
Abstract
Aims:
Cluster of differentiation 36 (CD36) is involved in the development of nonalcoholic steatohepatitis (NASH). Excess CD36 facilitates liver cells taking fatty acid and activates inflammatory signals to promote hepatic steatosis and inflammation. However, CD36 deficiency paradoxically promotes nonalcoholic fatty liver disease by unknown mechanisms. We explored the probable molecular mechanism of hepatic inflammation induced by CD36 deficiency.
Results:
CD36 deletion in mice (CD36−/− mice) specifically increased monocyte chemotactic protein-1 (MCP-1) in hepatocytes, promoted macrophage migration to the liver, and aggravated hepatic inflammatory response and fibrosis. The nuclear expression of histone deacetylase 2 (HDAC2), which highly expresses in wild-type hepatocytes and has an inhibitory effect on acetyl histone 3 (H3), was reduced in CD36-deficient hepatocytes. Consequently, the level of acetyl H3 binding to MCP-1 promoters was increased in CD36-deficient hepatocytes, causing hepatic-specific MCP-1 transcriptional activation. Reduction of nuclear HDAC2 in both CD36−/− mice liver and cultured hepatocytes was due to reduction of intracellular reactive oxygen species (ROS) level, while supplement of low-concentration hydrogen peroxide (H2O2) overcame the suppression of HDAC2 caused by CD36 deficiency, decreasing MCP-1 gene transcription and microphage migration.
Innovation:
Our results provide first evidence that decreased ROS production by CD36 deletion was also harmful for livers. The fine balance of CD36 plays an important role in maintaining balances of hepatic ROS and nuclear HDAC2, which could be a potential new therapeutic strategy for the prevention of NASH development.
Conclusion:
CD36 deficiency promoted the development of NASH by facilitating the transcription of MCP-1 in hepatocytes due to the reduction of ROS and nuclear HDAC2. Antioxid. Redox Signal. 00, 000–000.
It is generally accepted that overproduction of reactive oxygen species (ROS) has been regarded to be a deleterious process that induces an inflammatory response, causing the second hit for nonalcoholic steatohepatitis (NASH) development in the “two-hit model.” However, we demonstrated that decreased ROS production by cluster of differentiation 36 (CD36) deletion was also harmful for livers. The fine balance of CD36 plays an important role in maintaining balances of hepatic ROS and nuclear histone deacetylase 2 (HDAC2), which could be a potential new therapeutic strategy for the prevention of NASH development.
Introduction
C
Although the high expression of hepatic CD36 is closely related to NAFLD, CD36 deficiency paradoxically promotes the development of NAFLD. In the clinic, patients with genetic CD36 deficiency, which is relatively frequent in the Asian and African populations, have been reported to exhibit hyperlipidemia, insulin resistance, and a propensity to develop symptoms of “metabolic syndrome,” including fatty liver and atherosclerosis (18). When administered a high-glucose or high-fat diet (HFD), mice lacking CD36 exhibit increased plasma-free fatty acid and TG levels and decreased hepatic insulin sensitivities (15). CD36 deletion exacerbates the steatosis by impairing hepatic TG and apolipoprotein B (ApoB) secretion in homozygous ob/ob mice (31). These studies suggest that either CD36 overexpression or deletion causes hepatic steatosis and that the function of CD36 as a fatty acid transporter may not explain the conflicting results regarding hepatic steatosis.
CD36 has recently been identified as an important regulator of inflammatory response and functions as a pattern recognition receptor (PRR) that conducts signals and activates inflammatory pathways such as Toll-like receptor (TLR), c-Jun N-terminal kinase (JNK), and nuclear factor-κB (NF-κB) signals (17, 22, 36). Studies have shown that an increased expression of macrophage CD36 and other scavenger receptors contributes to hepatic macrophage infiltration and the development of nonalcoholic steatohepatitis (NASH) (5, 6). However, in the present study, we found that livers of mice with global CD36 gene knockout exhibit an enigmatic increase of macrophage infiltration and inflammation, indicating a largely unknown role for CD36 in regulating macrophage migration.
Macrophage migration and infiltration are regulated by chemokines and their receptors, particularly monocyte chemotactic protein-1 (MCP-1). MCP-1, also called CCL2, is a key chemokine in the development of NASH, and its upregulation promotes macrophage accumulation, inflammation, fibrosis, and steatosis (2). The gene expression of cytokines, including MCP-1, is usually regulated by transcription factors such as NF-κB and repressors on the gene promoter and enhancer regions (37). In addition to NF-κB, epigenetic modification, especially histone acetylation, is the most common and important mechanism regulating the gene transcription of chemokines. It has been demonstrated that the NF-κB signal is inhibited by CD36 deficiency (19), suggesting that increased macrophage migration and hepatic inflammation in CD36 deficiency are caused by NF-κB-independent pathways.
Acetyl histone 3 (H3), which is usually inhibited by histone deacetylases (HDACs), is a transcription factor that binds to MCP-1 promoters. Cooperation of histone acetyl transferases (HATs) and HDACs keeps the balance of histone acetylation: activation of HATs or inhibition of HDACs promotes gene transcription by inducing hyperacetylation of core histones. The activities of HDACs are regulated by intracellular reactive oxygen species (ROS) levels (4), and CD36 has been reported to participate in the production of intracellular ROS. Holloway and colleagues suggested that CD36 is positioned on the outer mitochondrial membrane, upstream of long-chain acyl-CoA synthetase, thereby contributing to the regulation of mitochondrial fatty acid transport and beta-oxidation (38). CD36 is also involved in transduction of intracellular signals such as mitogen-activated protein kinase (MAPK) signals to regulate ROS formation (27). Deletion of CD36 decreases intracellular ROS levels (9).
In a “two-hit model” of NAFLD development, ROS-mediated inflammation has been considered to be the second hit, which has been proposed to cause the transition of hepatic steatosis to more severe NASH (10). The accumulation of lipids in hepatocytes impairs the oxidative capacity of mitochondria and increases the generation of ROS, which triggers cell death and the production of inflammatory cytokines, ultimately resulting in the development of NASH (35). However, accumulating evidence has now indicated that ROS at low/moderate levels, especially the relatively stable hydrogen peroxide (H2O2) molecule, can function as an intracellular second messenger (39). Many ROS-mediated responses protect cells against oxidative stress and maintain “redox homeostasis.” In addition, physiological ROS levels are necessary for the prevention of hepatic steatosis in zebrafish larvae (32). The dual roles of ROS in the development of NAFLD are similar to that of CD36, suggesting that the development of NASH in CD36 knockout mice is probably related to the decreased ROS production induced by CD36 deficiency.
In this study, we explored whether CD36 deficiency inhibited hepatic HDACs by reducing ROS levels and whether the decrease of HDACs increased acetyl H3 binding to MCP-1 promoters, consequently enhancing MCP-1 expression and increasing hepatic macrophage infiltration as well as promoting NASH development in murine models and in vitro cellular experiments.
Results
CD36 deletion promoted the development of NASH in mouse livers
Age- and weight-matched wild-type (WT) mice and CD36 knockout (CD36−/− ) mice were fed with a normal chow diet (NCD) or HFD for 14 weeks. Data of liver sections showed that there was substantially more ballooning degeneration, inflammatory infiltration (hematoxylin and eosin [HE] staining), lipid deposits (Oil Red O [ORO] staining), and fibrosis (Sirius Red [SR] staining) in CD36−/− mouse livers than in WT mouse livers in both the NCD and HFD groups (Fig. 1A). The messenger RNA (mRNA) expression of cytokines, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6) (Fig. 1B), TG content (Fig. 1C), mRNA of fibrosis markers, including collagen I (col I) and collagen IV (col IV) (Fig. 1D), in CD36−/− mouse livers was much higher than in WT mouse livers, in both the NCD and HFD groups. These data indicated that the deletion of CD36 in mice promoted liver inflammation and fibrosis, which contributed to the development of NASH, regardless of the diet.
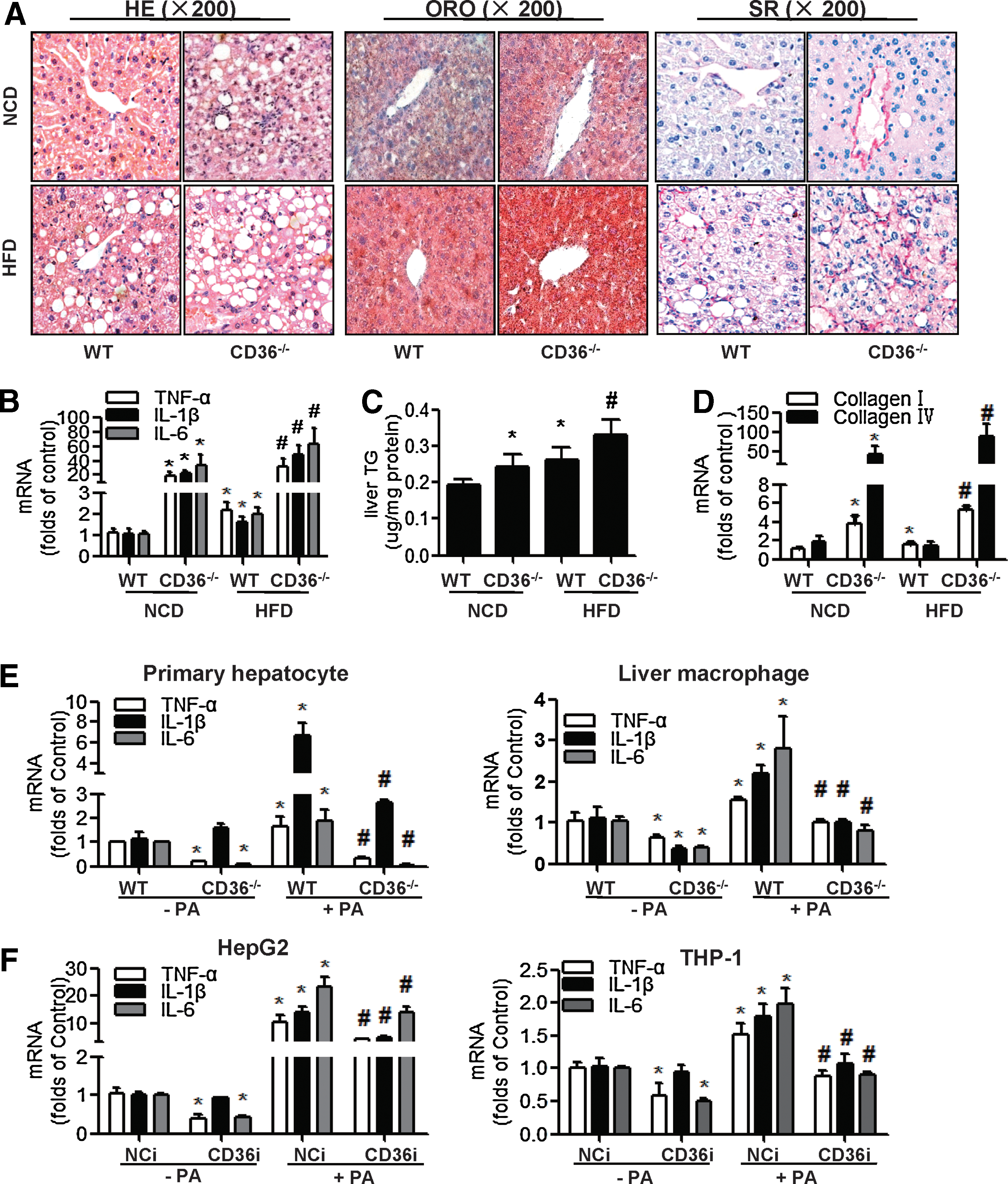
In contrast, results from cell culture experiments were markedly different from the in vivo data. The mRNA levels of cytokines (TNF-α, IL-1β, IL-6) in both the primary hepatocytes and liver macrophages were decreased in the CD36−/− group compared to WT group in the absence and presence of palmitate (PA) (Fig. 1E). HepG2 cells and THP-1 cells were transfected with negative control (NCi) or CD36 small interfering RNA (siRNA) (CD36i) and treated with or without PA for 24 h. PA treatment promoted the mRNA expression of cytokines (TNF-α, IL-1β, IL-6), which was attenuated by CD36 RNA interference (RNAi), both in HepG2 cells and THP-1 cells (Fig. 1F). These results demonstrated that a deficiency of CD36 in cultured hepatocytes or macrophages alleviated PA-induced inflammation.
Increased macrophage infiltration is responsible for the elevated inflammation and fibrosis of CD36−/− mouse livers
The contrary results from liver and cultured cells indicated that the likely reason for elevated inflammation in CD36-deficient livers was due to the interaction between hepatocytes and macrophages. The immunohistochemistry (IHC) staining showed that there were many more F4/80 (marker of macrophages)-positive cells in CD36−/− mouse livers (Fig. 2A) than the WT mouse livers, in the absence and presence of HFD. The “crown-like” structure (indicated by the arrow in Fig. 2A), which was considered a common histological feature for steatohepatitis, was increased in the CD36−/−- mouse livers compared to the WT mouse livers. The mRNA expression of F4/80 was also increased in livers of CD36−/− mice (Fig. 2A).
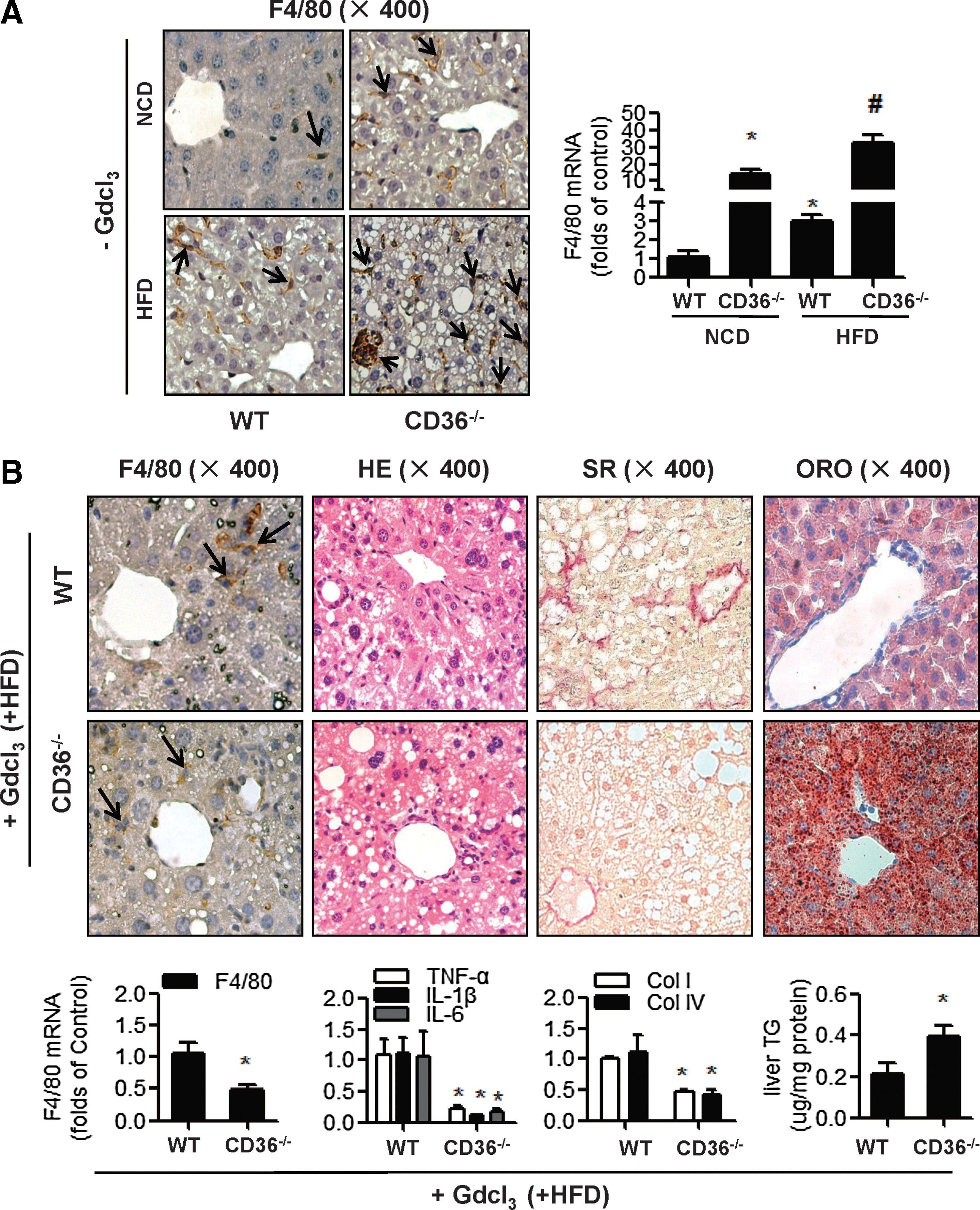
We then administered gadolinium chloride (GdCl3), which inhibits macrophage migration to the liver, to mice that were fed with HFD for 10 weeks. GdCl3 is an earth metal salt that is widely used as an in vivo macrophage selective inhibitor (1) and has been shown to depress macrophage numbers and phagocytic activity in the livers of experimental animals (17, 30). Interestingly, GdCl3 administration decreased macrophage infiltration (F4/80 staining and F4/80 mRNA levels), cytokine expression (mRNA of TNF-α, IL-1β, and IL-6), and fibrosis (SR staining and mRNA levels of col I, col IV) in CD36−/− mouse livers compared to WT mouse livers, whereas the lipid deposit (ORO staining and TG level) was still higher in CD36−/-− mouse livers than that in WT mouse livers (Fig. 2B). These results indicated that the inhibition of hepatic macrophage migration could alleviate the liver inflammation and fibrosis, induced by CD36 deletion in vivo, but it was not able to improve the steatosis in CD36−/− mouse livers.
In an in vitro coculture system, increasing macrophage migration induced by CD36 deletion in hepatocytes accounts for an increase in cytokine expression
Next, we set up a coculture system of HepG2 and phorbol 12-myristate 13-acetate (PMA) differentiated THP-1 cells in transwell chambers to study two cell interactions without (−PA) or with palmitate treatment (+PA). Treatment of PA increased THP-1 migration (Fig. 3A: I vs. V, II vs. VI, III vs. VII, IV vs. VIII), which may be due to increased release of various monocyte/macrophage chemoattracting agents (e.g., MCP-1, macrophage inflammatory protein-1 [MIP-1] macrophage inflammatory protein-2 [ MIP-2], and nucleotides) in PA-treated cells (14, 34). Interestingly, in the absence and presence of PA, CD36 RNAi in THP-1 cells decreased THP-1 migration in the coculture system (Fig. 3A, III vs. I, VII vs. V), whereas CD36 RNAi in HepG2 increased THP-1 migration in the coculture system (Fig. 3A, II vs. I, VI vs. V). CD36 RNAi in HepG2 overrode the suppression caused by CD36 RNAi in THP-1 (Fig. 3A, IV vs. I, VIII vs. V), suggesting that the overall biological effect of CD36 knockdown in both hepatocytes and THP-1 was to increase macrophage migration. The same conclusion could be obtained from coculture experiments of primary hepatocytes and liver macrophages isolated from WT and CD36−/−
mice livers (Supplementary Fig. S1; Supplementary Data are available online at
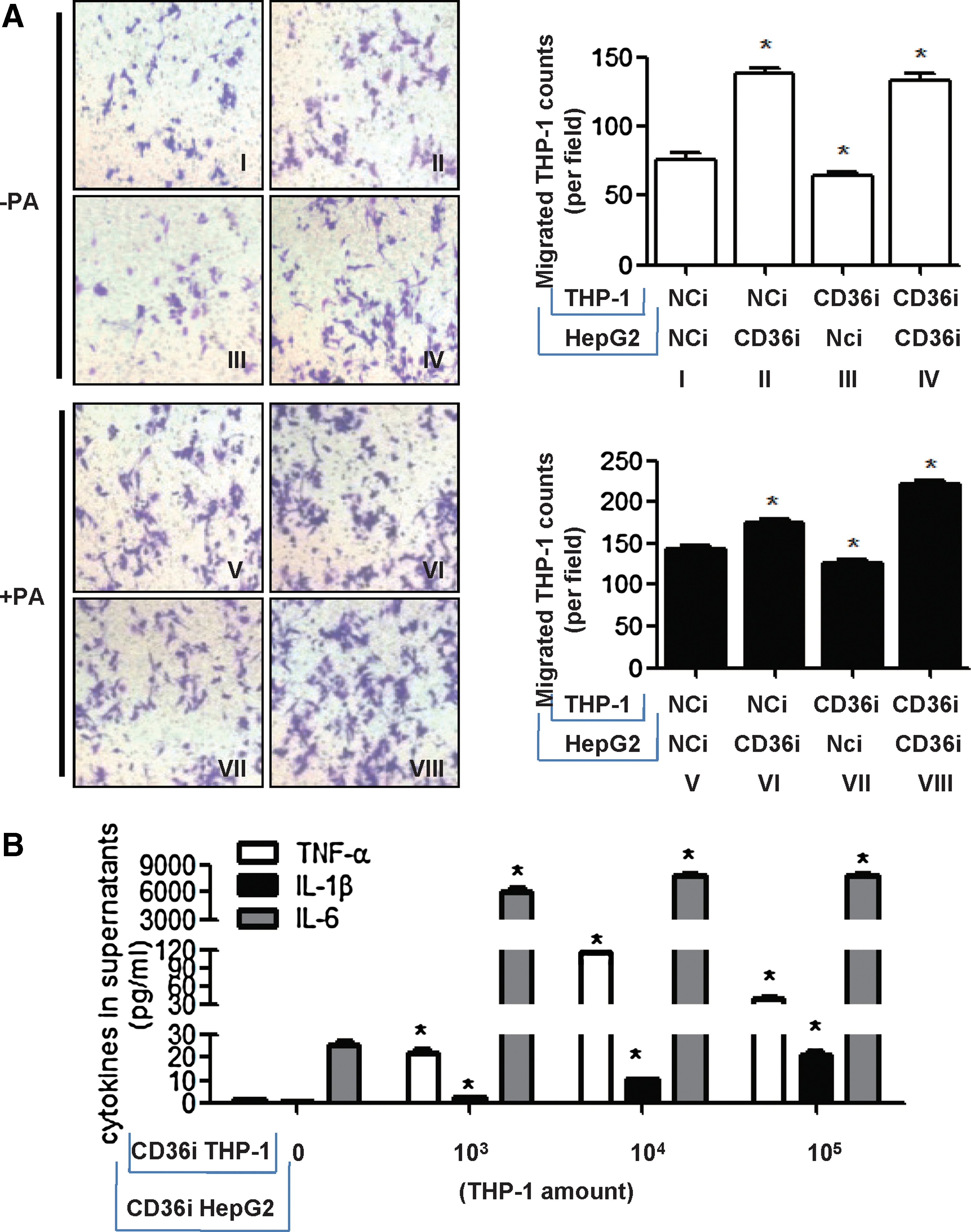
We then cultured HepG2 cells with different amounts of THP-1 cells, and both were transfected with CD36 siRNA to mimic the situation of increasing macrophage infiltration in CD36-deficient livers. The concentrations of cytokines in the supernatant of the coculture system increased as the amounts of THP-1 cells in the upper chamber increased (Fig. 3B). This suggested that the overall effect of CD36 deficiency is an inflammatory response due to large numbers of macrophage migration. Even the inflammatory responses of each cell (HepG2 and THP-1 cells) were diminished in CD36 deficiency conditions, as shown in Figure 1F.
Elevated MCP-1 expression in CD36-deficient hepatocytes should be responsible for the increased macrophage infiltration induced by CD36 deletion
We tested chemokine expression in different models. We found that the levels of chemokines (including MCP-1, MIP-1, and MIP-2) were higher in CD36−/-− mouse livers than in WT mouse livers, regardless of the diets administered to the mice (Fig. 4A). After suppression of macrophage migration by GdCl3, MIP-1 and MIP-2 mRNA in the liver decreased, whereas the MCP-1 mRNA was still higher in CD36−/− mouse livers than that in WT mouse livers (Fig. 4B). In HepG2 cells (Fig. 4C), MCP-1 mRNA was increased in the CD36i group compared with the NCi group in the absence or presence of PA, whereas MIP-2 mRNA was not different between the NCi and CD36i groups. In THP-1 cells (Fig. 4D), the mRNA of both MCP-1 and MIP-2 was decreased in the CD36i group compared to the NCi group. These in vitro results indicated that the elevated MCP-1 was specifically derived from CD36-deficient hepatocytes.
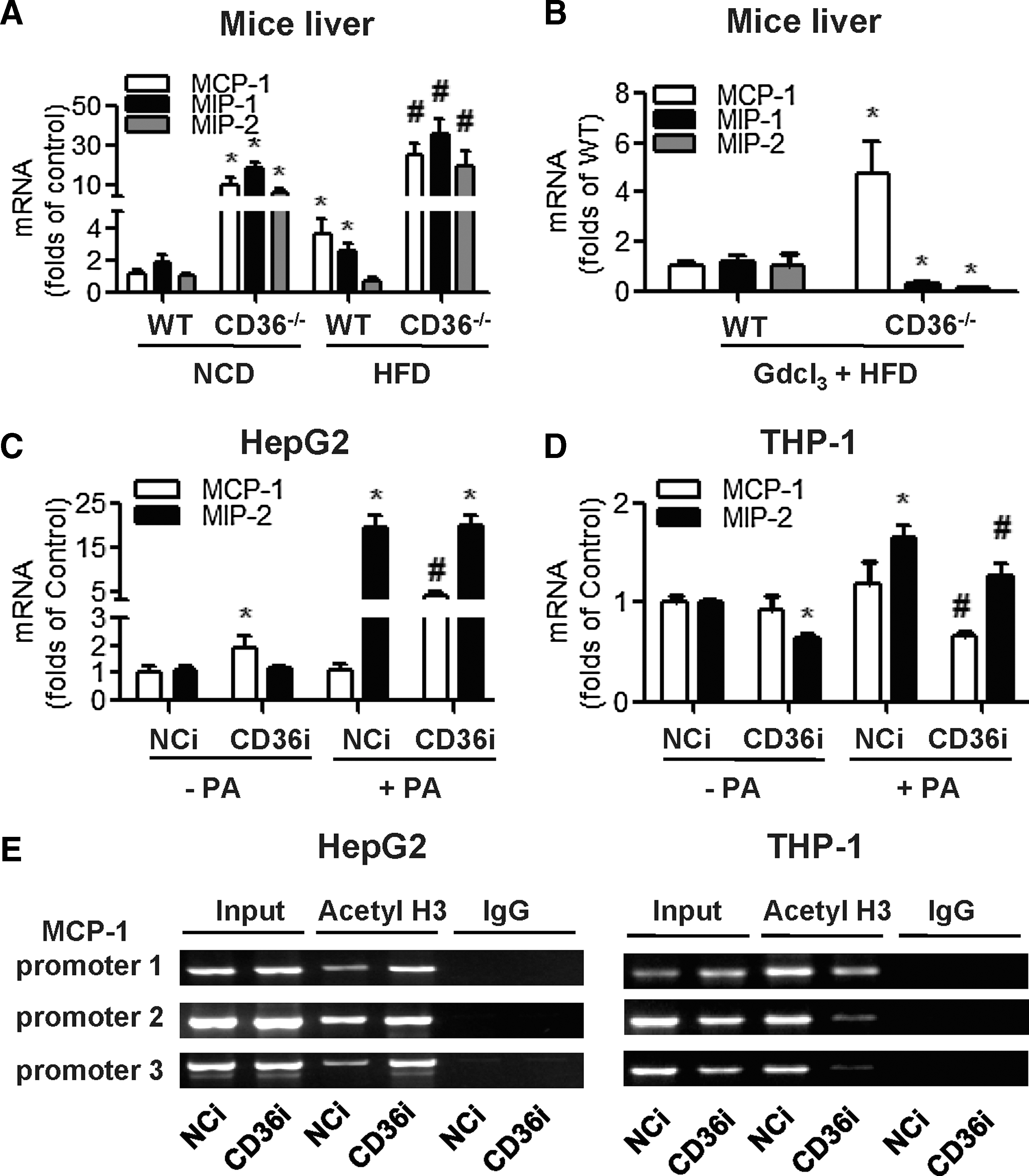
We then designed a chromatin immunoprecipitation (ChIP) assay to test whether the expression of CD36 affected the level of acetyl H3 binding to MCP-1 promoters (Fig. 4E). In HepG2 cells, the level of acetyl H3 binding to MCP-1 promoters was increased in the CD36i group compared to the NCi group. In contrast, in THP-1 cells, the level of acetyl H3 binding to MCP-1 promoters was decreased in the CD36i group. This corresponded to the changes of MCP-1 mRNA expression, indicating that CD36 tissue deletion specifically regulated MCP-1 expression through histone acetylation.
CD36 deletion inhibited nuclear HDAC2 expression, which regulated MCP-1 expression in hepatocytes but not in macrophages
Next, we screened the expression of HDACs in mouse livers and cultured hepatocytes. HDAC2 mRNA expression decreased both in CD36−/− mouse livers and CD36 RNAi HepG2 cells (Supplementary Fig. S2), indicating that increased MCP-1 expression was probably regulated by HDAC2.
We then determined the expression and distribution of HDAC2 in mouse livers and cultured cells by IHC staining. We found that in WT mouse livers, HDAC2-positive signals were located in hepatocytes, especially in hepatocellular nuclei (as indicated by the solid line arrow in Fig. 5A), with no signals in nonparenchymal cells (as indicated by the dashed arrow in Fig. 5A). The positive HDAC2 staining was decreased in the hepatocellular nuclei of CD36−/− mouse livers, indicating that CD36 deficiency attenuated the active expression of HDAC2 in hepatocytes (Fig. 5A). In primary cultures of mouse hepatocytes and HepG2 cells, an HDAC2-positive stain was observed in both the nuclei and cytoplasm of WT hepatocytes (Fig. 5B, I) and in NCi HepG2 cells (Fig. 5C, I) but was decreased in the nuclei of CD36−/− hepatocytes (Fig. 5B, II) and CD36i HepG2 cells (Fig. 5C, II). However, in mouse liver macrophages and THP-1 cells, the HDAC2 staining was very weak in the nuclei of all groups, including WT and CD36−/− liver macrophages (Fig. 5B, III, IV), NCi, and CD36i THP-1 cells (Fig. 5C, III, IV), indicating that the levels of HDAC2 in macrophages were too low to be altered by CD36 deficiency. These observations were also confirmed by the results of Western blotting for nuclear HDAC2 in HepG2 and THP1 cells (Fig. 5D and Supplementary Fig. S3).
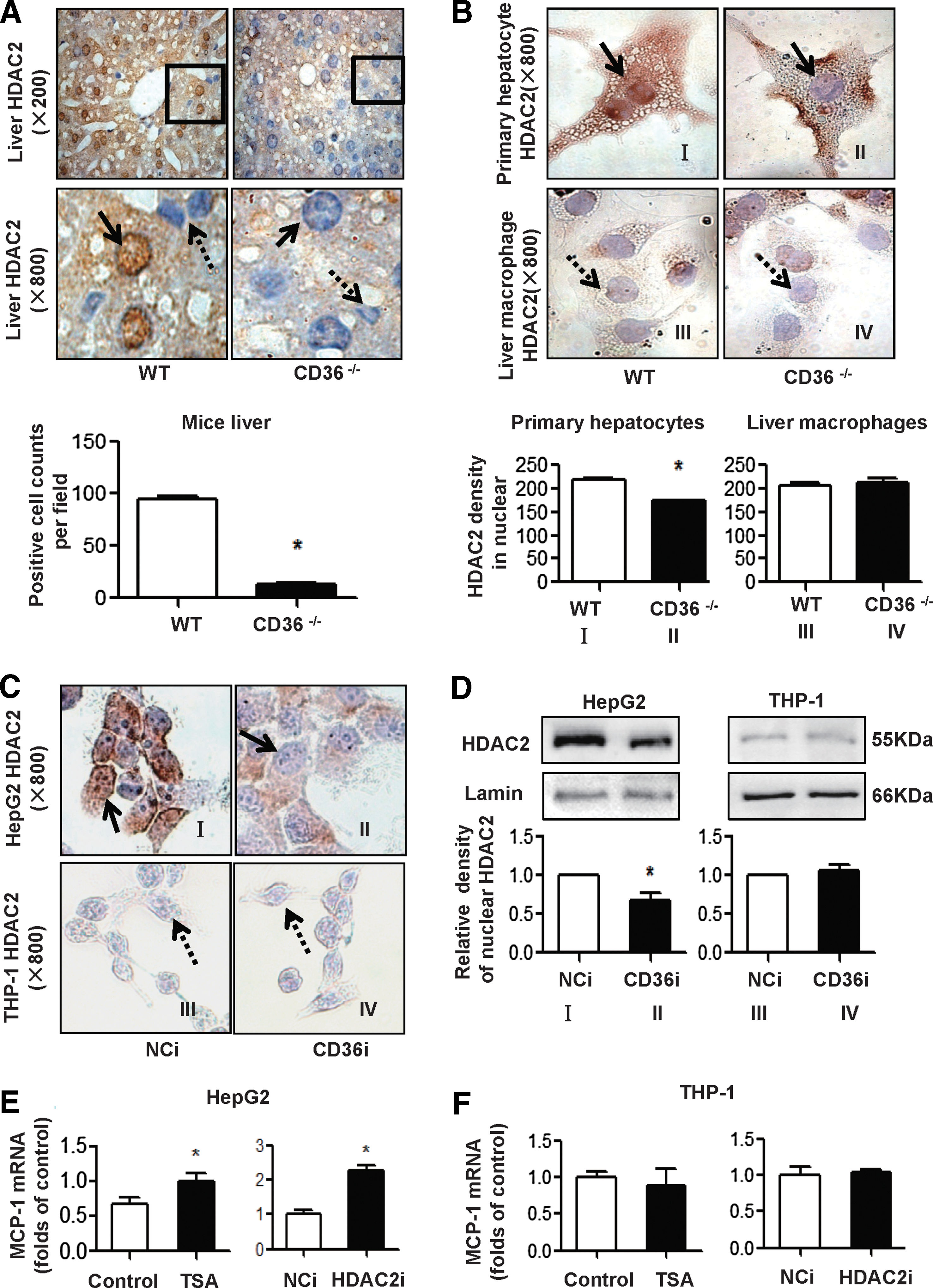
Next, we utilized the HDAC inhibitor trichostatin A (TSA) and HDAC2 RNAi (HDAC2i) to downregulate HDAC2 activities in HepG2 and THP-1 cells. In HepG2 cells, MCP-1 mRNA was increased in the TSA or HDAC2i groups compared to the corresponding controls (Fig. 5E). In THP-1 cells, MCP-1 mRNA was not changed in the TSA or HDAC2i groups, compared to the control or NCi (Fig. 5F). Collectively, these results indicated that suppressed nuclear HDAC2, especially in hepatocytes, promoted MCP-1 expression.
The disturbance of ROS production in CD36-deficient hepatocytes is responsible for the suppressed nuclear HDAC2 and increased expression of MCP-1
Because HDAC activity was reported to be closely related to intracellular ROS levels, we examined H2O2 levels in the liver tissue and the ROS content of cultured HepG2 cells. The H2O2 level in mouse livers in the HFD group was much higher than that in the NCD group. However, the H2O2 level in CD36−/− mouse livers was much lower than that in WT mouse livers in both the NCD and HFD groups, indicating that CD36 deletion attenuated H2O2 production in the liver (Fig. 6A). In HepG2 cells, the ROS content was elevated in cells that were treated with PA and was decreased in CD36i HepG2 compared with NCi HepG2 (Fig. 6B).
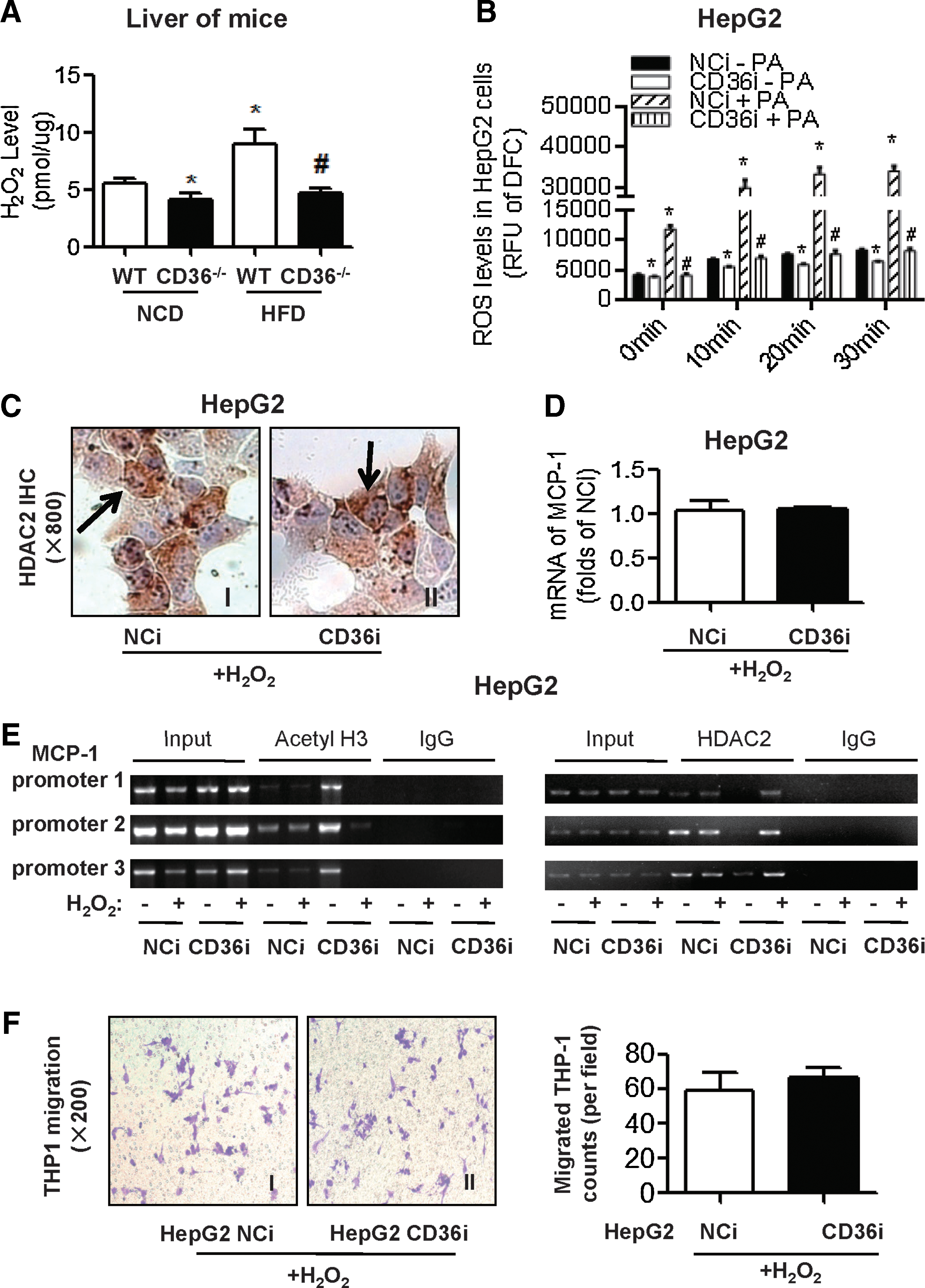
We then treated HepG2 cells with the supplement of a low concentration (50 μM) of H2O2. Under the treatment of H2O2, nuclear HDAC2 staining appeared again in CD36i HepG2 cells (Fig. 6C, II). Moreover, with the supplement of H2O2, MCP-1 mRNA expression was no longer increased in CD36i HepG2 cells (Fig. 6D). ChIP assays showed that level of acetyl H3 binding to MCP-1 promoters was clearly decreased, which was consistent with increased level of HDAC2 binding to MCP-1 promoters, after treatment with 50 μM H2O2 in CD36 RNAi HepG2 cells (Fig. 6E). Furthermore, CD36RNAi in HepG2 cells no longer promoted differentiated THP1 cell migration with the supplement of 50 μM H2O2 (Fig. 6F). All these results suggested that CD36 deficiency promotes MCP-1 expression by inhibiting ROS production and HDAC2 and that maintaining a balance of hepatic ROS could prevent macrophage migration induced by CD36 deficiency.
Discussion
Liver lobules are formed by parenchymal cells, such as hepatocytes, and nonparenchymal cells, including Kupffer cells, sinusoidal endothelial cells, and stellate cells. In NASH, the defining pathological element is hepatocellular injury, as evidenced by ballooning, Mallory bodies, and apoptosis. Hepatocytes are considered to be a major source of the inflammatory response in NASH-affected liver (13). In addition to hepatocytes, activated Kupffer cells can launch a biochemical attack and initiate interactions with hepatocytes and other liver cells by releasing a variety of biologically active mediators, including cytokines, chemokines, eicosanoids, proteolytic enzymes, ROS, and nitric oxide (3). An enlarged Kupffer cell pool, which is usually expanded by the migrated monocytes/macrophages from circulation, is believed to contribute to the onset of NASH by interacting with hepatocytes (24). However, the role of hepatocytes on macrophage migration remains unclear.
In this study, we demonstrated that CD36 deletion attenuated the expression of inflammatory cytokines both in hepatocytes and macrophages when they were cultured alone. However, when hepatocytes and macrophages were together in liver tissue or in an in vitro coculture system, CD36 deletion could not attenuate but instead promoted cytokine secretion as a consequence of increased macrophage infiltration. CD36 deletion in hepatocytes alone or both in hepatocytes and macrophages increased macrophage migration and inflammatory cytokine secretion in a coculture system of hepatocytes and macrophages. Targeted inhibition of macrophage infiltration into the liver by GdCl3 administration effectively relieved the increased hepatic inflammation and fibrosis induced by CD36 deletion. These results suggest that CD36 deletion could induce an increased macrophage pool in the liver and increase the interaction between hepatocytes and macrophages. This could completely compensate for the decrease in cytokine secretion by CD36 deletion in hepatocytes and macrophages. In addition, CD36−/− decreased fatty acid uptake, but CD36−/-− did not reduce lipid accumulation in mouse livers, probably because of a deficiency in very low-density lipoprotein (VLDL) excretion by hepatocytes (31). The overall effect of CD36 deficiency promotes hepatic inflammatory response, lipid accumulation, and liver fibrosis.
The role of CD36 in the regulation of macrophage migration has been studied extensively in models of arthrosclerosis; however, the results are controversial (20). A study from Harb et al. (16) and Kuchibhotla et al. (25) has suggested that CD36 expression promotes macrophage migration, whereas the results from Park et al. (33) suggest that the engagement of CD36 by ox-LDL inhibits macrophage migration. One potential explanation for these apparently conflicting results is that different ligands binding to CD36 may promote different intracellular signaling pathways related to cellular migration and inflammation. Other than the recognition of different ligands, CD36 is also expressed in different cells and tissues. In this study, we first demonstrated that CD36 deletion in hepatocytes promoted macrophage migration, whereas CD36 deletion in hepatic macrophages inhibited macrophage migration, indicating that the CD36 of parenchymal cells or nonparenchymal cells might play different roles in hepatic inflammation. The different regulation of macrophage migration by CD36 depends on the differential expression of MCP-1 in different cells.
MCP-1 expression can usually be stimulated by cytokines such as TNF-α (24), but in this study, MCP-1 was elevated, whereas cytokines and other chemokines such as MIPs were decreased in CD36−/-− mouse livers after the inhibition of liver macrophages. This indicates that MCP-1 in CD36−/− mouse livers was elevated before other cytokines and was derived from hepatocytes. The result was confirmed by in vitro experiments showing that MCP-1 was elevated, whereas cytokines and MIPs were decreased in CD36 RNAi HepG2 cells. These changes were consistent with the results of a macrophage migration assay. Thus, this is the first demonstration that MCP-1 derived from hepatocytes plays a key role in hepatic macrophage infiltration and hepatic inflammation in CD36-deficient models.
Furthermore, we explored the mechanism by which CD36 tissue deletion specifically induced the expression of MCP-1 in hepatocytes but not in macrophages. We demonstrated that CD36 deletion regulated MCP-1 expression at the transcriptional level by changing the acetylation of histones binding to the MCP-1 promoters. A balance of HATs and HDACs controls the histone acetylation. Approximately 18 different HDACs have been identified and grouped into 2 families and 4 classes in eukaryotic cells: Zn-dependent HDACs, including HDAC1 through 11, and NAD-dependent Sirtuins (8). Many studies have shown that MCP-1 expression is regulated by HDACs. In hepatic stellate cells, HDAC1 is recruited to specific regulator regions of MCP-1 and suppresses MCP-1 expression (12); HDAC3 has been reported to mediate allergic skin inflammation by regulating MCP-1 expression (23). Many HDACs are involved in the development of NASH, such as HDAC3 and SIRT1 (26). However, the regulation of MCP-1 by HDACs and CD36 deficiency remains unclear. We found that HDAC2 mRNA was decreased after screening for the expression of 11 classic HDACs in CD36−/− mouse livers and CD36-deficient hepatocytes. HDAC2 belongs to class I HDAC (comprising HDAC1, HDAC2, HDAC3, and HDAC8), which is ubiquitously expressed in all tissue types (7). HDAC2, with its highly related sister protein HDAC1, is present in the mammalian nucleus as part of stable multiprotein complexes, such as Sin3A, NuRD, and CoREST complexes (21). Simultaneous deletion of HDAC1 and HDAC2 in T cells and embryonic stem cells causes a 50% decrease in total HDAC activity, and therefore, they are recognized as the predominant HDACs in the mammalian nucleus (11). We demonstrated that in the mouse liver, HDAC2 is primarily expressed in hepatocytes and only rarely in nonparenchymal cells. CD36 deletion clearly inhibited nuclear expression of HDAC2 in hepatocytes but had no impact on the expression of HDAC2 in macrophages. MCP-1 mRNA expression was promoted by the treatment of the HDAC inhibitor TSA or HDAC2 RNAi in hepatocytes rather than in macrophages. Furthermore, CD36 deletion in hepatocytes promoted MCP-1 expression by suppressing the nuclear expression of HDAC2, which does not apply to macrophages because of the extremely low expression of HDAC2.
Numerous studies have shown that HDACs are redox sensitive. The relationship between ROS, HDAC activity, and acetylation status may depend on the intensity of ROS, endogenous HDAC activity, and the experimental cell types used (29). ROS has a bidirectional impact on the activity of HDACs: strong oxidative stress induces hypoacetylation, while weak oxidative stress induces hyperacetylation, even in the same cell line (4). A decreased oxygen environment inhibits the activities of HDACs (40).
CD36 is important to maintain intracellular ROS homeostasis, as it could modulate ROS production through mediating lipid uptake, facilitating fatty acid into mitochondria for oxidation, and activating redox signaling such as MAPK (27). In this study, we found that CD36 deletion decreased the hepatic ROS levels in vivo and in vitro. Supplementation with ROS (H2O2) improved the nuclear expression of HDAC2, decreased acetyl H3 binding to MCP-1 promoters, and eliminated the elevation of MCP-1 expression in CD36-deficient hepatocytes. This suggests that the decreased ROS level by CD36 deletion contributes to the suppressed nuclear HDAC2 and elevated MCP-1 in CD36-deficient hepatocytes, which promoted hepatic macrophage infiltration and the development of NASH in CD36−/− mice. The overproduction of ROS has been regarded to be a deleterious process that induces an inflammatory response and pathological conditions. Our data suggest that decreased ROS production by CD36 deletion was also harmful for mouse livers, making them susceptible to developing NASH by suppressing the expression of HDAC2 and promoting the expression of MCP-1 in hepatocytes.
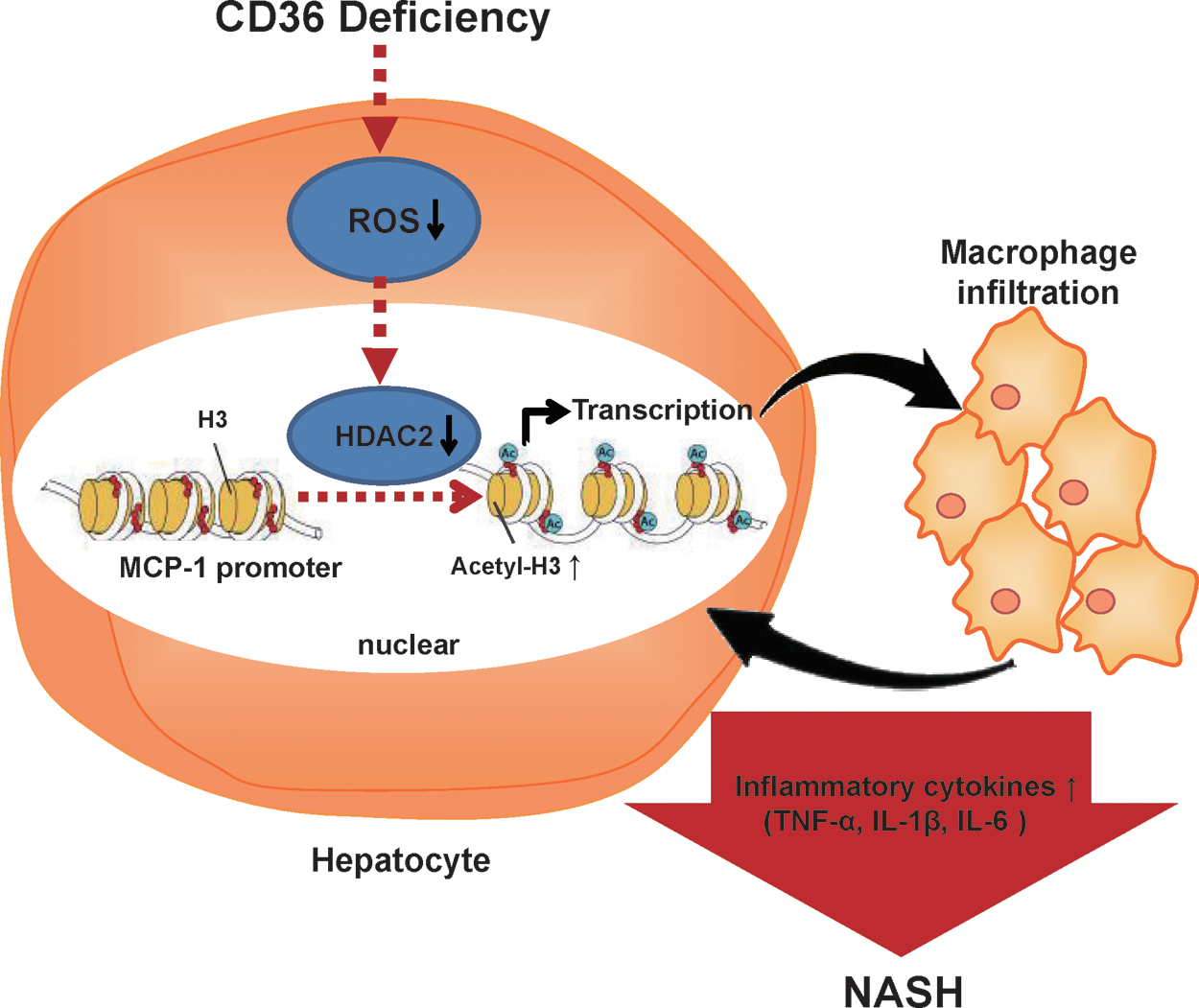
In conclusion, excess CD36 facilitates the transport by liver cells of fatty acids and activates inflammatory signals, thereby promoting hepatic steatosis and inflammation; whereas deletion of CD36 induces hepatic macrophage infiltration and inflammation by increasing the expression of MCP-1 in hepatocytes because of reducing ROS production and suppressing nuclear HDAC2 (Fig. 7). CD36 deficiency cannot alleviate the development of NASH in mouse models. The physiological levels of CD36 in hepatocytes are important to keep a balance of ROS, macrophage migration, and the inflammatory response. Maintaining a balance of hepatic ROS and nuclear HDAC2 could be a potential new therapeutic strategy for the prevention of NASH development in CD36-deficient individuals.
Materials and Methods
Animals and diets
CD36 knockout mice created on a C57BL/6J background were kindly provided by Dr. Maria Febbraio (Lerner Research Institute). Mice were randomly fed an NCD composed of 10% kcal% fat (Research Diets, Inc.) or an HFD with 1.25% cholesterol and 0.5% cholic acid (HFD) containing 40% kcal% fat (Research Diets Inc.) for 14 weeks before sacrifice. To inhibit the function of liver macrophages, WT and CD36−/− mice were administered 1% GdCl3 (Sigma) solution (10 mg/kg) twice a week through intraperitoneal injection and fed with HFD for 10 weeks before sacrifice. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985).
Cell culture and treatments
HepG2 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS; Equitech Bio, Inc.), and THP-1 cells were cultured in RPMI 1640 medium containing 10% FBS, in an incubator kept at 37°C with 5% CO2. Before experiments, THP-1 cells were differentiated into macrophages by incubation with 200 nM PMA (Sigma) for 48 h. The preparation of primary hepatocytes and liver macrophages from mouse livers followed the protocol in the supplementary material. Before harvesting, the cells were preincubated for at least 12 h in serum-free medium and then incubated for 24 h in serum-free medium (control) or serum-free medium containing 0.16 mM sodium PA (Sigma), or 100 μM TSA (Millipore), or 50 μM H2O2 solution (Sigma). siRNA was transfected using the X-tremeGENE siRNA Transfection Reagent (Roche) according to the manufacturer's instructions.
Coculture system and migration assay
HepG2 cells and differentiated THP-1 cells were cocultured using six-well transwell plate inserts with a 0.4 μm porous membrane (Corning) to separate the lower chamber of the serum-starved HepG2 from the upper chamber of serum-starved THP-1. For the in vitro migration assay, transwell inserts with an 8 μm porous membrane (Corning) were used. After allowing migration for 16 h, the migratory cells on the lower side of the membrane were stained with 4,6-diamidino-2-phenylindole. The average number of migratory cells in each well was counted from four random fields under the microscope. The cytokine content in the supernatants of the coculture system was determined by MILLIPLEX Analyst (Millipore).
Preparation of primary hepatocytes and macrophages from mouse livers
We anesthetized 8-week-old WT or CD36−/− mice and opened the abdominal cavity. We cannulated the portal vein and immediately perfused it with modified Hanks' buffered saline. The liver was infused with a collagenase solution (100 mg collagenase in 150 ml Hank's buffer) by a peristaltic pump at a rate of 1.5 ml/min for 20 min. The liver was removed and sliced into small pieces, which were placed in collagenase solution and filtered with a 100-mm cell filter. The cell suspension was centrifuged at 800 rpm for 5 min, and the deposits were primary hepatocytes. The supernatant was placed into another tube and centrifuged twice at 2000 rpm for 5 min. The deposits were macrophages. The hepatocytes and macrophages were cultured in DMEM containing 20% fetal calf serum (FCS) and maintained in an incubator at 37°C, 5% CO2.
Liver histology
Paraffin-embed liver sections (4 μm) were stained with HE. For IHC, the sections were incubated in 3% H2O2 followed by 1% bovine serum albumin in phosphate-buffered saline and then overnight (4°C) with anti-CD36 (1:100; Santa Cruz) or anti-F4/80 (1:50; BioLegend) or anti-HDAC2 (1:100; Millipore). A commercial kit (Zsbio) was used to perform the histochemical reaction and was counterstained with hematoxylin. For ORO staining, frozen sections were stained for 30 min and then counterstained with hematoxylin. For Picro-SR staining, sections were incubated with 0.5% SR F3B (Sigma) in saturated aqueous picric acid for at least 1 h and then washed with 0.1% acetic acid solution three times. Results were examined by light microscopy.
Real-time polymerase chain reaction
Total RNA was extracted from the liver of mice or cultured cells by RNAiso Plus reagent (Takara). By using a complementary DNA (cDNA) synthesis kit (Takara), 1.0 μg of total RNA was converted to first-strand complementary DNA in a 20 μl reaction system. Real-time polymerase chain reaction (PCR) was performed in a real-time PCR machine (Bio-Rad) using SYBR Green dye. The thermal cycling program was 5 min at 95°C for enzyme activation and 40 cycles of denaturation for 15 s at 95°C, 15 s annealing at 55°C, and 15 s extension at 72°C. Beta-actin was used as an internal control gene. All the primers were designed by Primer Express Software V2.0 (Applied Biosystems; see also Supplementary Tables S1 and S2).
ChIP assay
The procedure was performed according to the manufacturer's instructions with a Magna ChIP G kit (Millipore). The MCP-1 promoter-specific primers are shown in Supplementary Table S1.
TG content, ROS, and H2O2 assays
These assays were performed according to the manufacturer's instructions with a TG Assay kit (Millipore) and hydrogen peroxide assay kit (Beyotime). For ROS assays, cells were incubated with 2′,7′-Dichlorofluorescin diacetate for 40 min, and relative fluorescence unit (RFU) of 2′,7′-Dichlorofluorescin was measured at indicated time using an ROS assay kit (Beyotime).
Western blotting
Nuclear protein was extracted using a nuclear extraction kit (Abcam). Equal amounts of nuclear protein were resolved on sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). The blots were incubated with the primary antibodies anti-HDAC2 (Millipore) and anti-Lamin (Proteintech) and secondary antibody horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Zsbio). Blotted proteins were detected with the Odyssey Imaging System (LI-COR Biosciences). Quantification was performed with ImageJ software.
Statistical analysis
All the data were analyzed by GraphPad Prism 5.0. A t-test was used to compare the statistical relevance of the two groups. For groups of three or more, analysis of variance (one-way ANOVA) with post-test using Tukey was performed. Two-way ANOVA was used to determine the interactions between two factors. Data are expressed as the mean ± standard error of the mean (SEM). p < 0.05 was considered to be significant. All the data were from at least three separate experiments.
Footnotes
Acknowledgments
We thank Dr. Maria Febbraio (Lerner Research Institute) for providing the CD36 knockout mice. This study was supported by the Major State Basic Research Development Program of China (973 Program Nos. 2012CB517700 and 2012CB517500), the National Natural Science Foundation of China (81570517, 81500442, 81270789, 81270493, 31540029, 31640043, and Key Program No. 81390354), and the Chongqing Research Program of Basic Research and Frontier Technology (Nos. cstc2015jcyjBX0044 and cstc2016jcyjA0545).
Author Disclosure Statement
No competing financial interests exit.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.