
Abstract
Significance:
Breast cancer is the second leading cause of cancer-related deaths among women in the United States. Development and progression of malignancy are associated with diverse cell signaling pathways that control cell proliferation, survival, motility, invasion, and metastasis.
Recent Advances:
An increasing number of clinical studies have implicated a strong relationship between elevated tumor nitric oxide synthase-2 (NOS2) expression and poor patient survival.
Critical Issues:
Herein, we review what we believe to be key mechanisms in the role(s) of NOS2-derived nitric oxide (NO) as a driver of breast cancer disease progression. High NO increases cyclooxygenase-2 activity, hypoxia inducible factor-1 alpha protein stabilization, and activation of important cell signaling pathways, including phosphoinositide 3-kinase/protein kinase B, mitogen-activated protein kinase, epidermal growth factor receptor, and Ras, through post-translational protein modifications. Moreover, dysregulated NO flux within the tumor microenvironment has other important roles, including the promotion of angiogenesis and modulation of matrix metalloproteinase/tissue inhibitor matrix metalloproteinase associated with tumor progression.
Future Directions:
The elucidation of these and other NO-driven pathways implicates NOS2 as a key driver of breast cancer disease progression and provides a new perspective in the identification of novel targets that may be therapeutically beneficial in the treatment of estrogen receptor-negative disease. Antioxid. Redox Signal. 26, 1044–1058.
Introduction
B
Clinical management distinguishes disease subtypes according to estrogen (ER) and progesterone (PR) hormone receptor status, as well as human epidermal growth factor receptor-2 (HER2) status. ER status is defined by the presence (ER+) or absence (ER−) of the alpha form of the receptor. Approximately 70% of breast cancer patients are diagnosed with having ER+ status, while 30% present with the more aggressive ER− subtype. ER+ tumors can be successfully treated with hormone-based therapies, including antiestrogens and aromatase inhibitors, while patients with triple-negative (ER−/PR−/HER2−) breast cancer (TNBC) have fewer options. Toward this end, the identification of novel molecular targets can improve therapeutic response and survival in TNBC patients.
Nitric oxide (NO) is released intracellularly during the oxidation of
Recently, NOS2 was identified as a biomarker of breast cancer disease progression and patient survival (16, 36, 44, 75). Moreover, NOS2-derived NO can alter the redox state of cells, induce DNA, lipid, and protein modifications, promote an immunosuppressive microenvironment, and mediate angiogenesis and wound response, which are all key events in cancer disease progression (1). Previous work from our laboratory has elucidated mechanisms of feed-forward tumor NOS2 regulation by components of the tumor microenvironment, including nutrient deprivation, inflammatory cytokines, and hypoxia (44, 76). In this review, we will discuss our current understanding of NOS2-derived NO mechanisms associated with breast cancer disease progression as well as therapeutic implications.
NOS2 and P53 Mutation
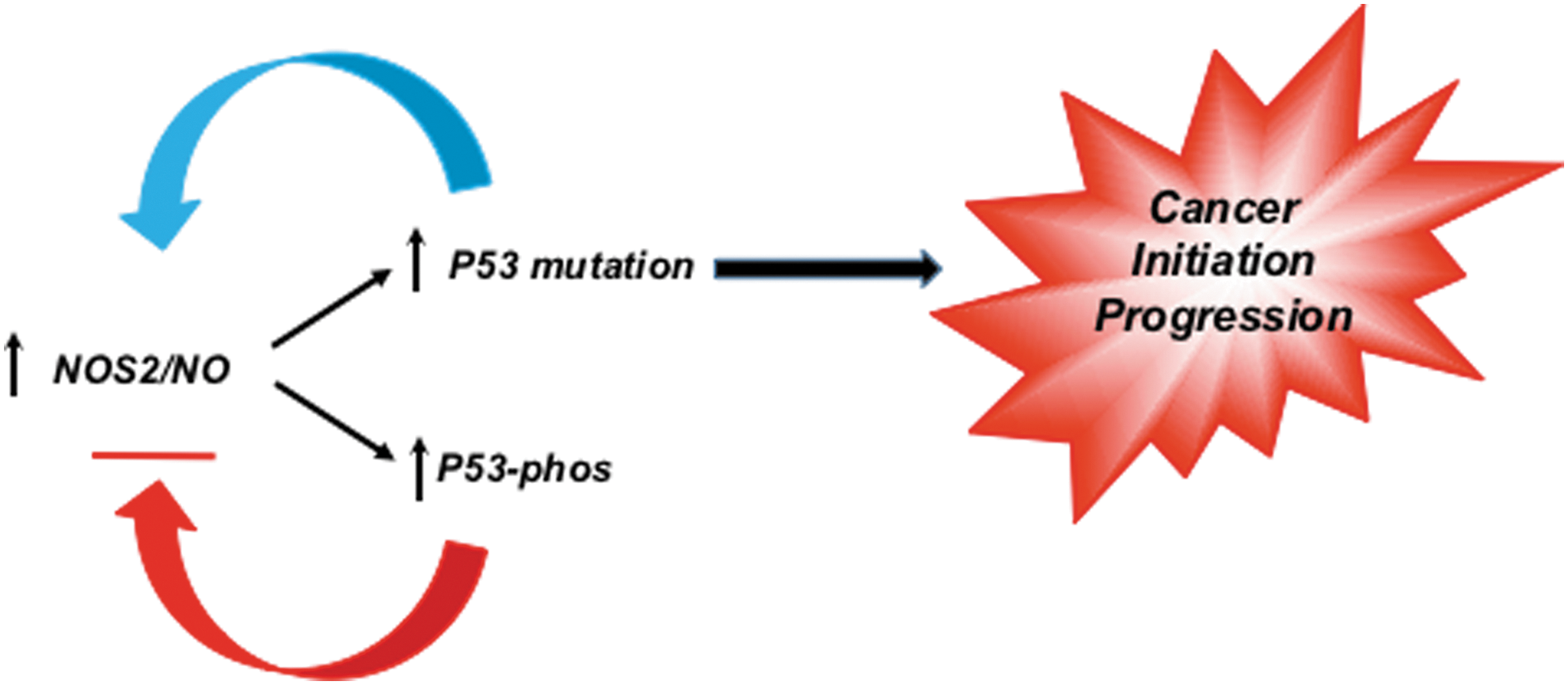
The activation of p53 is a critical component of cell cycle arrest, DNA repair, senescence, and apoptosis (69, 70). While P53 is not required for cell viability, the loss of its functions allows the accumulation of genetically damaged cells, which precedes the development of neoplastic lesions (69). Toward this end, mutations in p53 are among the most common changes found in human cancers (69, 70). The more aggressive TNBC accounts for a high percentage of death among breast cancer patients and p53 mutations are reported in 60–80% of these cases (149). Our recent breast cancer study found a significant correlation between increased p53 mutation and high tumor NOS2 expression (odds ratio [OR] 3.02; 95% confidence interval 1.19–7.66; p-value 0.020) (36).
Nitric oxide is a key bioactive modulator of several processes, including angiogenesis and host defense, which are dysregulated in cancer (52). Given that p53 is a transrepressor of NOS2 gene expression, these observations suggest that the loss of this negative feedback loop may provide selection pressure for tumor initiation and progression (Fig. 1) (2, 3, 34). Inflammatory factors and components of the tumor microenvironment increase NOS2 expression, which include hypoxia, nutrient deprivation, prostaglandin E2 (PGE2), interleukin-6 (IL-6), and interferon gamma (44, 129, 144). PGE2 can increase NOS2, while IL-6 induction of signal transducer and activator of transcription 3 leads to further activation of NOS2 (129, 144). These pathways conspire to form protumorigenic, feed-forward autocrine loops leading to increased metastasis (44). Furthermore, numerous factors from immune cells and stroma lead to acceleration of these loops, suggesting that NOS2 feed-forward signaling can perpetuate these mutation pathways (60, 81).
Tumor Hypoxia
Hypoxia is a common characteristic of the tumor microenvironment that drives disease progression and is associated with oxygen deficit in avascular tumors leading to metastasis (48). Multiple studies have identified associations between reduced intratumoral pO2 and decreased disease-free survival in cancer patients (153). Uncontrolled tumor cell proliferation leads to nutrient depletion and hypoxia, which are also major contributors of chronic inflammation within the tumor microenvironment. Hypoxic/necrotic regions within the tumor induce proinflammatory immune mediators that culminate in a local immunosuppressive microenvironment, which induces angiogenesis, tumor cell proliferation, migration, and invasion (79, 91). These events are mediated by hypoxia-inducible factor-1 alpha (HIF-1α) adaptive signaling that promotes chemoresistance, metastasis, and poor patient survival (5, 21, 77).
HIF-1α can be upregulated by other factors, including insulin, insulin-like growth factor (IGF-1 or IGF-2), v-src proto-oncogene, nonreceptor tyrosine kinase (Src), lactate, pyruvate, and tumor inflammation, as well as genetic alterations, including activation of oncogenes or inactivation of tumor suppressor genes (5). In addition, under normoxia, NO mediates HIF-1α protein stabilization through nitrosative mechanisms that block its proteasomal degradation (14, 72, 82, 139). NOS2 has emerged as a biomarker of poor survival in patients with aggressive tumors, suggesting that nitrosative/nitrosylative mechanisms that promote HIF-1α protein stability may be important in protumorigenic signaling associated with high NOS2 tumors (139).
Another impact of altered oxygen gradient and tumor hypoxia involves the generation of reactive oxygen species (ROS) and altered redox status. In addition to ROS, redox status is influenced by other small reactive molecules, including NO, and other nitrogen oxides, as well as the eicosanoids (i.e., cyclooxygenase-2 [COX2] and lipoxygenase) (140). Molecules, including carbon monoxide (CO) derived from heme oxygenase and hydrogen sulfide (H2S), a product of thiol metabolism, are components of redox inflammation. These molecules whose metabolism is either directly related to O2 or arise in response play critical roles in oxidative stress. Toward this end, O2 tension is a major determinant of NOS2-derived NO and downstream signaling because in addition to arginine, O2 is also a substrate of NOS.
The O2 availability within a tissue bed is a function of the rate of arterial delivery versus that of mitochondrial O2 consumption. Michaelis-Menten enzyme kinetics is a mathematical model that predicts the amount of product formed upon the binding interaction of an enzyme with its substrate (83). The equation employs the Michaelis constant (K M) substrate concentration where the reaction rate is at half-maximal and is an inverse measure of the substrates’ affinity to the enzyme (83). Based upon the NOS2 K M for O2 (135 μM), the work of Hickok et al. has shown a requirement of 3–5% O2 for maximum NO flux derived from the NOS2 enzyme (46). Moreover, NO consumption is also O2 dependent; therefore, steady-state NO flux and downstream signaling depend on the relative rates of these variables (46).
Chronic Inflammation, NOS2, and the Tumor Microenvironment
Current statistics estimate that chronic inflammation associated with inflammatory diseases contributes to a 25% increased risk of cancer occurrence (6, 80). Beyond the increased risk of occurrence, chronic inflammation within the tumor microenvironment has long been proposed as a contributing factor in tumor promotion and disease progression (6, 101, 113). These findings are supported, in part, by observations that modest intake of nonsteroidal anti-inflammatory drugs (NSAIDs) reduces cancer growth and recurrence (12, 23, 121, 122). The inflammatory composition of the tumor microenvironment has long been compared with nonhealing wounds (6). Dvorak observed a striking resemblance between the tumor stroma and tissue granulation of healing wounds, which implicated a role of host wound response in the formation of tumor stroma and disease progression (28).
Cancer inflammation involves the subtle coordination between tumor cells, activated stromal cells, including endothelial cells, fibroblasts, stem cells, and immune cell mediators. Together, this network provides an immunosuppressive environment rich in growth factors and cytokines that promote uncontrolled, sustained tumor cell proliferation and survival with proangiogenic and metastatic capabilities (40, 41). Importantly, this process is not self-limiting in the tumor microenvironment, thus implicating a key role of the presence of unresolved chronic inflammation in the promotion of metastatic disease progression and therapeutic resistance.
Cancerous tissue overexpresses COX2, NOS2, and ROS, which are associated with disease progression and reduced patient survival. In lung and gastric cancer, COX2 inhibition has positive therapeutic effects, while elevated COX2 expression is a characteristic of aggressive tumors (8, 37, 121, 122). Similarly, nicotinamide adenine dinucleotide phosphate oxidase and Duox expression in ovarian and pancreatic cancer drives mechanisms associated with disease progression (123). CO produced from heme oxygenase-1 (HO-1) has been shown to suppress T-cell proliferation by inhibiting IL-2 production (100). Recently, the involvement of the H2S/persulfide-producing enzymes, cystathionine β-synthase and cystathionine γ-lyase, in disease progression of colon cancer was shown (18, 137). These and other reports demonstrate the utility of the redox inflammation profile for elucidation of pathways that drive cancer progression, which can be therapeutically exploited.
While many cancer studies have focused on COX2 and ROS, NOS2 has recently emerged as a predictive biomarker in many solid tumors. Several reports have correlated high tumor NOS2 expression with reduced patient survival (29, 30, 33, 36, 65, 73, 75, 125, 166). Traditionally, NOS2 has been associated with immune activation (78, 158). NOS2 has an important role in murine biology and murine leukocytes can produce high concentrations of NO (upto 0.15 μM for 4 h) for prolonged periods of time (32). However, human NOS2 does not seem to have the same role and is expressed in a surprising number of epithelial cells (1).
An earlier study revealed moderate to high NOS2 expression within tumor epithelium in 73% of all patients with breast cancer regardless of ER status (36, 108). The same study identified a positive correlation between tumor NOS2 expression and protein kinase B (Akt) pathway activation, suggesting a mechanistic link with prosurvival signaling within the tumor (108). Exposure of breast cancer cells to NO donors further supported NO activation of Akt (106, 108). This study also revealed a positive association between NOS2 and p53 mutation frequency (36). Moreover, both NOS2 and COX2 predicted poor breast cancer survival in ER−, but not ER+, patients (36, 37). Together, these studies implicate a key role for NOS2 in breast cancer disease progression, which is supported by the finding that 92% of deceased patients in this cohort exhibited elevated tumor NOS2 expression (36).
Importantly, only 4 of 247 patients presented with lymph node-positive disease, which suggests that elevated tumor NOS2 expression may predict clinically undetected metastasis. Other studies have shown upregulation of NOS3 and NOS2 by mutated myeloid leukemia factor 2 (MLF2) and ribosomal protein L39 (RPL39), which correlated with poor disease-specific survival in patients with ER– breast cancer (22, 24, 38).
The mechanistic role of NOS2 in cancer progression has been examined using cell culture models exposed to NONOate donor agents. NONOates, or diazenium diolates, release NO in a defined manner at neutral pH and provide powerful tools for controlling the flux of NO in biological experiments (59, 142). The variety of different structures having pH- and time-dependent rates of NO release allows the generation of specific flux profiles, which can be compared with NOS2-derived NO.
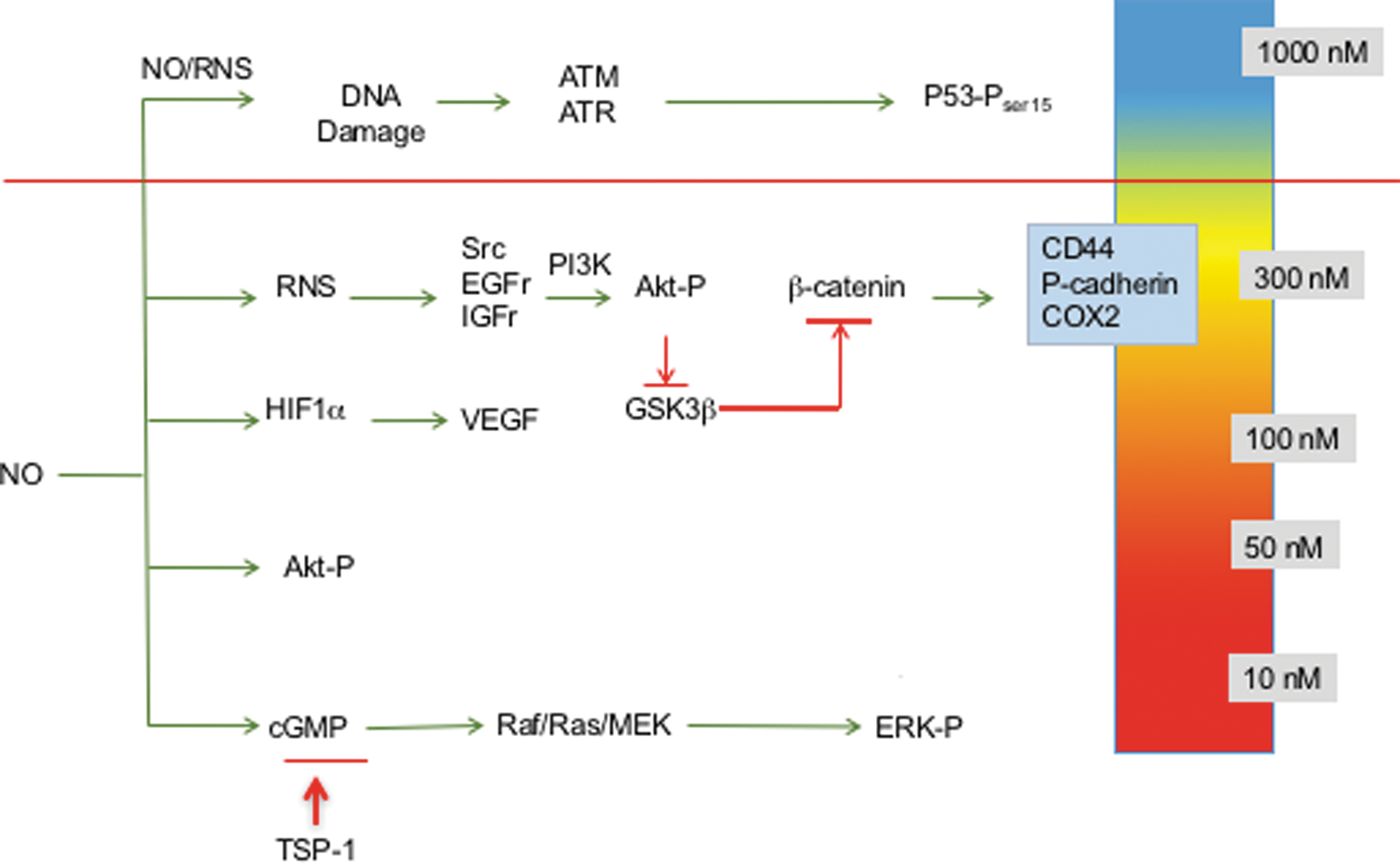
Using this strategy, NO flux-dependent activation of specific signaling cascades has been identified in breast cancer cells (36, 108, 112, 134, 135, 139). The concentration- and temporally dependent NO activation of extracellular signal-regulated kinase (ERK) and Akt, as well as HIF-1α stabilization, occurs at levels ranging from 200 to 500 nM steady-state NO, while phosphorylation of p53 occurs at higher levels of 700–800 nM NO (139). Inhibition of enzymes involved in DNA repair also occurred at higher NO flux (71), while modification of transforming growth factor-beta (TGF-β) (154) and matrix metalloproteinase (MMP) (11, 119) occurred at lower levels. Moreover, low NO donor concentrations that produce pM NO flux mediate cyclic guanosine monophosphate (cGMP)-dependent downregulation of the antiangiogenic molecule, thrombospondin-1 (50, 116).
These NO concentrations and downstream signaling effects can be achieved using activated murine macrophages as well as NONOate donors (108, 135, 139). Thus, NONOate donors can be effectively and reproducibly employed to examine NO mechanisms in cancer (139).
Three distinct NO flux ranges define NO-mediated signaling; low NO <100 nM cGMP-dependent signaling, higher levels ranging from 200 to 600 nM NO involve nitrosative signaling that is cGMP independent, and >600 nM generally involves stress response as well as antiproliferation mechanisms (Fig. 2) (46). Observations of elevated tumor NOS2 expression in breast cancer patients suggest that targeted pathway activations can affect patient outcome (16, 36, 75, 108). The role of NO in promoting or inhibiting cancer progression has been controversial. However, clarification of various roles of NO can arise by discussing phenotypes in the context of these different levels of NO and the respective signaling effects. We will begin with a discussion of the role of NOS2-derived NO in poor outcome of ER− breast cancer and the range of NO flux that upregulates predictive biomarkers identified in high NOS2-expressing breast tumors (36). In addition, the influence of cGMP-dependent processes will be discussed as well as higher levels of NO that affect p53-dependent signaling and other cell growth inhibitory pathways.
ER− Breast Cancer, NOS2, and Nitrosative Signaling
Elevated tumor NOS2 expression predicts poor outcome in ER− breast cancer patients (36). In addition, high NOS2 tumors exhibited elevated expression of predictive basal-like and stem cell biomarkers, including P-cadherin, IL-8, and cluster of differentiation 44; hyaluronic acid receptor (CD44) (36). Protein levels of these biomarkers were significantly enhanced in ER− breast cancer cells exposed to the NO donor Diethylenetriamine NONOate (DETA/NO) (∼600 nM steady-state NO flux), which further supports a mechanistic role for NO as a driver of breast cancer disease progression (36). Importantly, phenotypic and predictive biomarker analyses of patient tumors combined with the assessment of NO-induced protein regulation of these predictive biomarkers in breast cancer cells provide an invaluable tool for estimating the steady-state NO flux generated in high NOS2 tumors.
Pathway activation of phosphoinositide 3-kinase (PI3k)/Akt, mitogen-activated protein kinase (MAPK), epidermal growth factor receptor (EGFR), and protein c-ets-1 (Ets-1) signaling cascades promotes breast cancer disease progression and they are targets of NO (36, 108, 112, 134, 135, 139). Earlier findings of Prueitt et al. identified a strong correlation between tumor NOS2 expression and phosphorylated Akt, which suggested the increased occurrence of Akt pathway activation in high NOS2-expressing breast tumors (108). This report was further supported by observations of NO-induced Akt phosphorylation in breast cancer cells treated with NO donors (106, 108, 112, 135).
Similarly, a strong correlation between EGFR tyrosine phosphorylation and elevated tumor NOS2 expression was also identified in breast tumors as well as NO-induced EGFR phosphorylation in cells grown in culture (36). A subsequent study demonstrated nitrosation of EGFR, which mediated ligand-independent activation of its kinase receptor (86, 105). Earlier studies have also identified S-nitrosylation as a mediator of EGFR and Src activation (86, 105, 109). Moreover, high-flux NO decreased kinase activity of these membrane proteins, indicating concentration-dependent biphasic effects of NO (86), which was supported by Switzer et al. who showed that peak activation of these signaling pathways occurred at ∼400 nM steady-state NO (135). This intermediate level of NO also upregulates COX2 expression in breast cancer cells (135). Similarly, COX2 correlates with Akt pathway activation and predicts poor outcome in breast cancer patients (37).
Nitrosative signaling is mediated by reactive nitrogen species (RNS) such as N2O3 (118). Further analysis of nitrosative EGFR and Src activation implicated the requirement of a nitrosative species such as N2O3 (135). Addition of superoxide dismutase (SOD) to scavenge O2 − and prevent peroxynitrite formation actually increased Akt phosphorylation (unpublished results). Moreover, Thomas et al. demonstrated an antagonistic relationship between O2 − and NO during nitrosative signaling in breast cancer cells (141). Thus, NO and O2 − are mutual antagonists of their respective signaling pathways, which may be exploited by tumor cells for maintenance of optimal redox signaling conditions that promote their survival and growth.
Interestingly, during inflammatory response, ROS, NO, and even CO seem to be temporally distinct. There are additional mechanisms of NO beyond nitrosation involving N2O3; however, antioxidants and inhibitors of nitrosation such as azide, reduced glutathione, urate, and ascorbate abate NO signaling (135). While we have long proposed that NO/O2 − can lead to N2O3 and other RNS, in our hands, the addition of SOD enhanced Spermine NONOate (SPER/NO)-induced cGMP output by 10-fold in MCF-7 cells (141). Importantly, the titration of SPER/NO in the presence of hypoxanthine/xanthine oxidase did not mediate cytotoxicity, suggesting that ROS such as superoxide controls NO bioavailability (141).
NO signaling influences nonheme metal chemistry, including HIF-1α stabilization and prolyl hydroxylase activity. Moreover, NO signaling through PI3K and RAS appears to involve a nitrosating species such as N2O3. These and other observations suggest that targeting specific redox species may be therapeutically beneficial.
Prosurvival signaling mediated by MAPK is also involved in cancer progression. Pathway activation of ERK initiates many pathways associated with cancer such as c-Myc and activator protein-1. RAS and raf-1 proto-oncogene serine/threonine protein kinase (RAF-1) are key mediators upstream of ERK. Toward this end, cGMP-dependent signaling through RAF-1 increased ERK phosphorylation in MCF-7 breast cancer cells (106). Moreover, RAS inhibitors prevented ERK-dependent Ets-1 activation in NO-treated MB-231 and MB-468 cells (134). Many mutations are found in HRAS and KRAS that are thought to be important drivers in cancer. However, NO activates p21ras through S-nitrosation of a key cysteine residue (67). In addition, S-NO and sulfonic acid post-translational modifications have been shown, which regulate enzymatic activities (84). Ets activation can also occur through B-Raf and Raf-1, cAMP, or via PGE2 (51, 155). Thus, ERK pathway activation can go through several routes that circumvent direct inhibition of RAF.
NO and cGMP Signaling
The guanylyl cyclase (GC) enzymes catalyze the conversion of guanosine triphosphate to the second messenger cGMP. The soluble (sGC) and particulate isoforms are ligand activated by NO and hormones/natriuretic peptides, respectively (160). Downstream cGMP effectors include cyclic nucleotide-gated ion channels and cGMP-dependent protein kinases, as well as phosphodiesterase (PDE) enzymes, which promote cGMP degradation to control its intracellular levels. In addition, multidrug-resistant proteins (MRP4, MRP5, and MRP8) also regulate intracellular cGMP levels (54, 61, 120, 159). Early studies have implicated aberrant cGMP regulation in breast and other cancers where low cGMP levels were identified in neoplastic tissue when compared with normal tissue regions. These observations coincided with increased expression and altered compartmentalization of MRPs and PDE enzymes, which correlated with tumor grade, stage, and lymph node metastasis (47, 58, 85, 92, 132).
Mechanistically, cGMP-mediated activation of protein kinase G (PKG) leads to the phosphorylation and subsequent degradation of the oncogenic transactivator β-catenin, which culminates in the downregulation of growth-promoting and apoptosis-inhibiting proteins, including cyclin D1, c-myc, and survivin (143, 145, 146). Toward this end, PDE inhibition by sulindac sulfide elevated cGMP levels, inhibited growth, abated Wnt/β-catenin prosurvival signaling, and induced apoptosis in breast and colon cancer cells (145 –147). Interestingly, PDE inhibition is a secondary COX-independent target of clinically available NSAIDs, which have demonstrated chemopreventive and chemotherapeutic activities (145, 146). While COX inhibition is generally thought to be the primary antitumor mechanism of NSAIDs, other studies have shown (i) that the growth inhibitory activity of NSAIDs is not reversed by exogenous prostaglandins (ii) discrepancy between NSAID IC50 concentrations associated with COX inhibition and abated tumor cell proliferation, and (iii) cGMP activation (39, 42, 143, 145, 146).
These results implicate other targets, including PDEs in the antitumor effects of NSAIDs. Toward this end, the PDE5-selective inhibitors, sildenafil, tadalafil, and MY5445, enhanced intracellular cGMP/PKG signaling, which correlated with abated cancer cell proliferation and increased apoptosis (145). Interestingly, cytokine-induced NOS2 led to S-nitrosylation and inhibition of sGC activity, as well as reduced formation of cGMP and increased PDE1 in smooth muscle cells (110). These results suggest that high-flux NO derived from NOS2 abates cGMP signaling through S-nitrosylation and PDE mechanisms.
NO Regulation of HIF-1α
The HIF-1 transcriptional pathway is activated under conditions of reduced O2 bioavailability, which initiates physiological processes that when dysregulated can become pathological. These responses include angiogenesis, erythropoiesis, and vasomotor control, as well as modulation of energy metabolism and cell survival. The regulation of O2 gradients by HIF-1 is precisely controlled for ATP synthesis as well as prevention of excess O2 toxicity (56, 128). Under normoxic conditions, HIF-1 levels are regulated by the turnover of HIF-1α subunit by E3 ligase where hydroxylation of two proline residues (Pro402 and Pro564) by prolyl hydroxylase (PHD) targets the protein for ubiquitination and proteasomal degradation (57, 124).
In addition, normoxic conditions mediate the hydroxylation of Asn803 and Asn851 on HIF-1α and HIF-2α, respectively, which silences their COOH-terminal transactivation domains by abating HIF-1 interactions with coactivator protein p300/CREB-binding protein during transcriptional activation of target genes (68). Under hypoxic conditions, these protein modifications are attenuated, thus allowing HIF-1α protein stabilization and HIF-1 pathway activation.
In addition to hypoxia, NO also stabilizes HIF-1α by abated PHD-mediated HIF-1α ubiquitination and turnover (15, 57, 124). PHD is a nonheme Fe2+ enzyme that utilizes oxygen, α-ketoglutarate, and ascorbic acid to hydroxylate HIF-1α and is part of a larger family of oxygenases that includes ten-eleven translocation and Jumonji family that regulates DNA and histone demethylation (45, 64, 88). NO directly interacts with the nonheme Fe2+ site to inhibit PHD activity (82). In addition, S-nitrosylation of Cys533 within the oxygen-dependent degradation domain has been shown to stabilize HIF-1α protein levels in a manner independent of PHD activity (72). HIF-1α stabilization by NO activates a number of signaling pathways mediated by the promoter hypoxia response element (HRE) to confer survival and growth advantage, angiogenesis, wound repair, and tumor development (130, 163).
Moreover, HIF-1α stabilization promotes self-renewal of bone marrow-derived mesenchymal stromal cells, which involves the induction of pluripotent genes, including octamer-binding transcription factor 4 and kruppel-like factor-4, to abate terminal differentiation pathways (102). HIF-1α can activate multiple genes associated with such diverse functions as cell proliferation, cell survival, apoptosis, motility, invasion, cytoskeletal structure, cell adhesion, erythropoiesis, angiogenesis, vascular tone, transcriptional regulation, drug resistance, and metabolism (127).
HIF-1α also plays an important role in the expression of proteins associated with tumor development and progression, including erythropoietin (EPO), glucose transporter 1 (GLUT1), epithelial–mesenchymal transition (EMT), and vascular endothelial growth factor (VEGF) (148). EPO is a glycoprotein hormone and main regulator of red blood cell production and is associated with hematological malignancies. The EPO receptor is expressed in many organs and may function as an antiapoptotic factor; it is overexpressed in multiple cancers, including breast cancer, and mediates cell proliferation and angiogenesis (99, 161). Increased VEGF promotes angiogenesis, while elevated GLUT1 modulates metabolism to favor glycolysis. HIF-1α also modulates the EMT markers class A basic helix-loop-helix transcription factor (TWIST), vimentin, and E-cadherin in clinical samples and nonsmall cell lung cancer cells, implicating a role of TWIST in hypoxia-induced invasion and metastasis (157). The immune checkpoint inhibitor, programmed death ligand 1 (PD-L1), contains an HRE in its promoter and is a direct target of HIF-1α (89, 90). Importantly, blockade of PD-L1 enhances myeloid-derived suppressor cell (MDSC)-mediated T-cell activation and potentiates radiation therapeutic efficacy (90, 152). Thus, HIF-1α stabilization promotes EMT (metastasis), angiogenesis through VEGF, and PD-L1-mediated immunosuppression, indicating that HIF-1α is a key mediator of processes common to the most aggressive tumors.
NO and Mitochondrial Targets
Several metabolic enzymes are targeted by NO and S-nitrosation, including complex I and complex IV of the mitochondrial electron transport chain (13, 17, 20, 27). Targeted S-nitrosation of complex I has been shown to limit electron flux and minimize oxidative damage during reperfusion injury (107). NO regulation of O2 consumption by direct binding and reversible inhibition at the ferrous heme site of complex IV has been well documented (20). In addition, inhibition of the glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), by NO has been shown (13, 53). Other studies have employed extracellular flux technology to show distinct mechanisms of NO and S-nitrosation in the regulation of glycolysis and oxidative phosphorylation (25).
The NO donor, DETA/NO, stimulated glycolysis while impairing mitochondrial reserve capacity with no impact on basal respiration, which was reversed by the NO scavenger, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO), thus suggesting direct NO-heme interaction (25). Similar to the effects of DETA/NO, low (50 μM) concentrations of
Angiogenesis Versus Immunosuppression
The endothelial barrier maintains vascular and tissue homeostasis and is a key modulator of processes, including angiogenesis and immune response. In cancer, the endothelial barrier is disorganized, which leads to permeable or leaky vasculature that drives tumor-induced angiogenesis, altered blood flow, leukocyte infiltration, and tumor extravasation (4).
The angiogenic cytokine, VEGF, produced within the tumor microenvironment is a key driver of tumor angiogenesis and vascular permeability by Src-dependent VE-cadherin adhesion destabilization (31, 66). In addition, VEGF promotes immune suppression by disrupting the maturation of dendritic precursor cells and tumor-activated CD8+ T-cell function, thus limiting the efficacy of immunotherapies (49, 133, 138). Moreover, the VEGF antibody Avastin can reverse VEGF-mediated disruption of dendritic cell maturation and T-cell proliferation, recruitment, and infiltration at the tumor site (95, 97).
Similarly, the CD40 antitumor immune response was also potentiated by a neutralizing anti-VEGF antibody (126). Toward this end, an emerging paradigm suggests that improved tumor response to therapy requires normalized vasculature, a responsive endothelium, and correctly polarized immune mediators. In fact, some articles have shown that immune modulation is far more important for improved tumor response to radiation therapy (114, 152).
Increased tumor angiogenesis and increased cluster of differentiation 31; platelet endothelial cell adhesion molecule (CD31) leads to bidirectional flow where tumor vasculature lacks the ability to produce intracellular adhesion molecule and vascular cell adhesion molecule, which are both critical for the recruitment of cytotoxic leukocytes. VEGF can activate MDSCs as well as suppress T-cell expansion. Elevated IL-8 promotes tumor angiogenesis and vascular permeability, as well as the expansion of MDSCs. IL-10 and nuclear factor (erythroid-derived 2)-like 2 increased HO-1 and CO, which promotes angiogenesis and inhibits T-cell expansion. Thus, the promotion of tumor angiogenesis and immune suppression go hand-in-hand as it occurs during wound response, which may, at least in part, explain why putative antiangiogenic agents have multiple beneficial effects in cancer therapy.
Extracellular Matrix MMPs and Tissue Inhibitor of Matrix Metalloproteinases
The importance of the tumor microenvironment during cancer progression has become increasingly evident. The tumor microenvironment comprises immune cells, fibroblasts, endothelial cells, adipocytes, and extracellular matrix (ECM). Importantly, the ECM encompasses a complex network, which transmits biochemical and biomechanical cues to tumor cells that are actively involved in disease progression and metastasis. Moreover, the ECM in breast cancer is similar to that of mammary gland involution and wound response, which is characterized by the upregulation of fibrillar collagens, fibronectin, and matricellular proteins. In addition, ECM remodeling enzymes are aberrantly upregulated in advanced tumors (98). ECM remodeling enzymes that contribute to breast cancer progression include MMPs as well endogenous tissue inhibitor of matrix metalloproteinases (TIMPs) (7, 74, 111, 112, 162).
MMPs comprise a family of structurally similar endopeptidases with zinc ions at the active site. MMPs process components of ECM, which impacts the biological and functional properties of the targeted proteins, including cytokines. For example, MMP-9 truncates IL-8, which potentiates its biological activity and binding of its cell surface receptor in neutrophils (150, 151). In addition, MMP-9 has been shown to process IL-1β, leading to its activation and feed-forward regulation of MMP-9 expression (96). Similarly, MMP-9 processes IL-2 receptor α (CD25), which abates the function of tumor-reactive T cells and cytotoxic lymphocytes (131). Active MMP-9 has been shown to augment the release of VEGF from ECM stores as well as activate latent TGF-β via degradation of the latency-associated peptide to facilitate tumor angiogenesis and invasion (9, 165). Collectively, these observations provide evidence that MMP-9 modulates immune function to promote immunosuppression, tumor angiogenesis, and invasion within the tumor microenvironment (96, 131).
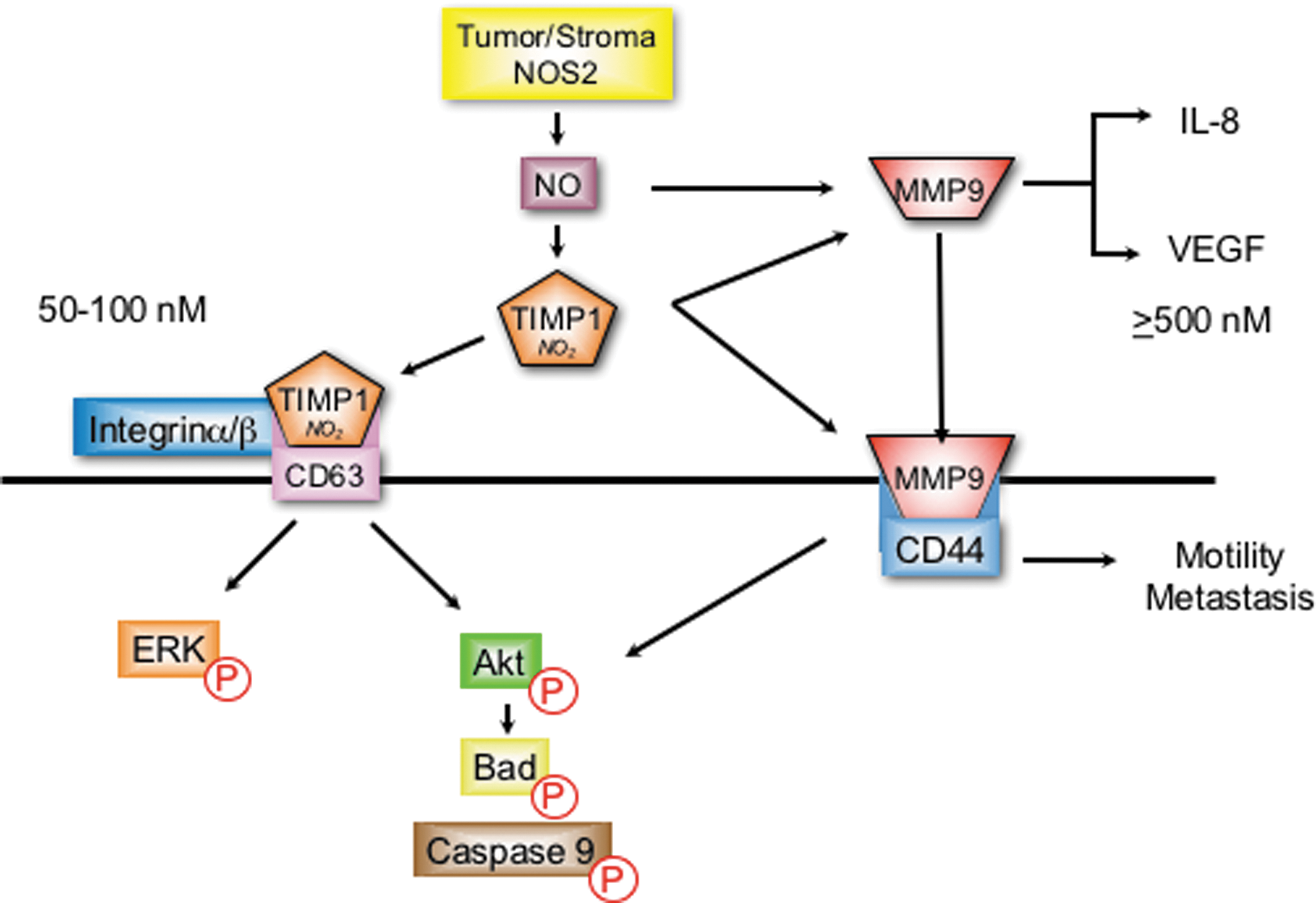
NO has multiple roles in the regulation of MMP-9 (94). NO/RNS and other electrophiles can activate MMPs through attacking the Zn thiolate bond of the latent protein (62, 63, 167). This activation has been shown at NO flux between 300 and 500 nM (119). In macrophages and microglia cells, NO increased MMP-9 activity via cGMP-dependent suppression of TIMP-1 (115, 119). Interestingly, as NO flux increased to higher levels that activate p53, MMP-9 activity diminished (119). These results demonstrate biphasic regulation of MMP-9 by NO, which is consistent with other progrowth signaling pathways that are similarly regulated by 300–500 steady-state nM NO flux (136). Interestingly, colocalization of NOS2 with MMP-9 was observed at the leading edge of migrating cells (93). Moreover, tyrosine nitration of MMP-9 resulted in enzyme activation in migrating astrocytes and reduced MMP-9 activity was observed in NOS2−/− mice (115, 156).
Protein localization through receptor binding provides an additional mechanism of MMP-9 regulation that mediates cell migration and invasion. Toward this end, the cell surface hyaluronan receptor, CD44, has been identified as a receptor for MMP-9 (96). This MMP-9/CD44 complex has been identified as a mechanism of localizing or concentrating MMP-9 at the leading edge of invasive breast cancer cells to facilitate enhanced metastasis during disease progression (10, 104, 164). NOS2 upregulates CD44 as well as IL-8 and correlates with MMP-9, TIMP-1, and enhanced tumor vascularization in ER− breast tumors (36, 112).
A recent mechanism for NO modulation of MMP-9 activity involves TIMP-1 protein nitration of key tyrosine residues that interfere with TIMP-1/MMP-9 binding, which abates TIMP-1 inhibition of active MMP-9 (103). Molecular modeling predicted two key tyrosine residues (Y95 and Y143) in loop structures that are critical for TIMP-1 inhibition of active MMP-9 (103). Interestingly, tyrosine nitration of these specific residues was later identified by mass spectrometry in recombinant human TIMP-1 protein following overnight exposure to the NO donor, DETA/NO (112).
In addition to its MMP inhibitory function, TIMP-1 also facilitates MMP-independent prosurvival PI3k/Akt/BAD and ERK pathway activation via interaction with the cell surface protein cluster of differentiation 63 (CD63) (19, 55). Toward this end, TIMP-1/CD63 colocalization and PI3k/Akt/BAD prosurvival signaling were enhanced in MB-231 breast cancer cells by NO concentrations that were optimal for TIMP-1 nitration (112). Importantly, TIMP-1 predicted poor breast cancer disease-specific survival, which was restricted to patients with high NOS2 tumor expression (112). Moreover, a direct correlation between NOS2 and pAkt (OR 4.5) was dramatically augmented (OR 12.7) in breast tumors expressing elevated TIMP-1, but reduced (OR 2.5) in tumors with low TIMP-1 expression (112).
Together, these results suggest a plausible mechanism for NO during breast cancer progression, where TIMP-1 nitration abates it MMP inhibitory function, which may favor TIMP-1/CD63 interaction and downstream PI3k/Akt/BAD prosurvival signaling while preserving MMP-9 activity to facilitate tumor angiogenesis, migration, and invasion, as summarized in Figure 3 (36, 112).
Conclusion
NO performs distinct and vastly different functions, which are concentration, spatially, and temporally dependent. Low nM NO produced by NOS1 and NOS3 regulates neuronal and vascular processes. In contrast, inflammatory processes generate NOS2-derived NO (ranging between 100 and 600 nM) that promotes nitrosative signaling. High tumor NOS2 expression has significantly correlated with increased p53 mutations, the vascular marker, CD31, and poor survival among breast cancer patients with the more aggressive ER− phenotype. Regarding disease progression, NOS2-derived NO upregulates prosurvival signaling pathways, including PI3k/Akt and ERK, promotes HIF1α protein stabilization, and induces NOS2 and COX2 (summarized in Fig. 4). NO-stabilized HIF1α helps the tumor as well as stromal cells cope with hypoxic stress by inducing angiogenesis, immunosuppression, chemoresistance, proliferation, and metastasis, which implicates the targeted inhibition of tumor NOS2 as a novel therapeutic strategy.
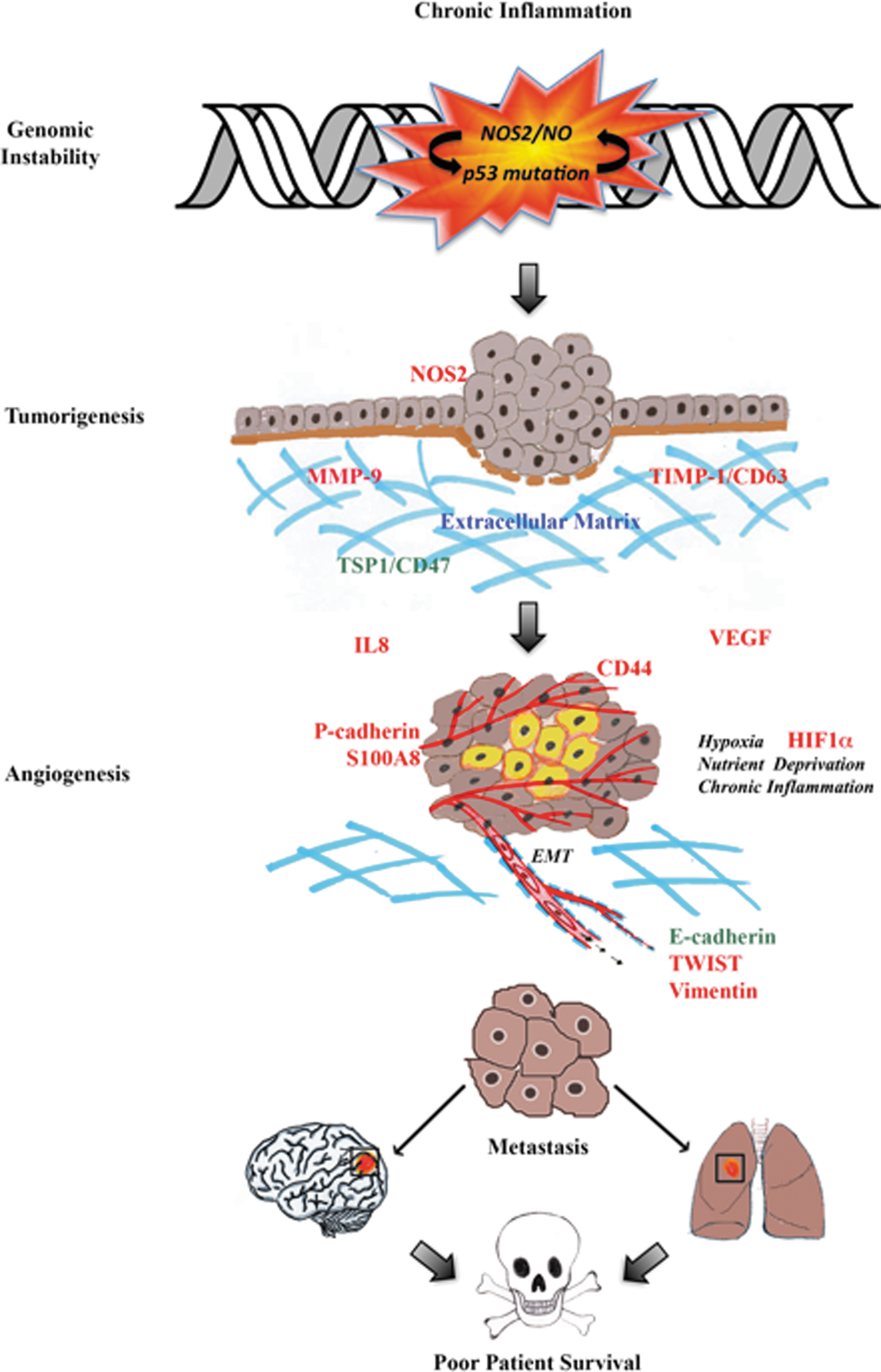
Toward this end, two novel cancer genes (RPL39 and MLF2) were recently identified in breast tumors that are regulated by hypoxia and NOS2 signaling (24). Mutational analysis identified gain-of-function effects in RPL39 (A14V and G50S) and MLF2 (D12H and R158 W) in a wound assay and significantly shorter time to relapse (p = 0.0259, χ2 test) in patients (24). Selective NOS2 inhibition abated RPL39 and MLF2 protein expression in breast cancer cells and siRNA targeting reduced tumor growth and improved median survival of mice treated with docetaxel (24). These promising findings warrant further clinical investigation of therapeutic applications of NOS inhibitors in breast cancer treatment.
Footnotes
Acknowledgments
This research was supported, in part, by the Intramural Research Program of the NIH, Cancer and Inflammation Program. G.A.de.O. is supported by the program Science Without Borders—CNPq process number 205342/2014-0.