
Abstract
Significance:
Oxidative stress and generation of free radicals are fundamental in initiating pathophysiological mechanisms leading to an inflammatory cascade resulting in high rates of morbidity and death from many inherited point mutation-derived hemoglobinopathies. Hemoglobin (Hb)E is the most common point mutation worldwide. The βE-globin gene is found in greatest frequency in Southeast Asia, including Thailand, Malaysia, Indonesia, Vietnam, Cambodia, and Laos. With the wave of worldwide migration, it is entering the gene pool of diverse populations with greater consequences than expected.
Critical Issues:
While HbE by itself presents as a mild anemia and a single gene for β-thalassemia is not serious, it remains unexplained why HbE/β-thalassemia (HbE/β-thal) is a grave disease with high morbidity and mortality. Patients often exhibit defective physical development, severe chronic anemia, and often die of cardiovascular disease and severe infections.
Recent Advances:
This article presents an overview of HbE/β-thal disease with an emphasis on new findings pointing to pathophysiological mechanisms derived from and initiated by the dysfunctional property of HbE as a reduced nitrite reductase concomitant with excess α-chains exacerbating unstable HbE, leading to a combination of nitric oxide imbalance, oxidative stress, and proinflammatory events.
Future Directions:
Additionally, we present new therapeutic strategies that are based on the emerging molecular-level understanding of the pathophysiology of this and other hemoglobinopathies. These strategies are designed to short-circuit the inflammatory cascade leading to devastating chronic morbidity and fatal consequences. Antioxid. Redox Signal. 26, 794–813.
Introduction
O


To date, more than 1679 hemoglobinopathies and thalassemias have been identified and listed in the HbVar database [
HbE/β-thalassemia (HbE/β-thal)
HbE/β-thal is a grave disease with high morbidity and mortality [e.g., (63, 128, 205)]. In the United States, the βE-globin gene is entering the gene pool of diverse populations with greater consequences than expected (112). The βE-globin gene is found in greatest frequency in Southeast Asia, including Thailand, Malaysia, Indonesia, Vietnam, Cambodia, and Laos. Of the 300 million carriers of hemoglobinopathies in the world, HbE/β-thal affects 1,000,000 people worldwide (127). In Thailand, approximately 3000 newborns per year are born with HbE/β-thal and unknown numbers in other parts of the world (159). HbE attains a frequency of 50–60% in Southeast Asia, and in Indonesia, the gene frequency may be as high as10-fold (65). People whose ancestors are from south China, Philippines, India, Pakistan, and Turkey may also present with HbE. HbE is the most common point mutation worldwide (HbE>HbS>HbC). HbE/β-thal will likely be the cause of the growing worldwide clinically severe thalassemia crisis. [See recent reviews by Fucharoen and Weatherall (64) and Vichinsky (192).]
The wave of migration to North America has resulted in the HbE carrier frequency becoming almost equal to that of the HbS gene (190), with HbE as the third most prevalent variant in the United States (after HbS and HbC). In the last decade, 75% of immigrants to North America came from areas where thalassemia mutations are prevalent. HbE/β-thal and α-thal disorders are now the most common thalassemias in California. In California, 1 in 4 Cambodian births and 1/9 Thai/Laotian births are HbE carriers. This development stands out in comparison with sickle cell trait (allele) (∼ 2 million Americans or 1 in 12 African Americans); sickle cell disease (∼1 in every 500 African American births and ∼1 in every 1000–1400 Hispanic American births); Hemoglobin C disease (∼1 of 5000–10,000 individuals); and Hemoglobin C trait (∼2–3% of all African Americans) (127, 128, 190). Thus, HbE diseases are becoming a national health concern due to increasing immigration from these Asian countries (NIH, National Healthcare Alert).
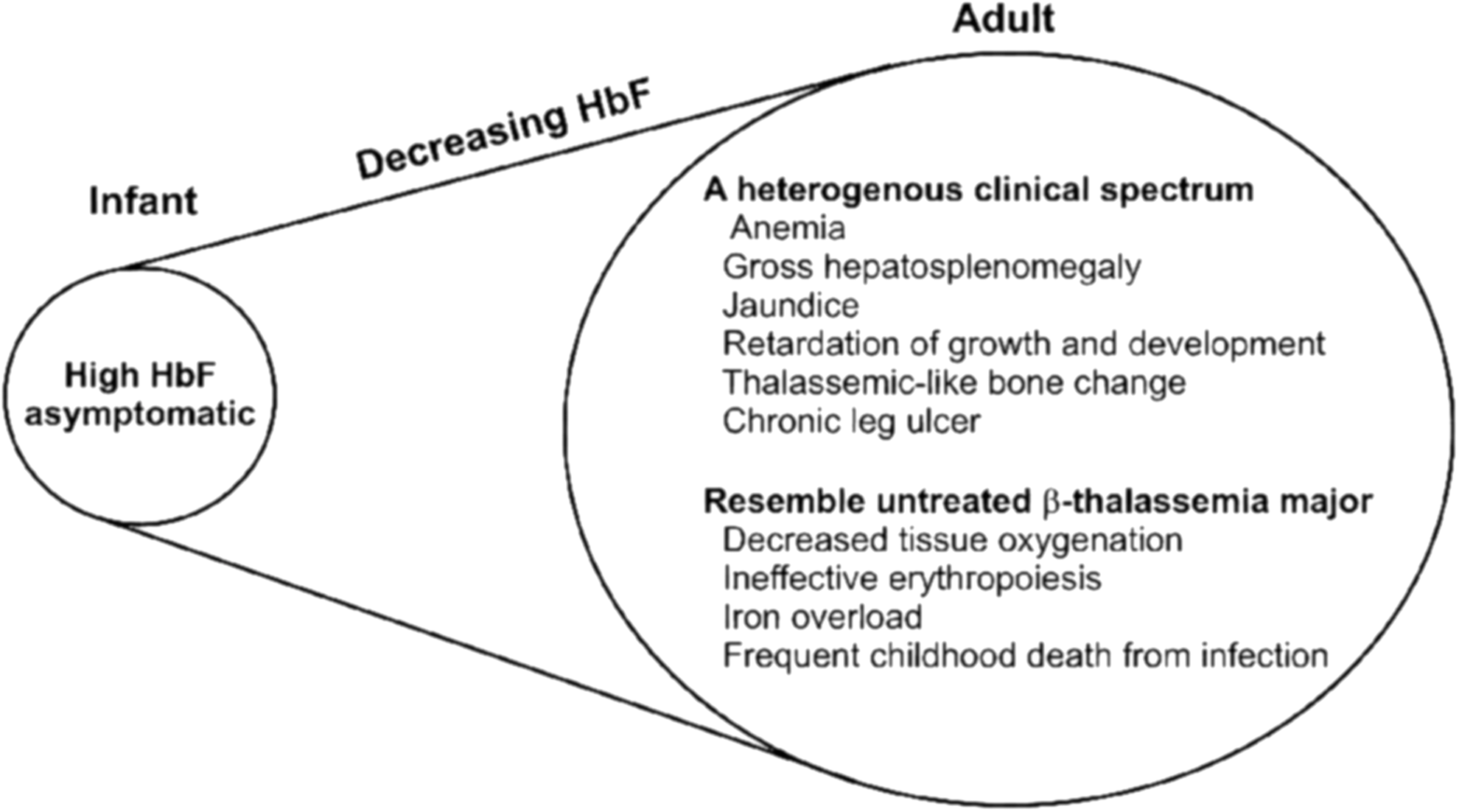
Table 1 briefly summarizes different HbE diseases posing a challenge to the United States and other countries. Infants are asymptomatic because HbF levels are high at birth (141). Adults with higher levels of HbF exhibit more moderate clinical manifestations. The higher levels of HbF appear to be due to decreased β-globin expression (e.g., a β-thalassemia) (103, 133). HbE appears as a β+-thalassemia because the synthesis of the βE-globin chain is reduced due to the mutation, GAG to AAG at codon 26 of the β-globin gene, which activates a cryptic splice site. As shown in Figure 2, the G to A mutation at codon 26 (GAG to AAG) in exon 1 of the β-globin gene leads to the Glu to Lys substitution in βE-globin. This mutation also activates a cryptic 5′ splice site at codon 25 of the βE-globin pre-mRNA, leading to a reduction in the correctly spliced βE-globin mRNA. Thus, the abnormal gene also results in both reduced amounts of βE-mRNA and in synthesis of βE-globin chains. The reduced amounts are also attributed to the demonstrated instability of βE-mRNA (186). It is suggested that different degrees of alternate splicing in the cryptic 5′ splice site could affect the severity of HbE/β-thal disease (203, 207).
With time and in the absence of transfusions, serious clinical symptoms appear similar to that seen in untreated β-thal major (43, 60, 62, 74) (Fig. 3). Both HbE/β0-thal transfusion-dependent and nontransfusion patients commonly die of heart disease (74). The dogma points to iron overload and chronic anemia as the primary cause of cardiac pathophysiology (125). The mechanisms that give rise to the pathophysiology and variability in HbE/β-thal have yet to be fully defined.
The enigma
It is unclear how the presence of the relatively benign HbE converts the relatively benign β-thal minor condition into a serious potential lethal condition. Patients homozygous for HbE exhibit minor clinical manifestations. HbE has normal oxygen binding properties. The well-documented HbE instability in vitro (33, 106, 163, 164, 178) could explain increased oxidative damage in HbE/β-thal red cell membranes. A model of double oxidative jeopardy has been proposed (39) (Fig. 4). In contrast to expectation, aged HbE/β-thal RBCs show no decrease in HbE content (143). Moreover, to date, conflicting evidence exists regarding the clinical relevance of HbE intrinsic instability since RBC oxidative stress is only observed in vitro when HbE/β-thal red cells were incubated above 37°C (142).

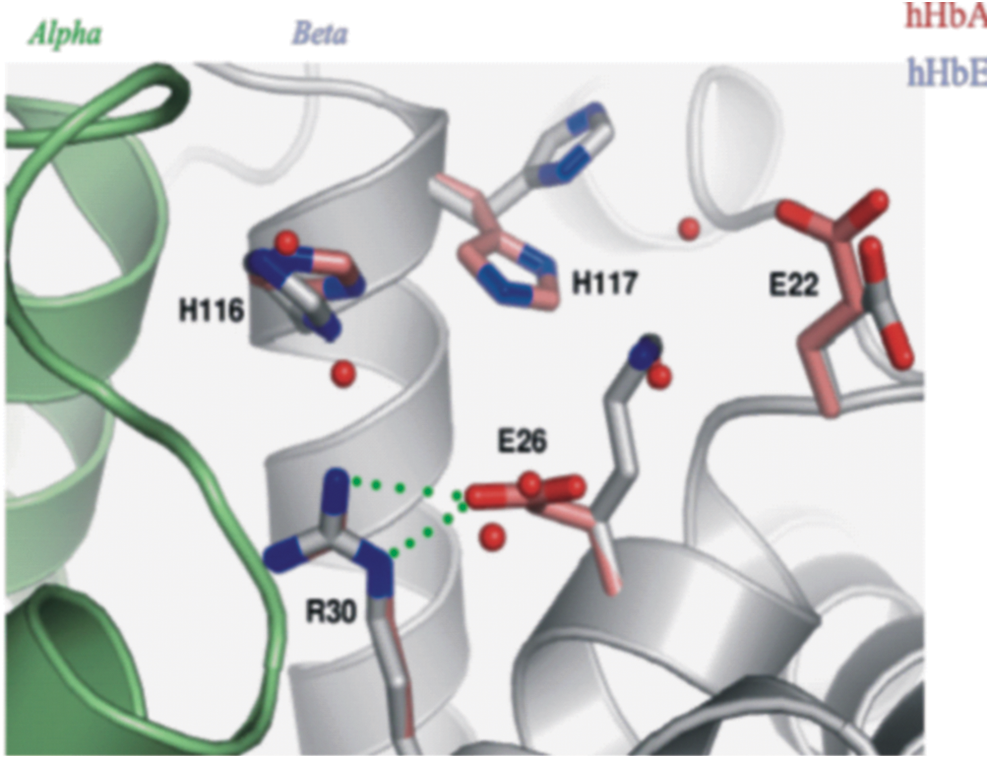
On a molecular level, the β26 Lys substitution in HbE is distant from the heme pocket accounting for normal oxygen affinity (61). The location of the mutation at the α1β1 dimer interface (a key flex point for the α1β1 dimer) may explain the instability. High-resolution (1.8Å) crystallographic findings, with a crystal grown close to physiological conditions (pH 7.35), show two noticeable alterations: (i) the βLys26 side chain exposed to solvent with loss of stabilizing H-bonds to βArg30 [in contrast to the normal βArg30-βGlu26 stabilizing bond in normal HbA], resulting in significantly modified α1β1/α2β2 interfaces; and (ii) the conformations, βHis116 and βHis117, differ in HbE compared with HbA (152) (Fig. 5). These latter residues are critical for Hb assembly, stability, and oxidation state (1, 2). A plausible mechanism for the functional impact of this local mutation at the homodimer interface is through an alteration of the flexing dynamics of homodimers, which could alter the accessibility and stability of bound and free ligands, including water within the distal heme pocket, through the modified dynamics of the nearby amino acid side chains. Overall, the greater mechanical, oxidative (106), and thermal instability (33, 142) of HbE as reported in vitro may be attributed, at least in part, to the conformational changes observed at βHis116 and βHis116, communicated by the βLys26 substitution with an ensuing electrostatic change (152).
Regardless of the mechanism, the biochemical studies in vitro, establishing a decreased stability of HbE, have yet to be linked to the circulating HbE RBC and its pathophysiology. Importantly, oxidative stress and reactive oxygen species (ROS) are reported to be increased in HbE/β-thal and in β-thal RBC due to excess free α-chains (35, 45, 160). How HbE exacerbates this oxidative stress is unknown. A recent in vitro study demonstrates for the first time that free α-globin chains are oxidatively unstable and damaging to HbA and HbE, with ferryl(Fe4+)HbE significantly less able to autoreduce than ferryl HbA (178). This should also be considered in light of the fact that HbE compared with HbA exhibits differences in its redox properties with indications of an increase in its redox potential (152).
This article presents an overview, without exhausting the literature, of HbE/β-thal clinical manifestations; current treatments; evidence of pathophysiological mechanisms involving oxidative stress; and new findings pointing to possible pathophysiological mechanisms involving the interplay between unstable HbE exacerbated by free α-globin chains, limited NO production as a result of HbE reduced in function as a nitrite reductase (NR) (152), enhanced population of ferryl Hb RBC damage, and lysis resulting in chronic inflammation and cardiovascular pathophysiology. Based on these insights, we present several innovative therapeutic approaches that address the proposed origins of the inflammatory cascade that ultimately leads to lethal cardiovascular consequences.
Clinical Manifestations
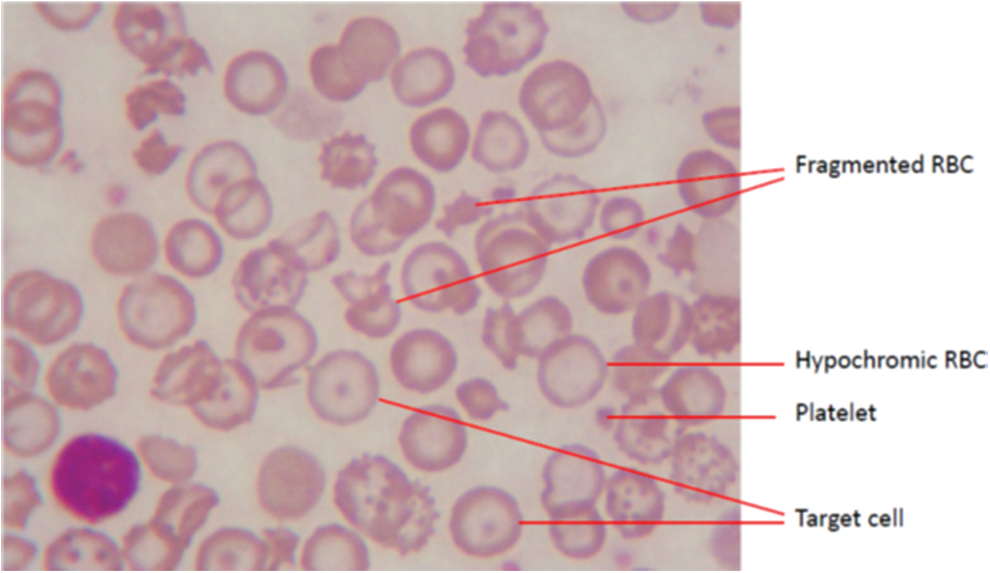
A wide spectrum of clinical manifestations presents in HbE/β-thal [for example, (63, 65, 127)] (Fig. 3). Diagnosis of HbE/β-thal is made by gel electrophoresis and blood smears (Fig. 6). Because of HbE comigration on gel electrophoresis with HbA2, HbC, and HbO-Arab, and it comigrates closely with other Hb variants on isoelectric focusing and high-performance liquid chromatography, conclusivity of the diagnosis of HbE requires using several methods (190). Hematological status is shown in Table 2. This clinical picture has been well characterized. Hb levels may vary from 30 to 130 g/L.
p < 0.05 compared with healthy individuals.
Courtesy of Nathawut Sibmooh and in close agreement with that reported in Ref. 190.
RDW data from Ref. 190.
Hb, hemoglobin; RBC, red blood cell; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; WBC, white blood cell.
A scoring system is used to classify severity differences in HbE/β-thal. One group described three distinctive categories, mild, moderate, and severe, based upon six independent parameters: hemoglobin level, age of disease presentation, age of receiving first blood transfusion, requirement for transfusion, spleen size, and growth and development (175). Another team categorized five distinct groupings for managing the diverse clinical spectrum (137). Regardless, patients with very low Hb levels manifest characteristics similar to β-thalassemia major with defective physical development; expansion of bone marrow cavity; jaundice; hepatosplenomegaly with extramedullary hematopoiesis; ineffective erythropoiesis; and increased hemolysis resulting in jaundice. The phenotype of this genetic disease (e.g., clinical presentation) is influenced by genetic and environmental factors such as, respectively, coinheritance of a modifying mutation, mutations associated with increased HbF synthesis, and malaria or splenectomy [For a complete discussion of factors affecting the phenotype, see, for example: (126, 127, 137, 208, 210)]. Increased fetal Hb and HbA2 levels are correlated with less severe disease (141, 210).
Cardiac failure and severe infections are the major leading causes of death (65, 74). Interestingly, the RBC distribution width (RDW) in HbE/β-thal individuals is significantly increased (Table 2). This parameter has been correlated and used as a marker for cardiac failure, inflammation, oxidative stress, and hemolysis (104). Iron overload was attributed to chronic hemolysis and/or frequent transfusions as one of the early mechanisms of disease. As with β-thalassemias, the primary mode of treatment for the severe HbE/β-thal patient has been iron chelation and transfusion (124). A recent review introduces the complex relationship between anemia, hypoxia, erythropoietin (EPO), erythropoiesis, and iron metabolism in β-thal, presenting new insight into mechanisms of iron overload that may involve hypoxia-inducible factor-2α, hepcidin, and other factors. These novel interactions provide the basis for the introduction of novel therapeutics directed at these particular pathophysiological mechanisms (147).
Platelet hyperactivity and thrombosis
Increased coagulation and platelet hyperactivation have been documented in HbE/β-thal, especially in splenectomized patients. Splenectomy results in an increase in phosphatidylserine (PS) surface-bearing erythrocytes, which promote coagulation, thrombin generation, and platelet activation (10, 21, 156). A model for platelet activation in HbE/β-thal is presented in Figure 7.
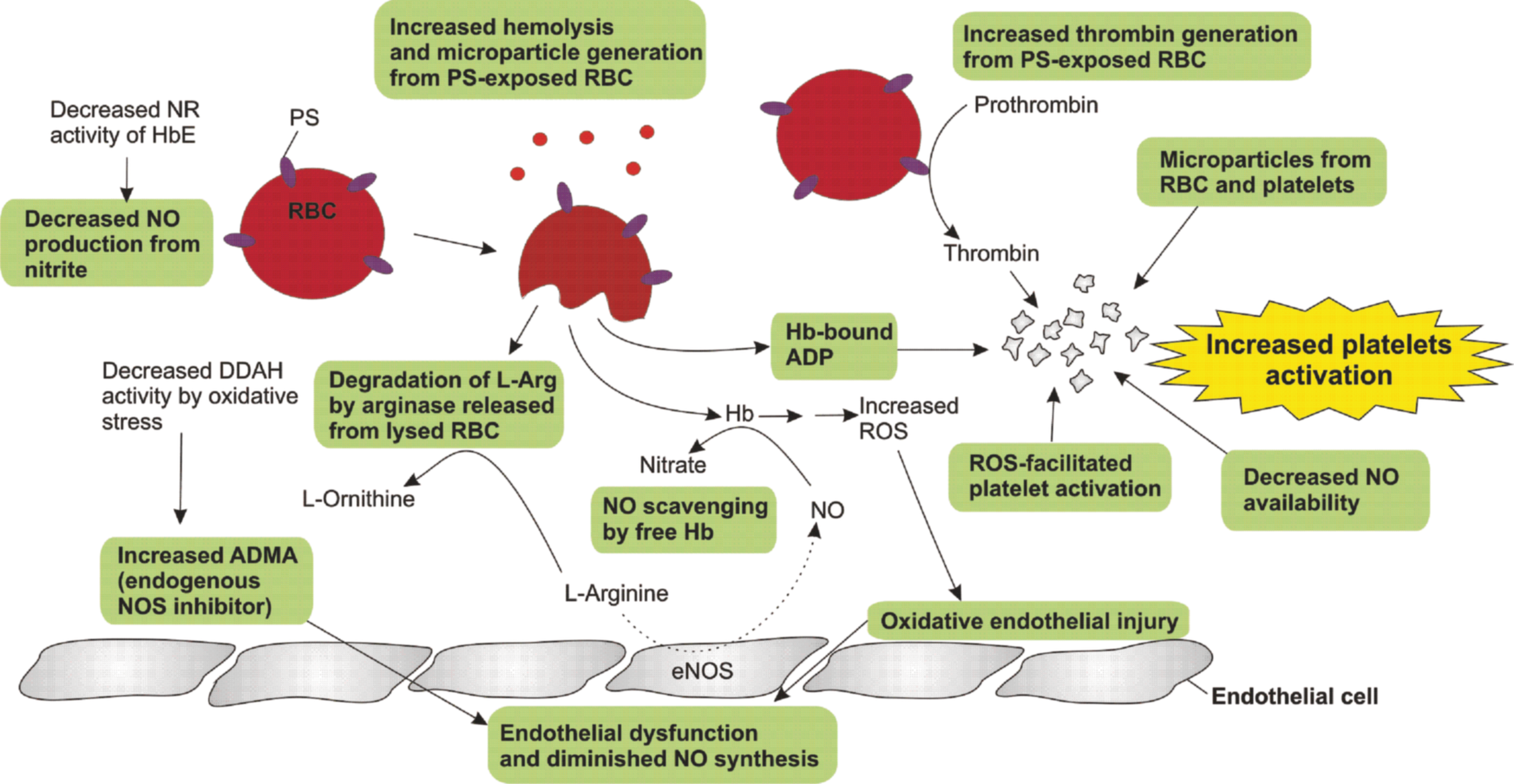
PS is a membrane phospholipid normally located at the inner side of the membrane bilayer; exposure to the outer membrane increases with aging and is known to be a catalyst in the conversion of prothrombin to thrombin (155, 161), which is a potent platelet agonist. Apart from PS-bearing erythrocytes, thalassemia patients have elevated levels of circulating PS-bearing microparticles mostly derived from platelets (132). Additionally, changes in membrane phospholipid asymmetry of thalassemic erythrocytes, as a consequence of oxidative stress and activation of other blood cells, including platelets, lead to high levels of microparticles that promote thrombosis (132). The increase in circulating soluble markers of platelet activation, such as P-selectin (170), CD40 L (169), and β2-thromboglobulin (9), and urinary excretion of thromboxane A2 metabolites (53) are indicators of increased platelet activation in thalassemia. Thalassemia patients also have an increased expression of P-selectin (degranulation marker) and activated glycoprotein IIb/IIIa (aggregation marker) on the platelet surface and increased platelet–leukocyte aggregates, which correlate with increased hemolysis (cell-free hemoglobin) and higher pulmonary arterial pressure (tricuspid regurgitant velocity) (90, 174).
ROS can facilitate platelet activation and play a role as second messengers to activate platelets (13, 85, 195). Following stimulation of platelet collagen receptor (glycoprotein VI), the superoxide anion is produced as a result of the reduction of molecular oxygen by an electron generated by NADPH oxidase or cyclooxygenase. Superoxide undergoes spontaneous or enzymatic dismutation to form H2O2 that can diffuse across the membrane. ROS can positively regulate signaling events in platelet activation, including activation of phospholipase C (134), inactivation of protein-tyrosine phosphatases (85), elevation of cytosolic calcium, degranulation (13), and aggregation (16). Interestingly, platelet hyperactivity in splenectomized HbE/β-thal patients is attenuated following oral administration of the antioxidant vitamin E (525 IU daily) for 3 months (188).
In summary, increased platelet activation in thalassemia is a consequence of combined factors, including oxidative stress, PS-exposed erythrocytes, high amounts of circulating microparticles, nitric oxide scavenging by cell-free hemoglobin (180), and endothelial dysfunction (99). Moreover, cell-free hemoglobin can activate platelets through the action of adenosine diphosphate-bound hemoglobin (79). Chronic platelet activation may, in part, contribute to an increased frequency of vascular complications in thalassemia such as pulmonary hypertension (3, 11), pulmonary embolism (173), cerebral thrombosis (209), and peripheral arterial and venous thrombosis (22, 30). Prevention of thrombosis, particularly in splenectomized patients, may be achieved by inhibition of platelet aggregation with aspirin and reduction of high platelet count by hydroxyurea (109, 171). Interestingly, stem cell transplantation can normalize the PS-bearing erythrocytes and levels of coagulation markers in thalassemia (172).
Evidence for RBC Oxidative Stress as the Origin of the Pathophysiology
The RBC contains a variety of self-maintaining redox systems that serve as specific antioxidants: redox enzymes (such as superoxide dismutase [SOD], catalase, glutathione peroxidase [GPx], glutathione reductase [GR], thioredoxin, and peroxyredoxin 2 [PRDX2]) and low-molecular-weight molecules such as reduced glutathione [GSH] and vitamins E and C (168). RBC anaerobic glycolysis yields reduced nicotiniamide adenine dinucleotide (NADH), which is the primary reducing agent of met (Fe3+)Hb (200). Oxidized Hb(Fe3+) results in the cascade of events leading to ferryl (Fe4+)Hb, free heme, ROS, and other reactive species (144) and ultimately endothelial dysfunction (Fig. 1).
RBC oxidative stress is indicated by enhanced RBC membrane lipid peroxidation, PS exposure, reduced glutathione:oxidized glutathione ratios (GSH:GSSG), and altered antioxidant enzyme activities. Compared with controls, RBCs from HbE/β-thal patients exhibited higher levels of G6PD, GR, GPx, and SOD (34). Low GSH levels associated with free iron in HbE/β-thal patients indicate that nonheme iron oxidative damage contributes to RBC destruction in these patients (35).
Significant iron-induced oxidative stress was shown in a large cohort of HbE/β-thal patients with increased serum ferritin levels, decreased GSH levels, and a 90% reduction of GSH:GSSG compared with controls (87). In contrast, these parameters of oxidative stress were unremarkable in HbE trait controls. It should be noted that a full knockout HbE transgenic mouse (HbEKO) was generated that expressed solely human HbE in a mouse RBC (38). The HbEKO mouse exhibited a mild oxidative stress similar to that seen in human HbEE individuals as indicated by RBC hematological indices and RBC characteristics (elevated zinc protoporphyrin [ZPP]; elevated RBC [ROS]; enhanced RBC membrane lipid peroxidation). Quite relevant is the observation that human HbE bound to the mouse RBC membrane mimics the human RBC membrane affinity of HbE > HbC > HbS (37). The pathophysiological relevance of human HbE bound with high affinity to the RBC membrane is discussed below in the section, “The Interplay of Reduced NOx Generation and Enhanced Ferryl Hb Lifetimes as a Model for RBC Instability Associated with HbE/β-thal Disease.”
The role of RBC oxidative stress, as a key player initiating the pathophysiological mechanisms of this disease, becomes convincing in a study of HbE/β-thal RBCs and a human plasma proteome analysis (76). Severe patients, compared with mild patients, had an increase in markers of RBC oxidative stress based on ineffective erythropoiesis, lower RBC counts, PS exposure, greater serum EPO and plasma malondialdehyde (MDA), and RBC SOD. Other laboratories have shown not only an increase in RBC redox enzymes, such as PRDX2, catalase, thioredoxin, and SOD, but also an increase in the alpha globin chaperone, alpha hemoglobin-stabilizing protein, negatively correlated with MCH and HbF (18, 102, 189).
Oxidative stress in hemolytic HbE/β-thal disease is a candidate for the origin of increased inflammatory cytokines (Fig. 1). Increased inflammatory cytokines in hemolytic RBC diseases may arise from endothelial activation, from oxidized hemoglobin and free hemin, reduction in NO, and/or other mechanisms (7, 17, 92, 144). The release into the circulation of free hemin and ROS upon RBC hemolysis activates NF-κB and AP-1, which in turn increase the expression of proinflammatory cytokines (IL1, IL6, TNFα) and endothelial adhesion molecules (L-selectin, P-selectin, VCAM-1, ICAM-1) (200 –202). A spiraling inflammatory process occurs as these cytokines trigger leukocyte infiltration that releases more ROS.
There is evidence mounting that the inflammatory cascade is initiated by RBC ROS-mediated oxidative stress that directly impacts Band 3 (aka AE1 anion exchanger), an important transmembrane protein in erythrocytes that plays a major role controlling ion transport, possibly NOx transport (71), glycolysis regulation by exchanging glycolytic enzymes with bound Hb or hemichromes (28, 101, 204), and maintains the linkage between the cytoskeleton and the membrane (54, 55, 116, 131, 146). The consequence of oxidative stress appears to originate from excess levels of H2O2 within the RBCs that originate from a progressive loss of the enzymatic detoxification machinery that occurs when RBCs deteriorate due to normal and oxidative damage-accelerated aging (113). Enhanced levels of H2O2 promote formation of ferryl derivatives of Hb (89).
In both sickle cell hemoglobin (HbS) (54, 55) and now HbE (178), the highly reactive ferryl derivative has an extended lifetime compared with HbA, which is proposed to contribute to the respective pathophysiologies. The prolonged presence of ferryl Hb appears to facilitate Hb modifications (including heme release and hemichrome formation) that alter the Band 3 transmembrane protein first via facilitating disulfide bond formation (130, 131), which then leads to enhanced phosphorylation of tyrosines and serines (54, 55, 105). These changes in Band 3 then initiate alterations in the local membrane environment surrounding Band 3. The result of excessive phosphorylation of Band 3 protein is correlated with an enhanced membrane degradation generating the production of proinflammatory microparticles and increasing the rate of hemolysis (42, 54, 55, 131).
There is evidence that the ferryl derivative promotes oxidation of the reactive thiols on the two β93 Cys of Hb (89). This oxidation is significant, in that the reduced state of these thiols plays a key role in protecting against oxidative stress (198). It is when the β93 Cys are oxidized that Hb alterations occur, including Hb dimer/hemichrome formation, which can disrupt the functional Band 3 protein complex under conditions of oxidative stress (110). Hemolysis and RBC-derived microparticles in turn create and exacerbate vascular inflammation, including platelet activation and endothelial dysfunction (42). All these vascular consequences will decrease tissue perfusion and, under conditions where the inflammation is chronic, will likely lead to organ damage/failure.
HbE/β-thal patients exhibit increased RBC microparticles (32). RBC and platelet-derived microparticles not only promote platelet activation, as discussed above, but also initiate inflammation that is reflected in a profile of increased inflammatory cytokines (129). In addition, ineffective erythropoiesis in HbE/β-thal has been attributed to overactive EPO-mediated signal transduction correlated with high levels of ROS emanating from an increased oxidative metabolism (100) and also correlated with increased levels of specific inflammatory cytokines (181). It was demonstrated that addition of IL-1β, TNF-α, or IFN-γ to cell cultures resulted in a decrease in erythroid progenitor cell numbers compared with untreated cells, with cytokine-treated cells from HbE/β-thal patients being more affected. Inflammatory cytokines appear to play a role in ineffective erythropoiesis (26). Moreover, platelet hyperactivity and platelet-RBC interactions, as discussed above, coupled with increased levels of specific cytokines in HbE/β-thal patients partake in signaling hypererythropoiesis and set the stage for inflammation as an upstream pathophysiological mechanism in this disease (31, 90).
RBCs, Nitric Oxide, and Vascular Homeostasis
New roles for RBCs are becoming ever more apparent. Multiple studies show that RBCs under oxidative stress are now linked to the initiation of a cascade of inflammatory events (25, 107, 138). Additionally, RBCs are emerging as a major participant in the maintenance of vascular homeostasis. Its ability to generate, transport, and deliver bioactive forms of NO is seemingly a critical factor in facilitating the maintenance of vascular health. Concomitantly, we propose that the loss or decrease in the capability of the RBC to produce, transport, and export the required bioactive forms of NO can account, in part, for many of the events in HbE/β-thal triggering of the inflammatory cascade that (when chronically persistent) results in the potentially lethal clinical picture associated with this hemoglobinopathy. In the previous paragraphs, the point was made that the RBC can trigger vascular inflammatory cascades through hemolysis, microparticle formation, and release of free Hb and free heme. As will be discussed below, many of these processes and consequences associated with these processes are likely to be inhibited, slowed, or negated through NO-related reactions.
On a fundamental level, hemoglobin and the RBC play the vital role of sustaining adequate tissue perfusion and oxygenation. Chronic conditions of decreased tissue perfusion and oxygenation (i.e., hypoxia) are likely contributors to organ damage and eventual organ failure. The role of Hb in transporting and delivering oxygen is obvious and well understood. However, adequate oxygen delivery by Hb to oxygen-sensitive organs and tissues also requires that the microvasculature (e.g., functional capillary density) allows for adequate tissue perfusion, which is often compromised under conditions of chronic vascular inflammation.
The RBC plays a role in controlling tissue perfusion by controlling vascular tone as a function of oxygen tension in a process called hypoxic vasodilation (6, 44, 51, 68, 69, 72, 94, 177, 187). Although the mechanism is not completely understood, it appears that the ability of RBCs to transport and deliver some form of bioactive NO in an oxygen tension-dependent manner is at the center of this process. While the endothelium is considered to be the primary source of local NO production, transport and delivery of NO or a related NO-derived/related molecule with similar bioactivity (NOx) by the RBC is emerging as a physiological process that can impact platelet activation and vascular tone.
Hemoglobin within the RBC has been implicated in the hypoxic vasodilation mechanism through two possible, but not mutually exclusive, reactions: (i) the NR reaction (Table 3, Reaction 1,) between Hb and nitrite (70, 83, 122) and (ii) the formation of SNOHb (Table 3, Reaction 3) where the two thiols on β93 Cys become S-nitrosated (51, 177).
FA, fatty acid; GSNO, S-nitrosoglutathione; NO, nitric oxide; NOD, nitric oxide dioxygenation; NR, nitrite reductase; SNO-Hb, S-nitrosohemoglobin
The chemistry of Hb-NOx generation
NR activity in Hb occurs via a five-coordinate ferrous heme in Hb (as in deoxyHb) reacting with nitrite to produce NO and ferric heme (Table 3, Reaction 1). The rate is at least an order of magnitude faster when the deoxygenated ferrous Hb tetramer has the R (relaxed) quaternary structure (83, 145, 151). For HbA, the T (tense, low ligand binding affinity) quaternary structure conformation is thermodynamically favored when the population of Hb is the fully deoxygenated ferrous derivative. As oxygen binds to the available ferrous heme sites, the equilibrium shifts in a pH and allosteric effector-dependent degree to the R quaternary state conformation (relaxed, high ligand binding affinity). The rate of this NR reaction is sensitive to the redox potential of heme, which accounts for the quaternary structure dependence of the reaction and hence a rate dependence per heme that depends on the degree of heme oxygenation within tetrameric Hb (73, 150, 151, 153), accounting for the quaternary structure dependence of the reaction and hence a rate dependence per heme that depends on the degree of oxygenation within tetrameric Hb (83). The hemes for the R state structure have, relative to the T state structure, a lower redox potential, which favors formation of the ferric heme product. Interestingly and highly relevant to the mechanistic enigma of HbE/β-thal are (i) HbE has a higher redox potential compared with HbA and (ii) HbE in vitro is less efficient, relative to HbA, at generating NO through the NR reaction (149, 152) (Fig. 8). The higher redox potential of HbE is consistent with its lower NR rates. The physiological implications of these relationships with respect to HbE/β-thal are discussed below.
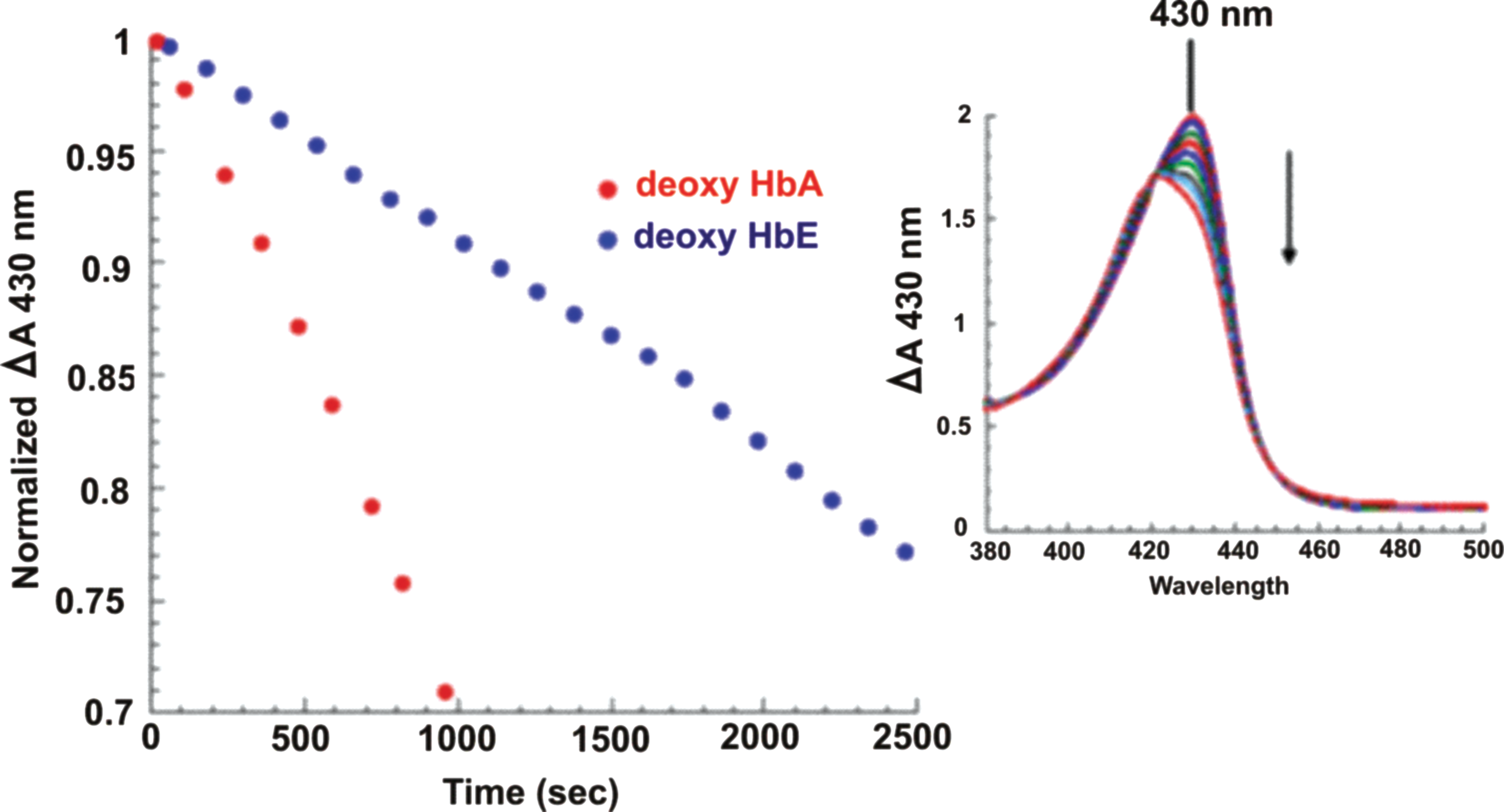
Preserving, transporting, and delivering Hb-derived NOx bioactivity
Many questions remain associated with how NR can ultimately produce a releasable bioactive form of NO. The following are two of the more obvious problems with this mechanism: (i) How does the NO generated within the RBC avoid being scavenged by HbO2 (primarily via the NO dioxygenation reaction, Table 3, Reaction 2)? (ii) What is the mechanism for exporting an NO bioactive species from an RBC?
If the NO generated through the NR reaction is a source of the NOx [designation for an NO-related molecule, for example, S-nitrosothiols (SNOs), N2O3] that ultimately transmits NO bioactivity from the RBC to other elements of the circulatory system, then there needs to be a mechanism to overcome the scavenging. The mechanism for overcoming the scavenging can be either through reactions that both preserve NOx species derived from NR and allow for a subsequent physiological signal-triggered export or through rapid export of the NO or NOx species upon formation in an oxygenation-dependent manner.
There are several possible mechanisms that have been proposed that could allow for preservation of NO bioactivity [e.g., (78)]. These mechanisms likely require converting NO to a more stable bioactive NOx that can eventually be utilized to export NO bioactivity. Converting NO to an SNO is a plausible mechanism since nitrosothiols are both relatively stable and can transfer NO (actually NO+) to other thiols (transnitrosation) as part of signaling, activation, and deactivation processes (23, 82, 176, 213). Given the high concentration of GSH within the RBC, S-nitrosoglutathione (GSNO) is an S-nitrosothiol that is likely to be a major participant in maintaining NO homeostasis in the RBC.
GSNO has been detected in RBCs and is likely associated with reactions and processes that are relevant to RBC integrity and RBC functionality. It has been postulated by many investigators that NO bioactivity via S-nitrosothiols can be exported into the plasma from the RBC through S-transnitrosation via GSNO and/or SNOHb transported by the RBC membrane Band 3. Most significant is the role that GSNO appears to play in modulating RBC metabolism through nitrosation of the transmembrane protein Band 3 associated with hemoglobin and hemichrome binding and glycolytic enzyme binding, for example, glyceraldehyde-3-phosphate dehydrogenase (GAPD) (28, 41, 66, 101). Within the RBC, GSNO is also going to be in equilibrium with SNOHb, both of which can clearly function as a reservoir for transnitrosable NOx (i.e., NO+). Yet, it is still ambiguous as to whether GSNO can be exported from the RBC. There is evidence that L-CysSNO, but not GSNO, can get transported through the RBC (158), but more recent data suggest that GSNO can also be exported (46, 167, 183). Regardless, whether GSNO does get out or not, it appears that the RBC can export NO bioactivity through SNO, either through direct export or transnitrosation (most likely via Band 3). Given that CysSNO does appear to get in and out of the RBC and is highly effective as a transnitrosating agent, it may be a mediator between the nitrosothiol populations within the plasma and RBC (158).
Hb-based production of GSNO, SNOHb, and other NOx and HbE/β-thal
Assuming the SNOs such as GSNO, CysSNO, and SNOHb are essential both for the export of NO bioactivity from the RBC and preservation of RBC integrity, the key questions are as follows: (i) How do these species get generated from the NO produced from NR? (ii) How do these processes get modified in HbE/β-Thal? NO production via NR is most effective when there is a substantial population of deoxyheme. Optimum production appears to occur for partially oxygenated Hb that is stabilized in the R state quaternary conformation (29, 83, 94, 199). Under these conditions, the resulting NO is likely to be captured by vacant deoxyheme sites to produce nitrosylated hemes (e.g., HbNO), which are very stable. When the blood becomes fully oxygenated, the reaction between nitrite and oxyHb becomes a source of the NO2 · radical (91, 135). Additionally, NO2 · can also be generated through the reaction of ferryl Hb with nitrite (81) (Table 3, Reaction 7). This radical can either oxidize ferrous nitrosylheme to yield the ferric NO-heme derivative (Reaction 4a, Table 3) or react directly with the ferrous heme-bound nitric oxide radical to produce N2O3 (Table 3, Reaction 4b) (78). Both ferric NOHb (via production of either NO+ or N2O3) (Table 3, Reactions 5b, 5c) (78, 153, 157) and N2O3 generated from NOHb (Table 3, Reaction 4b) are potential S-nitrosating agents that could generate SNOHb, GSNO, and CysSNO. These reactions, subsequent to the NR reaction, are still not fully tested and evaluated. However, this sequence of reactions does provide an oxygenation-dependent mechanism for generating, preserving, and exporting NO reactivity.
These NO2 and N2O3-generating reactions are also likely to produce nitro-fatty acids (14, 154, 193, 194) in the RBC membrane. Nitro-fatty acid derivatives of oleic acid have been found in RBC membranes (15). Formation of these NOx species is anticipated to be significantly enhanced in a lipophilic environment as would be the case for Hb molecules adjacent to the RBC membrane (197). Nitro-fatty acids could be another avenue for exporting NO bioactivity as well as preserving the integrity of the RBC membrane.
The Interplay of Reduced NOx Generation and Enhanced Ferryl Hb Lifetimes as a Model for RBC Instability Associated with HbE/β-thal Disease
A few years ago, the first functional abnormality for HbE was reported in vitro: relative to HbA, HbE is less efficient at generating NO through the NR reaction (Table 3, Reaction 1) as explained by the altered redox properties of HbE, relative to HbA, consistent with the slower rate of NR for HbE (152) (Fig. 8). Hence, in HbE/β-Thal disease, there are now two linked HbE-associated factors that could combine to exacerbate the impact of oxidative stress on Band 3 and membrane integrity: i) lower intracellular levels of NO and nitrite as a result of HbE dysfunction as an NR and ii) higher levels of ferryl due to enhanced HbE ferryl lifetimes (178).
The impact of the functional changes in HbE is likely magnified by HbE having an enhanced affinity for the RBC membrane [(HbE>HbC>HbS>HbA)] (37). Thus, HbE is poised to induce membrane dysfunction, especially in Band 3 (the anion transporter), which would further interfere with the normal NO RBC physiology and the RBC NO metabolin as proposed (95). There are several pathways whereby NO, nitrite, and ferryl Hb can intersect. Nitrite can react with ferryl Hb to yield met Hb plus the NO2 radical and thus lower the concentration of ferryl Hb (77, 80, 81). Met Hb in the presence of nitrite and NO can undergo rapid reductive nitrosylation to form HbNO (184). NO can also react with ferryl Hb to yield nitrate and met Hb (80, 81).
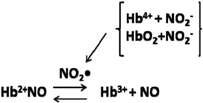
Production of the NO2 radical at a moderate rate is likely to be anti-inflammatory, in that it can both regenerate free NO by oxidizing HbNO to form met Hb plus NO (20) and help generate nitro-fatty acids, which have impressive anti-inflammatory properties (8, 58, 194). NO2 is also produced via the complex reaction of nitrite with oxyHb in a reaction that involves ferryl formation (77, 91, 135). Thus, low nitrite levels limit both the ferryl deactivation reaction and the oxyHb reactions. Both NO and NO2 are radicals that can react with other exposed radicals such as thiyl and tyrosine radicals and as such are likely to inhibit the progression of free radical-induced changes. The ferryl Hb cycle of formation and reformation includes the formation of a tyrosine radical that likely precedes the formation of a thiyl radical on the two β93 Cys. That radical migration to the thiol is the probable critical step in the forming of oxidized thiols on the β93Cys. These steps have been invoked (vide supra) as a key pathway through which the ferryl Hb modifies Band 3 via both hemichrome formation and disulfide formation in Band 3. Thus, those processes that both deactivate the ferryl-associated radicals and reduce the ferryl heme are likely to inhibit Band 3 degradation, hence the importance of NO and nitrite in modulating ferryl reactivity. The presence of nitrite and NO will also foster the formation of SNO-Hb, in which the thiols on β93 cysteine are S-nitrosated.
SNO Hb formation has been shown (198) to play a role in limiting oxidative stress and erythrocyte membrane damage by further supporting the concept that NO in many or all of its bioactive forms can function to maintain RBC homeostasis in the presence of oxidative stress. Furthermore, SNO Hb formation has also been shown to be a significant contributor to the mechanism for hypoxic vasodilation (212). It would therefore appear that SNOHb formation is significant for both Band 3 functionality and stability and for RBC functionality with respect to hypoxic vasodilation. The disruption of the complex and, as yet not fully worked out, interplay among such species and entities as Band 3, ferryl Hb, SNOHb, bioactive forms of NO, and ROS from oxidative stress are emerging as plausible underlying mechanisms for much of the chronic pathophysiology in hemoglobinopathies.
RBC instability with enhanced hemolysis and microparticle formation will trigger several vascular responses that can lead to decreased tissue perfusion and further enhancement of oxidative stress. Loss of hypoxic vasodilation functionality in RBCs will only further limit tissue perfusion and vascular homeostasis. The results point to the synergy between a decreased capacity to generate NO and enhanced ferryl Hb survival in a backdrop of enhanced oxidative stress as a plausible target for therapeutic intervention to limit chronic inflammatory consequences of HbE/β-thal. A model is presented to explain pathological mechanisms that give rise to the enigmatic HbE/β-thal disease (Fig. 1).
In addition to the proposed and anticipated reduced NO production in RBCs from HbE/β-thal patients, HbE has additional properties that can interfere with NO-related functionalities, and as pointed out earlier, HbE has an enhanced affinity for the RBC membrane (37). HbE-induced membrane dysfunction, including Band 3 (the anion transporter), would further interfere with normal NO RBC physiology and RBC NO metabolin, as proposed (95). Further compromise of the RBC membrane that is already corrupted with the high binding affinity of HbE can occur from the excess α-chains arising from heterozygous thalassemia. These excess chains produce heme, hemichromes, and labile ROS-producing iron, all of which can bind to and alter the lipid bilayer and Band 3 (35).
The backdrop of a stressed HbE/β-thal RBC coupled with the absence or reduction of the alleged protective attributes of NO should lead to chronic enhanced RBC hemolysis (resulting in platelet activation and vascular inflammation) and negative vascular consequences (e.g., loss of hypoxic vasodilation function). The model depicted in Figure 1 may be the basis or minimally a significant contributor for pathophysiological mechanisms of HbE/β-thal and other hemolytic hemoglobinopathies. This proposed NO-associated pathophysiology mechanism suggests therapeutic strategies to manage the chronic consequences of the disease by inhibiting RBC damage and limiting vascular inflammation.
Further Evidence for Oxidative Stress in HbE/β-thal: Therapeutic Antioxidants Ameliorate the Disease
Natural antioxidants appear to ameliorate the disease, providing further evidence that oxidative stress is a fundamental mechanism underlying the observed clinical manifestations. Fermented papaya appeared to reduce oxidative stress in thalassemic patients, including HbE/β-thal, and may also act by chelating iron (56, 139).
In 21 HbE/β-thal patients, the reduction of high levels of oxidants (MDA, SOD, GPx in RBC, serum nontransferrin-bound iron) and increased levels of RBC GSH were correlated with the administration of curcuminoids (a cooking spice) (86). More recently, antioxidant cocktails that include curcumin or vitamin E added to N-acetylcysteine and the iron chelator, deferiprone, administered to HbE/β-thal patients resulted in the first report of an increase in RBC hemoglobin concentration along with significantly decreased iron load, decreased oxidative stress with less RBC membrane damage, less deformability, increased membrane stability, and inhibition of PS externalization (211).
Curcumin is shown to limit lipid peroxidation and prevent oxidative damage to RBC Band 3 (115), which is quite relevant given the purported role of Band 3 as a mode of transport for NOx intermediates generated by hemoglobin (discussed above) and indications of possible GSH and GSSG efflux: for example, pharmacological inhibitors of this transport protein attenuated peroxide-induced ROS generation as well as osmotic fragility in RBCs of HbE/β-thal patients (119).
Another mechanism of curcumin action is proposed in a study of chronic iron-overloaded rats: curcumin attenuated iron accumulation and oxidative stress in the liver and spleen with indications that not only does curcumin work as an antioxidant, as noted in the above human studies, but additionally curcumin itself may bind labile iron (12). Curcumin is also a potent anti-inflammatory agent, an inhibitor of platelet activation, and possibly a clot-dissolving agent (88, 96, 123). Furthermore, there is growing evidence that curcumin can mimic the vasodilatory action of NO (50). All of these properties are of potential value in treating/limiting the chronic inflammatory consequences of the disease. The major challenges associated with curcumin as a vascular therapeutic are its in vivo bioavailability and short circulation time.
Curcumin toxicity appears not to be a significant factor since no side effects were reported in curcumin therapies administered to HbE/β-thal patients, as discussed above (86, 211). Moreover, the literature shows an extraordinary number of articles using curcumin, a natural therapeutic with low toxicity, as an antioxidant in both animal models and in clinical settings for a plethora of human diseases. Yet, some point to negative effects of curcumin and urge that the benefit–risk profile must still be established (24). As with all therapeutics (both natural and synthesized), efficacious dosages need to be determined as well as finding the threshold whereby concentrations or dosages may become toxic.
Current Therapeutics Addressing Oxidative Stress and Iron Overload in HbE/β-thal
Clinical management of HbE/β-thal by transfusions, splenectomy, and iron chelation therapy has been and is still the mainstay of treatment (124). With the unraveling of complex genetic and environmental factors modulating clinical severity, new potential therapeutic approaches are under development [e.g., (65, 147)]: gene switching (19) that includes HbF induction by hydroxyurea; stem cell transplants; gene therapy; pharmacological targeting of defective erythropoiesis; targeting free α-chains and with α-Hb chaperones; and countering the vasculature depletion of NO from chronic hemolysis (18, 19, 36, 102, 111, 133, 179, 189).
Therapies addressing NOx imbalance: NO inhalation therapy
With findings that NO is critical in vasodilation and maintaining a healthy cardiovascular system, NO inhalation therapy and NOx have been introduced in animal studies and clinical trials in attempts to ameliorate the depletion of vascular NO in hemolytic diseases, such as sickle cell disease and thalassemias (191). Most circulating NO is produced by vascular endothelium, a significant function of the endothelium (97). NO is oxidized to the nitrite anion by the oxidase activity of ceruloplasmin (165) and dehydroascorbic acid (166), which is primarily stored in erythrocytes (47). The biological action of nitrite is mediated through bioactivation to NO by deoxyhemoglobin, myoglobin, xanthine oxidoreductase, and nonenzymatic disproportionation (93).
Lower blood levels of the endogenous vasodilator NO are reported in thalassemia (162, 180). Plasma circulating cell-free hemoglobin decreases NO bioavailability from endothelial cells by oxidizing NO to nitrate. Arginase, the enzyme responsible for the conversion of L-arginine to L-ornithine and urea, is released from erythrocytes during hemolysis and increases in thalassemia (52). Endothelial dysfunction as a consequence of oxidative stress (99) is associated with vascular complications in thalassemia such as atherosclerosis, pulmonary hypertension, and thrombosis. These events lead to an imbalance of vasodilator and vasoconstrictor autacoids; for example, NO and prostacyclin vasodilators decrease, while endothelin-1 vasoconstrictor increases (169, 196). Thalassemia patients with pulmonary hypertension have increased markers of endothelial cell activation (endothelin-1), platelet activation (sCD40 L), and hemolysis (lactate dehydrogenase and cell-free Hb) (169).
Most pulmonary hypertension in thalassemia is precapillary pulmonary hypertension (pulmonary arterial hypertension) with the prevalence of 4.8% and 1.1% in β-thalassemia intermedia and β-thalassemia major, respectively (49). Age and splenectomy are significant risk factors, but there are no longitudinal studies evaluating the effect of pulmonary hypertension on survival in thalassemia patients (49, 118). Pulmonary hypertension in thalassemia has poor prognosis due to the lack of effective therapy. To date, there is no randomized control trial for pulmonary hypertension in thalassemia. Delivery of nitrite by inhalation can enhance therapeutic efficacy of NO with minimal systemic adverse effects such as hypotension and methemoglobinemia (148). NO gas therapy is difficult to administer and unsuitable in this chronic setting. Inhaled nebulized nitrite causes a sustained reduction in pulmonary arterial pressure in hypoxia-induced pulmonary hypertension in lambs (84) and hypoxia- or monocrotaline-induced pulmonary hypertension in mice and rats (215). Therefore, sodium nitrite solution, which is inexpensive and convenient to use by nebulization, has therapeutic potential for pulmonary hypertension and has entered phase II clinical trials (
Emerging Therapeutic Strategies
A limitation for many of the existing or in trial therapeutic strategies is sustained delivery and deployment. A clear challenge is the development of a drug delivery platform that can provide sustained delivery of therapeutic levels of anti-inflammatories, NO, and related molecules. Nanoparticle platforms have been developed, capable of sustained NO (27, 59, 75, 121, 182) and NOx (40, 117, 120) delivery, and have been shown to be highly effective for both topical and systemic (IV, IP) applications. The systemic applications clearly indicate that these IV-infused nanoparticles can ameliorate vascular inflammation and maintain normal function capillary density. Some of the topical indications illustrate that these nanoparticles effectively penetrate the skin and release NO in a controlled sustained manner. Using the same protocol, curcumin-releasing nanoparticles have also been prepared (98) and used topically with clear indications of transdermal delivery, release, and efficacy (214). Experiments are underway to evaluate whether sustained systemic delivery of NO and or curcumin can be achieved through topical application of drug-releasing nanoparticles. Preliminary results (Friedman, Cabrales, Davies unpublished results) indicate that systemic efficacy (vascular and inflammatory response) can be achieved via topical delivery of both NO and curcumin-releasing nanoparticles. In brief, the overall strategy is to minimize vascular inflammation, including RBC hemolysis and platelet activation, through facile sustained delivery of molecules that counter the processes that drive these deleterious states.
Conclusion
This overview of possible contributing mechanisms to the pathophysiology of HbE/β-thal disease points to two new targets to minimize the progression of chronic sequelae of this life-threatening hemoglobinopathy founded in major cardiovascular inflammatory consequences originating in the destabilized HbE/β-thal RBC. The first strategy would be based on the emerging picture where the combination of enhanced ferryl Hb populations along with low NO/nitrite levels promotes RBC instability primarily through disruption of the Band 3 transmembrane protein. Therapies that offset this process as a method to enhance RBC stability include strategies that deliver NO/nitrite and curcumin to the RBC in a sustained manner. Stabilizing the RBC and thereby reducing the rate of hemolysis and microparticle formation are expected to decrease vascular inflammation and iron overloading. Reducing/treating vascular inflammation is the second approach to offset the chronic consequences of this disease. Maintaining adequate tissue perfusion and reducing inflammatory markers could also be achieved using sustained vascular delivery of bioactive forms of NO and curcumin. Nanoparticle-based delivery systems are emerging as promising vehicles for achieving these therapeutic objectives. The concept of a slow sustained release and topically applied nanoparticle-containing patch would be relatively easy to deploy and a cost-effective method to reduce the severity of this disease.
Footnotes
Acknowledgments
Partial support is provided by Albert Einstein College of Medicine Global Health Center Microgrants to mPIs R.E.H. and J.M.F.; NIH 5P01 HL110900 to J.M.F., PI; NIH 1R21HL106421 to R.E. H. and J.M.F., mPIs; American Heart Association, Heritage Affiliate, Grant-in-Aid No. 0755906T to R.E.H., PI; and a Research Chair Grant from the National Science and Technology Development Agency (NSTDA) and from the Office of the Higher Education Commission and Mahidol University under the National Research University Initiative, Thailand Research Fund.
Author Disclosure Statement
J. M.F. is on the Science Advisory Board of Nano Biomed, Inc. R.E.H., S.F., and N.S. have no disclosures to report.