
Abstract
Protein Degradation
C
The proteasome and its regulators
The 20S Proteasome is one of the cell's primary adenosine triphosphate (ATP)-independent mechanisms that is used to break down oxidatively damaged proteins (30). The 20S comprises four stacked rings. The two external rings are made of seven α subunits, whereas each ring of the core is made of seven β subunits, with three of them responsible for the proteolytic activity (13, 43). The β1 subunit has caspase-like activity, the β2 provides trypsin-like activity, and the β5 has chymotrypsin-like activity (10). In addition, various regulatory subunits can attach to the α rings at each end of the 20S Proteasome, including the 11S (Pa28) regulator (Fig. 1), which, during periods of oxidative stress, can expedite the degradation of oxidized proteins (173).
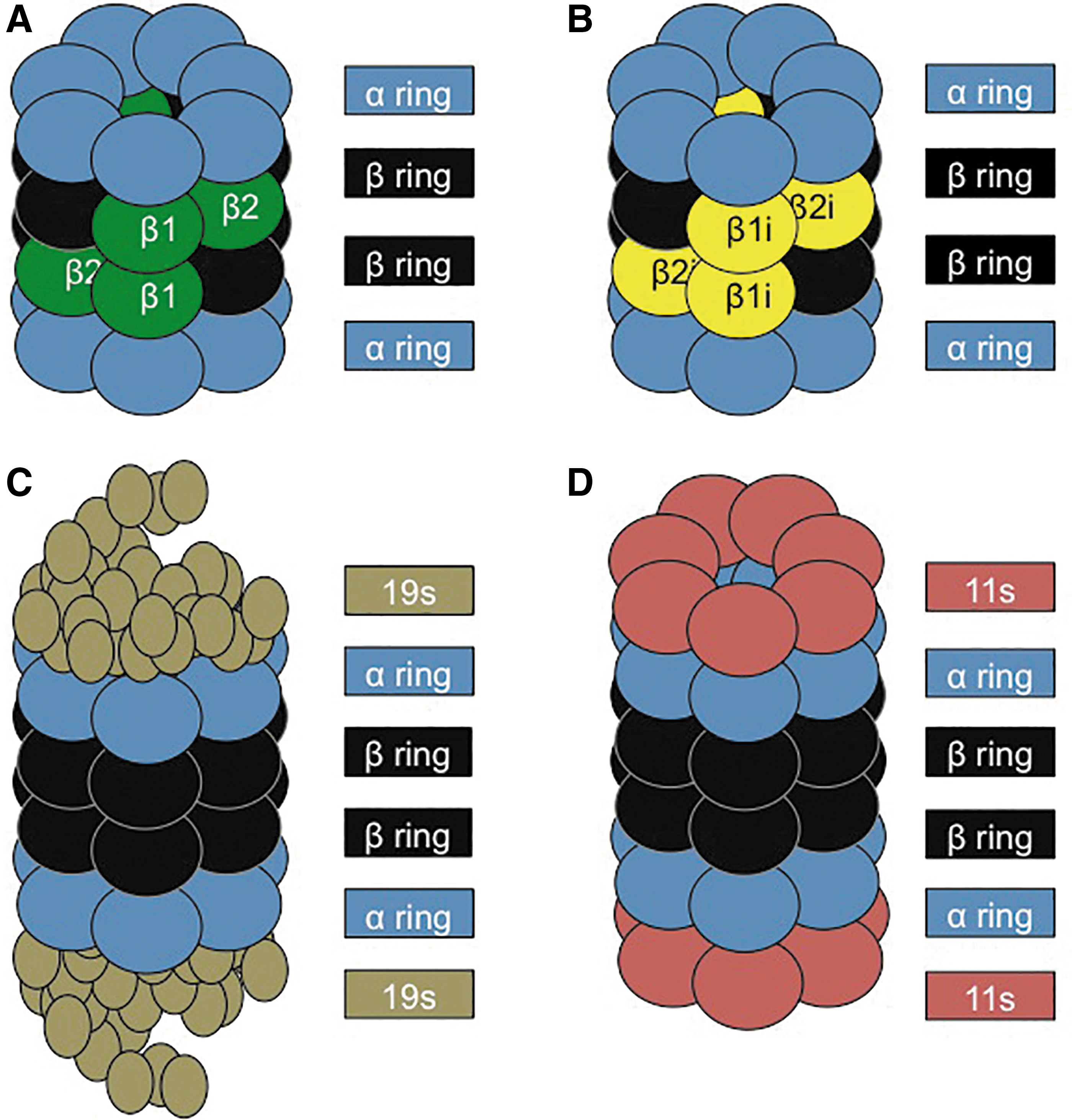
Although the focus of this article is on oxidative stress, the IP was actually discovered, and is most known, as a specialized immune-activated form of the 20S catalytic core. Specifically the β1, β2, and β5 subunits of the 20S core, which are constitutively expressed, are substituted with the catalytic subunits β1i, β2i, and β5i of the IP (41). The IP has primarily been implicated in mediating an immune response. Various studies have shown that cells that are exposed to Interferon-γ trigger upregulation of the IP (112). In addition, cells of the immune system typically have constitutively high levels of IP, in contrast to other cell types (112) (Fig. 1).
The 26S Proteasome is formed from the addition of a 19S regulatory subunit to each of the α rings of the 20S Proteasome, in an ATP-dependent manner (10). During periods of homeostasis, the cell relies on the ubiquitin-Proteasome system, with the 26S Proteasome as the predominant form for protein degradation (13, 89) (Fig. 1). Proteins marked for degradation first undergo ubiquitinylation, involving three different enzymes: the ubiquitin-activating enzyme (E1), which binds a ubiquitin molecule in an ATP-dependent manner and transfers it to an ubiquitin-conjugating enzyme (E2), which binds multiple ubiquitin monomers into a poly-ubiquitin chain, which is then transferred to a specific ubiquitin-ligase enzyme (E3), which links the poly-ubiquitin to a target protein (43). This poly-ubiquitin chain acts as a targeting sequence or “tag” to direct proteins to the 26S Proteasome for degradation. The 19S regulatory subunits of 26S Proteasomes first remove the poly-ubiquitin “tags” of targeted proteins, then unfold those proteins, and finally deliver them into the 20S core of the 26S Proteasome for proteolysis (160).
During periods of oxidative stress, however, critical sulfhydryl groups in the 19S regulatory subunits are highly susceptible to oxidative damage and many lose their proteolytic capacity (156). Additionally, most of the remaining 26S Proteasomes are disassembled by Ecm29, with the 19S caps being sequestered by heat shock protein 70 (HSP70). This, in turn, provides additional free 20S core Proteasomes to quickly degrade oxidized proteins by interacting with their hydrophobic patches, thus ensuring their rapid removal before they can aggregate and cross-link. Approximately 3–5 h after the initial stress, the 26S Proteasome is reassembled in a process that is catalyzed by HSP70 (53, 137).
One of the primary pathways that triggers increased expression of the 20S Proteasome (and that is also negatively impacted by Alzheimer's disease) is through the activation of the cap-n-collar transcriptional activator, the nuclear factor erythroid 2-like factor 2 (Nrf2) (127). Nrf2 has been shown to activate numerous antioxidant response genes, which together provide a significant cellular defense to combat an oxidative insult (74, 179). Under homeostatic conditions, Nrf2 is inhibited from translocating into the nucleus by its negative regulator Keap1, which also targets Nrf2 for poly-ubiquitinylation and subsequent degradation by the 26S Proteasome, thus keeping intracellular levels quite low (180). However, during periods of stress, Keap1 is phosphorylated, causing its dissociation from Nrf2. Oxidant-induced dissociation of the 26S proteasome (mediated by Ecm29 and HSP70) prevents further Nrf2 degradation and increases the Nrf2 pool size (53, 179). After phosphorylation by AKT and/or PKCγ, Nrf2 migrates into the nucleus, where it binds to various antioxidant response elements (ARE), also called electrophile responsive elements (EpRE), within target genes, thereby increasing their transcription (179). During periods of high oxidative stress, when the cellular proteome is the most vulnerable to oxidative damage, it is critical that the cell has an available, functioning pool of 20S Proteasome. Interestingly, if proteasomal activity becomes inhibited, the Nrf2 signal transduction pathway is used to increase transcriptional activity (109).
Oxidative stress has been associated with Alzheimer's disease (159) and in the triple-transgenic murine model for Alzheimer's disease, young mice exhibit increased phosphorylation of Nrf2, whereas older triple-transgenic mice show a sex-dependent decrease in Nrf2 phosphorylation (109). These findings suggest that responses to the early stages of Alzheimer's disease include an attempt by the Nrf2 system to combat oxidative damage. As the disease progresses, however, Nrf2 phosphorylation decreases and it remains in the cytoplasm (133), allowing greater Nrf2 ubiquitinylation and consequent degradation by the 26S Proteasome. As a result, many Nrf2 antioxidant targets are not expressed in the highly oxidative environment that accompanies Alzheimer's disease development (109). In turn, decreased amounts of active Nrf2 greatly hinder the de novo pool of 20S Proteasome that could help compensate for the already diminishing proteasomal activity. Due to the importance of Nrf2 in the upregulation of various stress response genes, it has become a focal point for neurodegenerative therapies. Exposure to various Nrf2 inducers has been shown to successfully ameliorate the accumulation of Aβ plaques (176), possibly through increased activation of ARE/EpRE elements.
Structure and function of the mitochondrial Lon protease
Although the 20S Proteasome is the major protease dealing with oxidized protein substrates in the cell cytoplasm, nuclear and endoplasmic reticulum, intracellular organelles such as mitochondria and peroxisomes do not have Proteasomes and have developed specialized proteases to cope with protein damage. This is especially important, since mitochondria and peroxisomes are major sources of superoxide or hydrogen peroxide production (105). Multiple proteases have been uncovered in mitochondria, but perhaps none have been as well investigated as the ATP-dependent Lon P1 protease. Lon was initially discovered in Escherichia coli and was subsequently found to be conserved in all types of mammalian tissues, indicating its importance in mitochondrial protein quality control (49, 171).
Lon P1 comprises six to seven monomeric subunits. Each subunit consists of three domains: an N-domain, which interacts with the hydrophobic regions of the substrate; an ATPase domain, which binds to ATP and facilitates the ring opening; and a serine proteolytic domain (115) (Fig. 2). Early studies showed Lon expression as highly controlled, with loss of this regulation being detrimental for the cell or organism. For example, in E. coli, continual overexpression of Lon is lethal (24), and in mammalian tissue, Lon P1 overexpression accelerates tumorigenesis (12, 68). In contrast, loss of Lon results in mitochondrial deficiency and cellular senescence (15, 114, 132). Lon P1 has been shown to be the primary mitochondrial protease that degrades mildly oxidatively damaged proteins, including aconitase, and other oxidized intramitochondrial proteins (14). During increased cellular stress, Lon is transiently induced to cope with elevated mitochondrial protein damage (67, 113). Peroxisomes, which generate significant amounts of H2O2 during metabolism of odd-chain fatty acids, have a different Lon variant called Lon P2 (129a).

Alzheimer's Disease
Alzheimer's disease is the most common type of dementia that targets higher cognitive processes, resulting in memory impairment and behavioral disorders (141). In the United States alone, it is estimated that ∼14 million people will suffer from this disease by 2050 (7, 65). Current costs to treat individuals with Alzheimer's disease are estimated at $400 billion per year, and the economic burden will clearly only increase, unless a cure is found (6, 7).
The two major hallmarks of the disease are the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs), comprising mainly hyperphosphorylated Tau protein. Both markers were originally detected by Alois Alzheimer, the disease's namesake and discoverer, in the early 20th century (3). Although the etiology of Alzheimer's disease remains elusive, scientists have developed several theories to explain its origins. Many fall within two scientific camps: those who believe Aβ accumulation is the central axis of the disease (“Baptists”) (59), and others who consider Tau to be the primary culprit (“Tauists”) (51).
Amyloid-β
The amyloid precursor protein (APP) functions as both a paracrine signaling peptide and a membrane-bound receptor protein. The secreted soluble APP fragment is critical in growth stimulation, cell adhesion, and blocking of various serine proteases of neighboring neurons (104, 148). APP is highly conserved, with homologs found in multiple species, including humans, mice, Drosophila melanogaster, and Caenorhabditis elegans (151). The importance of APP in neuronal health was explored in APP homozygous-deficient mice. These animals showed normal development, but they experienced increased reactive glial cell formation, coupled with decreased neuronal and muscular function in adulthood (182). Similarly, cultured neurons lacking APP exhibited decreased neurite outgrowth and decreased viability (122). Conversely, transgenic mice overexpressing APP show many of the pathological features of Alzheimer's disease, specifically the formation of Aβ plaques and a decline in neuronal function, highlighting the importance of a regulated amount of APP for proper neurological health with age (174).
APP can undergo cleavage (Fig. 3) by an α-secretase enzyme, forming two fragments: a large 100 kD soluble fragment and an 11 kD membrane-associated protein (167). Importantly, α-secretase cleavage results in disruption (separation) of the Aβ region, between lysine 16 and leucine 17 (78, 167). Consequently, α-secretase cleavage does not produce Aβ peptides, and it does not have a pathological impact. Instead, Aβ is formed by the sequential proteolytic cleavage of the APP by a β-secretase enzyme, referred to as “β-site APP-cleaving enzyme (BACE-1),” followed by a γ-secretase enzyme or presenilin (72, 152). Two isoforms of Aβ are actually formed, with 40 amino acids, and with a second containing 42 residues. This slight cleavage variation allows for the inclusion of two additional hydrophobic residues in the Aβ42 peptide (75). As a result, Aβ42 aggregates much more readily compared with the more soluble Aβ40 form. Studies that focused on plaque formation in Alzheimer's disease brains found these deposits to mainly comprise Aβ42 (147). This indicates that the loss of regulated processing of the amyloid-β protein accelerates plaque formation, with this insoluble Aβ42 being much more toxic to neurons than is the soluble Aβ40 (178).
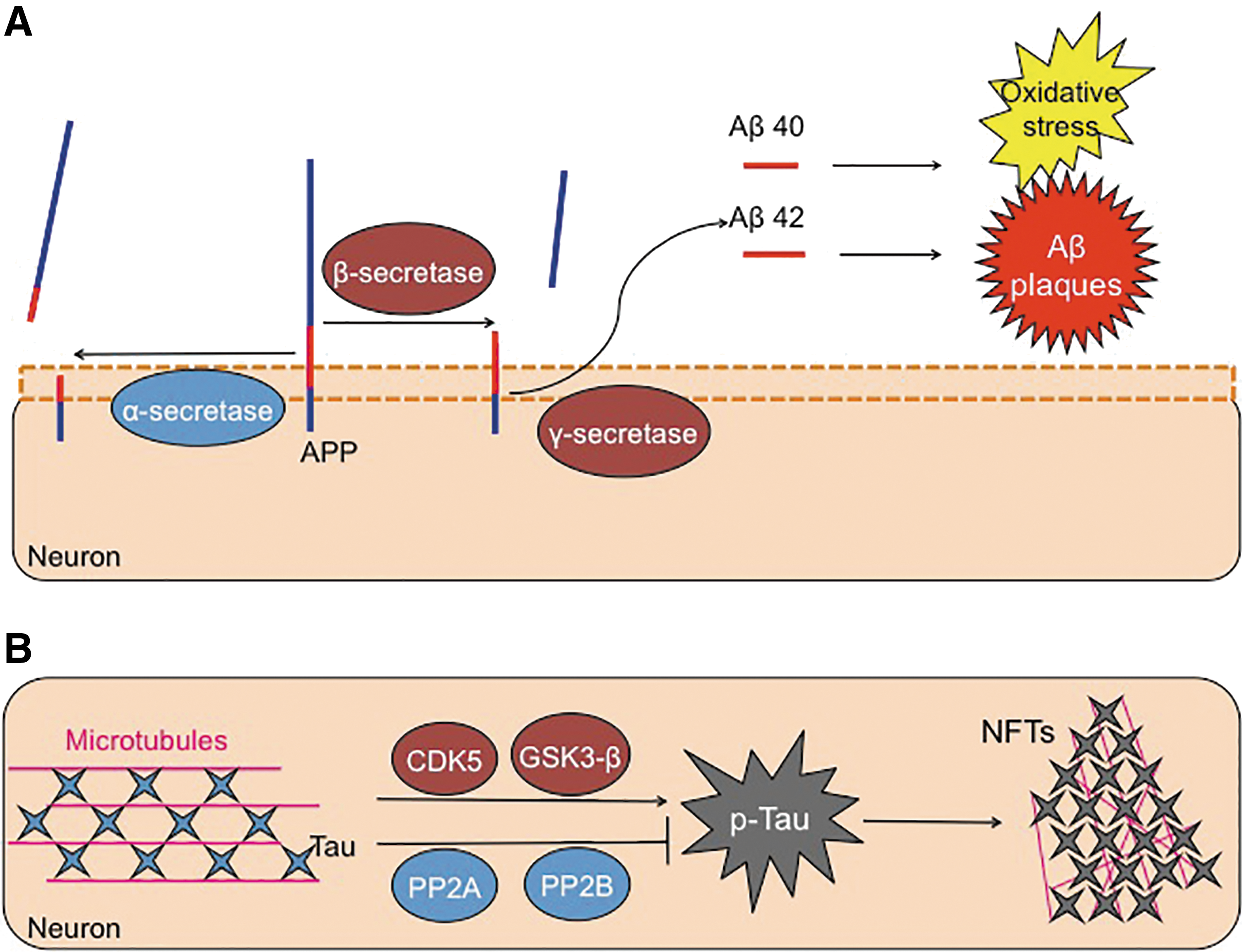
Interestingly, APP does not appear to be the preferred substrate of BACE-1, as APP lacks BACE-1's desired cleavage site. This indicates that the enzyme should serve a different, yet still unknown, role in the cell (91, 177). Studies in BACE-1 null mice showed decreased accumulation of amyloid-β and restoration of memory deficiencies that are common in the transgenic mouse model that overexpresses human-APP (118).
Aβ and mitochondria
The accumulation of Aβ not only hampers protein turnover but also contributes to the chronic elevation of oxidative damage, which is common in the Alzheimer's disease pathology. The brain is highly susceptible to oxidative damage due to its high lipid content and elevated energy demands (134). In turn, the high metabolic needs of the brain place a huge demand on the mitochondria, increasing their susceptibility to oxidative damage (19). In addition, postmortem studies of Alzheimer's disease brains show a decline in the enzymatic activity of both nuclear-derived and mitochondrial-encoded oxidative phosphorylation enzymes (46). In turn, the decline in ATP production may weaken the mitochondrial transmembrane electrochemical gradient, further contributing to oxidant formation (136). The close proximity of mitochondrial proteins to endogenously generated oxidants also makes them highly susceptible to damage.
To stave off the accumulation of oxidized proteins, mitochondria rely on various proteases to remove damaged proteins, with the most well studied being the Lon protease. Short-term exposure to various stresses (e.g., heat shock, serum starvation, hydrogen peroxide, peroxynitrite) has been shown to stimulate both Lon protein expression and proteolytic capacity (113, 162). However, long-term oxidant exposure or the aging process dampens Lon's ATP-dependent activity (16). Conversely, a decline in Lon expression has been associated with increased protein oxidation and decreased mitochondrial membrane potential (114).
Overall, mitochondrial dysfunction is a key feature of Alzheimer's disease pathology (22, 28, 135, 165), which is characterized by altered cellular metabolism. Recent studies have found that Aβ accumulation on mitochondrial membranes directly blocks the mitochondrial import of nuclear encoded proteins by the mitochondrial outer-membrane translocase (158). Additionally, Aβ has been shown to localize into the mitochondrial cristae, independent of the mitochondrial membrane potential (58). This poses two problems for mitochondria: (1) newly synthesized (and functioning) proteins are unable to enter; (2) localized Aβ aggregation hinders the electron transport chain and further increases free radical production (20, 21, 98, 169, 170). As Lon is the primary mitochondrial protease, the inability to import this nuclear-encoded protein could have dire consequences for mitochondrial function, and, in turn, further Alzheimer's disease progression (Fig. 4).
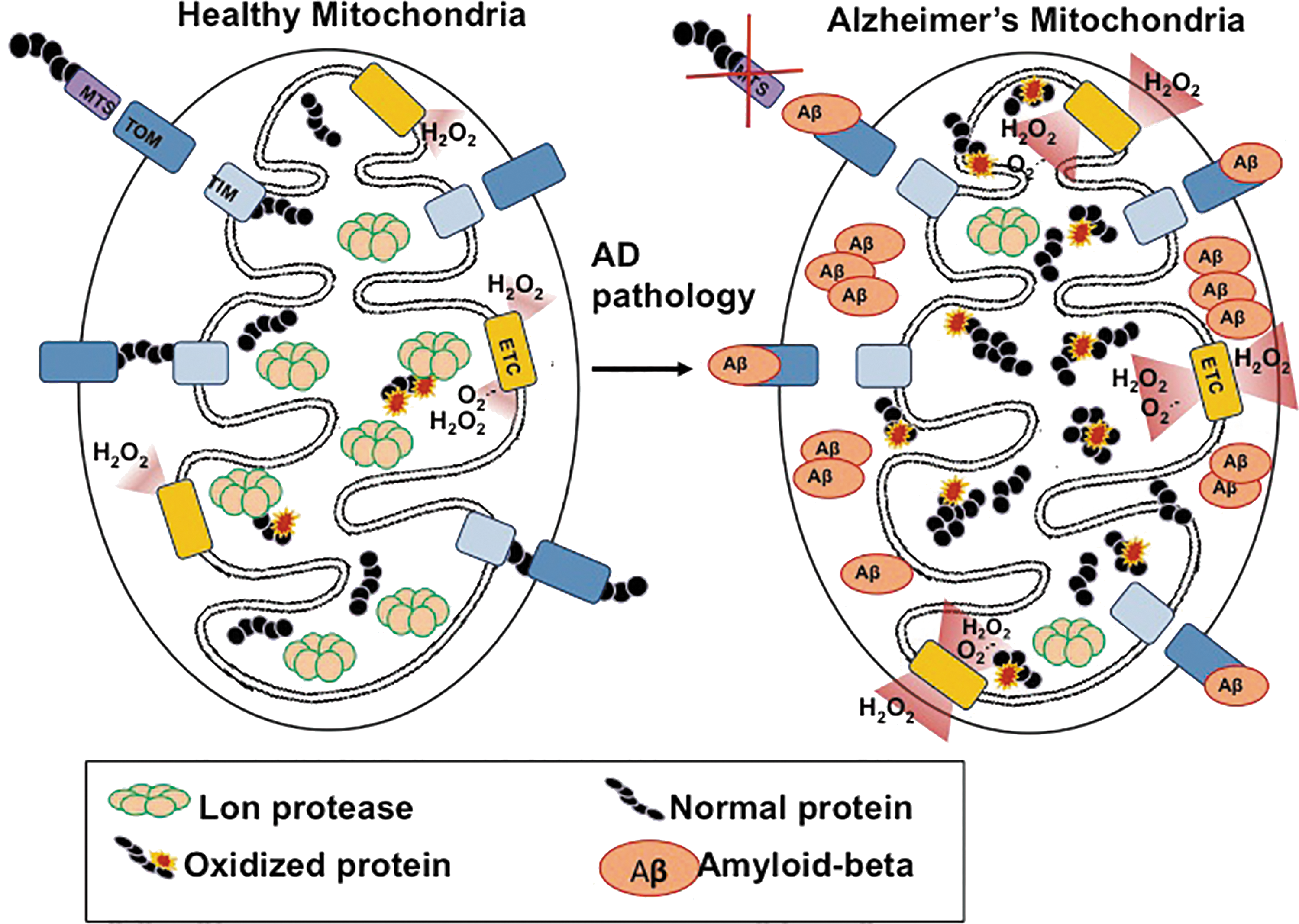
The importance of various mitochondrial metabolic enzymes (and their dysregulation) is readily evident in Alzheimer's disease pathology. Aconitase, a critical enzyme of the Krebs cycle, has been shown to become inactive during Alzheimer's disease (153). This is, in large part, due to the iron-sulfur cluster at its active site, which makes this enzyme highly susceptible to inactivation by oxidation (123). Under normal physiological conditions, mild oxidation of aconitase allows for rapid recognition and degradation by the Lon protease. However, in Alzheimer's disease pathology, which is characterized by a chronic elevation of oxidative stress, more severe aconitase oxidation occurs, making it a poor substrate for the Lon protease (14). The consequent accumulation and aggregation of inactive aconitase greatly decreases the metabolic production of NADH and FADH2 by the Krebs cycle (150). Both molecules are essential for the entry of reducing equivalents into the electron transport chain. Thus, a decline in Krebs cycle activity results in decreased activity of the respiratory chain, which further degrades the mitochondrial transmembrane potential and, ultimately, exacerbates the effects of the disease.
The Tau protein
Tau is a microtubule-associated protein that functions to stabilize microtubules, with the highest expression pattern found in the axons of neurons. It was originally characterized for its involvement and upregulation, along with tubulin, during neuronal differentiation (36). Due to Tau being critical for cellular morphology, its dynamic regulation is necessary to maintain neuronal structure and the migration of signaling vesicles (146). The six major isoforms of Tau, each varying in the number of microtubule-binding domains, are primarily regulated by phosphorylation, which is ideal due to the high amount of phosphorylation sites in Tau (45 serine, 35 threonines, and 4 tyrosines). Under Alzheimer's disease, phosphorylation of Tau is higher than in healthy people (85). Tau phosphorylation is also dependent on the developmental state, with peak levels occurring during fetal growth, and drastically tapering off in the adult brain (9, 102).
Tau activity is controlled by its phosphorylation state, which is modulated by two prominent kinases: cyclin-dependent kinase 5 and glycogen synthase kinase 3β (GSK3β), and phosphate removers: phosphatase 2A and 2B (17, 55). However, in the Alzheimer's brain, the highly orchestrated regulation of Tau is lost and Tau hyperphosphorylation occurs. Increased levels of unbound Tau (143), or declines in kinase activity (110, 121), may increase the likelihood of Tau misfolding. In turn, prevention of its normal turnover by the Proteasome leads to the disintegration of the microtubule skeleton and the formation of NFTs (25, 70).
The cause for Tau dysregulation is still unclear. Abnormal Tau phosphorylation may also be conducted by the phosphorylated protein itself, with hyperphosphorylated Tau sequestering away native Tau and resulting in microtubule disarrangement (70).
Linking Aβ and Tau
Though Tau hyperphosphorylation and Aβ aggregation have been linked to Alzheimer's disease development, the relationship between these two proteins remains unclear. The stress-inducible Regulator of Calcineurin gene (RCAN1) may be one of the bridges from one molecule to the other (93). Calcineurin is a serine/threonine phosphatase that is involved in p-Tau dephosphorylation. RCAN1 expression can result in synthesis of RCAN1-1L, RCAN1-1S, and/or RCAN1-4, proteins that inhibit calcineurin (44, 145). RCAN1 expression also causes an induction/activation of GSK3β, which is responsible for Tau phosphorylation (38). Thus, it can cause extensive Tau phosphorylation. During transient oxidative stress, RCAN1-4 is highly induced, causing beneficial short-term inhibition of calcineurin (29). However, chronic expression of the RCAN1-1L and RCAN1-1S isoforms, seen in Alzheimer's disease and Down syndrome, causes chronic inhibition of calcineurin and higher steady-state levels of phosphorylated Tau (97) (Fig. 5). This is further substantiated by studies conducted in RCAN1 knock-in mice, in which elevated levels of phosphorylated Tau are seen (39).
In parallel studies, Aβ42 has been shown to trigger the generation of oxidants in both cells and mitochondria, a characteristic of the Alzheimer's disease pathology (77). Specifically, Aβ42 can trigger the transcriptional increase of RCAN1 (60). In cultured rat neurons, the presence of Aβ42 resulted in the increase of steady-state levels of phosphorylated Tau, which was only reversed on silencing of RCAN1 (93). Hence, Aβ42, which is highly expressed in the Alzheimer's disease brain, triggers the chronic increase of RCAN1 and, therefore, the inhibition of calcineurin and activation of GSK3β, which accelerates the Tau hyperphosphorylation.
In humans, the apolipoprotein E (apoE) ɛ4 allele has been identified as a risk factor for developing sporadic Alzheimer's disease (128, 144). Potentially, this is because the apoE ɛ4 allele is not capable of exerting a neuroprotective effect, unlike the ɛ2 and ɛ3 alleles (64, 99). Individuals with the apoE ɛ4 allele have also been found to express higher levels of RCAN1 compared with noncarriers (8) and, consequently, more p-Tau (61, 93). Thus, this further cements a possible connection between Alzheimer's disease and RCAN1. Lloret and colleagues have also pointed out that various other molecules may represent potential links between Aβ and Tau, including Pin-1, p38 MAPK, or cyclin-dependent kinase 5-p25 (94).
Another possible link is the activation of the caspase pathway, which can be mediated by Aβ (143). In turn, this activates various kinases, such as MAP-kinase (110, 121) or GSK3β, all of which may contribute to Tau hyperphosphorylation (57).
Aβ, the Proteasome, and the IP
Under normal physiological conditions, Aβ is produced and secreted by neurons in response to synaptic activity. On secretion to the extracellular environment, Aβ is degraded by glial cells, which migrate toward amyloid deposits (76). Microglia can phagocytize and degrade Aβ (76), whereas astrocytes also degrade Aβ (175) and facilitate its clearance from the brain to the blood that is mediated by apoE (84, 131). Although the mechanism that causes the transition from normal physiological function to pathological Aβ accumulation is still unknown, several lines of evidence point to the involvement of a dysregulation of protein degradation (26, 73, 107, 168). The inability to maintain a homeostatic balance between amyloid production and degradation results in the damage from amyloid accumulation being twofold: direct inhibition of Proteasome activity (95, 117, 166), and the indirect elevation of oxidative stress (98, 159, 170) (Fig. 6), both of which contribute to protein malfunction. Aβ degradation by both the 20S Proteasome and the ATP-ubiquitin-dependent 26S Proteasome has been demonstrated in in vitro experiments (95, 100, 181), but there is insufficient evidence to tell which one may be more important in vivo.
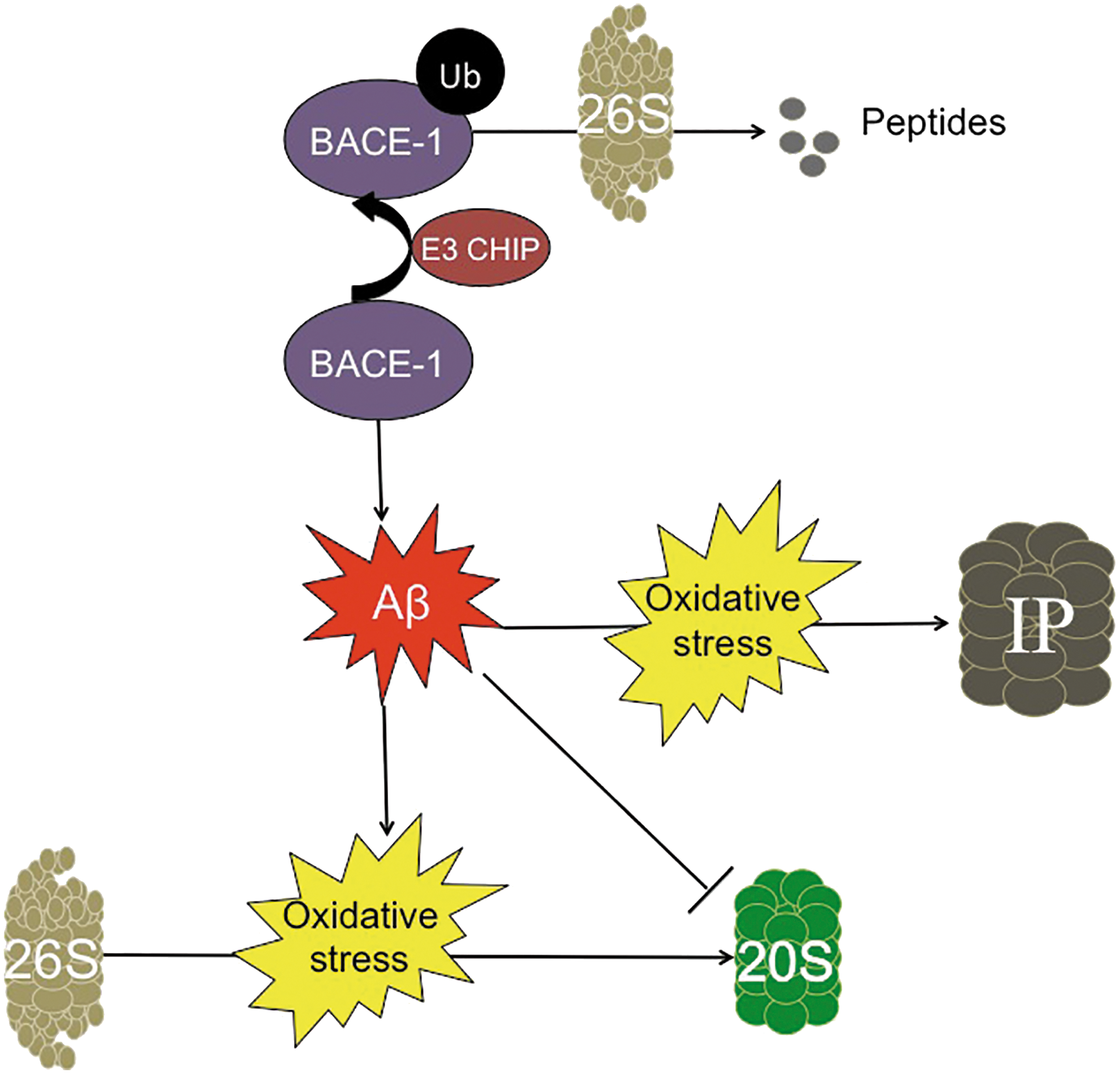
Initial studies into the role of the 20S Proteasome in neurodegenerative diseases found that inhibition of Proteasome activity was enough to accelerate neuronal death. Early in vitro studies showed that direct application of Aβ to neuronal cells triggers Proteasome inhibition (140) (Fig. 6). Application of pharmacological inhibitors of the Proteasome was separately shown to induce neuronal toxicity and eventual death (130). Later studies demonstrated a substantial decrease in proteolytic activity when crude lysates of (postmortem) brain tissue from Alzheimer's disease patients were compared with age-matched disease-free controls. Specifically, 20S Proteasome activity was dramatically suppressed in the hippocampus, the superior and middle temporal gyri, and the inferior parietal lobule (81). The study also concluded that Proteasome activity most likely declined as a result of post-translational modifications, as there was little difference in the overall amount of the proteasomal α and β subunits between Alzheimer's disease subjects and controls. These findings were extended by a later study, which showed that purification of 20S Proteasomes from crude lysates of Alzheimer's disease brains resulted in a substantial increase in proteolytic capacity (44). These results indicate that Alzheimer-related decreases in 20S core Proteasome activity are likely due to accumulation of damaged, aggregated, and cross-linked cytosolic proteins (including Aβ) that act as Proteasome inhibitors (47). Additional studies showed that removal of Aβ by treatment with a γ-secretase inhibitor in the triple-transgenic Alzheimer's disease mouse model restores Proteasome degradation capacity (2).
Activity of the ATP-ubiquitin-dependent 26S Proteasome greatly diminishes in oxidizing environments, including the Alzheimer's disease brain (140). Various studies have shown the importance of protein ubiquitinylation in modulating multiple cellular processes, including the ubiquitin-dependent degradation of transcriptional activators such as Nrf2 and NF-κB, and a plethora of kinases (66, 101, 179). In turn, degradation by the 26S Proteasome provides the cell with a controlled mechanism to turn on and off key regulators of these various cellular response pathways.
Interestingly, the normal proteolytic turnover conducted by the ATP-ubiquitin-dependent 26S Proteasome is diminished in the Alzheimer's disease state. This is seen through the accumulation of ubiquitin in amyloid deposits and NFTs. Additionally, a mutant form of ubiquitin has been observed in the brains of Alzheimer's patients. This mutant form of ubiquitin (Ub+1) not only evades disassembly by deubiquitinating enzymes but is also a potent inhibitor of degradation by the 26S Proteasome (87). Importantly, loss of 26S Proteasome activity appears to accelerate the Alzheimer's disease phenotype. One study utilized a conditional knockout against psmc1, an ATPase subunit of the 26S Proteasome, and it selectively targeted neurons of the substancia nigra and forebrain. The impairment of ubiquitin-dependent proteolysis triggered the formation of inclusion bodies and neurodegeneration within the nigrostriatial pathway and forebrain regions (11).
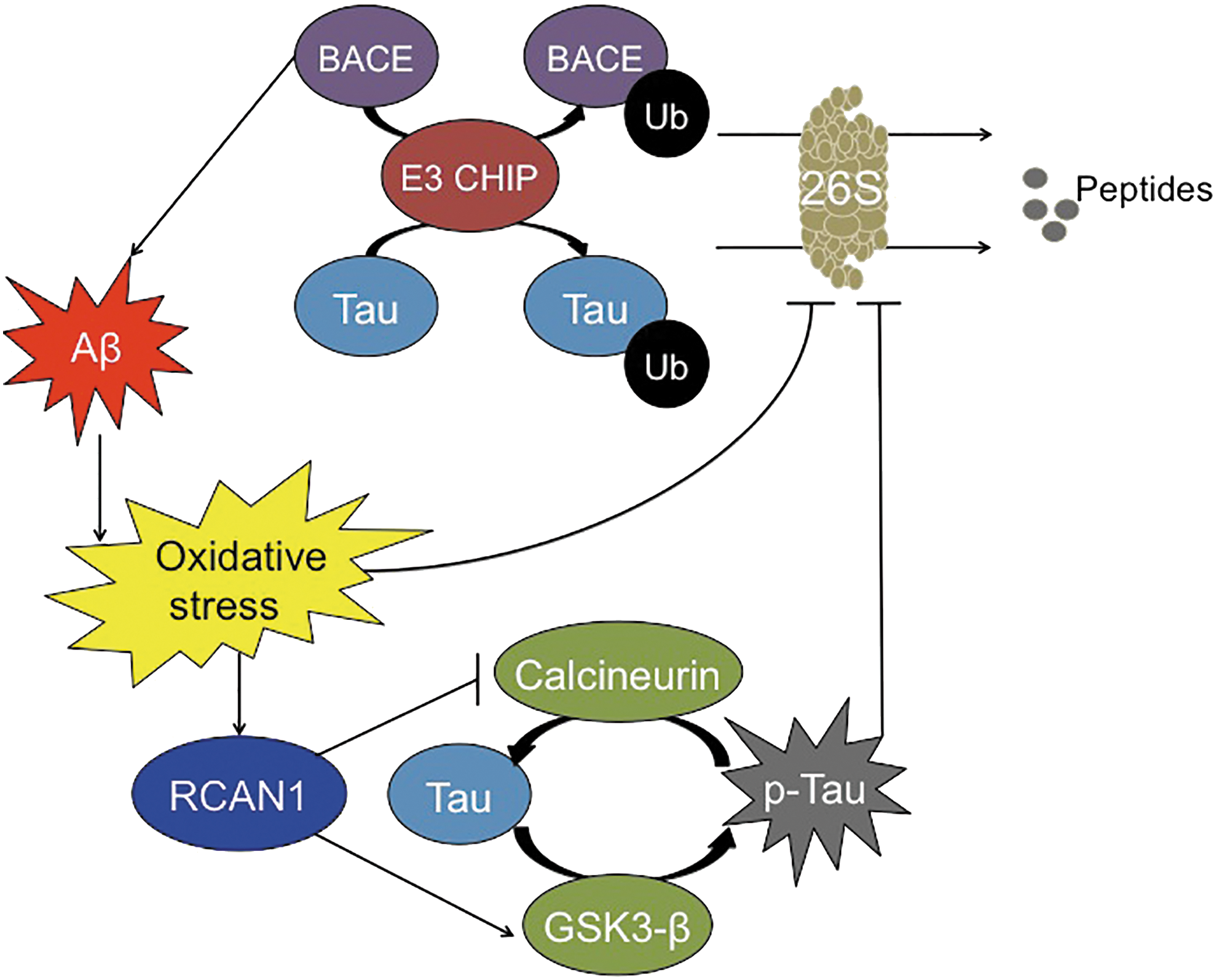
Various proteins involved in the ATP-ubiquitin 26S Proteasome system appear to become dysfunctional during Alzheimer's disease development. One such protein is the C terminus of HSP70 interacting protein (CHIP), which is an E3 ligase that has been recently implicated in degrading BACE-1 (157). Hence, an inverse relationship exists between CHIP and BACE-1; by decreasing BACE-1 expression, CHIP prevents the aberrant processing of APP and decreases the accumulation of Aβ (Fig. 6). This relationship was further supported in studies by Oddo and colleagues, who found that Aβ accumulation inhibits CHIP expression (116), hence demonstrating a positive feedback loop that would only promote further amyloid accumulation.
Less well studied but, perhaps, no less important is the IP, which was originally characterized for its role in the immune response (4). Prior studies have shown that the IP works not only to degrade proteins for antigen presentation for the major histocompatibility complex but also to degrade oxidized proteins with rates and selectivity that are comparable to, or better than, those of the 20S Proteasome (126). This similarity in degradation rate may be partially attributed to the key roles that both proteasomes play in highly oxidant-rich environments: The immune response triggers an elevation in cellular oxidants and, in consequence, an increase in oxidized proteins (124). Therefore, it is perhaps unsurprising that in the highly oxidative environment of the human Alzheimer's disease brain, the IP is activated (106). More importantly, unlike the 20S and the 26S Proteasomes, which show decreased activity, both the expression and proteolytic activity of the IP are elevated in transgenic mice models of Alzheimer's disease (5, 119) (Fig. 6).
Other animal models of amyloidosis, such as the nematode C. elegans, have also studied the interaction of protein degradation systems with Aβ. Overexpression of proteasome subunits such as AIP-1 (63) {an ortholog of the human AIRAP component of the 19S regulatory subunit (161)}, or pbs-5 (23) {an ortholog of the human β5 subunit of the 20S core (10)}, has been shown to increase 26S and 20S Proteasome activities, respectively. In both cases, reduced Aβ toxicity and aggregation were observed in C. elegans. Promotion of the activity of protein degradation with polyphenols has also been successful in alleviating amyloidosis in these worms (138, 139). Compounds that act against Aβ aggregation help to recover protein homeostasis (1), through the action of transcription factors involved in the stress response, such as the heat shock factor 1 and SKIN-1 (homolog of human Nrf2).
Because of the critical role that amyloid accumulation has been believed to exert on the development of pathology, various therapies targeting Aβ removal are currently under development or evaluation. Those that have gained the most traction are Aβ immunotherapies. These treatments rely on antibodies that have high specificity toward a disease-related antigen (50). Bapineuzumab, which is a humanized IgG monoclonal antibody derived from the murine antibody, 3D6, nonselectively targets both soluble and fibrillary Aβ. This approach relies on the hope that nonspecific targeting of Aβ may decrease the total amount of Aβ. Unfortunately, phase three clinical trials have shown no difference in cognitive abilities of Alzheimer's disease patients treated with the antibody compared with controls (149). Other efforts utilize more stringent approaches by targeting soluble Aβ specifically, rather than its insoluble counterpart. However, the low penetrance of the antibody across the blood–brain barrier has made it difficult to show significant efficacy in patients in the early phases of the disease. Solanezumab, a humanized IgG1 monoclonal antibody derived from the murine monoclonal antibody 266, has high binding affinity only to soluble Aβ. However, phase three clinical trials have shown poor results (34, 40).
At the present time, it is fair to say that anti-amyloid therapies have so far not been useful in human Alzheimer's disease treatment. This may be because once overt disease symptoms appear, it is already too late to act against the pathology; this interpretation is invoked by those who argue for the need to find early and sensitive biomarkers. It is also possible, however, that Aβ is not the only factor(s) responsible for Alzheimer's disease, and that a multifactorial approach will be necessary, including Aβ, Tau, oxidative stress, and the regulation of proteostasis at the very least (31, 70, 86, 88). Clearly, we are still quite a long way from having any effective therapy to offer Alzheimer's disease patients and their families.
Tau and the Proteasome
Under homeostatic conditions, endogenous Tau is usually turned over by the Proteasome (42) (Fig. 7). However, pharmacological inhibition of the Proteasome by lactacystin results in Tau accumulation, regardless of its phosphorylation state (52, 92, 166). It is important to note that this finding has been contested, as other groups have shown that Proteasome inhibition decreases Tau accumulation, potentially through the activation of pathways that are secondary to the Proteasome, such as other proteases or apoptosis (42, 45). Importantly, in vitro aggregation of Tau blocks proteolysis by the 20S core Proteasome (79). This demonstrates that the Proteasome interacts with Tau, primarily during the disease state. Since the 20S Proteasome usually removes oxidatively damaged proteins, the large protein aggregates formed during Tau accumulation become difficult to degrade. In consequence, Tau continues to accumulate and the Proteasome is further inhibited (Fig. 7).
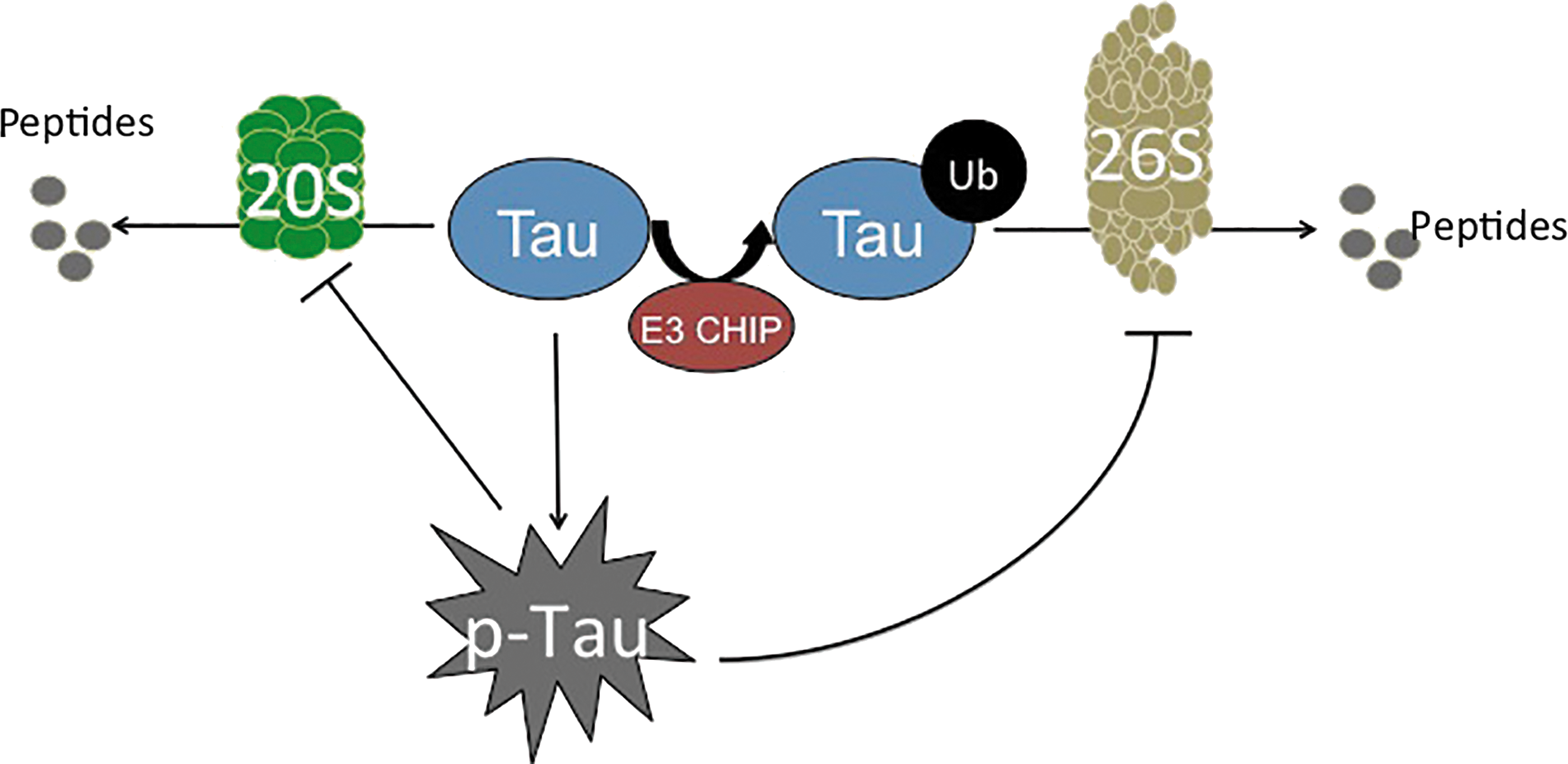
Not only is the 20S core Proteasome's proteolytic capacity limited, but also the ATP-ubiquitin-dependent 26S Proteasome system appears to be inhibited by p-Tau. The relationship between the ATP-ubiquitin-dependent 26S proteasomal system and Alzheimer's disease pathology was initially evident on the identification of ubiquitin within NFTs (108). Further studies showed that the phosphorylation state of Tau dictates the addition of ubiquitin. Specifically, toxic Tau binds to Hsc70, a constitutively expressed protein of the heat-shock family that facilitates the ubiquitin tagging of Tau by the E3 ligase, CHIP (154). CHIP seems to be implicated, as with Aβ, in the processing of Tau, providing another link between the two of them, as suggested by Oddo and colleagues (116) (Fig. 5). Interestingly, Metcalfe et al. found that the pharmacological elevation of cyclic AMP (cAMP) levels provides neuronal protection by preventing caspase activation, which is shown to be increased due to tau hyperphosphorylation (103). In concordance with this study, Myeku et al. showed that activation of cAMP-protein kinase A is able to reverse part of the repression of proteolysis caused by p-Tau, thus promoting the reduction of Tau aggregation and diminishing the decline of cognitive abilities (111).
CHIP has been identified as a chaperon-like E3 ligase, due to its interaction not only with Hsc70 but also with Hsp70 and Hsp90, both of which are critical in protein folding (125). Apart from this, these heat-shock chaperones have been shown to directly interact with Tau, although its phosphorylation status was unknown (35). Thus, CHIP may act to selectively ubiquitin-tag misfolded proteins that cannot be correctly folded by various chaperone proteins (i.e., the heat-shock chaperones) and target them for degradation by the 26S Proteasome. However, in the advanced states of Alzheimer's disease pathology, which is already heavily plagued by an accumulation of highly insoluble Tau, the targeting of these protein aggregates for degradation may be futile. Large protein aggregates become too cumbersome for Proteasome degradation (they cannot enter into the proteasome cylinder for degradation), and they only continue to accumulate.
Various strategies are currently under development, which will attempt to combat loss of proteolytic activities in Alzheimer's disease. To combat the decline of Proteasome function in the Alzheimer's disease brain, a novel exogenous delivery method of Proteasome via nanoparticles has been successfully implemented in cell culture (56) but now awaits verification of efficacy in vivo.
Aging, the Proteasome, and Alzheimer's Disease
The exact relationship between Alzheimer's disease and aging is highly controversial, due to the difficulty of separating normal “brain” aging from disease development. Nevertheless, a strong predictor of disease development is age (90). Many older adults develop dementias, which, based on postmortem studies, would classify them as having suffered Alzheimer's disease, as evidenced by a high abundance of amyloid plaques and Tau tangles (163). Women are also more susceptible to the disease development than men (120, 169). Many others, however, either experience minimal dementia with aging or suffer from other forms of dementia entirely.
In “normal aging,” clinical studies have shown that certain processes of the brain decline, whereas others remain functional. For example, autobiographical memory and semantic knowledge typically remain untouched, compared with working memory and processing, which appear to diminish (164). These findings have been further supported by neuroimaging studies that found atrophy in the prefrontal cortex, indicative of a decline in executive function, whereas the hippocampus shows no significant shrinkage (142).
In contrast, Alzheimer's disease pathology shows a much greater decline in cognitive functions and more marked molecular changes. Not only there is a physical atrophy of the white matter, but also there is shrinkage within the hippocampus. This results in a decline in CA1 hippocampal neurons that does not occur during normal aging (172). These neurons are critical in the output pathway from the hippocampus, which is involved in the formation of long-term memory and spatially oriented tasks (172). Also, postmortem studies show a high accumulation of NFTs and Aβ plaques. However, it should be noted that simply the presence of these plaques and tangles is no longer considered a definitive diagnosis of Alzheimer's disease, as postmortem brains of healthy individuals also show this morphology (80). Brain histology from cognitively healthy individuals 85 years and older typically shows at least some amount of plaque and tangle formation (129). Therefore, many clinicians believe that cognitive decline in Alzheimer's disease occurs much sooner than normal age-related dementia. Thus, early-age onset of dementia-like symptoms places these individuals at a high risk for developing the disease (37).
Both Alzheimer's disease and normal brain aging show a decline in protein turnover (62). This has been partially attributed to the increased oxidative stress that arises in the brain with age, due to the brain's already high metabolic rate, and the formation of potentially toxic byproducts from neurotransmitter signaling (18, 83). Concurrently, Proteasome activity declines, with mild inhibition causing up to 5% of total cellular proteins to aggregate, with a small portion showing heavy amounts of cross-linking (32, 82). This indicates that low levels of Proteasome inhibition are capable of triggering many of the phenotypes of neuronal aging, but can be rescued, on increased expression of various proteasomal subunits (33). In the Alzheimer's disease state, Proteasome inhibition is more pronounced, potentially due to the rise in protein oxidation, and Proteasome activity declines significantly below the age-related norms. Clearly, future work will be critical in deciphering whether the differences in Proteasome dysfunction during normal aging versus Alzheimer's disease are outcomes or contributors to the decline of neuronal function.
Conclusions
Alzheimer's disease is one of the most common neurodegenerative diseases whose prevalence is projected to continue increase significantly due to the world's aging population. As current treatments are able to do little to alleviate the disease or slow its progression, researchers must explore alternative approaches beyond those that target the disease characteristics: Aβ accumulation, Tau hyperphosphorylation, and oxidative stress. Since protein homeostasis is critical in the disease progression, this pathway offers a new avenue for exploration. Much work is still necessary to understand how the normal neuronal circuitry loses regulation of proteostasis.
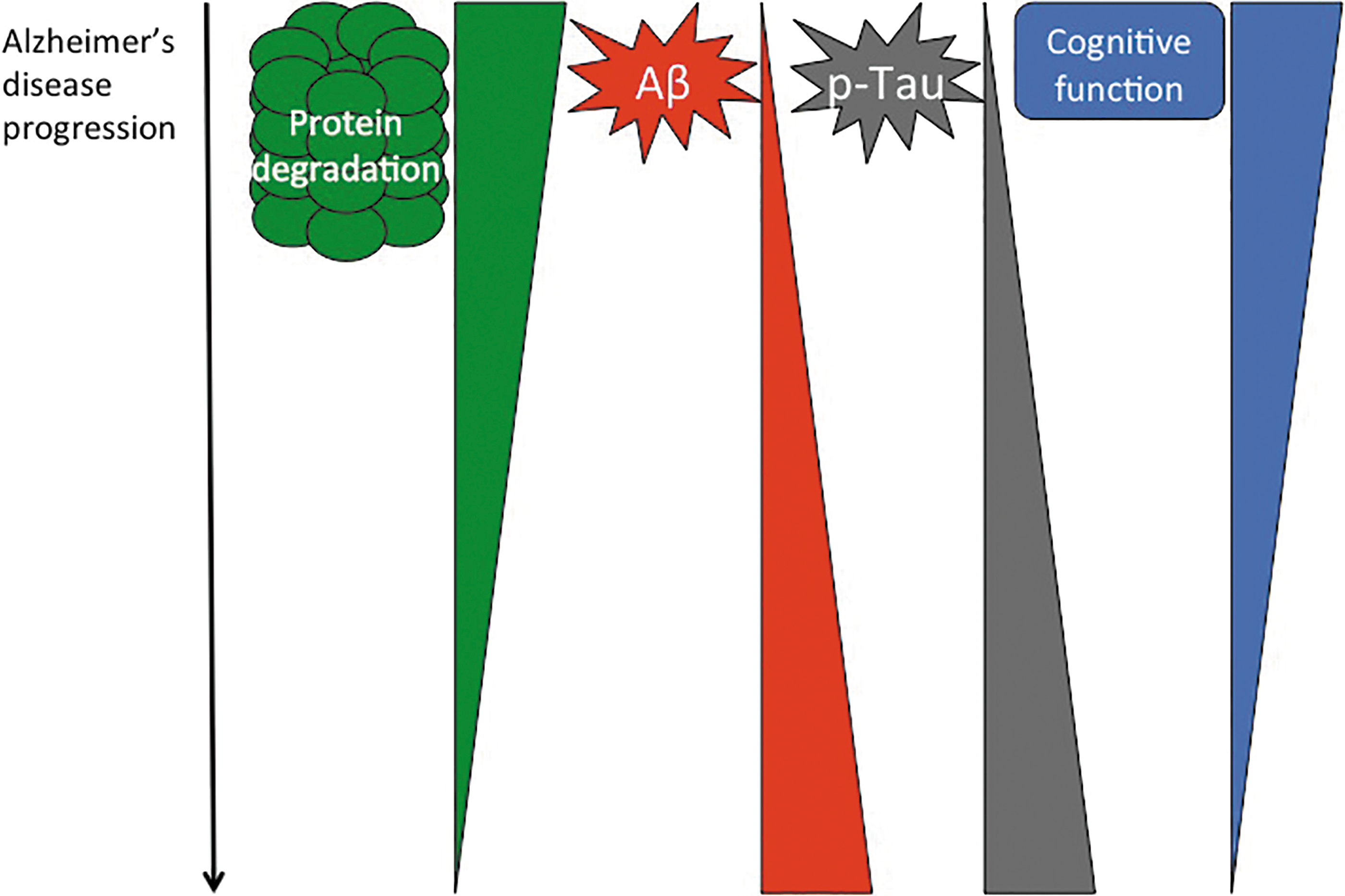
In many ways, Proteasome activity now appears to be at the tipping point for disease development. Regulated proteostasis is critical for proper cellular function, and loss of Proteasome activity is a feature of Alzheimer's disease pathology. Both amyloid accumulation and Tau hyperphosphorylation appear to negatively impact Proteasome function, placing it at a key point in disease progression (Fig. 8). Much prior work has focused on characterizing the dysregulation of the Proteasome in the disease state. However, we are only just beginning to unravel the underlying mechanism(s) behind Proteasome dysfunction. Such mechanistic understanding will be critical in unraveling Alzheimer's disease initiation and development, and in the targeted exploration of disease prevention or effective therapies.
Footnotes
Acknowledgments
The authors were supported by Grant No. ES003598 from the National Institute of Environmental Health Sciences, of the U.S. National Institutes of Health, to K.J.A.D.