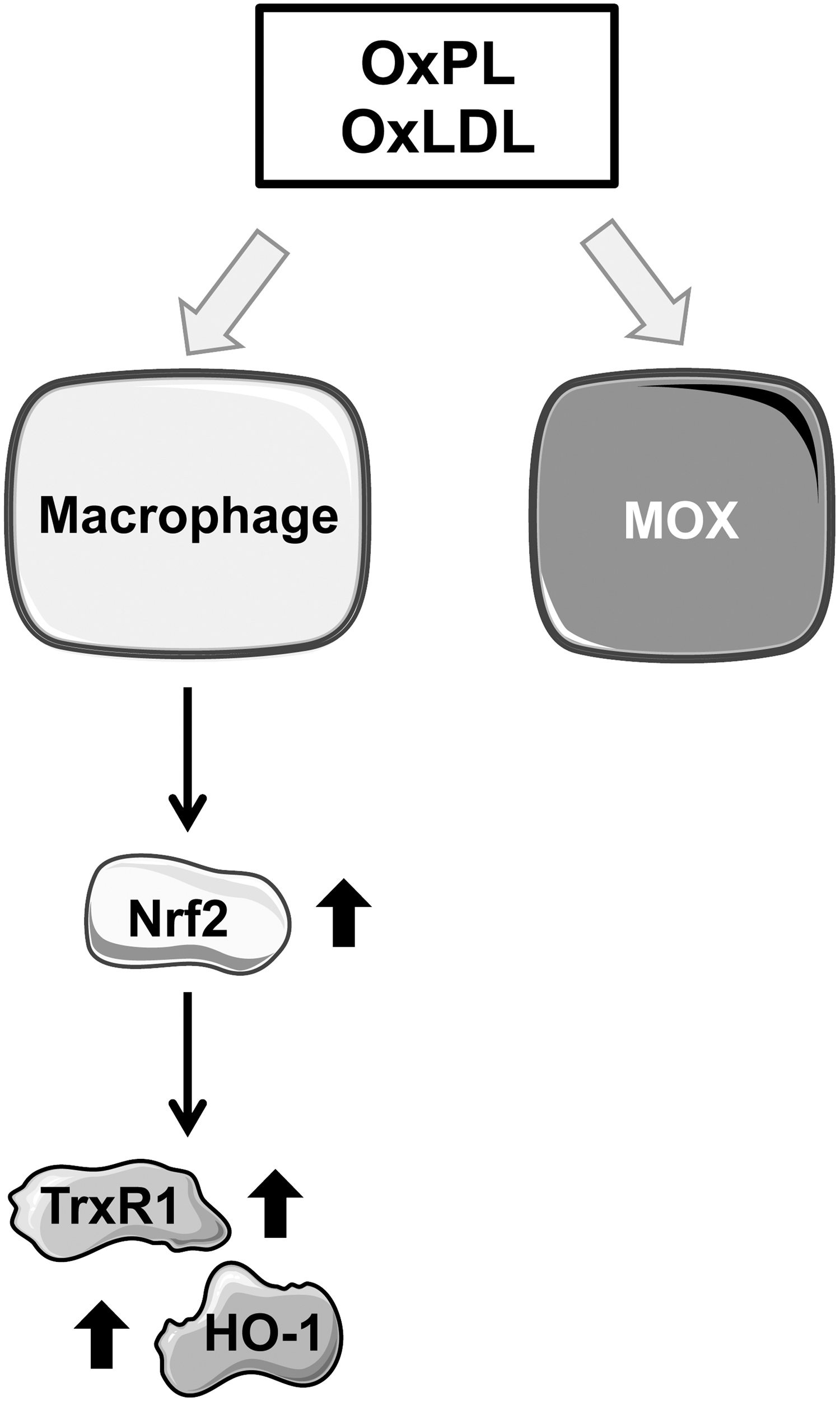
Trx-1 can be secreted by cells to promote M2 macrophage polarization, leading to atheroprotection (22) (Fig. 4). Full-length extracellular Trx-1 has also been shown to inhibit complement deposition on human EC, effectively competing with a complement for its binding site (54). A truncated form of Trx-1 shown to exist in vivo, known as Trx80, has the opposite effect, leading to complement activation via the classical and alternative pathways (54). Activation of Nrf2 has also been shown to attenuate inflammation and prevent tissue damage in a mouse model of sickle cell disease, a mechanism believed to be driven primarily by the heme-metabolizing action of HO-1 (51).

Polarized macrophages are associated with the development of atherosclerosis. M1 macrophages have pro-inflammatory and pro-atherogenic properties, whereas M2 macrophages show the opposite (66). Therefore, one could speculate that changing the balance between M1/M2 macrophages could lead to either progression or improvement of atherogenic events with the potential to treat atherosclerosis. As mentioned already in the Introduction section, Trx-1 has an anti-inflammatory potential. It was demonstrated that treatment with recombinant human Trx-1 in murine peritoneal isolated macrophages led to enhancement of the expression of CD206 and IL-10, which are markers of M2 macrophages. Moreover, LPS stimulation led to an increase in TNF-α expression, a marker for M1 macrophages, which was abrogated after treatment with Trx-1 (Fig. 4). As an in vivo atherosclerosis model, mice have been chosen in which the mouse ApoE gene is replaced by the human ApoE2. These mice virtually exhibit all characteristics of type III hyperlipoproteinemia in humans. Those mice are markedly defective in clearing β-migrating VLDL particles, and they spontaneously develop atherosclerotic plaques, even on a regular diet. Treatment with Trx-1 showed increased macrophage polarization in the animals (22).
Taken together, Nrf2 and Trx-1 signaling is crucial at the level of macrophage invasion as well, and it exerts protective functions in antagonizing progression of atherosclerosis.
Role of Nrf2 and Trx-1 in Atherosclerosis, Especially in VSMCs
Besides EC, a large part of the disease progression relies on the activity of VSMCs and macrophages. The intimal hyperplasia is mediated by growth factors such as platelet-derived growth factor (PDGF) (48). PDGF activation of SMCs induces ROS production in an NADPH-oxidase-dependent manner and disrupts cellular homeostasis (58). PDGF stimulates SMCs to migrate and proliferate, and a recent study using a lineage tracing approach identified a role for progenitor cells in the adventitia (94) in promoting arterial occlusion. It has been shown in SMCs that Nrf2 is a key regulator of cell migration and neointimal hyperplasia (2). PDGF activation of SMCs also leads to the activation of Nrf2 (2), possibly as a feedback mechanism, to counteract increased ROS production and reinstate a cellular redox balance.
The complex interplay between the endothelium and VSMCs has been demonstrated by analyzing changes of the redox balance in a co-culture model. In this approach, a reduction of superoxide and H2O2 levels was observed in VSMCs co-cultured with microvascular EC. These changes were independent of NADPH oxidase expression patterns, but rather due to upregulation of antioxidant systems such as Trx-1 and copper/zinc superoxide dismutase (Cu/Zn SOD). These findings highlight, at least under non-pathological conditions, that Trx-1 not only protects the endothelium itself but also that EC serve as a protector against ROS generation by upregulation of Trx-1 in VSMCs (110).
I/R Injury in the Heart
Ischemia is the result of the blockage of a vessel, for example, due to a blood clot, which can last from minutes to hours. After removal of the causal blockage physically or by drugs, blood flow is restored during the reperfusion phase. Tissue damage and the severity of cell dysfunction, injury, and/or death are, on one hand, dependent on the extent as well as on the duration of ischemia. Moreover, during reperfusion, the harmful event for cells is an oxidative burst (102, 115).
It has long been known that the dimensions of MI can be decreased by repetitive episodes of ischemia, even at the remote sites. Ischemic preconditioning (IPC) as well as remote preconditioning can improve heart function after MI. This remote preconditioning has been applied successfully before percutaneous coronary intervention (36, 39). In addition, ischemic postconditioning (POC) has been applied in humans as well. Excellent insights into the signaling events evoked by pre- or postconditioning events have been summarized elsewhere (37).
Role of Nrf2 in I/R Injury in the Heart
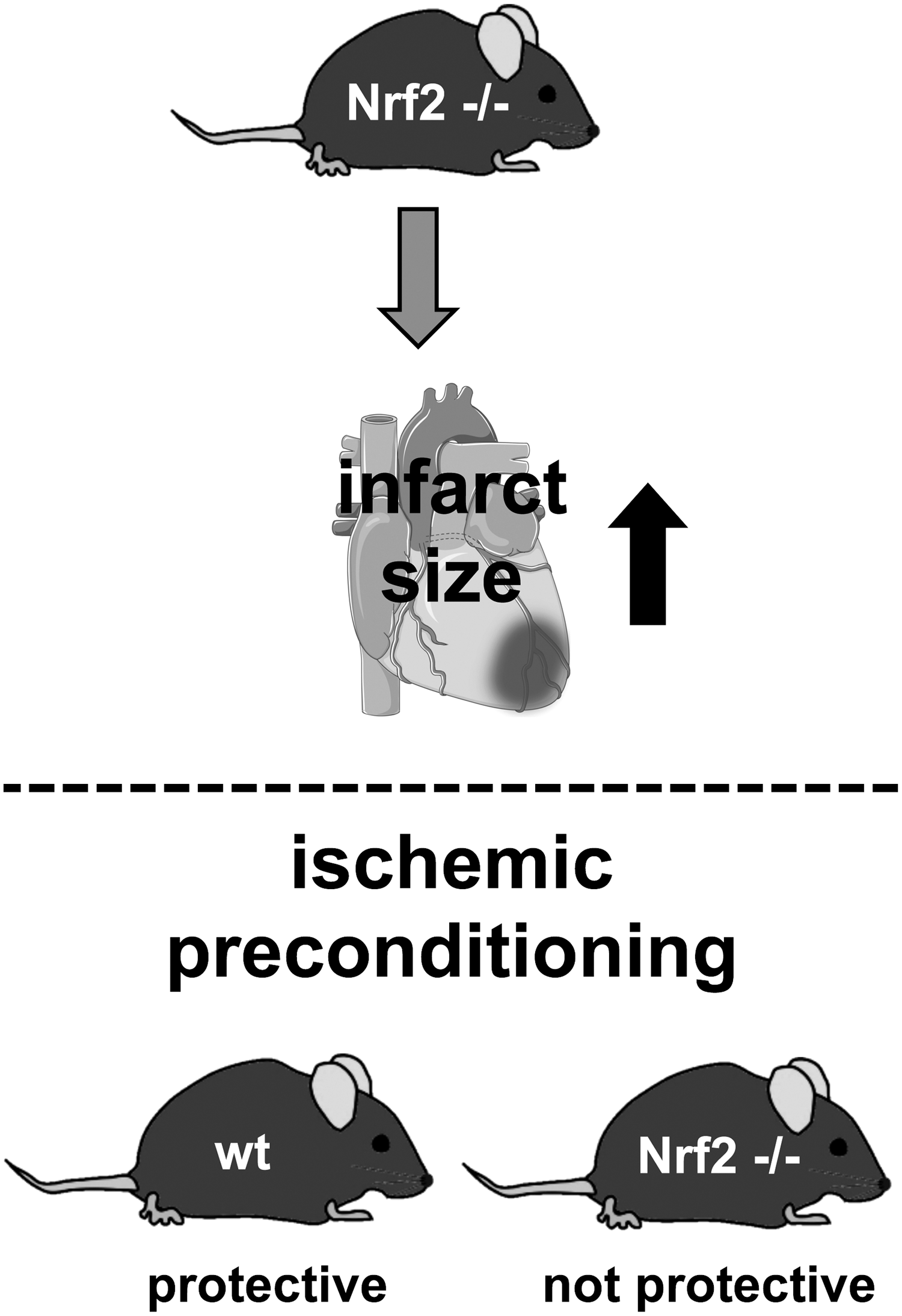
Involvement of Nrf2 in I/R-related processes has been shown by a comparison of WT mice with Nrf2-deficient mice. Of note, Nrf2 knockout mice have been reported to show an impaired cardiovascular phenotype, for example, due to left ventricular diastolic dysfunction and higher expression of eNOS (24). These findings underscore the important function of Nrf2 in the cardiovascular system. Occlusion of the left anterior descending (LAD) artery for 30 min revealed a twofold increase in infarct size in Nrf2−/− mice (Fig. 5) (109). In the same study, it was observed that Nrf2 is required for successful preconditioning, since two cycles of IPC did not result in cardiac protection in the absence of Nrf2 (Fig. 5). Since ROS levels are elevated after I/R, the oxidant-sensing Nrf2 transcription factor might be a promising target to ameliorate injuries after I/R. This is supported by several studies using potential Nrf2 activators.
Glucocorticoids induce Nrf2 target genes, for example, HO-1, in a lipocalin-type prostaglandin D synthase-dependent manner. Glucocorticoids are unable to act cardioprotective in the Nrf2-deficient heart after I/R injury. Moreover, endogenous prostaglandin D2 increased nuclear translocation of Nrf2, reduced infarct size, and partially preserved heart function (Fig. 6) (49). Alpha-lipoic acid, which can be found in food such as broccoli, spinach, and tomatoes, is clinically approved (17).
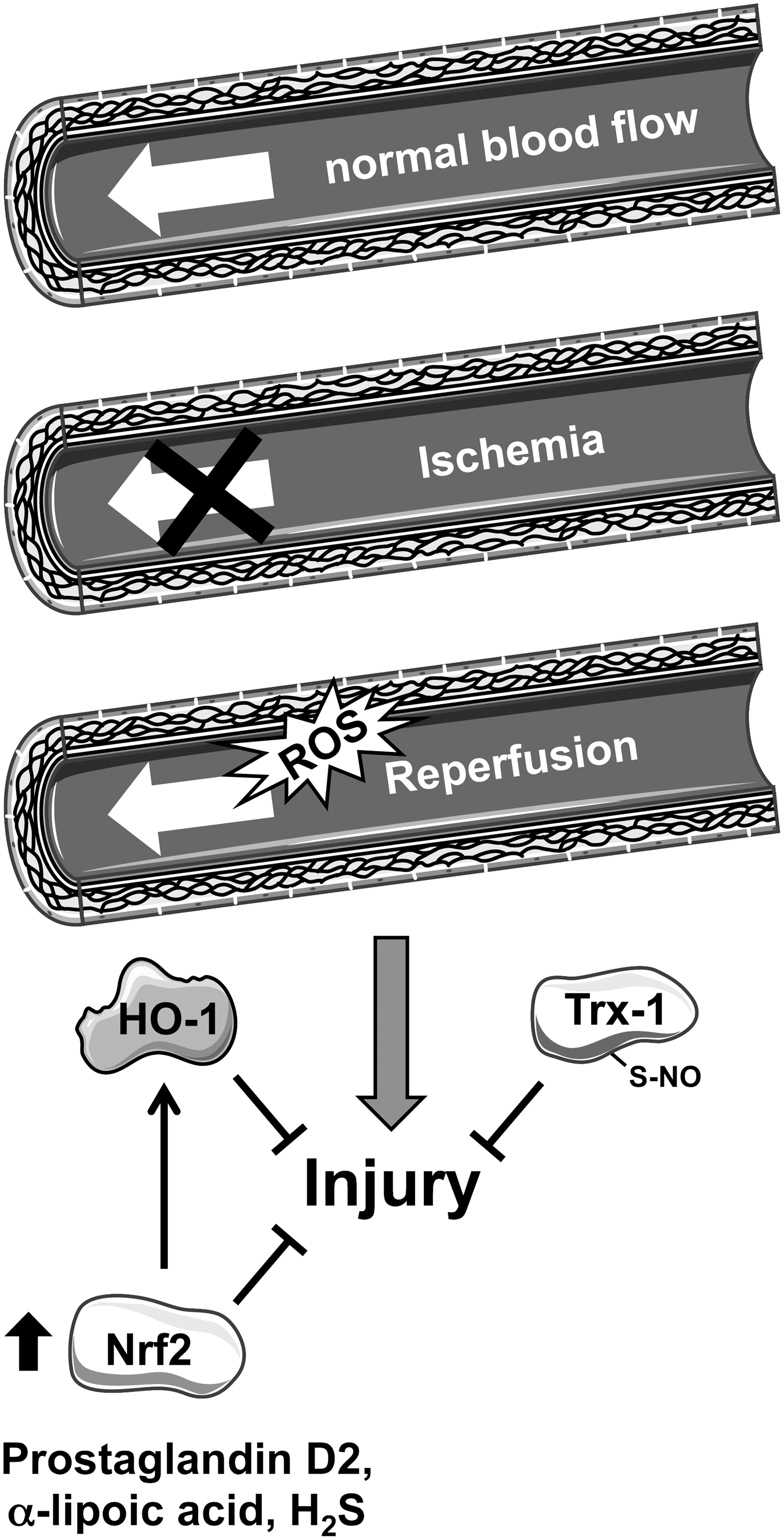
Preconditioning with alpha-lipoic acid partially preserved heart function by reducing myocardial infarct size, cardiomyocyte apoptosis, and inflammation in I/R injury in rats. This seems to be dependent on increased nuclear localization and activation of Nrf2 target genes such as HO-1 (Fig. 6) (20). Recently, hydrogen sulfide (H2S) has been suggested to be a cardioprotective molecule. Preconditioning with H2S for 24 h reduced infarct size after I/R injury in an Nrf2-dependent manner, since this protection was not seen in Nrf2-deficient mice (Fig. 6) (10). The Nrf2-dependent, protective effect of H2S preconditioning was also seen in diabetic mice (78). Resveratrol is a polyphenol that can be found in red grapes and wine. Resveratrol has been observed to exert antioxidative properties, for instance, by inducing the expression of enzymes such as manganese superoxide dismutase (MnSOD) and Catalase (44). Recently, Cheng et al. (14) demonstrated that resveratrol treatment before reperfusion after 30 min of ischemia improved cardiac function and reduced infarct size in the rat heart. This study is of potential interest, because the administration of resveratrol was 5 min before reperfusion and, thus, it could be of clinical significance.
The underlying mechanisms induced by resveratrol cannot be attributed to nuclear localization of Nrf2 alone, although an involvement of Nrf2 in the beneficial effects of resveratrol in mice and rats has been shown (14). However, the poor bioavailability and the concentrations of resveratrol, which would have to be used in humans, are critical. Moreover, the timing and the duration of treatment are unclear (84). Therefore, larger controlled human trials are needed to investigate those points and to then determine the contribution of Nrf2 (7).
Taken together, several natural molecules are cardioprotective in an Nrf2-dependent manner.
Role of Trx-1 in I/R in the Heart
With respect to Trx-1, it has been demonstrated that cardiac-specific overexpression of Trx-1 in mice improved systolic and diastolic post-ischemic ventricular function (99). Reduction of endogenous Trx-1 activity by using dominant negative-Trx-1 transgenic mice abolished this protective effect. Interestingly, this was accompanied by improved mitochondrial function measured in isolated mitochondria from those Trx-1 transgenic mice (79). Trx-1 protein levels are reduced after MI induced by I/R in young mice and could be partly restored after post-conditioning (Fig. 7). Middle-aged (12 months) and old (20 months) mice also showed lower Trx-1 levels after I/R; however, this could not be rescued by post-conditioning (Fig. 7). The protective effect of post-conditioning seems to decline with age, as was described already for preconditioning (1, 108). However, findings regarding age-dependent decline of ischemic conditioning are controversial. In a recent study, no association between age, gender or cardiovascular medication, and remote IPC could be observed in patients undergoing coronary artery bypass grafting (55).

As mentioned earlier, reperfusion leads to an overload of ROS due to an oxidative burst. High levels of ROS resulted in loss of Trx-1, which was demonstrated by Perez et al., 120 min after reperfusion. Loss of Trx-1 then leads to an “over”-adaptive response, which explains the opposite finding of an increase of Trx-1 24 h after the reperfusion phase (80). Analyzing Trx-1 levels in cardiac tissue biopsies collected from cardiopulmonary bypass surgeries revealed an increase of Trx-1 in patients who were preconditioned by four cycles of 5 min upper arm ischemia followed by a 5 min reperfusion phase (113). This reveals one possible molecular mechanism for the improved outcome, for example, in MI after applying IPC. However, injury induced by myocardial I/R is accompanied by apoptosis.
Since Trx-1 exerts anti-apoptotic functions, for example, due to nitrosation at cysteine 69 in EC (30), it is most likely that enhancement of nitrosated Trx-1 during MI results in decreased apoptosis. Indeed, a reduction of apoptosis was observed in I/R cardiac tissue of mice, which received human Trx-1 10 min before reperfusion. This effect was even more pronounced after using S-nitrosated Trx-1 (Fig. 6) (95). Interestingly, under conditions of oxidative stress, Trx-1 is degraded (32). This reduction of Trx-1 is also seen in mice subjected to a high-fat diet. As a consequence, increased apoptosis of cardiac myocytes is detected after ischemia in mice on a high-fat diet (92).
Taken together, these studies support the notion that Trx-1 is cardioprotective in I/R.
Role of Nrf2 and Trx-1 in I/R in the Heart
Nrf2 and Trx-1 have also been shown to act in concert in I/R injury. An APEX-1/Trx-1/Nrf2/NF-κB axis was proposed. Although no or low interaction of APEX-1 with NF-κB and Nrf2 in nuclear extracts of left ventricular tissues could be observed after I/R, increased association of NF-κB and Nrf2 with APEX-1 was found in preconditioned hearts using four cycles of ischemia. Immunoprecipitated NFκB pulled down Trx-1 in the preconditioned heart, which was dependent on the presence of APEX-1 as shown by application of antisense oligonucleotides for APEX-1. In general, the amount of nuclear Trx-1 and Nrf2 was reduced after I/R, which could be restored after preconditioning (29). Moreover, the beneficial effect of H2S was dependent not only on Nrf2 but also on Trx-1, as it was lost in mice with cardiac-specific overexpression of a dominant negative mutant of Trx-1 (74). These findings indicate a crucial role in IPC by crosstalk and association of these two proteins.
Therefore, the extent of injury after I/R, occurring in some of the most frequent causes of deaths such as MI, is dependent on both Nrf2 signaling and functional Trx-1. Interestingly, both proteins also play a role in pre- or post-conditioning. However, further studies regarding the impact of the Nrf2/Trx-1 crosstalk, elucidating detailed action on the molecular level, are necessary.
Conclusions/Future Directions
In summary, both Nrf2 and Trx-1 play an important role in atherosclerosis and ischemia and reperfusion injury of the heart alone as well as in concert (Fig. 8).
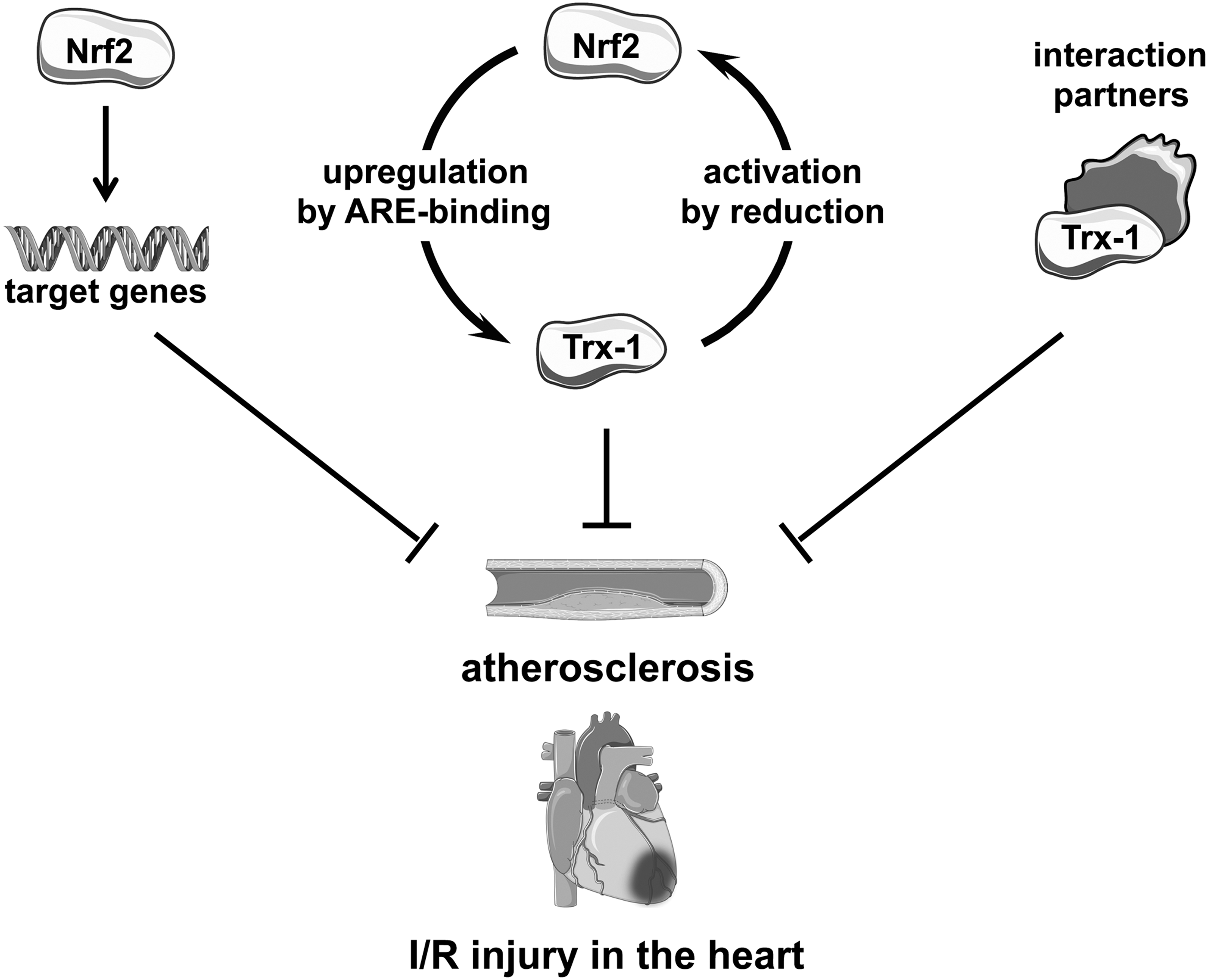
Interventions regarding nutritional and/or pharmacological activation of Nrf2 to improve heart and vessel function in atherosclerosis or ischemia/reperfusion should be well reflected. Since Nrf2 is a transcription factor, it regulates the expression of multiple genes, which even vary from organ to organ. Thus, pleiotropic effects cannot be overseen when modulating Nrf2 activity therapeutically. Moreover, overactivation of Nrf2 may be deleterious; length and starting point of treatment should be deliberate.
Treatment with Trx-1 could be beneficial even after MI. However, the most relevant patients who one has to deal with over the next decades are people over 75. From the studies in mice, Trx-1 alone will probably not be protective.
Since Nrf2 can increase Trx-1 mRNA and protein levels may be a combination therapy leading to increased and enhanced nuclear Nrf2, active Trx-1 protein could be of potential interest. Nevertheless, more studies in mice and humans are needed to address those potential therapeutic options.
Footnotes
Acknowledgments
P.J. is a scholarship holder of the IRTG1902. This work was, in part, supported by the Deutsche Forschungsgemeinschaft (HA2868/9-1 and 10-1, IRTG1902 P2); by the Forschungskommission of the Medical Faculty, University of Duesseldorf (28/2014) to J.H.; and by an NIH grant R01 DK096076 to N.L.V.S. was supported by a training grant NIH T32 GM007055-42 and an American Heart Association pre-doctoral fellowship 15PRE25560036. Single elements for figures were taken from the Powerpoint image bank of Servier Medical Art (