
Abstract
Significance:
Worldwide demand has driven the development of hemoglobin (Hb)-based oxygen carriers (HBOCs) as potential acellular oxygen therapeutics. HBOCs have the potential to provide an oxygen bridge to patients and minimize current problems associated with supply and storage of donated blood. However, to date, safety and efficacy issues have hampered the approval of viable HBOCs in the United States. These previous efforts have underscored the need for a better molecular understanding of toxicity to design safe and oxidatively stable HBOCs.
Recent Advances:
High-resolution accurate mass (HRAM) mass spectrometry (MS) has recently become a versatile tool in characterizing oxidative post-translational modifications that occur in Hb. When integrated with other analytical techniques, HRAM data have been invaluable in providing mechanistic insight into the extent of oxidative modification by quantifying oxidation in amino acids near the reactive heme or at specific “oxidative hotspots.”
Critical Issues:
In addition to providing a deeper understanding of Hb oxidative toxicity, HRAM MS studies are currently being used toward developing suitable HBOCs using a “two-prong” strategy that involves (i) understanding the mechanism of Hb toxicity by evaluating mutant Hbs identified in patients with hemoglobinopathies and (ii) utilizing this information toward designing against (or for) these reactions in acellular oxygen therapeutics that will result in oxidatively stable protein.
Future Directions:
Future HRAM studies are aimed at fully characterizing engineered candidate HBOCs to determine the most oxidatively stable protein while retaining oxygen carrying function in vivo. Antioxid. Redox Signal. 26, 777–793.
Introduction
C
The demand for blood donation alternatives has driven the commercial development of Hb-based oxygen carriers (HBOCs) or “blood substitutes” over the past 30 years (43). Initially, there was a high level of optimism for HBOCs to replace blood transfusion requirements. The benefits of such an accomplishment would potentially eliminate the problems associated with blood availability, reduce the risk of disease transmission, and improve oxygen delivery and need for long-term blood storage (34, 66, 88).
Despite these advantages, no viable HBOCs have been approved in the United States. The primary reason for this stems from multiple reports of adverse cardiovascular and cerebrovascular events associated with hypertension in individuals transfused with these products (79, 86). In hindsight, this should not be surprising since HBOCs (studied to date) are specifically acellular Hb-derived proteins; because they are not “actual blood” they do not perform other blood functions such as nutrient transport, immune response, and coagulation. Preliminary evaluation using animal models suggests that some HBOCs do oxygenate tissues shortly after infusion (3, 15, 86). Current-generation HBOCs are intended therefore to complement standard blood transfusions by acting as an “oxygen bridge” in extreme life-threatening situations, such as battlefield trauma and in surgical settings and/or where patients refuse blood transfusions due to religious reasons (3).
Historical Overview of HBOC Design Strategies
The first attempt to produce the so-called blood substitutes involved transfusion of stroma-free, unmodified acellular Hb into humans, which resulted in renal dysfunction, abdominal pain, and hypertension (64, 66, 81). Additional analysis of these early studies indicated that diluting Hb into blood plasma resulted in the accumulation of Hb dimers due to rapid tetrameric dissociation; dimeric Hb readily passed through the kidneys resulting in rapid clearance, which can be damaging to kidneys. In addition to this pathology, Hb dimers were characterized to be unstable and therefore susceptible to oxidation, heme loss, and precipitation; these events ultimately led to oxidative stress, blockage of kidney tubules, and ultimately renal failure. Finally, it was also observed that these acellular Hbs scavenge endothelial NO (a vessel dilator) thus negatively impacting vasoregulation and possess oxygen binding behavior vastly different than what occurs in the protective environment of red blood cells (5, 21, 45, 64).
To address these drawbacks, several different biochemical and recombinant strategies (Fig. 1) have been attempted with varying success. Among these included chemically crosslinking human- or bovine-derived Hb using a site-specific bifunctional crosslinker such as di-bromo bis-fumarate (DBBF/also known as diaspirin crosslinked Hb [DCLHb]), polymerization/conjugation with polyethylene glycol polymers (PEGylation), and genetically engineering α-crosslinked recombinant Hb (expressed in bacteria or yeast host systems) (3, 16, 36, 40, 47, 73, 97, 98). These modifications aim at either covalently linking dimers in a stable tetrameric form or in the case of polymerization/conjugation (such as PEGylation) to increase the surface area or hydrodynamic volume; all three modification strategies have been shown to increase intravascular retention and substantially reduce renal filtration of Hb dimers (4, 17, 25, 80).
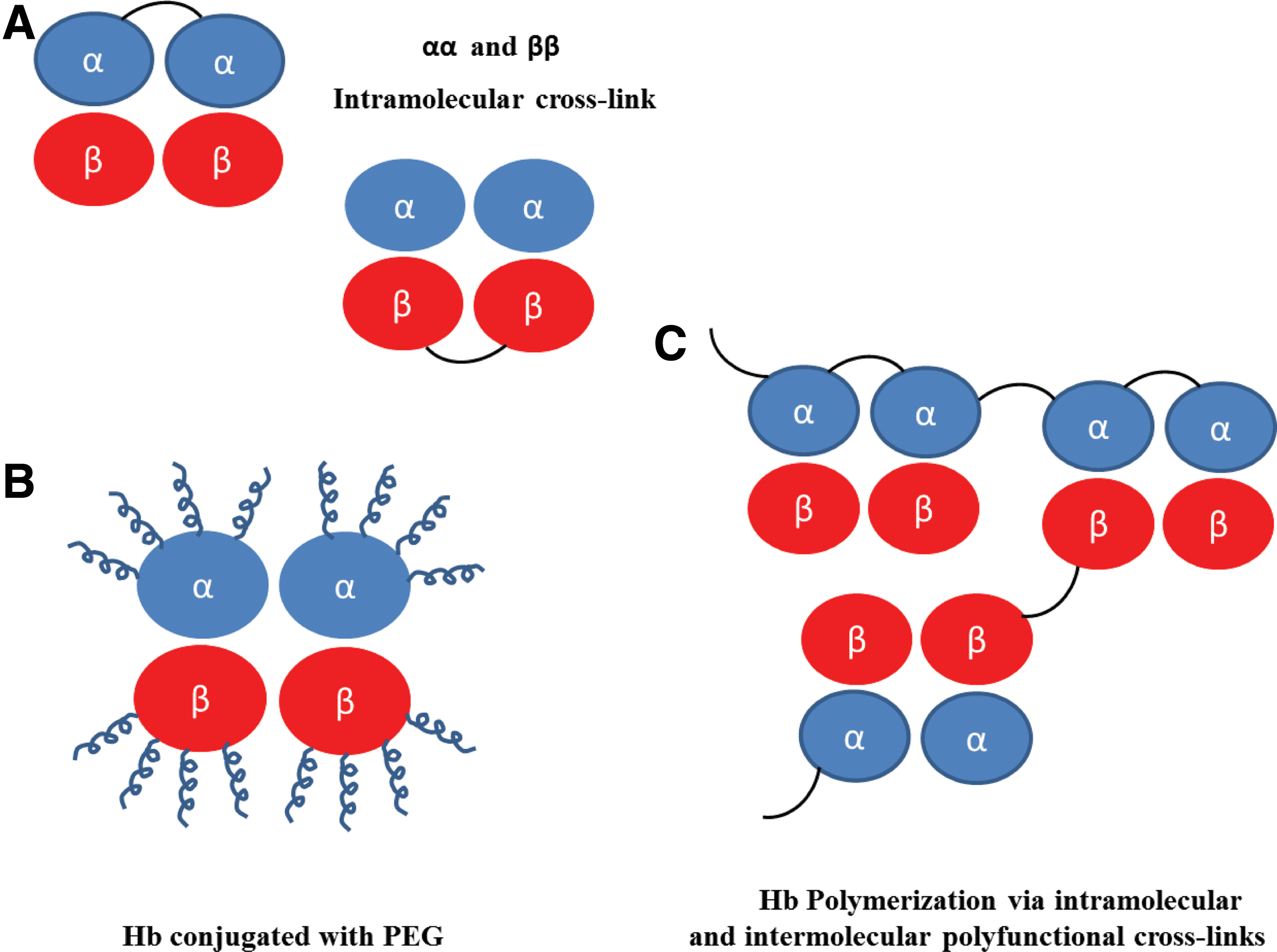
To date, several different commercially developed HBOCs (using anyone of the methods in Fig. 1) have been tested in animal and human subjects. These Hbs, in general, demonstrated reasonable preclinical safety and efficacy in healthy volunteers in Phase 1 trials. However, as clinical development advanced into later stages, an undesirable safety and efficacy profile became clearer in the patient population studied leading to the termination of the clinical development of many first-generation HBOCs (8, 35, 52, 59, 73, 75, 83, 87). These HBOCs failed clinically, in large part, due to serious adverse events associated with their infusion in humans attributed to nitric oxide (NO) scavenging by extracellular Hb, heme-mediated oxidative reactions, and oxygen oversupply (100).
The Basis of HBOC Toxicity
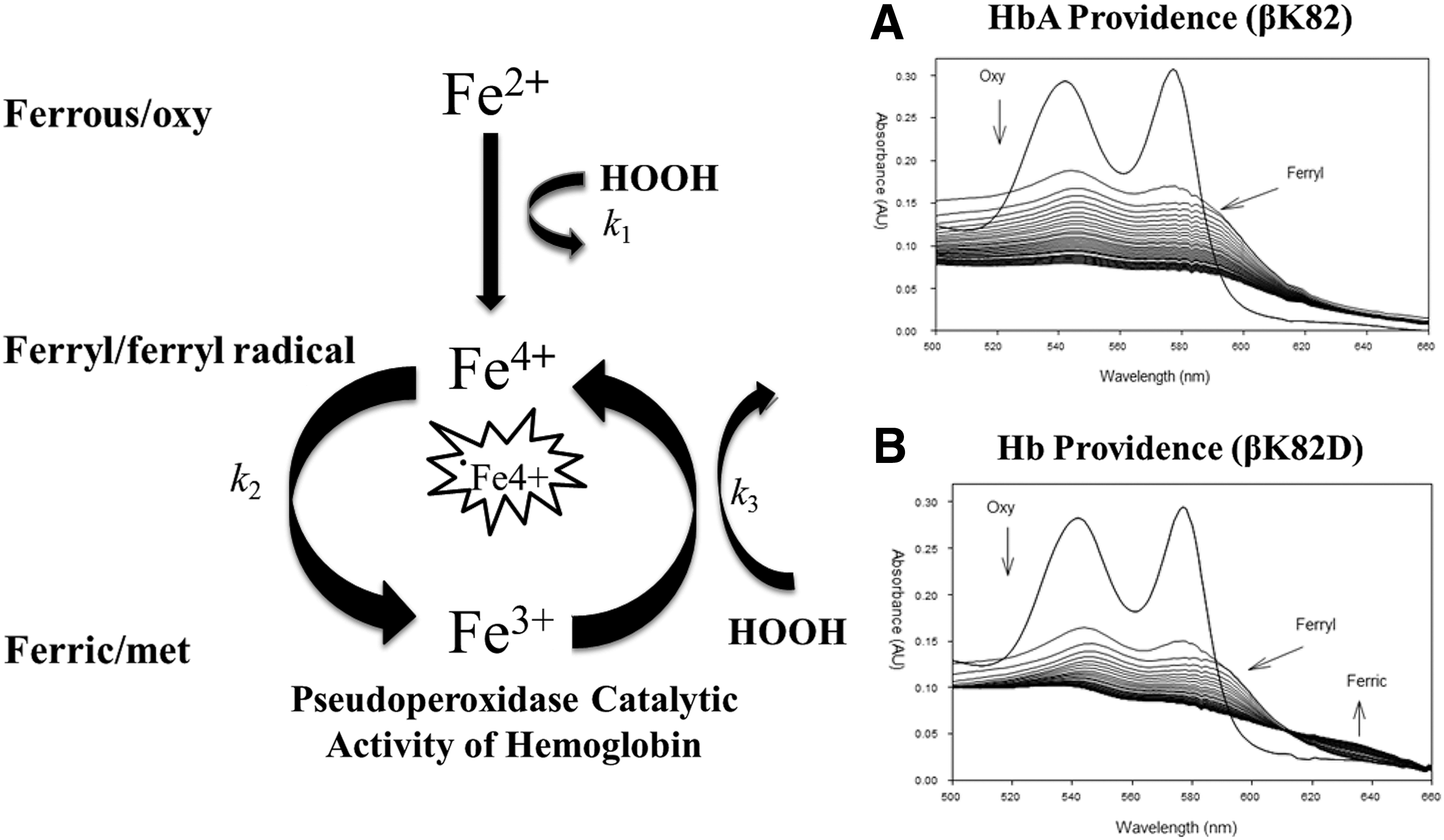
Several recent in vitro and animal studies showed that Hb oxidation plays a key role in the genesis of many adverse events associated with Hb (chemically and nonchemically modified) (16, 64). Outside the RBC, acellular HBOCs are oxidatively toxic due to heme redox reactivity, which is ironically essential for the oxygen (O2) carrying function. On O2 binding, ferrous heme iron (HbFe2+-O2) spontaneously auto-oxidizes to form a non-O2 carrying ferric heme iron (HbFe3+ aka metHb). During this naturally occurring process, reactive oxygen species such as superoxide (O2·−) and hydrogen peroxide (H2O2) are formed, which (in the case of H2O2) drives a catalytic pseudoperoxidase cycle that includes the transformation of HbFe2+-O2 to a transient reactive oxyferryl Hb (HbFe4+ = O) intermediate. The HbFe4+ = O species autoreduces back to HbFe3 and in the presence of additional H2O2, the ferryl Fe4+ iron is regenerated. However, if the reaction with H2O2 begins with the HbFe3+, a ferryl protein cation radical (ċHbFe4+ = O) is formed (Equation 1
–3) (76). These oxidative processes are well controlled within the RBCs where Hb is continuously reduced to the functional ferrous form by several reductive enzymes (catalase, superoxide dismutase, methemoglobin reductase, etc.). However, when left unchecked (i.e., in noncompartmentalized environment), acellular Hb becomes oxidants that damage themselves and nearby biological molecules. These reactions are summarized by the following equations:
This oxidative toxicity is primarily due to the high midpoint redox potentials (E°1/2∼1.0 V) of the ferryl heme and its associated protein cation radical (76). These internal reactions result in irreversible oxidation of amino acids, areas in the protein known as the “oxidative hotspots” (particularly the β Cys93 side chain) that cause the collapse of β subunits, Hb degradation, and the ultimate release of heme (39). Recent animal studies from sickle cell mice suggest that this resulting heme moiety gives rise to serious consequences, including endothelial cell activation and vaso-occlusion (9, 14). These studies have also shown that heme released from oxidizing Hb binds to Toll-like receptors (TLR4) that trigger a cascade of inflammatory reactions (9). Consequently, heme has been recognized as a “damage-associated molecular pattern” (DAMP) molecule on par with molecules such as lipopolysaccharides and other inflammatory stimulants (28). Clearly, these internal oxidative pathways (leading to heme release) would have to be curtailed to minimize these inflammatory reactions.
Two-Prong Strategy Toward Engineering Oxidatively Stable HBOCs
The challenge for HBOC developers is to design stable Hbs with optimal biochemical properties to reduce these oxidative toxicities. These include (i) enhancing globin stability for storage by reducing the rate of auto-oxidation and heme loss and (ii) adjusting oxygen affinity; this optimization will also reduce the oxidant potency of HBOCs. To date, one of the most versatile approaches for making candidate HBOCs has been a recombinant strategy which involves (in one case) genetically linked in-frame tandem copies of the α subunit gene to produce a di-α gene with a single glycine linker between the C-terminal end of one α subunit and the N-terminal end of the other in a low oxygen affinity variant (Presbyterian βN108K). This di-α gene is then coexpressed with V1 M β-subunit genes in Escherichia coli to produce the recombinant Hb rHb1.1 (47). Crystal structural data of rHb1.1 have indicated that the glycine linker does not perturb the overall tertiary, quaternary, or even local structure of Hb (Fig. 2) (13). The major advantage of using this recombinant strategy is that one can potentially engineer HBOCs with beneficial amino acid substitutions using mutagenesis strategies (98). To date, rational mutagenesis approaches developed from a “two-prong strategy” (as opposed to random mutagenesis where screening for multiple properties is not feasible) have provided some promising results (98). However, well-documented engineering problems associated with recombinant proteins, such as correct protein folding and heme orientation within the heme pocket, present additional hurdles that protein engineers have to overcome (25).

In the search for favorable amino acid substitutions, our laboratory has relied on studying Hb variants identified in patients with hemoglobinopathies. Naturally occurring human Hb mutants potentially result in protein resistance to oxidative challenges, while others develop into a full circulatory disorder; in this regard, these mutants can be viewed as “Experiments in Nature” (27, 95). Exploring subunit-specific oxidative stability within these mutants may therefore lead to re-engineering of oxidatively stable HBOCs based on a combination of evolutionary stable mutations and rationally designed prototypes (Table 1 for summary of Hb mutants reviewed here). Our “two-prong” strategy involves (i) understanding the mechanism of Hb toxicity by evaluating mutant Hbs identified in patients with hemoglobinopathies and (ii) utilizing this information toward engineering acellular blood substitutes that are more oxidatively stable than native HbA. The implementation of this two-prong strategy for studying Hb variants will be highlighted in the section describing quantitative mass spectrometry for characterizing oxidative post-translational modifications (PTMs).
Approaches to Characterizing HBOCs
Several analytical approaches are used to characterize Hb variants and candidate HBOCs when implementing this strategy. One example of the classic methods for assaying auto-oxidation and denaturation involves incubating rHb variants in an air equilibrated buffer and spectrophotometrically following auto-oxidation and precipitation over time and/or by chemically inducing oxidation (at pH 7 at 37°C) (11, 39, 63, 84). This review will focus on the use of high-resolution accurate mass (HRAM) mass spectrometry (MS) for characterizing oxidative PTMs that occur in Hb. HRAM analysis is new to the field of characterizing Hb oxidative stability and when integrated with other more familiar methods (i.e., spectrophotometric, stopped-flow spectrophotometric assays) is invaluable in providing mechanistic insight into the extent of oxidative toxicity by quantifying oxidation in amino acids near the reactive heme or at specific oxidative hotspots. A basic introduction to mass spectrometry will be presented followed by examples of HRAM characterization of Hb variants. This review will also discuss promising results for a candidate HBOC designed from principles learned by studying Hb variants.
Introduction to HRAM Mass Spectrometry for the Study of Oxidative Changes and PTMs in Hemoglobins
The postgenomic era has had a huge impact on how biologists and biochemists study proteins. Scientists are now using high-throughput strategies that are versatile for characterizing enriched proteins, protein complexes, and whole proteomes, which not only identify and quantify proteins but can also identify a wide variety of PTMs, under a variety of experimental conditions (54). The most commonly used and extremely powerful technique for the high-throughput characterization of proteins, peptides, and PTMs is liquid chromatography coupled online with mass spectrometry (LC-MS) (38, 54, 85). By combining the power of bioinformatics, chromatography, and HRAM mass spectrometry, LC-MS is a versatile tool capable of comprehensive protein identification and in-depth characterization of PTM changes from complex biological samples (55).
Major Components of Mass Spectrometer
Mass spectrometers (MS) are analytical instruments that measure ionized analytes in the gas phase. The basic setup for a mass spectrometer consists of an ion source, a mass analyzer that measures the mass-to-charge ratio (m/z) of gas-phase ions, and a detector that registers the number of ions at each m/z value (Fig. 3A). For electrospray ionization (ESI-MS), this MS configuration is generally coupled online to an ultra-performance or high-performance liquid chromatography system (UPLC and HPLC, respectively), which separates the peptide analyte; as peptides are eluted from the column (see section “HPLC Online Peptide Separation”), they are ionized by the electrospray source and then directly emitted into the mass spectrometer (at the spray needle or emitter tip).

Electrospray and Matrix-Assisted Laser Desorption/Ionization
The most common ion sources for peptide and protein identification utilize either ESI, which is displayed in Figure 3A, or matrix-assisted laser desorption/ionization (MALDI). These soft ionization methods (developed in the 90s and early 2000s) revolutionized the field of mass spectrometry as they allowed for the nondestructive ionization of macromolecular structures (i.e., peptides or proteins) (23, 41). Before their development, hard ionization (i.e., electron and chemical ionization) limited the utility of mass spectrometry to the analysis of small volatile molecules stable at temperatures required for vaporization (99). However, ESI and MALDI ionization convert macromolecular structures (including peptides, proteins, lipids, carbohydrates, and nucleic acids) into ions with minimal chemical degradation (23, 41, 99).
Specifically, ESI involves applying high voltage, via ionization source at spray needle (Fig. 3A), to a liquid aqueous/organic/acid mixture to create an aerosol of droplets that are then emitted into the mass spectrometer through a heated capillary carrying a potential difference in atmospheric pressure (due to vacuum). Immediately after emission, the droplet surface area decreases due to solvent evaporation; as the droplet surface area decreases, the positively charged peptide ions eventually experience a columbic explosion (due to charge repulsion) resulting in desorption of solvated ions (67). MALDI, in contrast, desorbs (via sublimation) and ionizes the analyte from a crystalline matrix using a directed laser pulse. Traditionally, MALDI is used to analyze relatively simple peptide mixtures. The application of ESI for studying proteins far exceeds the use of MALDI because ESI ionizes analytes out of a solution and as a result is more amenable to liquid-based chromatographic separation tools. The primary focus of this review will, therefore, be on MS methods that utilize ESI coupled online with liquid chromatography (LC-MS).
Mass Analyzers
The mass analyzer is a central component for HRAM MS methods. In the context of peptide/protein and PTM identifications, its key parameters are sensitivity, dynamic range, duty cycle, resolution, mass accuracy, and the ability to generate sequence information by virtue of fragmenting peptides (tandem or MS/MS spectra) (2, 70). There are four major types of mass analyzers currently used with ESI-based mass spectrometers. These include the linear ion trap, time-of-flight (TOF), Fourier transform ion cyclotron (FT-ICR), and the Orbitrap analyzers. All four (uniquely different in design and performance) can be the principle analyzer but are generally featured in tandem as part of hybrid mass spectrometers that combine the strengths of each analyzer.
Commonly used mass spectrometer configurations (for protein/PTM identifications) with one or more of these analyzers are illustrated in Figure 3B. Briefly, the linear ion-trap (Fig. 3B-1) first captures ions (for a short time interval) and subjects them to either MS or fragmentation analysis (MS/MS) via collisional-induced dissociation (CID) (32). Linear ion traps are a common component of HRAM hybrid mass spectrometers such as the LTQ-Orbitrap (50, 51, 82). TOF analyzers (traditionally coupled with MALDI ion sources) are most often used with the HRAM hybrid Q-TOF (quadrupole-time-of-flight). A typical configuration for a Q-TOF mass spectrometer (Fig. 3B-2) involves ion selection via a quadrupole mass filter (Q) followed by fragmentation in a collisional cell (q2); the fragment ions then travel through the TOF analyzer at a rate proportional to their m/z ratio (19, 46). The FT-ICR analyzer captures ions under high vacuum in a high magnetic field (similar to an NMR instrument); the ions orbit within the cyclotron at a frequency proportional to their m/z ratio (56, 57). The FT-ICR mass spectrometer (Fig. 3B-3) has the best resolving power for analyzing intact proteins. Finally, the Orbitrap analyzer (type of ion trap analyzer) consists of an outer barrel-like electrode and a coaxial inner spindle-like electrode that trap ions in an orbital motion around the spindle (49). Two major Orbitrap configurations include coupling the Orbitrap to a linear ion trap analyzer (LTQ-Orbitrap) or quadrupole mass filter (Q-Exactive) (50). A typical LTQ-Orbitrap configuration involves ion capture in the linear trap, analysis in the Orbitrap analyzer, and then ion fragmentation (MS/MS) via CID in the linear trap (50). The Q-Exactive mass spectrometer (Fig. 3B-4) involves ion selection (and isolation) with a quadruple mass filter, ion capture in the Orbitrap analyzer followed by fragmentation in an HCD cell, and analysis in the Orbitrap (26).
Step 1: LC/MS/MS Analysis: The Bottom-Up Approach (LC/MS/MS) for Identifying Proteins and PTMs
Two HRAM methods are the frequently applied “bottom-up” (also known as shotgun proteomics) and the less widely used “top-down” strategies. An essential component of the quantitative methods described in this review, the bottom-up strategy involves enzymatic digestion of intact proteins into peptides that are then analyzed by LC-MS. The resulting peptides are fragmented by CID or HCD (higher energy C-trap dissociation) to generate MS/MS spectra that are then analyzed by database search algorithms (such as Mascot/SEQUEST) for sequence information (22, 71, 103). The ion current from reproducibly matched peptides (or fragment ions) can then be used for label-free quantification of Hb oxidation (discussed in Step 2). Key features of the bottom-up strategy (Fig. 4) are discussed in the following sections. The alternative top-down strategy involves direct analysis and/or fragmentation of intact proteins by the mass spectrometer to potentially identify protein isoforms (from complex mixtures) resulting from translational start and stop sites, mRNA splice variants, and PTMs of expressed gene products.
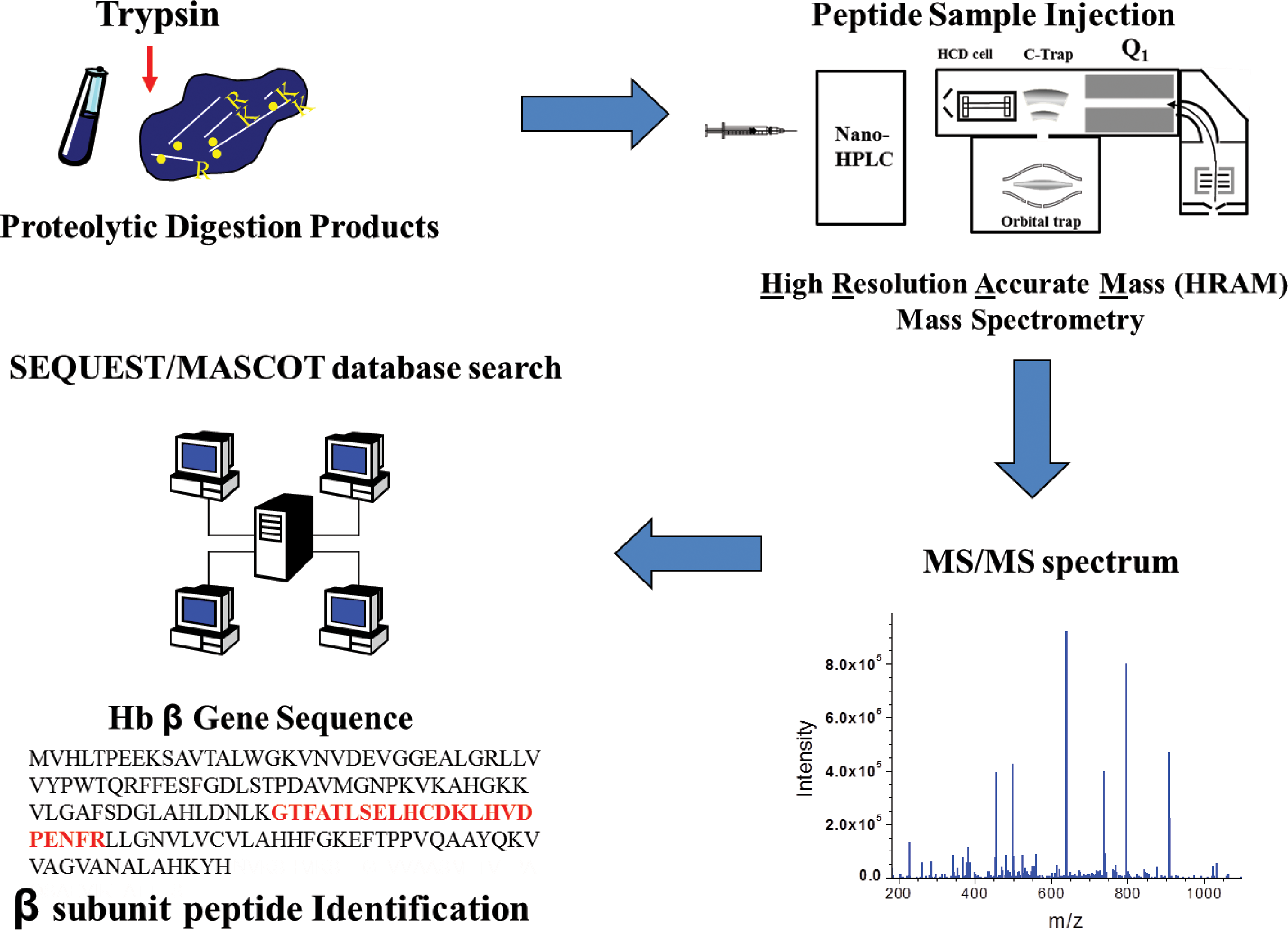
A major advantage of the bottom-up method is that peptide MS/MS spectra are much more efficiently analyzed than top-down fragmentation spectra of intact proteins; to date, the bioinformatic software required to interpret top-down spectra lags far behind algorithms designed for interpreting peptide MS/MS spectra. The bottom-up strategy is therefore much more effective at systematically identifying a large number of proteins (via identified peptides) and their corresponding PTMs from complex biological samples. There are two different methods for conducting bottom-up MS proteomic measurement. The first implementation uses one-dimensional (1D) or two-dimensional (2D) gel electrophoresis followed by in-gel digestion and either peptide mass fingerprinting or tandem mass spectrometry (1D LC MS/MS) to identify peptides that are matched to their corresponding proteins. The second more high-throughput approach involves in solution direct proteolytic digestion of complex mixtures (enriched protein, protein complexes, or whole proteomes) followed by 1D or 2D liquid chromatography coupled online with ESI MS/MS. The latter 1D LC MS/MS implementation is more commonly used in the quantitative examples described later in this report.
Enzymatic Digestion of Intact Proteins into Peptides
As stated above, peptide MS/MS spectra are more efficiently analyzed (relative to fragmentation spectra of intact proteins) for PTM identifications. Before LC-MS analysis, it is therefore critical to optimally digest protein samples toward maximizing the number of identifiable peptide isoforms (a peptide can exist unmodified or contain PTMs) in a given sample (Fig. 4). Important to MS/MS fragmentation is the use of site-specific proteases during the enzymatic digestion of a protein sample. Although a variety of different digestive enzymes are available for generating peptides, trypsin is the most commonly used protease because it cleaves C-terminally at lysine and arginine with a remarkable degree of specificity. The frequency of lysine and arginine in proteins generally results in a decent number of peptides that are the ideal size for gas-phase fragmentation (between 5 and 30 amino acids) (92). Furthermore, trypsin cleavage results in peptides with positive C-terminally protonated ends, beneficial for CID fragmentation; trypsin digestion results in a large frequency of +2 charged peptides (and +3 charge) that fragment well to produce product ion spectra that are very informative. These characteristics of trypsin generally increase the number of identifiable peptides (reflected by the primary sequence covered) per LC-MS analysis.
While LC-MS analysis of trypsin digestions most often reveal oxidative hotspots containing Hb peptides (see section “Database Searches of MS/MS Fragmentation Spectra”), alternative digestion strategies are commonly utilized for the purpose of mapping PTM identifications. One such strategy involves digestion with multiple enzymes; this approach increases the likelihood of generating additional peptides that are missed by one of the other proteases and increases the confidence of PTM identification when the same PTM is identified by overlapping peptides generating from more than one protease (48). In scenarios where the protein of interest has a low frequency of lysine or arginine residues (i.e., the transmembrane spanning domains), other cleavage strategies can be used, including the use of cyanogen bromide cleavage or the use of other proteases such chymotrypsin (cleaves C-terminally at aromatic amino acid residues) or pepsin (cleaves nonspecifically) (92).
HPLC Online Peptide Separation
Due to the complex nature of biological samples, online chromatographic separation is important for improving sensitivity and dynamic range for bottom-up peptide identifications. Dynamic range here refers to the ability to detect low-abundance peptides/proteins in the presence of high-abundance peptides/proteins. The goal of HPLC separation (before MS and MS/MS analyses) is to therefore simplify the mixture of peptides presented to the mass spectrometer at any given point in time. Indeed, direct infusion (without online chromatography) of a peptide mixture into the mass spectrometer would result in limited detection of only the most abundant peptides and those with the highest ionization efficiency (53). The separation of this complex mixture of peptides via HPLC allows for the mass spectrometer to interrogate a much wider range of peptides and thus provide sequencing and PTM detection of low-abundance peptides (from low-abundance proteins or low-abundance PTM containing peptides).
In a typical LC/MS/MS experiment, the peptide mixture is loaded onto an HPLC column (typically a C18 reversed-phase column that separates peptides based on hydrophobicity) via an autosampler. Peptides are eluted from the column via gradient elution and subsequently ionized into the mass spectrometer via an electrospray emitter (directly connected to the HPLC column or part of the HPLC column to minimize dead volume). During the entire chromatographic separation, the mass spectrometer is acquiring full-scan intact peptide masses and subsequently isolating and fragmenting the peptides to produce MS/MS spectra. Because ESI efficiency is greatly enhanced at lower flow rates (low flow rates generate much smaller initial droplets), modern online HPLC system components are scaled down to provide flow rates at the optimal nl/min range (61, 102). For single-dimension chromatography (1D), online separation columns most often consist of reversed-phase (RP) stationary phases. RP columns (typically C18) are hydrophobic; 1D peptide separations therefore involve the use of an organic mobile phase (acetonitrile) to elute peptides as a function of hydrophobicity. For 2D chromatographic separation, the 2nd dimension column generally consists of a strong cation exchange (SCX) column and RP in the 1st. A 2D configuration (generally only used for extremely complex samples) requires incremental ionic mobile-phase elution (off the SCX column) followed by organic mobile-phase elution for the 1D.
Database Searches of MS/MS Fragmentation Spectra
MS/MS fragmentation spectra provide sequence information that is searched against genomic databases (described in the next section). As stated, HRAM mass spectrometers utilize collision-induced dissociation CID (or HCD) to fragment peptides. CID fragmentation energizes peptide ions (precursor ion) via collisions with gas molecules (such as helium or nitrogen) to produce predominantly y and b ions (although fragmentation also results in some a/x and c/z ions) (69, 94). A typical MS/MS spectrum therefore represents y and b ion series (among other less populated ion types) of a fragmented peptide at various positions along the peptide backbone (103). Experimentally derived MS/MS fragmentation spectra (Fig. 4) are searched by database algorithms such as SEQUEST, Mascot, MyriMatch, and X-tandem (just to name a few) against predicted protein databases (generally created from genome annotation) to identify which peptide they represent (18, 22, 71, 91). The basic function of a database algorithm is to interrogate an MS/MS spectrum and identify the most likely peptide sequence matching (based on the y and b fragment ions) the spectrum from a provided translated genomic database.
There are four main processes involved in peptide identification from MS/MS spectra via database search algorithms: in silico peptide list generation from the database, candidate sequence generation, theoretical spectrum prediction, and scoring. Depending on the database and enzyme specificity, the above database algorithms first create a list of peptide sequences. For example, if the researchers used trypsin on a human blood sample (to generate a complex mixture of tryptic peptides), they would configure the algorithm to generate a complete list of all tryptic peptides from the predicted human proteome database (such databases can be easily downloaded from UniProt and other online genome databases). The algorithm next takes MS/MS spectra, extracts m/z values, and then compares these m/z values to calculated m/z values derived from peptides produced by the in silico list. A theoretical spectrum is then generated and compared to the experimentally derived spectrum and a score is assigned (18, 93). It is up to the researcher to decide what scores are considered probable matches. The researcher then utilizes additional filtering software (i.e., Scaffold software) that groups all high-scoring peptide matches to their corresponding protein.
An important step before PTM quantification (discussed in the next sections) is to first reproducibly identify which peptides contain PTMs; this of course will reflect where on the protein sequence the modification occurs. Database search algorithms are also used to identify peptides with PTMs; to do this the researcher specifies the change in mass associated with the modified form on a particular peptide residue (such as 48 Da shift in mass associated with tri-oxidation of cysteine residues). When the algorithm generates candidate sequences, it considers either of two masses possible for the specified residues. This allows the algorithm to match and score peptides that would otherwise be missed by looking for MS/MS that matched to peptides predicted by genomic sequence. Peptides identified with PTMs can then be further studied using quantitative methods.
Step 2: Quantitative Approach for Characterizing Hb Oxidative PTMs
This section illustrates how quantitative HRAM mass spectrometry has recently been used to provide mechanistic insight into the extent of oxidative changes within hemoglobin variants. First, we describe recent studies evaluating several mutants identified in patients with hemoglobinopathies and show how information gleaned from these studies was utilized toward the design of oxidatively stable HBOCs (blood substitutes). Table 1 provides a list of Hb variants recently investigated in our laboratory (1, 42, 62, 89). Information from several of these studies has been utilized toward the design of candidate acellular blood substitutes (discussed at the end of this section). To date, Hb mass spectrometry laboratories (with an interest in Hb oxidation studies) utilize a quantitative strategy similar to the one shown in Figure 5 (or some minor variation of it) (10, 74, 89, 96, 104). For example, our laboratory generates extracted ion chromatograms of oxidative hotspot containing peptides directly from full-scan data; however, a targeted approach using select ion monitoring can also be used to quantify peptides identified from the bottom-up approach (96). As previously stated, the first step in all Hb quantitative experiments involves implementation of the bottom-up strategy (LC/MS/MS analysis) to identify modified/oxidized Hb residues. After identifying oxidatively modified peptides, the second step involves a label-free method of quantifying the abundance of the oxidized peptide relative to the unmodified peptide; comparative differences in this ratio (relative to native HbA) provide insight into oxidative stability.
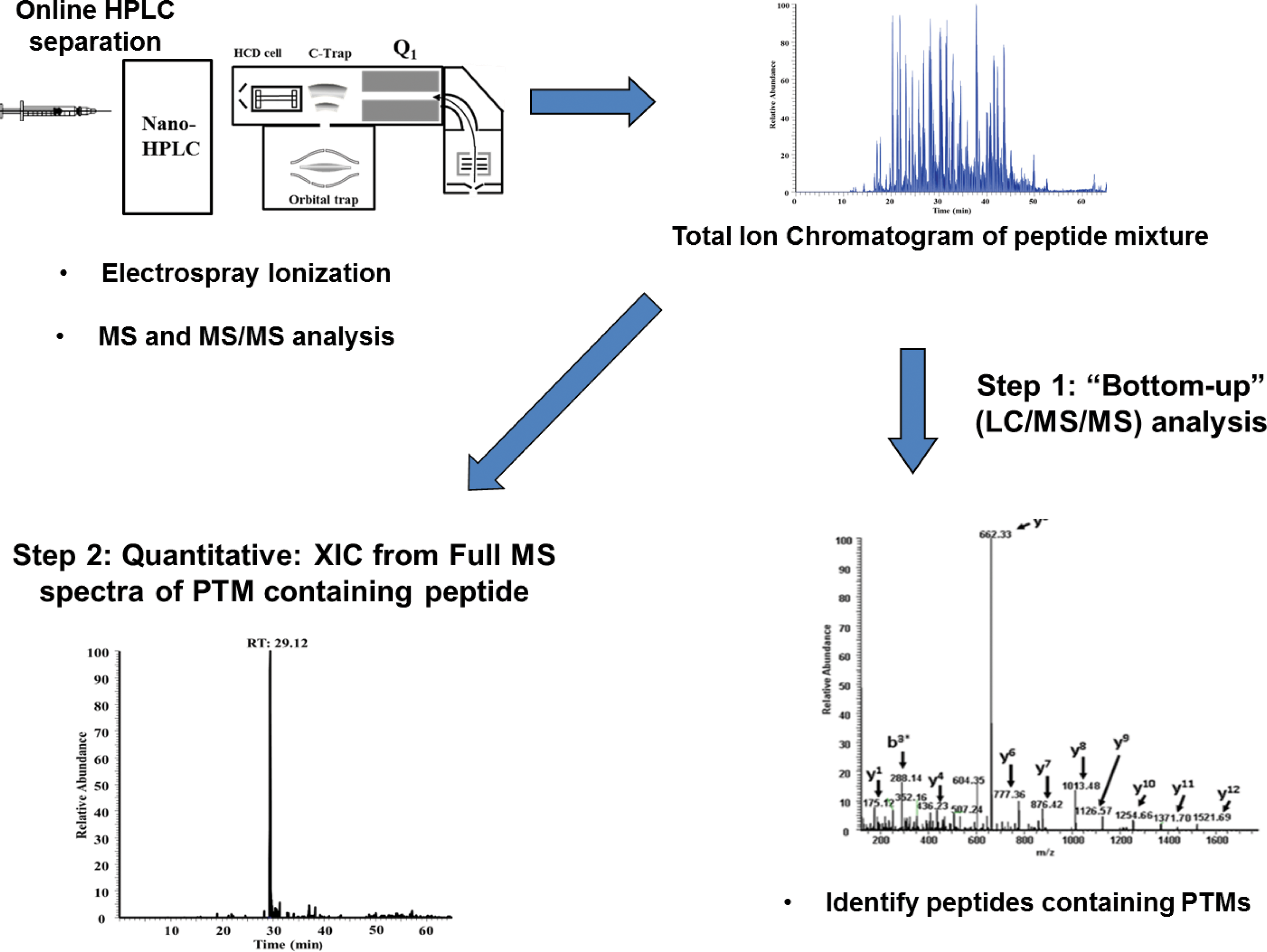
HRAM Analysis Provides Mechanistic Insight into Subunit-Specific Post-Translational Transformation of Methionine to Aspartate
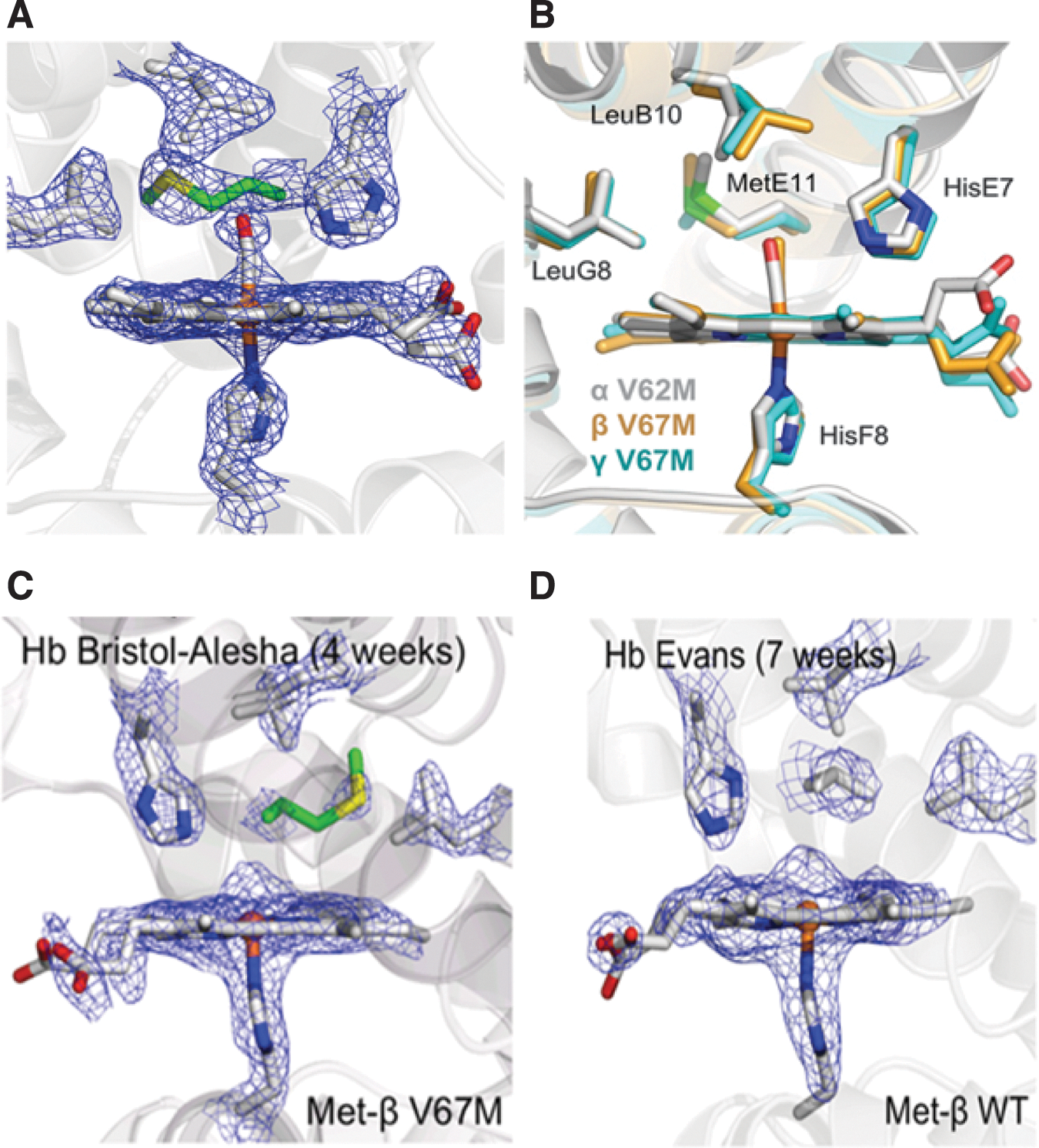
In a recent report from our laboratory, Strader et al. (89) provide an example of how this quantitative mass spectrometric approach was used to characterize the impact of positionally equivalent E11 methionine (E-helix is adjacent to the reactive iron in the distal heme pocket) mutations in three recombinant Hb variants; Toms River (γVal67Met) (γV67 M), Alisha Bristol (βVal67Met) (βV67 M), and Hb Evans (αVal67Met) (αV62 M) (89) (Table 1). The origin of this study began with a focus on characterizing the fetal Hb variant Hb Toms River (γV67 M) from an infant girl in Toms River, New Jersey; the patient was heterozygous for the V67 M mutant allele. In addition to identifying the γV67 M isoform, 1D/LC/MS/MS bottom-up analysis (step one in Fig. 5) also confirmed the presence of an unexpected γV67D variant in the patient hemolysate (Fig. 6A, B); based on DNA sequencing, the presence of this variant could only occur as a consequence of post-translational oxidation of γM67.
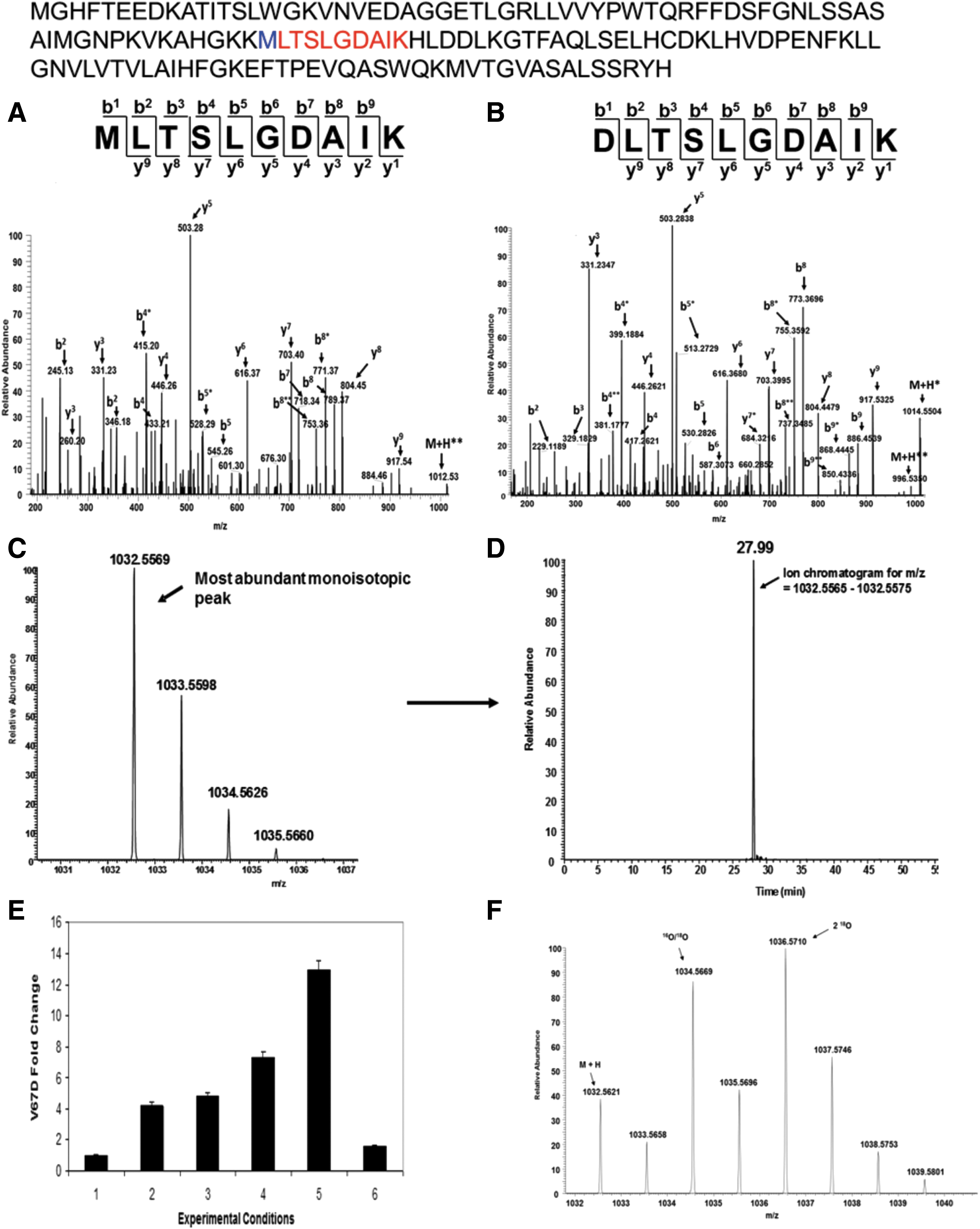
We reasoned that Met transformation to Asp was driven by an oxidative event that emanated from the nearby heme group. As outlined in the second step of Figure 5, an integrated quantitative mass spectrometric approach was then utilized to explore the relationship between increasing H2O2 levels and γMet-67 post-translational conversion to V67D; the results established a direct linkage between accelerated auto-oxidation (the rate was seven times faster than native HbA) and the Met(E11) modification. Figure 6C and D shows how HRAM data from a hybrid Q-Exactive Orbitrap mass spectrometer (illustrated in Fig. 3B-4) were utilized to generated extracted ion chromatograms (XICs) for quantifying the impact of increasing H2O2 on γMet-67 (the example in Fig. 3C, D corresponds to the singly charge D67 containing peptide DLTSLGDAIK). Specifically, XICs were generated from the most abundant monoisotopic peak of all Met-67 (thioether, sulfoxide, and sulfone) and Asp-67 peptide isotopic profiles using a designated mass tolerance of 0.01 Da, and mass precision set to three decimals. For relative quantification, the ratio of each Asp-67 or Met-67 peptide isoform was calculated based on the sum of the XIC peak area from all forms, which was normalized to be 100%. The experimental conditions and results from these XIC studies show that increasing H2O2 concentrations resulted in higher V67D levels (Fig. 6E). It is important to note that precursor isotopic resolution (that HRAM instruments provide) is required for the type of XIC construction described above; generating XICs from the most abundant isotopic peak results in XICs with superb signal to noise ratio and eliminates the potential for ion current from coeluting ions to interfere with data analysis. High-resolution data were also required in H2 18O2 follow-up experiments aimed at further examining the direct role of H2O2 in V67D formation. Impressively, experimental conditions with labeled H2 18O2 and HbF γV67 M resulted in Met-67 and Asp-67 (peptide isotopic profile shown in Fig. 6F) containing peptides with incorporated 18O, which correlates with the results shown in Figure 6E and confirms that the source of oxygen for the Asp modification comes from H2O2 (a proposed mechanism is discussed in the article (89).
This study was expanded to characterize the impact of positionally equivalent E11 methionine mutations in recombinant βV67 M (Hb Bristol-Alesha) and αV62 M (Hb Evans) variants. Met→Asp conversion was identified in the β(E11) but not in the α(E11) variants under similar experimental conditions. The initial crystal structures of the three recombinant Met(E11) HbCO forms show similar active site thioether side-chain orientations (Fig. 7A, B), with the γ side-chain carbon atoms all being roughly 5.0–5.3 Å from the heme iron. However, in agreement with solution experiments, auto-oxidation of the crystals over a period of weeks revealed electron density maps indicative of Asp(E11) formation in the active sites of the HbA β V67 M mutant but not in the active site of the HbA αV62 M variant (Fig. 7C, D). These findings demonstrate the unique redox reactivity of the γ and β heme iron atoms, which leads to post-translational oxidative conversion of amino acid side chains within these subunits, but not within the α subunits of human Hb. We went on to show that Asp was formed by an oxidative mechanism involving the ferryl heme complexes. Interestingly, ferryl Hb complexes were seen in Hb Bristol-Alesha (Fig. 7C, D), however, in the case of Hb Evans, the internal electron pathway may have minimized the accumulation of sufficient quantities of ferryl heme iron to drive this transformation.
Comparative HRAM Analysis of Oxidation in HbS and HbA
As previously stated, internal oxidative pathway reactions result in irreversible oxidation of amino acid in oxidation hotspots, regions (particularly the β Cys93 side chain) that cause the collapse of β subunits, Hb degradation, and the ultimate release of heme (39, 72). The quantitative strategy outlined in Figure 5 has also been recently used to quantify the levels of irreversible oxidation of hotspot residues, including βCys93 in the presence of H2O2 (10, 72, 74, 96, 104). This surface amino acid has previously been shown to be an important endpoint for free radical-induced protein oxidation (72).
HbS (β6Glu→Val substitution, linked to sickle cell disease [SCD]) is known to be unstable in vitro, is less stable than HbA (on exposure to heat, oxidants), and autoxidizes at faster rates than HbA (7, 33, 58). In a recent study by our group, Kassa et al. (42) indicated that chemically induced oxidation of HbS resulted in higher accumulation of the highly reactive ferryl HbFe4+ (due to slower autoreduction of ferryl to the ferric form) in HbS solutions (42). HRAM quantitative MS analysis was also used in this HbS study to compare differences in the post-translational oxidation of β subunit hotspot residues for both HbS and HbA under increasing H2O2 conditions (42). Amino acid residues identified by LC-MS/MS analysis in this study correlated well with previously published data; the most prevalent oxidative changes were found to be restricted to peptides containing βCys93 (10, 72, 74, 96, 104). XICs for the βCys93 containing peptide GTFATLSELHCDKLHVDPENFR (Fig. 8A) were used to construct the XIC in Figure 8B. Because βCys93 residue can either be oxidized or unoxidized, the percentages of both isoforms were calculated based on the sum of the XIC peak area from all charged forms of βCys93 peptides. The HRAM quantitative results from this study indicated that HbS βCys93 is substantially more oxidized (>2-fold difference when the ratio of H2O2 to heme equivalent is 5:1) in the presence of excess H2O2 compared to HbA. This dose-dependent increase correlated (with previous reports) with the oxidation kinetic data providing further evidence that HbS oxidation results in a higher accumulation of the highly reactive ferryl HbFe4+ (and protein radical).

To further confirm that ferryl ions and globin-centered radicals are more abundant in HbS, HRAM quantitative experiments were performed using the spin trap label 5,5-dimethyl-1-pyrroline N-oxide (DMPO). The exposure of Hbs to H2O2 has previously been shown to initially produce a porphyrin cation radical (and ferryl ion) that oxidizes cysteinyl and tyrosyl amino acids to form globin-centered radicals (20, 31, 60, 96). The spin trap DMPO reacts with these modified amino acids to form a nitroxide radical that is further oxidized to the corresponding globin radical-derived nitrone adduct by the ferryl moiety (20, 31, 60). These DMPO-derived adducts are stable under fragmentation conditions used in LC-MS/MS analysis. Analyses of LC/MS/MS and full MS data in the presence of H2O2 (5- and 10-fold excess relative to heme) identified DMPO-labeled Cys112 and Tyr42α peptides with the addition of H2O2. Among the three DMPO-labeled amino acids, only Cys93 was substantially different between HbA and HbS. DMPO Cys93 labeling (Fig. 8C) in the absence of H2O2 was at negligible levels (∼1% for both HbA and HbS). The degree of DMPO labeling was considerably more abundant for HbS than HbA (>2-fold when the ratio of H2O2 to heme equivalent is 5:1). These labeling results further substantiate the observation that ferryl levels are more prevalent in HbS and likely account for the observed accelerated oxidation and oxidative changes within the protein. We have postulated that dominant Cys93 oxidation in sickle cell Hb is due, in part, to the persistence of its ferryl and radical species. Thus, highly oxidizing ferryl Hb and heme may be central to the evolution of vasculopathy in SCD and suggest therapeutic modalities that interrupt heme-mediated inflammation (42).
The Role of Unpaired Alpha Subunits in Destabilizing HbE
β-thalassemia (caused by chromosome 11 β-globin gene alterations) is characterized by reduction or absence of β-globin chain synthesis (101). HbE (β26Glu→Lys) is an oxidatively unstable human Hb often coinherited together with a β-thalassemia allele; the resulting HbE/β-thalassemia condition is often severe requiring multiple transfusions (24, 37). Because of HbE instability, a higher level of unpaired α subunits within HbE containing red cells has been proposed to cause the pathogenesis associated with HbE/β thalassemia (77, 78). However, it is experimentally challenging to determine the effects of free uncomplexed α subunits in the presence of another Hb tetramer (i.e., pairs of α2β2 of either HbA or HbE). Because we have previously identified βCys93, as the main target of heme radical chemistry, we recently performed a comparative study that monitors the effects of excess α subunits on βCys93 oxidation within both HbE and HbA (90). Using the same HRAM quantitative mass spectrometric strategies described for HbS characterization (including the spin trap, DMPO), we quantified irreversible oxidization of βCys93 to reflect oxidative instability of β subunits. In the presence of free α subunits and H2O2, βCys93 oxidation increased with higher H2O2 concentrations for both HbA and HbE. In the presence of alpha-hemoglobin stabilizing protein (AHSP), which stabilizes the α-subunit in a redox-inactive hexacoordinate conformation (thus the α subunit is unable to undergo the redox ferric/ferryl transition), Cys93 oxidation was substantially reduced in both proteins. These experiments establish two important features that may have relevance to the mechanistic understanding of these two inherited hemoglobinopathies, that is, HbE/β thalassemia: First, a ferryl and ferryl protein radical target βCys93 of both proteins but results in the destabilization of HbE due to its persistent ferryl species. Second, in the presence of excess free α-subunit and under the same oxidative conditions, these events are substantially increased for HbE compared to HbA and may therefore create an oxidative milieu affecting the already unstable HbE.
Step 3: Engineering Oxidative Stability in Human Hemoglobin Based on Rationally Designed Proteins
As stated, the first part of the two-prong strategy is to utilize information from Hb variant studies toward the rational design of acellular blood substitutes that are more oxidatively stable than native HbA (and chemically modified Hbs). We have begun to engineer HBOC prototypes based on naturally occurring human mutant Hbs and on rational design (65, 98).
The first of these variant studies involves the variant Hb Providence (Table 1); a mutant identified in a patient at Providence hospital in Washington DC. The patient was heterozygous for the allele expressing the βK82 N variant; due to spontaneous deamidation of asparagine to aspartic acid, two β-subunit variants with the single amino acid mutations βLys82→Asn (βK82 N) and βLys82→Asp (βK82D) were identified in the patient hemolysate. We have previously separated Hb Providence into its three component fractions and contrasted oxidative reactions of these β-mutant fractions with HbA. The Hb Prov. βK82 N variant oxidation reaction kinetics and ligand binding properties were similar to HbA; however, the βK82D fraction was more effective in consuming H2O2 and appeared more oxidatively stable. The spectrophotometric data in Figure 9 show that ferryl βK82D, unlike native HbA, reverts back to the ferric state more readily (HbA shows oxidative damage), suggesting that βK82D safely internalizes radicals (through the ferric/ferryl pseudoperoxidase cycle) better than HbA (1). The βK82D amino acid substitution has recently been reported to improve expression levels of soluble recombinant Hb in E. coli (30). Taken together, the structural and oxidative stability of Hb Prov. βK82D may assist in reducing auto-oxidation, oxidative modifications, and heme loss; such features are important goals in the design of oxidatively stable HBOCs. In collaboration with Dr. John Olson at Rice University in Houston, TX (using the rational engineering approach discussed earlier in this review), a genetically crosslinked di-α gene (see section describing two-prong strategy) βK82D Hb (rHb0.1) has recently been engineered. Preliminary kinetic, crystal structure and HRAM quantitative mass spectrometry data of βK82D rHb0.1 indicate that this candidate HBOC is structurally similar to and autooxidizes at similar rates to native HbA yet maintains exceptionally higher oxidative stability in the face of higher levels of peroxide (Strader M, unpublished data). Additional studies are underway to determine how the βK82D substitution impacts oxygen carrying function and if the observed oxidative stability reduces the rate of heme loss relative to control HbA.
Conclusion
Blood substitutes have long been a dream for physicians, researchers, and industrial scientists alike, because they can be stored for a long time, potentially save lives without waiting for typing, and substantially reduce the risk of infection. Despite these advantages, no viable HBOCs have been approved in the United States due, in large part, to clinical trial reports of serious adverse events (79, 86). To address these toxicological responses, several different biochemical and recombinant strategies have been attempted with limited success. A major factor for this limited success is that outside the RBC, acellular HBOCs are oxidatively toxic due to the reactive heme, which is ironically essential for oxygen binding. The two-prong strategy that we have described here is an approach aimed at exploring ways of mitigating Hb oxidative toxicity. Studying oxidative pathways in select Hb variants (with diverse oxidative profiles) has enabled us to develop criteria toward the rational design of a new series of oxidatively stable HBOC candidates. Future HRAM studies will therefore be aimed at characterizing these new candidate HBOCs to determine which are the most oxidatively resistant toward losing heme, while retaining oxygen carrying function.
Footnotes
Acknowledgments
We thank Dr. Tigist Kassa (Laboratory of Biochemistry and Vascular Biology, Center for Biologics Evaluation and Research, Food and Drug Administration) for assistance in designing the molecular graphics illustrated in ![]() . We thank Dr. Nathan C. Verberkmoes (University of Texas, El Paso) for reviewing and suggesting edits to the article. Work in this review was supported by the National Institutes of Health (NIH/NHLBI) grant P01-HL110900 and a grant from the U.S. Food and Drug Administration (MODSCI).
. We thank Dr. Nathan C. Verberkmoes (University of Texas, El Paso) for reviewing and suggesting edits to the article. Work in this review was supported by the National Institutes of Health (NIH/NHLBI) grant P01-HL110900 and a grant from the U.S. Food and Drug Administration (MODSCI).